
Boronic esters as protective groups in carbohydrate chemistry: processes for acylation, silylation and alkylation of glycoside-derived boronates†
Ross S.
Mancini
,
Jessica B.
Lee
and
Mark S.
Taylor
*
Department of Chemistry, University of Toronto, Toronto, ON M5S 3H6, Canada. E-mail: mtaylor@chem.utoronto.ca
First published on 8th November 2016
Abstract
Procedures for selective installation of acyl, silyl ether and para-methoxybenzyl (PMB) ether groups to glycoside substrates have been developed, employing phenylboronic esters as protected intermediates. The sequence of boronic ester formation, functionalization and deprotection can be accomplished with only a single purification step, and the boronic acid component can be recovered and reused after deprotection. Key advances include the identification of reaction conditions for installation of PMB groups in the presence of boronic esters, and the use of the ‘phase switching’ protocol developed by Hall and co-workers as an efficient method for boronic ester cleavage. The relatively mild conditions for boronate deprotection are tolerant of several functional groups, including esters, silyl ethers, ketals and thioglycosides.
Introduction
The formation of cyclic boronic esters from boronic acids and carbohydrate derivatives was first reported more than 60 years ago.1,2 Condensations of this type have been applied extensively in glycoscience, including in drug delivery and in the sensing and separation of carbohydrate derivatives.3 Boronic ester formation generally occurs at 1,2- or 1,3-diol groups, the same motifs that engage in cyclic ketal and acetal formation. However, whereas acetals and ketals are employed routinely as protective groups for such motifs,4–6 applications of boronic acids for this purpose are relatively uncommon. Among the factors that may have contributed to this difference are the rather sensitive nature of boronic esters, not only to hydrolysis under mild acidic or basic conditions, but also to proteodeboronation, oxidation and nucleophilic attack; and the cost of arylboronic acids, which generally exceeds those of the reagents used for acetal and ketal formation. Although the fragility of boronic esters is a disadvantage for certain applications, it could also represent an opportunity to develop operationally simple reaction sequences that occur under mild conditions and with high functional group tolerance. The second issue – the cost of boronic acids – could be mitigated by recovering and recycling this reagent. Based on these considerations, and as part of a broader program focused on applications of organoboron compounds in carbohydrate synthesis,7 we set out to determine whether efficient and practical protocols for selective functionalization of carbohydrates could be developed based on the use of boronic esters as temporary blocking groups for 1,2- or 1,3-diol groups.Protection of carbohydrate-derived diols as cyclic boronic esters has been explored in several previous studies.8 Boronic ester protective groups have been used to facilitate the synthesis of mannofuranosides,9 β-mannopyranosides,10 functionalized hexitols and sialic acid derivatives.11 They have been combined with stannylene acetal activating groups to achieve selective acylations and sulfations of pyranosides.12 The facile formation and cleavage of boronic esters are advantageous features in polymer-supported synthesis, as demonstrated by applications of boronic acid-functionalized, cross-linked polystyrene as a protective group for pyranosides.13,14 Recently, selective sulfations15 and glycosylations16 of pyranosides and free sugars have been achieved using arylboronic acids as transient protective groups. Despite these significant advances, boronic esters are not generally viewed as being viable alternatives to acetals and ketals for partial installation of widely employed protective groups such as esters, silyl ethers and benzyl ethers. Here, we describe procedures for selective installation of acyl, silyl ether and para-methoxybenzyl (PMB) ether groups to pyranoside and furanoside substrates, employing phenylboronic esters as protected intermediates. The sequence of boronic ester formation, functionalization and deprotection can be accomplished with only a single purification step, and the boronic acid component can be recovered and reused after deprotection. Key advances include the identification of reaction conditions for installation of PMB groups in the presence of boronic esters, and the use of the ‘phase switching’ protocol developed by Hall and co-workers17 as an efficient method for boronic ester hydrolysis.
Results and discussion
Acylation and silylation of sugar-derived boronates
We selected 2,3-bis-benzoylation of methyl α-D-glucopyranoside as a model reaction for our initial studies (Scheme 1). Acylation of sugar-derived boronates is well established,8,13 meaning that we could focus on developing an operationally simple protection–acylation–deprotection protocol that would facilitate isolation of the sugar-derived product and recovery of the boronic acid component. Condensation with phenylboronic acid generated the 4,6-O-boronate quantitatively and without need for purification, by heating the reaction components to reflux in toluene, followed by evaporation of the solvent. The resulting material was subjected to benzoyl chloride (BzCl) in pyridine at 0 °C. As anticipated, analysis of an aliquot of the reaction mixture by 1H NMR spectroscopy showed clean conversion to the 2,3-benzoylated boronic ester intermediate 2a after 1 hour. | ||
| Scheme 1 Bis-benzoylation of methyl α-D-glucopyranoside by protection as a phenylboronic ester. Phase-switching workup is used to deprotect the boronic ester and to separate the carbohydrate product from the boronic acid component. | ||
Hydrolysis of the boronic ester proved to be more challenging. Solutions of boronate 2a in ethyl acetate could be recovered unchanged after washing with aqueous solutions of hydrochloric acid (1 M) or sodium bicarbonate (saturated). The boronate was also stable towards flash column chromatography on silica gel, eluting with 10→20% acetone in dichloromethane. Stirring in an acetone/water mixture at room temperature, conditions that have been used to cleave functionalized carbohydrate derivatives from a polystyreneboronic acid solid support,13,14 resulted in recovery of the boronic ester. Similar results were obtained upon treatment with a methanol/water mixture and with a 1 M aqueous hydrochloric acid/acetone mixture. Hydrolysis could be accomplished using a 1 M aqueous sodium hydroxide solution, but it was accompanied by saponification of the ester groups.
It has been observed that carbohydrate-derived boronic esters can show appreciable stability under aqueous conditions, and that their rates of hydrolysis appear to depend on the configuration and substitution pattern of the carbohydrate moiety.8,11,15,18 It is presumably for this reason that most authors have opted for ‘non-hydrolytic’ methods such as oxidation16 or transesterification9,15 to liberate boronic ester-protected diols in carbohydrate chemistry. Indeed, treatment of 2a with pinacol in refluxing toluene overnight yielded the desired diol 3a and phenylboronic acid pinacol ester, which could be separated by flash column chromatography. Although this approach was effective, the requirement for a prolonged heating step and the inability to directly recover the phenylboronic acid component were drawbacks.
We speculated that an efficient method for transesterification-based cleavage of sugar-derived boronic esters could be developed based on the ‘phase switching’ concept pioneered by Hall and co-workers.17 Hall's group showed that boronic acids can be separated from other organic compounds by liquid–liquid extraction, taking advantage of the pH-switchable complexation of the boronic acid with sorbitol, a sugar alcohol. Extraction with a basic solution of sorbitol in water draws the boronic acid into the aqueous phase through formation of a tetracoordinate boronate. Acidification of this phase releases the boronic acid, which can be extracted into organic solvent. This method has not been applied to boronic esters, but we anticipated that transesterification of the sugar-derived boronates with sorbitol could be relatively rapid under the basic conditions of the liquid–liquid extraction. Indeed, washing an ethyl acetate solution of 2a with a 1 M aqueous solution of sorbitol and sodium carbonate resulted in quantitative cleavage of the boronic ester to give 3a, which was partitioned to organic solvent, while phenylboronic acid was sequestered in the aqueous phase as the sorbitol-bound boronate. Diester 3a was isolated in 80% yield (from 1a, on 5 mmol scale) after purification by flash column chromatography on silica gel, while phenylboronic acid was recovered in 77% yield after acidification of the aqueous extract followed by extraction with ethyl acetate. Overall, the preparation of 3a from 1a was accomplished in a ‘one-pot’ operation: diol protection and acylation were conducted in the same flask and without purification of intermediates, while deprotection was carried out during the phase-switching aqueous workup.
The protocol depicted in Scheme 1 proved to be useful for the synthesis of a variety of mono- or dibenzoylated glycosides (Fig. 1). The reactions were generally conducted on 5 mmol scale, leading to the isolation of gram quantities of the products shown. Recovery of the phenylboronic acid reagent was carried out in each case, and the recovery yields (which ranged from 67% to 80%) are listed in the Experimental section. The method could be applied to gluco-, galacto- and mannopyranosides, but with these last substrates, prior protection of the 6-OH group was necessary. Condensation of phenylboronic acid with ‘free’ methyl α-D-mannopyranoside led to the formation of the bis-boronate and recovery of unprotected pyranoside, regardless of the stoichiometry used. It appears that the rate of formation of the second boronic ester exceeds that of the first. Consistent with previous observations,18 this method was applicable to the synthesis of a partially acylated thioglycoside (3g). The synthesis of 3f demonstrates that an isopropylidene ketal group is tolerant of the conditions for formation and cleavage of boronic esters. It should also be noted that the 3,5-O-boronate was formed selectively from glucofuranoside 1f, thus leading to benzoylation of the free 6-OH group.
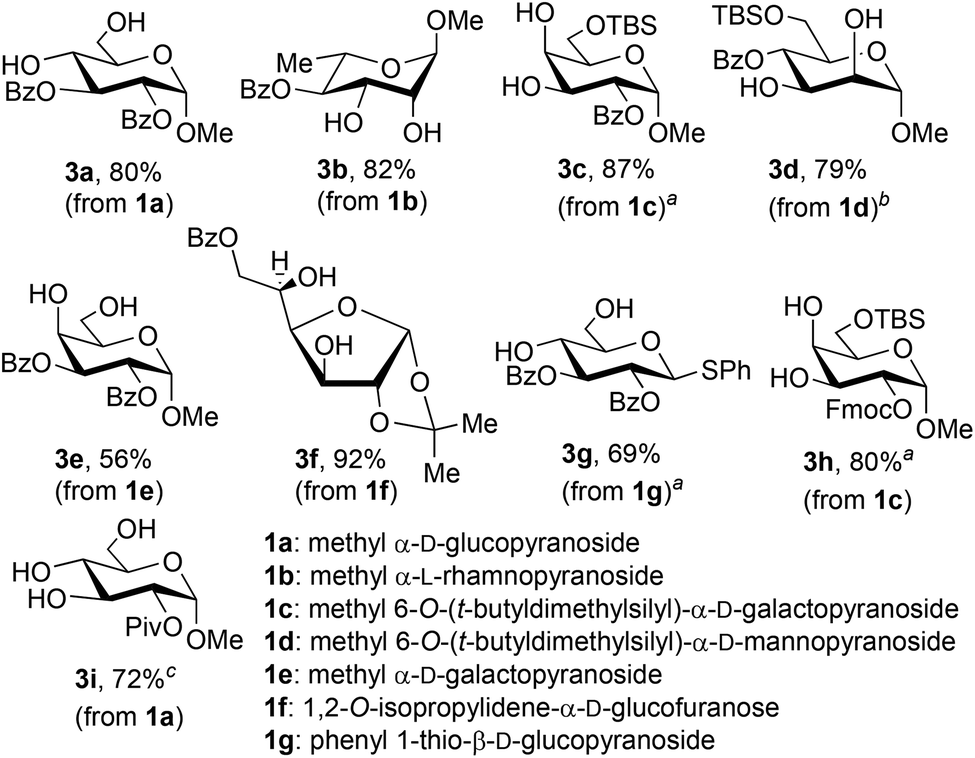 | ||
Fig. 1 Partially acylated carbohydrate derivatives prepared using boronic ester protection. Reaction conditions: (1) substrate (5.0 mmol), PhB(OH)2 (1.0 equiv.), toluene, 110 °C; (2) acylating agent, pyridine, 0 °C; (3) phase-switching workup with 1 M aqueous sorbitol/Na2CO3. For details, see the Experimental section. Yields after purification by flash chromatography on silica gel are listed. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction was carried out on 2.0 mmol scale. bPhB(OH)2 recovered from a previous experiment was used in the reaction. cDeprotection was conducted by transesterification with pinacol (toluene, 110 °C). Reaction was carried out on 2.0 mmol scale. bPhB(OH)2 recovered from a previous experiment was used in the reaction. cDeprotection was conducted by transesterification with pinacol (toluene, 110 °C). | ||
Fluorenylmethyl carbonate (OFmoc) and pivaloyl ester (OPiv) protective groups could also be installed efficiently using this protocol (products 3h, 3i). Selective installation of the pivaloyl group19 occurred at the less sterically hindered 2-OH group of the α-D-glucopyranoside-derived boronic ester. However, acyl migration was observed under the basic conditions of the phase-switch deprotection, leading to the isolation of both 2-O-Piv and 3-O-Piv products in 66% and 15% yield, respectively. Migration of the pivaloyl group was suppressed by changing the conditions for cleavage of the boronic ester, using transesterification with pinacol in refluxing toluene in place of the phase-switch workup. In this way, the yield of 3i was improved to 72%.
In a similar way, partially silylated pyranosides were prepared using this protocol (Fig. 2). The formation of the bis-silylated isomers 4e and 4e′ from methyl α-D-galactopyranoside 1e merits further comment. Condensation of 1e with phenylboronic acid resulted in the clean formation of the 4,6-O-boronate, and yet the 2,6-bis-silylated derivative was the predominant product, indicating that migration of the boronic ester group takes place under the silylation conditions. Indeed, 1H NMR spectroscopic analysis of the unpurified reaction mixture after the silylation step showed the presence of the isomeric boronates 2e and 2e′ in a 3.6:1 ratio, which is in good agreement with the ratio of isolated yields of 4e and 4e′ (Scheme 2). Prolonging the reaction time for silylation from 30 minutes to two hours caused an increase in the amount of 2e′, and the 2,3-bis-O-silylated boronate could be favored by conducting the silylation for 18 hours at 50 °C. After phase-switch workup, the latter silylation protocol led to the isolation of 4e′ in 50% yield. These results suggest that 2e is the kinetically preferred product, while 2e′ is thermodynamically favored. Kinetically controlled formation of 2e could arise under a Curtin–Hammett scenario wherein the 4,6-O-boronate rearranges to a less stable 3,4-O-boronate, which undergoes rapid silylation at the primary 6-OH group. The ability to convert 2e to 2e′ by prolonged heating suggests that migration of both the boronic ester and silyl groups can occur under these conditions, leading to the more stable 4,6-O-boronate product.20 Evidence of boronate migration was also observed in the synthesis of bis-benzoylated galactopyranoside 3e (Fig. 1): the 2,6-bis-benzoylated regioisomer was obtained as a side product in 15% yield. However, in this case, heating and/or prolonged reaction times did not lead to a change in the distribution of isomers. The benzoylation reaction is likely under kinetic control, with migration of benzoyl groups being less rapid than that of triethylsilyl groups in this system. The galactopyranoside is unique among substrates 1a–1g in having a triol moiety that can form two relatively stable boronic ester isomers, and analogous isomerizations were not observed in the other reactions studied here.
 | ||
| Fig. 2 Partially silylated carbohydrate derivatives prepared using boronic ester protection. Reaction conditions: (1) substrate (5.0 mmol), PhB(OH)2 (1.0 equiv.), toluene, 110 °C; (2) chlorotriethylsilane (TESCl), pyridine, 0 °C; (3) phase-switching workup with 1 M aqueous sorbitol/Na2CO3. For details, see the Experimental section. Yields after purification by flash chromatography on silica gel are listed. aReaction was carried out using tert-butyl(chloro)dimethylsilane (TBSCl) at 60 °C. | ||
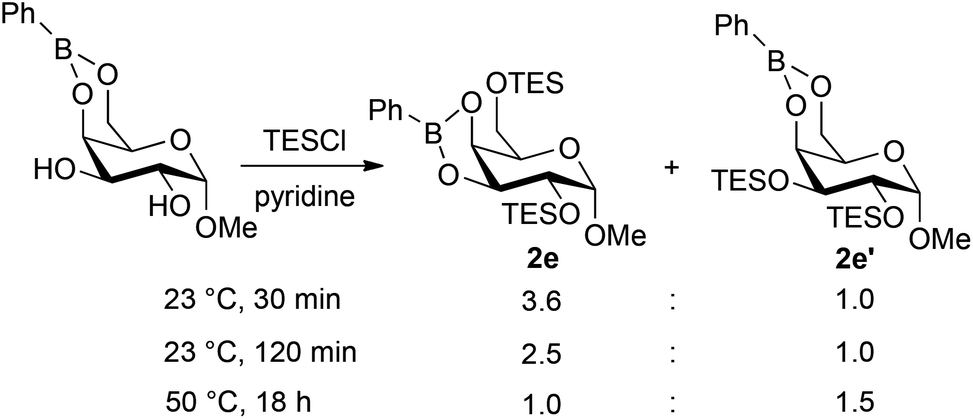 | ||
| Scheme 2 Formation of isomeric bis-silylated boronates 2e and 2e′. Isomer ratios were determined by integration of the signals corresponding to the anomeric OCH3 groups in the 1H NMR spectrum of unpurified reaction mixtures. | ||
Alkylation of sugar-derived boronates
Although ethers (especially benzyl ethers) are useful protective groups in carbohydrate chemistry, alkylation reactions of hydroxy groups in glycoside-derived boronates have not been reported. (It should be noted, however, that regioselective O-alkylations of pyranosides have been achieved by activation as tetracoordinate boronic ester–amine adducts.21) Duggan and co-workers prepared the 1,6-bis-benzyl ether of mannitol by treating the hexitol-derived bis-boronic ester with benzyl bromide and sodium hydride in DMF solvent.11 However, upon subjecting the boronic ester derived from PhB(OH)2 and 1a to these conditions, we obtained a complex mixture. We speculated that the boronic ester group might undergo side reactions with alkoxides, and turned our attention to trichloroacetimidates, alkylating agents that are reactive under Lewis or Brønsted acidic conditions. Our initial attempts at using benzyl trichloroacetimidate for bis-alkylation of 1a led to incomplete conversion under relatively mild activation conditions (10 mol% BF3·OEt2, dichloromethane, 4 Å molecular sieves, 0 °C), and decomposition under more forcing activation conditions (50 mol% trifluoromethanesulfonic acid). However, promising results were obtained using the more reactive para-methoxybenzyl trichloroacetimidate, which gave a 28% yield of bis-alkylated product 5a (Table 1, entry 1). Variation of the boronic ester component revealed that 4-methoxyphenylboronic acid gave inferior results, while 4-(trifluoromethyl)phenylboronic acid resulted in a higher yield (entries 2 and 3). This trend may reflect the higher stability of the electron-deficient boronic ester under the alkylation conditions.22,23 A further improvement was achieved by using lanthanum(III) trifluoromethanesulfonate (La(OTf)3) as the Lewis acid activator in place of BF3·OEt2.24 Using a mixture of dichloromethane and toluene as the solvent, 5a was obtained in 60% yield, as determined by 1H NMR using an internal standard.|
|
||||
|---|---|---|---|---|
| Entry | Boronic acid | Lewis acid | Solvent | Yieldb |
|
a Reaction conditions: (1) 1a (0.1 mmol), ArB(OH)2 (1.0 equiv.), toluene, 110 °C; (2) 4-(methoxy)benzyl trichloroacetimidate (6.0 equiv.), Lewis acid (5.0 mol%), solvent, 0 °C; (3) phase-switch workup.
b NMR yield using 1,3,5-trimethoxybenzene as internal standard.
c A 1:1 mixture of the two solvents was used.
|
||||
| 1 | C6H5B(OH)2 | BF3·OEt2 | CH2Cl2 | 30% |
| 2 | 4-(OCH3)-C6H4B(OH)2 | BF3·OEt2 | CH2Cl2 | 15% |
| 3 | 4-(CF3)-C6H4B(OH)2 | BF3·OEt2 | CH2Cl2 | 35% |
| 4 | 4-(CF3)-C6H4B(OH)2 | La(OTf)3 | CH2Cl2 | 45% |
| 5 | 4-(CF3)-C6H4B(OH)2 | La(OTf)3 | PhCH3 | 45% |
| 6 | 4-(CF3)-C6H4B(OH)2 | La(OTf)3 | CH2Cl2/PhCH3c |
60% |
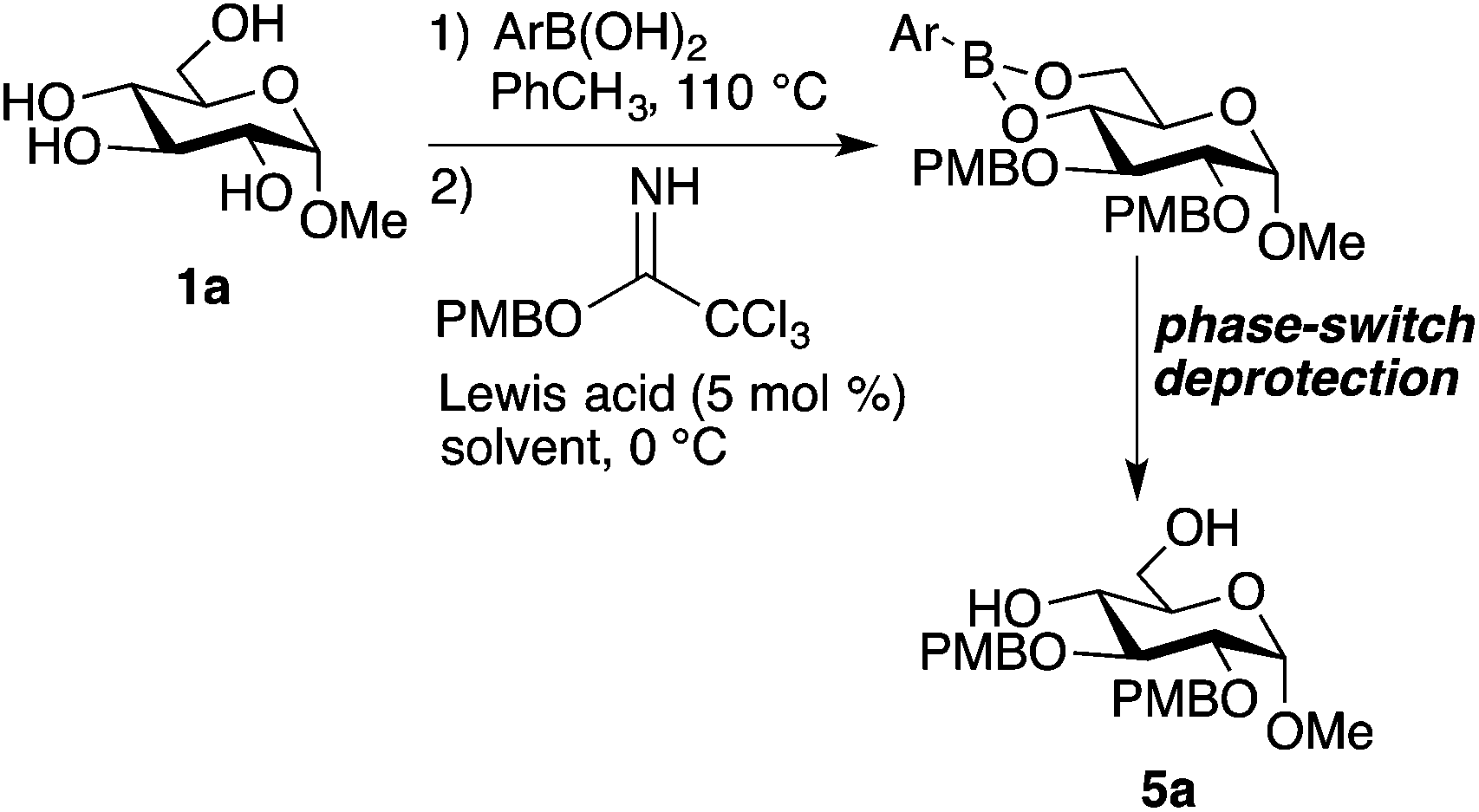
The optimized conditions for installation of PMB groups were carried out on preparative (5 mmol) scale for a range of glycoside substrates, using the phase-switch deprotection described above (Fig. 3). The protocol is compatible with silyl groups (products 5c and 5d) and an isopropylidene ketal (product 5f), but could not be applied to thioglycoside 1g, presumably due to competing S-alkylation and subsequent decomposition. Given that 4-(trifluoromethyl)phenylboronic acid is more expensive than PhB(OH)2, the ability to recover this reagent after the phase-switching deprotection (78–91% recovered yields: see the Experimental section) is noteworthy.
 | ||
Fig. 3 Carbohydrate-derived para-methoxybenzyl (PMB) ethers prepared using boronic ester protection. Reaction conditions: (1) substrate (5.0 mmol), 4-(trifluoromethyl)phenylboronic acid (1.0 equiv.), toluene, 110 °C; (2) PMBOC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)CCl3, La(OTf)3 (1 mol%), toluene, 0 °C; (3) phase-switching workup with 1 M aqueous sorbitol/Na2CO3. For details, see the Experimental section. Yields after purification by flash chromatography on silica gel are listed. a1:1 Toluene/dichloromethane was used as the solvent. bReaction was carried out on 1.6 mmol scale using ArB(OH)2 recovered from a previous experiment. NH)CCl3, La(OTf)3 (1 mol%), toluene, 0 °C; (3) phase-switching workup with 1 M aqueous sorbitol/Na2CO3. For details, see the Experimental section. Yields after purification by flash chromatography on silica gel are listed. a1:1 Toluene/dichloromethane was used as the solvent. bReaction was carried out on 1.6 mmol scale using ArB(OH)2 recovered from a previous experiment. | ||
Sequential functionalizations of methyl α-D-glucopyranoside
The ability to install a pivaloyl group selectively at O-2 of the phenylboronate of methyl α-D-glucopyranoside (Fig. 1, product 3i) suggested the possibility of generating differentially protected products by further functionalization of the free 3-OH group. Indeed, sequential addition of PivCl and BzCl in pyridine, without any workup or purification of the intermediate monoacylated boronic ester, resulted in the efficient formation of the expected differentially acylated product (Scheme 3). After phase-switch deprotection and purification by flash chromatography on silica gel, pyranoside 6a was isolated in 80% yield.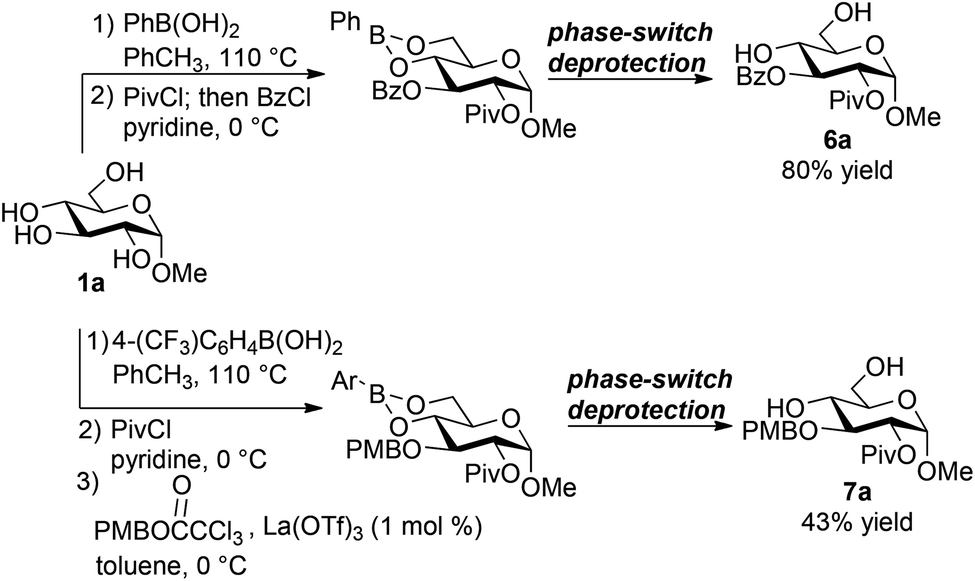 | ||
| Scheme 3 Sequential functionalizations of a boronic ester-protected glucopyranoside. | ||
Sequential installation of pivaloyl and para-methoxybenzyl groups was also achieved. 4-(Trifluoromethyl)phenylboronic acid, the optimal protecting group for the PMB installation protocol (see above), was used in this case. After the reaction with PivCl was complete, the pyridinium chloride byproduct was removed by precipitation and filtration, and a solvent switch from pyridine to toluene was conducted. Without further purification, the intermediate arylboronate was subjected to the trichloroacetimidate and La(OTf)3, followed by phase-switch deprotection. Product 7a was obtained in 43% yield over this reaction sequence.
Conclusions
In summary, installation of acyl, silyl and para-methoxybenzyl groups to glycoside substrates can be accomplished in a preparatively useful way by employing boronic esters as protective groups for cis-1,2- or 1,3-diol motifs. The sequence of protection, functionalization and deprotection can be conducted in one pot, which is not typically the case for similar operations using acetal or ketal protective groups. For the most part, the boronic esters are sufficiently robust to enable efficient acylation and silylation under standard conditions, although boronic ester migration led to mixtures of products for the α-D-galactopyranoside substrate. Using PMB trichloroacetimidate and La(OTf)3, we were able to expand the scope of available transformations of glycoside-derived boronic esters to include alkylation. The use of the phase-switching workup to enable both the deprotection of the boronic ester and the separation of the glycoside and boronic acid components contributed in an important way to the overall efficiency of the processes described here. These relatively mild conditions for boronic ester hydrolysis are tolerant of several functional groups, including esters, silyl ethers, ketals and thioglycosides. Overall, our results suggest that boronic esters have features that may be complementary to those of acetals and ketals. Boronic esters are arguably less well suited to multistep reaction sequences, but their relatively facile deprotection lends itself to streamlined operations wherein isolation and purification of intermediates can be minimized or avoided.Experimental
General methods
Stainless steel syringes were used to transfer air- and moisture sensitive liquids. Flash chromatography was performed using silica gel (60 Å, 230–400 mesh). Thin-layer chromatography (TLC) was performed using aluminum-backed silica gel plates, products were visualized by UV irradiation at 254 nm and by staining with KMnO4 solution. Toluene and dichloromethane used in alkylation chemistry was purified by passing through two columns of activated alumina under nitrogen. Deionized water was acquired from an in-house supply. Pyridine used for regioselective reactions with PivCl was dried over calcium hydride with stirring at 80 °C prior to use. The remainder of reagents and solvents were purchased from Sigma-Aldrich, Caledon or Alfa Aesar and were used without further purification. Nuclear magnetic resonance (NMR) solvents were purchased from Cambridge Isotope Laboratories. Proton nuclear magnetic resonance (1H NMR) spectra and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a 400 MHz Varian Mercury spectrometer or 400 MHz Bruker spectrometer. Chemical shifts for protons are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CDCl3: δ 7.26; DMSO-d6: δ 2.50). Chemical shifts for carbon (13C NMR) are reported in parts per million downfield from tetramethylsilane and are referenced to the carbon resonances of the solvent (CDCl3: δ 77.16; DMSO-d6: δ 39.52). Data are represented as follows: chemical shift (δ, ppm); multiplicity (s, singlet; d, doublet; app t, apparent triplet; br s, broad singlet; m, multiplet); integration; coupling constant (J, Hz), proton assignment. Proton resonances were assigned based on 2-D COSY and HSQC experiments. High-resolution mass spectra (HRMS) were obtained on a VS 70-250S (double focusing) mass spectrometer at 70 eV using either electrospray ionization (ESI) or direct analysis in real time (DART) methods. Infrared (IR) spectra were obtained on a Fourier-transform IR instrument equipped with a single-bounce diamond/ZnSe ATR accessory. Spectral features are represented as follows: wavenumber (cm−1); intensity (s, strong; m, medium; w, weak; br, broad). Optical rotations were measured on an automatic polarimeter. Specific rotation values are reported in 10−1 deg cm2 g−1 (c g per 100 mL in CHCl3).:4). [α]20D = 41.5 (c 0.92 in CHCl3). FTIR (powder, cm−1): 3420 (br), 2954 (w), 2935 (w), 2857 (w), 1718 (m), 1316 (w), 1261 (s), 1106 (s), 1065 (s), 1027 (s), 970 (m), 834 (s), 776 (s), 708 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.06–8.03 (m, 2H, o-ArH), 7.61–7.57 (m, 1H, p-ArH), 7.48–7.43 (m, 2H, m-ArH), 5.22 (app t, 1H, J = 9.6 Hz, H-4), 4.82 (d, 1H, J = 1.5 Hz, H-1), 4.05 (dd, 1H, J = 9.4, 3.5 Hz, H-3), 3.98 (dd, 1H, J = 3.5, 1.5 Hz, H-2), 3.92–3.88 (m, 1H, H-5), 3.80 (d, 2H, J = 4.2 Hz, H-6, H-6), 3.43 (s, 3H, OCH3), 0.85 (s, 9H, (CH3)3CSi), 0.00 (s, 3H, CH3Si), 0.00 (s, 3H, CH3Si). 13C NMR (100 MHz, CDCl3): δ (ppm) 167.4, 133.6, 130.0, 129.6, 128.6, 100.5, 71.4, 70.89, 70.87, 70.8, 63.0, 55.1, 26.0, 18.4, −5.25, −5.29. HRMS (DART, m/z) Calculated for [C20H33O7Si] (M + H)+ 413.19955; Found: 413.19978.
:4). [α]20D = 2.7 (c 1.27 in CHCl3). FTIR (powder, cm−1): 3487 (m), 3458 (w), 2945 (w), 2897 (w), 1687 (s), 1456 (w), 1378 (w), 1322 (m), 1288 (s), 1213 (m), 1138 (m), 1064 (s), 1005 (s), 952 (m), 881 (m), 782 (m), 713 (s), 619 (m). 1H NMR (400 MHz, DMSO-d6): δ (ppm) 8.03–8.00 (m, 2H, o-ArH), 7.68–7.64 (m, 1H, p-ArH), 7.55–7.51 (m, 2H, m-ArH), 5.82 (d, 1H, J = 3.7 Hz, H-1), 5.30 (d, 1H, J = 4.9 Hz, OH), 5.21 (d, 1H, J = 5.9 Hz, OH), 4.46 (dd, 1H, J = 11.3, 2.0 Hz, H-6), 4.41 (d, 1H, J = 3.7 Hz, H-2), 4.20 (dd, 1H, J = 11.3, 5.5 Hz, H-6), 4.09–3.98 (m, 3H, H-3, H-4, H-5), 1.37 (s, 3H, CH3), 1.23 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δ (ppm) 165.7, 133.3, 129.9, 129.2, 128.6, 110.6, 104.5, 84.7, 80.1, 72.9, 67.5, 65.2, 26.6, 26.2. HRMS (DART, m/z) Calculated for [C16H21O7] (M + H)+ 325.12873; Found: 325.12870. Phenylboronic acid was recovered according to general procedure C (0.485 g, 80%).
:4). [α]20D = 35.4 (c 1.10 in CHCl3). FTIR (powder, cm−1): 3432 (br), 2958 (w), 2937 (w), 2856 (w), 1743 (m), 1450 (m), 1388 (w), 1256 (s), 1097 (s), 1035 (s), 988 (m), 836 (s), 734 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.78–7.75 (m, 2H, ArH), 7.65–7.62 (m, 2H, ArH), 7.43–7.39 (m, 2H, ArH), 7.34–7.30 (m, 2H, ArH), 4.99 (d, 1H, J = 3.8 Hz, H-1), 4.95 (dd, 1H, J = 9.9, 3.8 Hz, H-2), 4.48–4.40 (m, 2H, (O-2)CH2), 4.29 (app t, 1H, J = 7.4 Hz, (O-2)CH2CH), 4.17 (dd, 1H, J = 3.4, 1.2 Hz, H-4), 4.06 (dd, 1H, J = 9.9, 3.4 Hz, H-3), 3.95 (dd, 1H, J = 10.8, 5.2 Hz, H-6), 3.91 (dd, 1H, J = 10.8, 4.5 Hz, H-6), 3.82–3.79 (m, 1H, H-5), 3.41 (s, 3H, OCH3), 3.41 (br s, 1H, OH), 2.66 (br s, 1H, OH), 0.92 (s, 9H, (CH3)3CSi), 0.11 (s, 3H, CH3Si), 0.11 (s, 3H, CH3Si). 13C NMR (100 MHz, CDCl3) δ (ppm) 155.4, 143.5, 143.4, 141.44, 141.41, 128.02, 128.01, 127.33, 127.28, 125.4, 120.17, 120.16, 97.5, 75.5, 70.6, 70.4, 69.1, 68.7, 63.7, 55.5, 46.8, 26.0, 18.4, −5.3, −5.4. HRMS (ESI, m/z) Calculated for [C28H42O8Si] (M + NH4)+ 548.2667; Found: 548.2674.
:4). [α]20D = 31.6 (c 1.65 in CHCl3). FTIR (neat, cm−1): 3385 (br), 2953 (m), 2911 (m), 2877 (m), 1460 (w), 1413 (w), 1238 (w), 1194 (w), 1150 (s) 1116 (s), 1090 (m), 1046 (s), 1010 (s), 836 (m), 732 (s), 674 (m). 1H NMR (400 MHz, CDCl3): δ (ppm) 4.62 (d, 1H, J = 3.5 Hz), 3.83–3.77 (m, 3H, H-3, H-6, H-6), 3.63–3.58 (m, 1H, H-5), 3.50 (dd, 1H, J = 9.0, 3.5 Hz, H-2), 3.41 (dd, 1H, J = 9.8, 8.4 Hz, H-4), 3.36 (s, 3H, OCH3), 2.30 (br s, OH), 2.09 (br s, OH), 0.99–0.95 (m, 18H, 6 × (CH3CH2Si)), 0.70–0.59 (m, 12H, 6 × (CH2Si)). 13C NMR (100 MHz, CDCl3): δ (ppm) 100.6, 75.3, 73.8, 72.2, 70.8, 62.8, 55.1, 7.1, 6.9, 5.5, 5.2. HRMS (ESI, m/z) Calculated for [C19H42NaO6Si2] (M + Na)+ 445.2412; Found: 445.2416. Phenylboronic acid was recovered according to general procedure C (0.491 g, 81%).
:4). [α]20D = −32.1 (c 1.25 in CHCl3). FTIR (powder, cm−1): 3465 (br), 2936 (m), 2858 (m), 1462 (w), 1405 (w), 1249 (m), 1114 (s), 1061 (s) 976 (m), 880 (s), 833 (s), 774 (s), 673 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 4.65 (d, 1H, J = 1.6 Hz, H-1), 3.90 (dd, 1H, J = 3.5, 1.6 Hz, H-2), 3.69 (dd, 1H, J = 8.8, 3.5 Hz, H-3), 3.63–3.56 (m, 1H, H-5), 3.44 (app t, 1H, J = 9.0 Hz, H-4), 3.35 (s, 3H, OCH3), 2.33 (br s, 2H, 2-OH, 3-OH), 1.26 (d, 3H, J = 6.3 Hz, CH3), 0.90 (s, 9H, (CH3)3CSi), 0.13 (s, 3H, CH3Si), 0.10 (s, 3H, CH3Si). 13C NMR (100 MHz, CDCl3): δ (ppm) 100.6, 75.0, 72.3, 71.4, 68.2, 55.0, 26.1, 18.4, 18.2, −3.7, −4.3. HRMS (DART, m/z) Calculated for [C13H32NO5Si] (M + NH4)+ 310.20497; Found: 310.20448. Phenylboronic acid was recovered according to general procedure C (0.469 g, 77%).
:4). [α]20D = 32.1 (c 1.06 in CHCl3). FTIR (powder, cm−1): 3453 (br), 2953 (m), 2911 (m), 2877 (m), 1458 (w), 1414 (w), 1355 (w), 1238 (w), 1192 (w), 1131 (s), 1095 (s), 1049 (s), 1005 (s), 937 (m), 831 (m), 728 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 4.68 (d, 1H, J = 3.6 Hz, H-1), 4.11 (dd, 1H, J = 3.3, 1.2 Hz, H-4), 3.95 (dd, 1H, J = 9.6, 3.6 Hz, H-2), 3.91 (dd, 1H, J = 10.3, 5.7 Hz, H-6), 3.87–3.80 (m, 2H, H-3, H-6), 3.80–3.77 (m, 1H, H-5), 3.39 (s, 3H, OCH3), 2.88 (s, 1H, 4-OH), 2.37 (s, 1H, 3-OH), 0.99–0.95 (m, 18H, 6 × (CH3CH2Si)), 0.67–0.56 (m, 12H, 6 × (CH2Si)). 13C NMR (100 MHz, CDCl3): δ (ppm) 100.3, 70.8, 70.6, 69.9, 69.6, 62.8, 55.3, 6.82, 6.81, 5.0, 4.4. HRMS (ESI, m/z) calc'd for C19H42NaO6Si2 [M + Na]+ 445.2412; found 445.2415.
:9). [α]20D = 38.0 (c 1.06 in CHCl3). FTIR (powder, cm−1): 3445 (br), 29.53 (m), 2912 (m), 2877 (m), 1458 (w), 1413 (w), 1355 (w), 1238 (w), 1194 (w), 1138 (s), 1100 (s), 1040 (s), 1005 (s), 849 (s), 728 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 4.69 (d, 1H, J = 3.5 Hz, H-1), 3.98–3.92 (m, 2H, H-3, H-6), 3.88–3.78 (m, 4H, H-2, H-4, H-5, H-6), 3.38 (s, 3H, OCH3), 2.44 (br s, 2H, 4-OH, 6-OH), 0.99–0.94 (m, 18H, 6 × (CH3CH2Si)), 0.69–0.61 (m, 12H, 6 × (CH2Si)). 13C NMR (100 MHz, CDCl3): δ (ppm) 100.7, 71.9, 71.3, 70.3, 68.9, 63.3, 55.4, 7.0, 6.9, 5.2, 5.1. HRMS (ESI, m/z) Calculated for [C19H42NaO6Si2] (M + Na)+ 445.2412; Found: 445.2410.
:1 mixture of toluene and dichloromethane (by volume) was used as the solvent, and 4-methoxybenzyl trichloroacetimidate (6.20 mL, 30 mmol, 6.00 equiv.) was added to the reaction in one portion prior to the addition of lanthanum(III) trifluoromethanesulfonate at 0 °C. The title compound was isolated as a colorless syrup (1.112 g, 51%). NMR data were consistent with previous literature reports.291H NMR (400 MHz, CDCl3): δ (ppm) 7.31–7.27 (m, 4H, o-ArH), 6.91–6.86 (m, 4H, m-ArH), 4.94 (d, 1H, J = 11.2 Hz, ArCH), 4.72 (d, 1H, J = 11.8 Hz, ArCH), 4.62 (d, 1H, J = 11.2 Hz, ArCH), 4.61 (d, 1H, J = 11.8 Hz, ArCH), 4.55 (d, 1H, J = 3.6 Hz, H-1), 3.81 (s, 3H, ArOCH3), 3.80 (s, 3H, ArOCH3), 3.80–3.70 (m, 3H, H-3, H-6, H-6), 3.62–3.58 (m, 1H, H-5), 3.49–3.44 (m, 2H, H-2, H-4), 3.37 (s, 3H, OCH3). 4-(Trifluoromethyl)phenylboronic acid was recovered according to general procedure C (0.857 g, 90%).
:4). [α]20D = 35.1 (c 1.19 in CHCl3). FTIR (powder, cm−1): 3420 (br), 2933 (w), 2859 (w), 1612 (w), 1515 (m), 1250 (s), 1079 (s), 1048 (s), 1017 (s), 965 (m), 833 (s), 772 (s), 750 (m). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.29–7.26 (m, 2H, o-ArH), 6.88–6.85 (m, 2H, m-ArH), 4.65 (d, 1H, J = 3.5 Hz, H-1), 4.60 (s, 2H, ArCH2), 4.07–4.05 (m, 1H, H-4), 3.95–3.90 (m, 1H, H-3), 3.85 (dd, 1H, J = 10.3, 5.8 Hz, H-6), 3.81–3.73 (m, 5H, H-5, H-6, ArOCH3), 3.71 (dd, 1H, J = 9.7, 3.8 Hz, H-2), 3.33 (s, 3H, OCH3), 2.91 (d, 1H, J = 1.8 Hz, 4-OH), 2.61 (d, 1H, J = 3.8 Hz, 3-OH), 0.88 (s, 9H, (CH3)3CSi), 0.07 (s, 3H, CH3Si), 0.07 (s, 3H, CH3Si). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.6, 130.3, 129.8, 114.1, 98.0, 76.6, 72.7, 69.7, 69.6, 69.4, 63.0, 55.4, 55.3, 25.9, 18.4, −5.3, −5.4. HRMS (ESI, m/z) Calculated for [C21H36NaO7Si] (M + Na)+ 451.2123; Found: 451.2120.
:4). [α]20D = 23.36 (c 1.37 in CHCl3). FTIR (powder, cm−1): 3335 (br), 2952 (w), 2929 (w), 2856 (w), 1615 (w), 1515 (m), 1249 (m), 1094 (s), 1063 (s), 1026 (s), 965 (m), 832 (s), 779 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.30–7.27 (m, 2H, o-ArH), 6.91–6.87 (m, 2H, m-ArH), 4.70–4.63 (m, 3H, H-1, ArCH2), 3.90–3.85 (m, 4H, H-2, H-3, H-6, H-6), 3.81 (s, 3H, ArOCH3), 3.64 (dd, 1H, J = 9.8, 8.5 Hz, H-4), 3.57–3.52 (m, 1H, H-5), 3.34 (s, 3H, OCH3), 1.97 (br s, 2H, 2-OH, 3-OH), 0.92 (s, 9H, (CH3)3CSi), 0.10 (s, 3H, CH3Si), 0.09 (s, 3H, CH3Si). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.6, 130.7, 129.8, 114.2, 100.5, 75.5, 74.4, 72.1, 71.8, 71.1, 62.6, 55.4, 54.9, 26.1, 18.5, −5.0, −5.2. HRMS (ESI, m/z) Calculated for [C21H36NaO7Si] (M + Na)+ 451.2123; Found: 451.2124. 4-(Trifluoromethyl)phenylboronic acid was recovered according to general procedure C (0.860 g, 91%).
:1 mixture of toluene and dichloromethane (by volume) was used as the solvent, and 4-methoxybenzyl trichloroacetimidate (6.20 mL, 30 mmol, 6.00 equiv.) was added to the reaction in one portion prior to the addition of lanthanum(III) trifluoromethanesulfonate at 0 °C. The title compound was isolated as a colorless syrup (1.270 g, 58%). TLC Rf = 0.34 (acetone/dichloromethane 1:4). [α]20D = 11.7 (c 1.75 in CHCl3). FTIR (powder, cm−1): 3460 (br), 2940 (w), 2909 (w), 2840 (w), 1612 (m), 1512 (s), 1466 (w), 1244 (s), 1088 (s), 1032 (s), 961 (m), 819 (m). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.30–7.26 (m, 4H, o-ArH), 6.90 (m, 4H, m-ArH), 4.74 (d, 1H, J = 11.8 Hz, ArCH), 4.73 (d, 1H, J = 11.2 Hz, ArCH), 4.63–4.57 (m, 3H, H-1, ArCH, ArCH), 4.01 (dd, 1H, J = 3.2, 1.1 Hz, H-4), 3.92–3.87 (m, 1H, H-6), 3.85–3.72 (m, 10H, H-2, H-3, H-5, H-6, ArOCH3, ArOCH3), 3.36 (s, 3H, OCH3). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.6, 159.5, 130.6, 130.3, 129.8, 129.6, 114.1, 114.0, 98.7, 77.1, 75.3, 73.3, 72.8, 69.4, 69.0, 63.2, 55.51, 55.43, 55.42. HRMS (ESI, m/z) Calculated for [C23H30NaO8] (M + Na)+ 457.1833; Found: 457.1838. 4-(Trifluoromethyl)phenylboronic acid was recovered according to general procedure C (0.865 g, 91%).
:4). [α]20D = −0.89 (c 1.01 in CHCl3). FTIR (powder, cm−1): 3396 (br), 2994 (w), 2933 (w), 2867 (w), 2839 (w), 1612 (w), 1513 (m), 1376 (w), 1246 (s), 1215 (m), 1167 (m), 1071 (s), 1017 (s), 956 (m), 885 (m), 851 (m), 818 (m). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.28–7.24 (m, 2H, ArH), 6.90–6.87 (m, 2H, ArH), 5.96 (d, 1H, J = 3.7 Hz, H-1), 4.53–4.51 (m, 3H, H-2, ArCH2), 4.32–4.31 (m, 1H, H-3), 4.21–4.18 (m, 1H, H-5), 4.09 (dd, 1H, J = 6.2, 2.7 Hz, H-4), 3.81 (s, 3H, OCH3), 3.75 (dd, 1H, J = 9.9, 3.4 Hz, H-6), 3.60 (dd, 1H, J = 9.9, 6.0 Hz, H-6), 1.48 (s, 3H, CH3), 1.32 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ (ppm) 159.6, 129.7, 129.6, 114.1, 111.8, 105.1, 85.3, 80.0, 76.1, 73.5, 70.9, 69.8, 55.4, 27.0, 26.3. HRMS (DART, m/z) Calculated for [C17H24NO7] (M + NH4)+ 358.18658; Found: 358.18674. 4-(Trifluoromethyl)phenylboronic acid was recovered according to general procedure C (0.746 g, 79%).
:4). [α]20D = 64.7 (c = 15.8 mg mL−1, CHCl3). FTIR (powder, cm−1): 3415 (br), 2970 (w) 2935 (w), 1721 (s), 1452 (w), 1270 (s), 1156 (s), 1120 (s), 1038 (s), 1026 (s), 916 (m), 710 (s). 1H NMR (400 MHz, CDCl3): δ (ppm) 8.01–7.98 (m, 2H, o-ArH), 7.57–7.55 (m, 1H, p-ArH), 7.46–7.41 (m, 2H, m-ArH), 5.56 (dd, 1H, J = 10.1, 9.0 Hz, H-3), 4.99 (dd, 1H, J = 10.1, 3.7 Hz, H-2), 4.92 (d, 1H, J = 3.7 Hz, H-1), 3.95–3.78 (m, 4H, H-4, H-5, H-6, H-6), 3.42 (s, 3H, OCH3), 3.06 (br s, 1H, 6-OH), 2.05 (br s, 1H, 4-OH), 1.06 (s, 9H, (CH3)3CCO). 13C NMR (100 MHz, CDCl3): δ (ppm) 178.1, 167.6, 133.7, 130.0, 129.3, 128.6, 97.3, 74.6, 71.5, 70.6, 70.2, 62.3, 55.7, 38.9, 27.0. HRMS (DART, m/z) Calculated for [C19H27O8] (M + H)+ 383.17059; Found: 383.17129. Phenylboronic acid was recovered according to general procedure C (0.488 g, 80%).
:4). [α]20D = 29.3 (c 1.01 in CHCl3). FTIR (powder, cm−1): 3435 (br), 2959 (w), 2934 (w), 2917 (w), 2837 (w), 1728 (m), 1614 (w), 1514 (m), 1246 (m), 1157 (s), 1120 (s), 1031 (s), 912 (m), 820 (m). 1H NMR (400 MHz, CDCl3): δ (ppm) 7.26–7.22 (m, 2H, o-ArH), 6.89–6.85 (m, 2H, m-ArH), 4.92 (d, 1H, J = 3.7 Hz, H-1), 4.80 (d, 1H, J = 11.1 Hz, ArCH), 4.71 (dd, 1H, J = 9.9, 3.7 Hz, H-2), 4.56 (d, 1H, J = 11.1 Hz, ArCH), 3.86–3.74 (m, 6H, H-3, H-6, H-6, ArOCH3), 3.66–3.56 (m, 2H, H-4, H-5), 3.36 (s, 3H, OCH3), 2.57 (br s, 1H, 4-OH), 2.20 (br s, 1H, 6-OH), 1.25 (s, 9H, (CH3)3CCO). 13C NMR (100 MHz, CDCl3): δ (ppm) 178.1, 159.5, 130.5, 129.5, 114.2, 97.2, 79.4, 74.9, 73.9, 70.9, 70.4, 62.4, 55.5, 55.4, 38.9, 27.2. HRMS (DART, m/z) Calculated for [C20H34NO8] (M + NH4)+ 416.22844; Found: 416.22950.
Acknowledgements
This work was supported by NSERC (Discovery Grant and Canada Research Chairs Programs), the McLean Endowment, the Canada Foundation for Innovation (projects 17545 and 19119) and the Ontario Ministry of Research and Innovation.Notes and references
- H. G. Kuivila, A. H. Keough and E. J. Soboczenski, J. Org. Chem., 1954, 19, 780–783 CrossRef CAS.
- R. J. Ferrier and D. Prasad, J. Chem. Soc., 1965, 7425–7428 RSC; R. J. Ferrier and D. Prasad, J. Chem. Soc., 1965, 7429–7432 RSC; R. J. Ferrier and D. Prasad, J. Chem. Soc., 1965, 858–863 RSC; R. J. Ferrier, J. Chem. Soc., 1961, 2325–2330 RSC.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1106–1123 RSC; C. A. McClary and M. S. Taylor, Carbohydr. Res., 2013, 381, 112–122 CrossRef CAS PubMed; S. D. Bull, M. G. Davidson, M. H. van den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef PubMed.
- P. G. M. Wuts and T. W. Greene, Greene's Protective Groups in Organic Synthesis, John Wiley and Sons, New York, 4th edn, 2007 Search PubMed.
- J. Robertson and P. M. Staffort, Best Synthetic Methods: Carbohydrates, Academic Press, Massachusetts, 2003 Search PubMed.
- D. M. Clode, Chem. Rev., 1979, 79, 491–513 CrossRef CAS.
- M. S. Taylor, Acc. Chem. Res., 2015, 48, 295–305 CrossRef CAS PubMed.
- R. J. Ferrier, Adv. Carbohydr. Chem. Biochem., 1978, 35, 31–80 CrossRef CAS; P. J. Duggan and E. M. Tyndall, J. Chem. Soc., Perkin Trans. 1, 2002, 1325–1339 RSC.
- W. V. Dahlhoff, Synthesis, 1987, 366–368 CrossRef CAS.
- D. Crich and M. Smith, J. Am. Chem. Soc., 2002, 124, 8867–8869 CrossRef CAS PubMed.
- K. Vijaya Bhaskar, P. J. Duggan, D. G. Humphrey, G. Y. Krippner, V. McCarl and D. A. Offermann, J. Chem. Soc., Perkin Trans. 1, 2001, 1098–1102 RSC.
- S. Langston, B. Bernet and A. Vasella, Helv. Chim. Acta, 1994, 77, 2341–2353 CrossRef CAS.
- J. M. J. Fréchet, L. J. Nuyens and E. Seymour, J. Am. Chem. Soc., 1979, 101, 432–436 CrossRef.
- G. Belogi, T. Zhu and G.-J. Boons, Tetrahedron Lett., 2000, 41, 6969–6972 CrossRef CAS; G. Belogi, T. Zhu and G.-J. Boons, Tetrahedron Lett., 2000, 41, 6965–6968 CrossRef.
- K. Fukuhara, N. Shimada, T. Nishino, E. Kaji and K. Makino, Eur. J. Org. Chem., 2016, 902–905 CrossRef CAS.
- E. Kaji, D. Yamamoto, Y. Shirai, K. Ishige, Y. Arai, T. Shirahata, K. Makino and T. Nishino, Eur. J. Org. Chem., 2014, 3536–3539 CrossRef CAS; T. H. Fenger and R. Madsen, Eur. J. Org. Chem., 2013, 5923–5933 CrossRef; T. Nishino, Y. Ohya, R. Murai, T. Shirahata, D. Yamamoto, K. Makino and E. Kaji, Heterocycles, 2012, 84, 1123–1140 CrossRef; E. Kaji, T. Nishino, K. Ishige, Y. Ohya and Y. Shirai, Tetrahedron Lett., 2010, 51, 1570–1573 CrossRef.
- S. Mothana, J.-M. Grassot and D. G. Hall, Angew. Chem., Int. Ed., 2010, 49, 2883–2887 CrossRef CAS PubMed.
- G. G. Cross and D. M. Whitfield, Synlett, 1998, 487–488 CrossRef CAS.
- L. Jiang and T.-H. Chan, J. Org. Chem., 1998, 63, 6035–6038 CrossRef CAS PubMed.
- Y. H. Ahn and Y.-T. Chang, J. Comb. Chem., 2004, 6, 293–296 CrossRef CAS PubMed.
- K. Oshima, E.-I. Kitazono and Y. Aoyama, Tetrahedron Lett., 1997, 38, 5001–5004 CrossRef CAS.
- H. G. Kuivila, J. F. Reuwer Jr. and J. A. Mangravite, J. Am. Chem. Soc., 1964, 86, 2666–2670 CrossRef CAS; H. G. Kuivila, J. F. Reuwer Jr. and J. A. Mangravite, Can. J. Chem., 1963, 41, 3081–3090 CrossRef; K. V. Nahabedian and H. G. Kuivila, J. Am. Chem. Soc., 1961, 83, 2167–2174 CrossRef; H. G. Kuivila and A. G. Armour, J. Am. Chem. Soc., 1957, 79, 5659–5662 CrossRef.
- M. A. Martinez-Aguirre, R. Villamil-Ramos, J. A. Guerrero-Alvarez and A. K. Yatsimirsky, J. Org. Chem., 2013, 78, 4674–4684 CrossRef CAS PubMed.
- A. N. Rai and A. Basu, Tetrahedron Lett., 2003, 44, 2267–2269 CrossRef CAS.
- I.-H. Chen, K. G. M. Kou, D. N. Le, C. M. Rathburn and V. M. Dong, Chem. – Eur. J., 2014, 20, 5013–5018 CrossRef CAS PubMed.
- K. Yamamoto, Y. Sato, A. Ishimori, K. Miyairi, T. Okuno, N. Nemoto, H. Shimizu, S. Kidokoro and M. Hashimoto, Biosci., Biotechnol., Biochem., 2008, 72, 2039–2048 CrossRef CAS PubMed.
- R. J. Linhardt and S. Bera, J. Org. Chem., 2011, 76, 3181–3193 CrossRef PubMed.
- M. Heuckendorff, C. M. Pedersen and M. Bols, Org. Lett., 2011, 22, 5956–5959 CrossRef PubMed.
- K. Yamamoto, S. Noguchi, N. Takada, K. Miyairi and M. Hashimoto, Carbohydr. Res., 2010, 345, 572–585 CrossRef CAS PubMed.
- A.-L. Chauvin, S. A. Nepogodiev and R. A. Field, J. Org. Chem., 2005, 70, 960–966 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR spectra for all new compounds. See DOI: 10.1039/c6ob02278b |
| This journal is © The Royal Society of Chemistry 2017 |