
Large-scale synthesis and structural analysis of a synthetic glycopeptide dendrimer as an anti-cancer vaccine candidate†
Christelle
Ganneau
ab,
Catherine
Simenel
cd,
Emeline
Emptas
ab,
Tiphanie
Courtiol
ab,
Yves-Marie
Coïc
 ab,
Cécile
Artaud
e,
Edith
Dériaud
fg,
Frédéric
Bonhomme
bh,
Muriel
Delepierre
cd,
Claude
Leclerc‡
fg,
Richard
Lo-Man§‡
fg and
Sylvie
Bay
*ab
ab,
Cécile
Artaud
e,
Edith
Dériaud
fg,
Frédéric
Bonhomme
bh,
Muriel
Delepierre
cd,
Claude
Leclerc‡
fg,
Richard
Lo-Man§‡
fg and
Sylvie
Bay
*ab
aInstitut Pasteur, Unité Chimie des Biomolécules, 75724 Paris Cedex 15, France. E-mail: sylvie.bay@pasteur.fr
bCNRS UMR 3523, 75724 Paris Cedex 15, France
cInstitut Pasteur, Unité Résonance Magnétique Nucléaire des Biomolécules, 75724 Paris Cedex 15, France
dCNRS UMR 3528, 75724 Paris Cedex 15, France
eInstitut Pasteur, Pôle Intégré de Recherche Clinique, 75724 Paris Cedex 15, France
fInstitut Pasteur, Unité Régulation Immunitaire et Vaccinologie, Equipe Labellisée Ligue Contre le Cancer, Paris, France
gINSERM U1041, 75724 Paris Cedex 15, France
hInstitut Pasteur, Unité de Chimie et Biocatalyse, 75724 Paris Cedex 15, France
First published on 20th October 2016
Abstract
Herein, we report a new process that enables the gram-scale production of a fully synthetic anti-cancer vaccine for human use. This therapeutic vaccine candidate, named MAG-Tn3, is a high-molecular-weight tetrameric glycopeptide encompassing carbohydrate tumor-associated Tn antigen clusters and peptidic CD4+ T-cell epitopes. The synthetic process involves (i) the stepwise solid-phase assembly of protected amino acids, including the high value-added Tn building blocks with only 1.5 equivalents, (ii) a single isolated intermediate, and (iii) the simultaneous deprotection of 36 hindered protective groups. The resulting MAG-Tn3 was unambiguously characterized using a combination of techniques, including a structural analysis by nuclear magnetic resonance spectroscopy. The four peptidic chains are flexible in solution, with a more constrained but extended conformation at the Tn3 antigen motif. Finally, we demonstrate that, when injected into HLA-DR1-expressing transgenic mice, this vaccine induces Tn-specific antibodies that mediate the killing of human Tn-positive tumor cells. These studies led to a clinical batch of the MAG-Tn3, currently investigated in breast cancer patients (phase I clinical trial). The current study demonstrates the feasibility of the multigram-scale synthesis of a highly pure complex glycopeptide, and it opens new avenues for the use of synthetic glycopeptides as drugs in humans.
Introduction
The tumor-associated Tn antigen (α-D-GalNAc-O-Ser/Thr or S*/T*) is expressed in several carcinomas (e.g., breast, ovarian, colon, lung, and prostate cancers).1 Therefore, the Tn and other carbohydrate antigens have attracted much interest as a target for the development of anti-tumor immunotherapy through the induction of a specific immune response against cancer cells.2–11 The induction of carbohydrate-specific immunity has primarily relied on glycan conjugation to an immunogenic carrier protein. Although this approach is very successful, it has major limitations (i.e., uncertainty in the vaccine structure and composition, and carrier-induced epitopic suppression12). As an alternative, we designed a novel fully synthetic immunogen, named MAG-Tn3 (MAG: multiple antigenic glycopeptide) (Fig. 1), that associates Tn antigen clusters with peptidic CD4+ T-cell epitopes on a tetravalent lysine backbone.13–15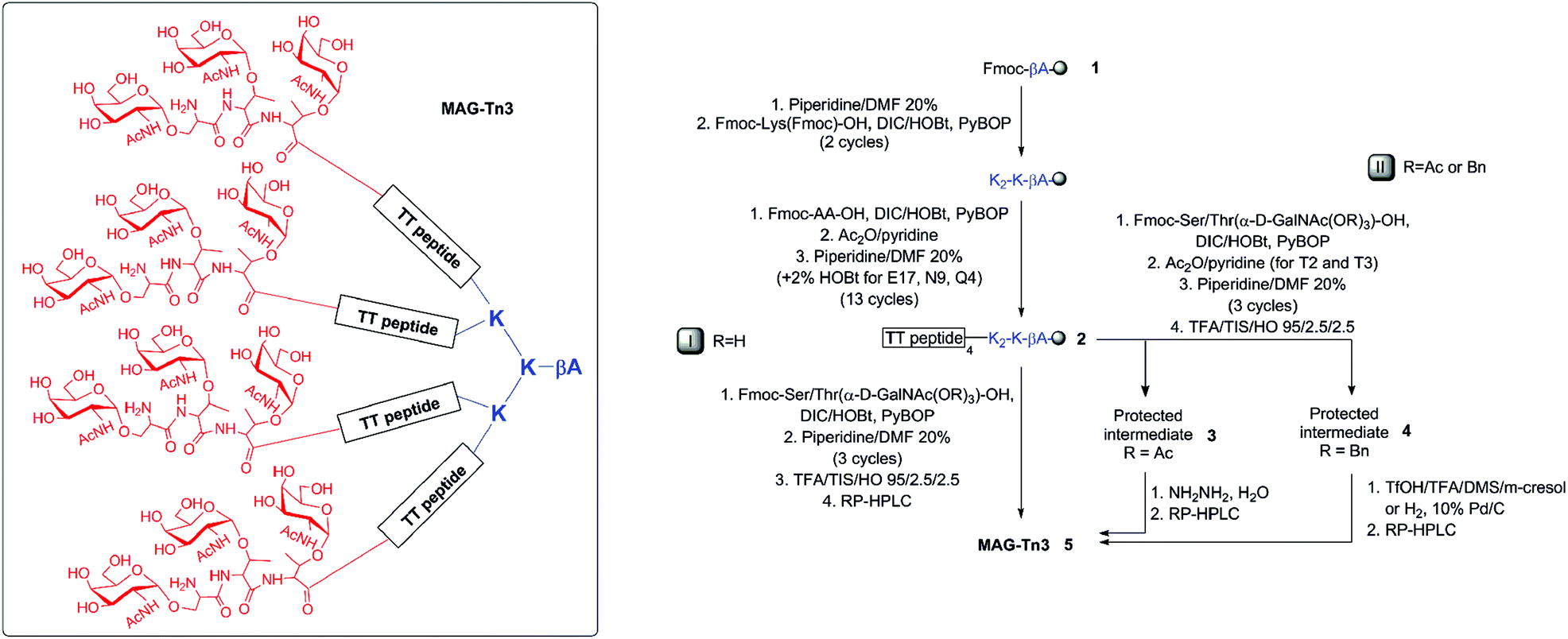 | ||
| Fig. 1 Synthesis of MAG-Tn3 (H-[S*T*T*-QYIKANSKFIGITEL]4-K2-K-βA-OH, 5, structural formula in the inset) with the unprotected (initial route I, R = H)15 or protected (new route II, R = Ac or R = Bn) carbohydrate building blocks. AA9–10 (NS) and AA15–16 (IT) were incorporated as pseudo-Pro dipeptides. See Table 1 for yield and purity. | ||
MAG-Tn3 is based on a multiple antigenic peptide (MAP) construct which has been introduced by Tam16 and widely used to present peptidic epitopes to the immune system.17 Only a few examples of MAGs that feature a multivalent display of both peptidic and carbohydrate epitopes have been reported by us and others.9,13,14,18,19 Notably, the characterization of previously reported MAP or MAG immunogens is typically incomplete. The analytical methods mainly include amino acid analysis (AAA) and, at best, partial data of reverse-phase high-performance liquid chromatography (RP-HPLC), mass spectrometry (MS) or nuclear magnetic resonance (NMR).19–23 In addition to allowing definitive assessment of the identity and purity of the MAG, NMR analysis has proven very useful for providing structural information.
We have previously demonstrated that MAG-Tn3 is an efficient strategy for inducing a high-level Tn-specific antibody response (both in mice and in non-human primates), and for increasing the survival of tumor-bearing mice when used as a therapeutic vaccine.14,15,24 On the basis of these successful pre-clinical studies, MAG-Tn3 was identified as a promising therapeutic vaccine candidate for the treatment of carcinomas.
As part of our efforts to investigate the efficiency of MAG-Tn3 in a phase I clinical trial, we report the development of an improved solid-phase synthetic process that allows for a gram-scale production of high-quality MAG-Tn3 for human use. MAG-Tn3 was extensively characterized using a combination of techniques, including NMR. In addition, we demonstrated that the four peptidic chains are flexible in solution, with a more constrained and extended conformation at the Tn3 antigen motif. Finally, this vaccine candidate was shown to induce Tn-specific antibodies in HLA-DR1-expressing transgenic mice. These antibodies not only recognize human Tn-positive tumor cells, but also mediate their killing.
Results and discussion
Synthesis and physicochemical characterization of MAG-Tn3 (5)
The preparation of MAG-Tn3 is challenging because of (i) its high molecular weight (Mr 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 897), (ii) its branched structure (steric hindrance), and (iii) the presence of clustered glycosylated AAs (four Tn trimer antigens). The synthetic process involves stepwise solid-phase peptide synthesis (SPPS) using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry, cleavage and a final deprotection step (Fig. 1). Because the intermediates could not be isolated and the purification of such large glycopeptides can be complex, the upstream process was precisely optimized for (i) peptide assembly and (ii) protection/deprotection steps.
897), (ii) its branched structure (steric hindrance), and (iii) the presence of clustered glycosylated AAs (four Tn trimer antigens). The synthetic process involves stepwise solid-phase peptide synthesis (SPPS) using 9-fluorenylmethoxycarbonyl (Fmoc) chemistry, cleavage and a final deprotection step (Fig. 1). Because the intermediates could not be isolated and the purification of such large glycopeptides can be complex, the upstream process was precisely optimized for (i) peptide assembly and (ii) protection/deprotection steps.
In a typical MAG/MAP stepwise SPPS, interchain (and/or intrachain) clustering occurs as the peptide chains grow, which leads to the accumulation of multiple deletion products and complex mixtures.25 A few strategies have been proposed to minimize these by-products.26 Convergent syntheses, which involve ligation of purified peptide fragments to a preformed MAP core, have been developed.22,27,28 However, they generate non-peptide bonds that may adversely affect the biological properties of the construct. In addition, when rigorously compared to identical peptide sequences, the stepwise approach was found to be more advantageous than the convergent approach.21 Other approaches have met with varying degrees of success. They include the introduction of flexible spacers, double couplings, capping and/or 1,8-diazabicyclo-[5.4.0] undecen-7-ene deprotection.21,29,30 To reduce the aggregation effect, Boykins et al. used N-[2-hydroxy-4-methoxybenzyl] (Hmb) derivatives in the growing peptide chain of a divalent MAP.31 Pseudoproline dipeptides are also known to minimize aggregation and to limit deletion by-products in synthetic peptides.32 Accordingly, the use of two pseudoproline dipeptides significantly improved the quality of crude MAG-Tn3, thereby facilitating the downstream purification.
 | ||
| Fig. 2 MS analysis (deconvoluted spectra) of crude deprotected glycopeptide 5 obtained directly from peptide 2 (route I, calcd Mr 10897.123) (A), crude protected glycopeptides 3 (route II, R = Ac, calcd Mr 12410.465) (B), and 4 (route II, R = Bn, calcd Mr 14141.610) (C). M ± S* and M ± T* were side-products due to incorporation defaults of the Tn building blocks. M-1Ac and M-1Bn, M-2Bn were partially deprotected compounds arising from resin cleavage under acidic conditions; they were not considered impurities because they all eventually led to 5. | ||
| Route I | Route II | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Substrate (protecting group R) | 2 (none, H) | 2 (none, H) | 3 (Ac) | 4 (Bn) | 4 (Bn) | 4 (Bn) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a Isolated final product (net peptide content). b After RP-HPLC purification, calculated on the basis of the isolated net peptide content and includes all of the synthesis steps from 1. c Corresponding average yield per each coupling step > 96%. d As determined by RP-HPLC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Deprotection method | — | — | NH2–NH2, H2O | TfOH | H2 | H2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Scalea (g) | 0.001–0.01 | > 0.01 | ∼0.005 | ∼0.005 | 0.225 | 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Overall yieldb (%) | 3 | < 1 | 2.7 | 1.6 | 1.5 | 6c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Purityd (%) | 94.5 | < 95 | 96.4 | 95.9 | 96.3 | 96.6 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Comment | Issues during scale-up | To be validated on a large scale | Partial degradation | Validated on a multigram scale | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Two different hydroxyl protecting groups (R) were evaluated (i.e., acetyl (Ac) and benzyl ether (Bn)). Notably, the molar excess of the high value-added glycosylated building blocks was minimized to 1.5 equivalents. After resin cleavage and AA side-chain deprotection, MS analysis of the crude products indicated that, in both cases, N-acetylgalactosamine (GalNAc) protection prevented the side-reactions observed in route I (Fig. 2B, C versus A). Protected glycopeptides 3 and 4 were obtained as the major compounds, with a significant improvement in the crude purity. Advantageously, each glycopeptide constitutes the only intermediate isolated in the process.
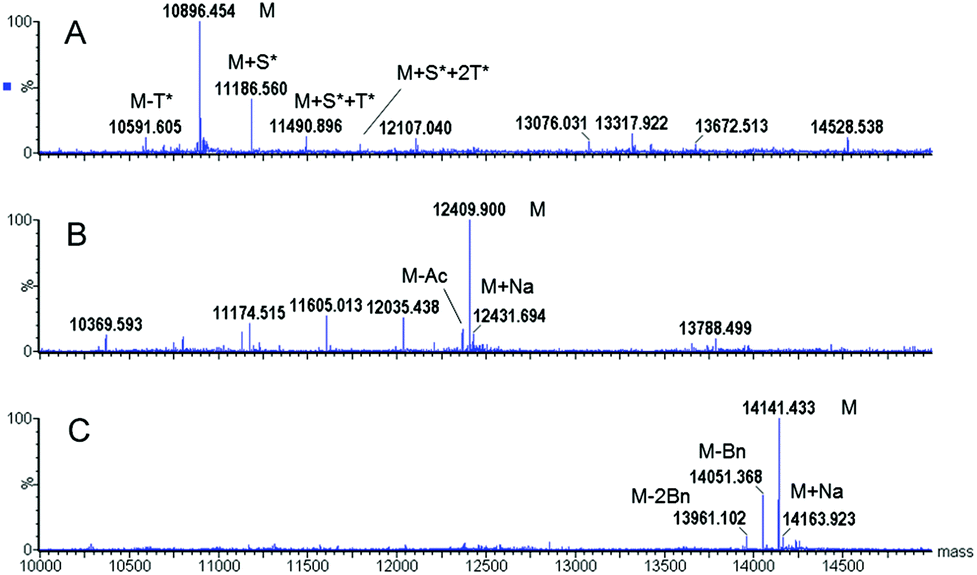
Several deprotection methods were evaluated for both protecting groups. The best results are summarized in Table 1. Treatment of compound 3 with MeONa afforded MAG-Tn3 5 after RP-HPLC purification but with a moderate purity (91.4%) and a 1.4% overall yield (data not shown). In addition, the reaction progress was difficult to monitor, which may affect the process reproducibility. When 3 was treated with hydrazine hydrate, HPLC/MS monitoring indicated that MAG-Tn3 was formed as the major product after 1 h. Surprisingly, complete deprotection could not be achieved despite an increase in pH or an extension of the reaction time. The crude profiles in MeONa and hydrazine were similar, showing that 1–2 acetates (from among 36) were resistant to basic conditions, irrespective of the reagent used. This partial deacetylation may be due to a sterically hindered environment. The partially deprotected intermediates were easily separated by RP-HPLC, and this method afforded 5.8 mg of MAG-Tn3 in 96.4% purity, with an overall yield of 2.7%. Compound 4 was treated with a solution of trifluoromethanesulfonic acid (TfOH)/TFA/dimethylsulfide (DMS)/m-cresol33 and HPLC/MS analysis indicated complete Bn removal after 1 h 15 min. RP-HPLC purification afforded 3.9 mg of MAG-Tn3 5 in 95.9% purity, with an overall yield of 1.6%. However, this method was not entirely satisfactory because a compromise is required between complete deprotection and degradation, which may adversely affect the reaction repeatability and result in scale-up issues. Catalytic hydrogenation is widely regarded as the method of choice for Bn deprotection because it typically proceeds under mild conditions and with limited degradation. The hydrogenolysis of 4 was performed using palladium catalysis under optimized deprotection conditions (10% Pd/C at 37 °C, 5 bar, 170 h, in NMP/H2O (87.5/12.5)) enabling the simultaneous deprotection of the 36 Bn. The crude product (Fig. 3A) was purified by RP-HPLC to give 225 mg of 5 in 96.3% purity, with an overall yield of 1.5%. This process was further validated on a multigram scale with a final purity of 96.6% and an overall yield of 6% (Table 1). It is worth noting that the corresponding average yield is over 96% when related to each individual coupling step involved in the synthesis of this high molecular weight molecule.
 | ||
| Fig. 3 Analysis of the compound 5 obtained by hydrogenolysis before (A) and after (B, C) purification. RP-HPLC (A, B) and ESMS (deconvoluted spectrum, calcd Mr 10897.123) (C) spectra. Magnification of the HPLC peak base is presented in the inset. The purity of 5 was calculated as the percentage of the total area under the curve at 220 nm (B). $ = fragmentation product. | ||
To ensure both its purity and its identity, the MAG-Tn3 5 was analyzed by RP-HPLC (Fig. 3B), ESMS (Fig. 3C), quantitative AAA, and N-ter sequencing (data not shown). MAG-Tn3 was also thoroughly characterized by NMR (assignments of 1H, 13C, and 15N nuclei in Table S1A, and in Fig. S2†). The four chains are equivalent even though small differences in the chemical shifts were observed in the C-ter part from Ile15 to Leu18. This result is explained by environmental differences due to the branching of Leu18 on either αNH (Leu18a and a′) or εNH (Leu18b and b′) of the lysine scaffold residues (Lys73 and Lys74) as well as by the steric hindrance close to the lysine core, as previously observed by Esposito and coworkers.20 All of the NMR data (Tables S1A, S2, and S3, and Fig. S2 and S3†) confirmed the sequence and the presence of three GalNAc residues α-linked to Ser1, Thr2 and Thr3.
Despite recent progress in glycopeptide and glycoprotein synthesis,11,34–37 access to such complex (macro)molecules remains a challenging task. Here, in a stepwise solid-phase process, we showed that the GalNAc protection was critical for minimizing the side reactions that occur during the Tn coupling, thereby allowing the scale-up. Two protocols emerged as convenient and repeatable strategies. The Ac/hydrazine method afforded the highest overall yield on a milligram scale. This process offers the advantages of short reaction time, and a lack of safety issues associated with the use of hydrogen. This method may be worthy of consideration in the future even though it remains to be validated on a large scale. The Bn/H2 method successfully afforded MAG-Tn3 on a multigram scale. Notably, this optimized process was further improved and yielded 11.3 g of a “Good Manufacturing Practice” (GMP) batch.
Until the late 1990s, the crude MAPs and glycodendrimers were typically partially purified and the resulting products were poorly characterized. Inherent problems have been highlighted with their purity and chemical definition, especially by MS.22,26,30 Improvements in both synthesis and analysis have allowed the more accurate characterization of MAPs/MAGs by MS, HPLC or NMR,18–23,26,28,31 even though only partial data are usually reported. Herein, we describe the complete and detailed characterization of the MAG-Tn3, with all of the results being consistent with a high quality product. As for the chemical characterization, literature reports have often lacked experimental data on the exact purity of the MAG/MAP, overall yield of the synthesis and the working scale. Nevertheless, by inference, most of the MAP/MAG syntheses have been performed on the 1–20 mg scale (Mr ranging from 4000 to 15000). To the best of our knowledge, our results provide the first process that enables the gram-scale synthesis of a large pure dendrimer glycopeptide with comprehensive characterization using stringent quality controls, including NMR.
Immunological properties of MAG-Tn3 (5)
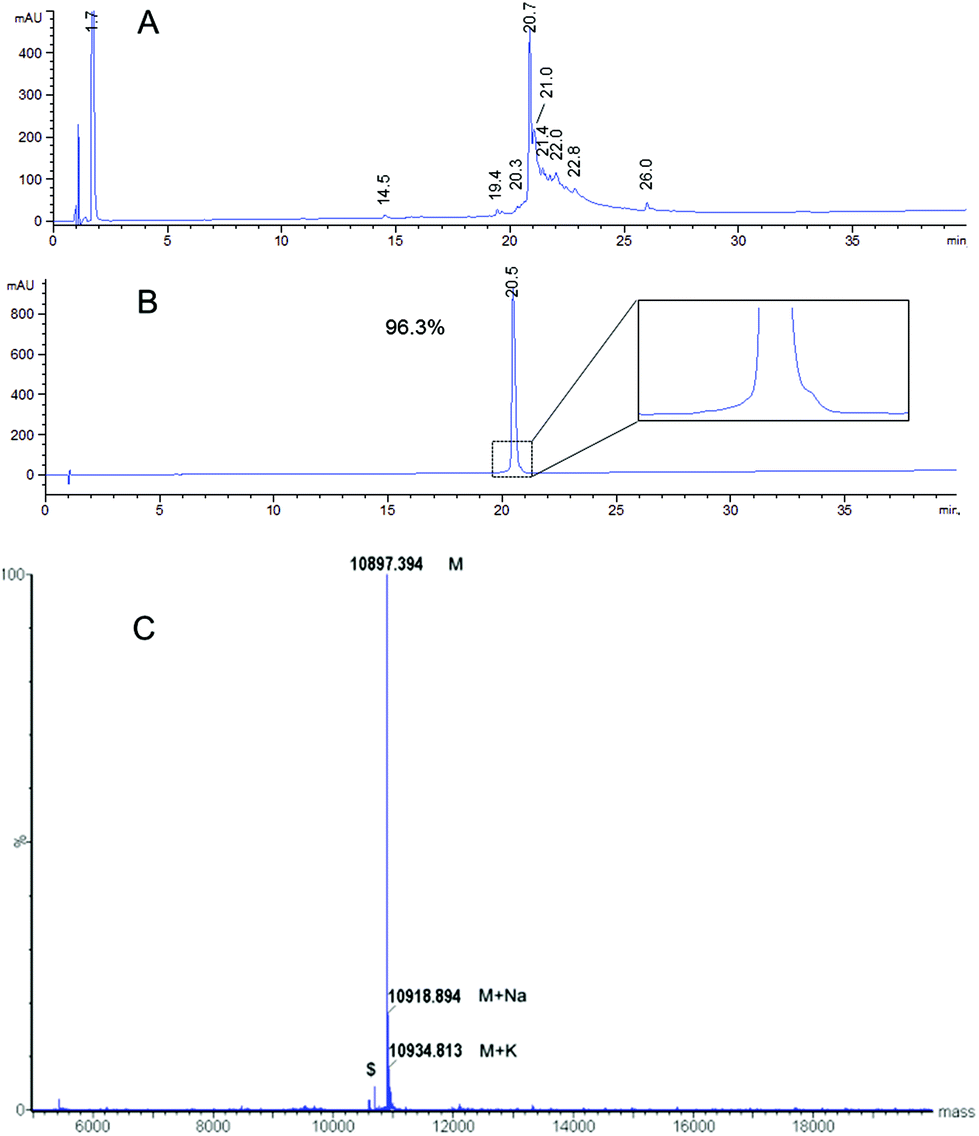
The MAG-Tn3 vaccine aims to induce specific anti-Tn antibody responses that are dependent on the activation of helper T-cells, specific for the inserted CD4+ T-cell peptidic epitope.14 The clinical-grade MAG-Tn3 designed for human use includes a pan-HLA-DR restricted TT peptide,38 which we previously evaluated in non-human primates.15 To formally demonstrate the functionality of this TT epitope in an HLA-DR context, we tested the immunogenicity of MAG-Tn3 in HLA transgenic mice expressing human DR1 or DR1*A2. In contrast to the control groups, mice immunized with the vaccine developed high levels of anti-Tn IgG antibody titers, as detected by ELISA on a synthetic Tn3 cluster attached to a poly-glycine sequence (Fig. 4A and B). Also, the antibodies from MAG-vaccinated mice recognized physiological forms of Tn as shown by their binding to Tn-positive Jurkat tumor cells (Fig. 4C), which demonstrated the immunogenic properties of MAG-Tn3. Finally, sera from MAG-vaccinated mice mediated antibody-dependent cytotoxicity as assessed by the killing of Jurkat cells (Fig. 4D). | ||
| Fig. 4 MAG-Tn3 induced anti-Tn antibodies in HLA-transgenic mice. (A) DR1*A2 and (B) DR1 mice were immunized with 10 μg of MAG-Tn3 (n = 4) in adjuvant (Alum/CpG1826) or with only adjuvant (n = 4) or with 10 μg of the non-glycosylated control MAP-TT15 (n = 3). Sera from these mice were collected at days 20, 28 and 49 post-immunization (D20, D28 and D49, respectively), and they were analyzed for Tn recognition by ELISA (A and B) and by FACS on Tn-positive Jurkat cells (C). The antibody-mediated cytotoxicity was assessed on Jurkat cells with sera from panel B (D). The statistical significance of differences was determined by the Student t test (*P < 0.05). | ||
Structural properties of MAG-Tn3 (5) by NMR
The NMR analyses were used not only as a quality control method but also for structural characterization. A detailed analysis of chemical shifts, coupling constants, and 1H, 1H NOEs is provided in the ESI.†The chemical shift analysis indicated that the peptide chains behave similarly and are flexible with motions that are gradually restricted at N-ter from Ser1* to Ile6 or Lys7 (Table S1, Fig. S4†). The glycosylation of the N-ter residues (Ser1*, Thr2* and Thr3*) affects the chemical shift values of these modified residues as well as those of their neighbors. However, because these glycosylated AAs are not taken into account in the chemical shift databases, other NMR approaches were employed to confirm the presence of the favored extended conformation of the N-ter residues.
The coupling constant analysis (Fig. S3, Table S1A†) revealed: (i) the preferred extended conformation of the Tn3 antigenic motif, (ii) the restricted rotation around the peptide-sugar linkage upon glycosylation of threonine and, to a lesser extent, serine residues, as previously described (see ref. 39 and 40 and references therein), and (iii) the preferred orientation of the N-acetyl group of the GalNAc residues.
The backbone 1H, 1H NOE intensities highlighted the preferred extended conformation at N-ter up to Ile6. This was confirmed by the presence of a set of 1H, 1H NOEs between the GalNAc residues and the AA residues up to Tyr5 (Fig. S3, Tables S2 and S3†). A similar behavior was observed for a small glycosylated peptide (Ser*)3 with strong dαN(i,i + 1) coupled to weak dNN(i,i + 1) NOEs, whereas medium dNN(i,i + 1) NOEs were obtained for the non-glycosylated peptide (Ser)3.41
The 15N longitudinal and transverse relaxation time (i.e., T1 and T2, respectively) measurements for MAG-Tn3 (Fig. 5) suggested a restriction in the flexibility at the N-ter including the first three glycosylated AAs and several of the following residues. Indeed large T1 and T2 values (ranging from 0.51 s to 0.76 s and from 0.14 s to 0.34 s, respectively) were measured from Thr2* to Leu18, showing a high flexibility characteristic of an unfolded molecule. However, smaller T1 values were observed from Thr2* to Lys7, especially for Thr2* (T1 = 0.51 ± 0.03 s). Differences were also observed for T2 along the sequence with larger values from Thr2* to approximately Asn9, followed by decreasing values up to Leu18, as expected for residues adjacent to the lysine core. For an unstructured peptide, larger T2 values due to fast internal motions are typically observed as a consequence of fraying at the free N-ter. In the case of MAG-Tn3, the plateau values observed for T2 from Thr2* to Ala8/Asn9 may result from restricted flexibility induced by the glycosylation of the first three residues. This result is in agreement with the decrease in the T1 values at N-ter. Similarly, Coltart et al. concluded that conformational rigidity was persistent through residues 1 to 4 of a mucin-derived tri-glycosylated peptide (STTAV).42 Here, for the longer peptide chains of MAG-Tn3, the stiffening effect induced by glycosylation is perceptible up to approximately Ala8.
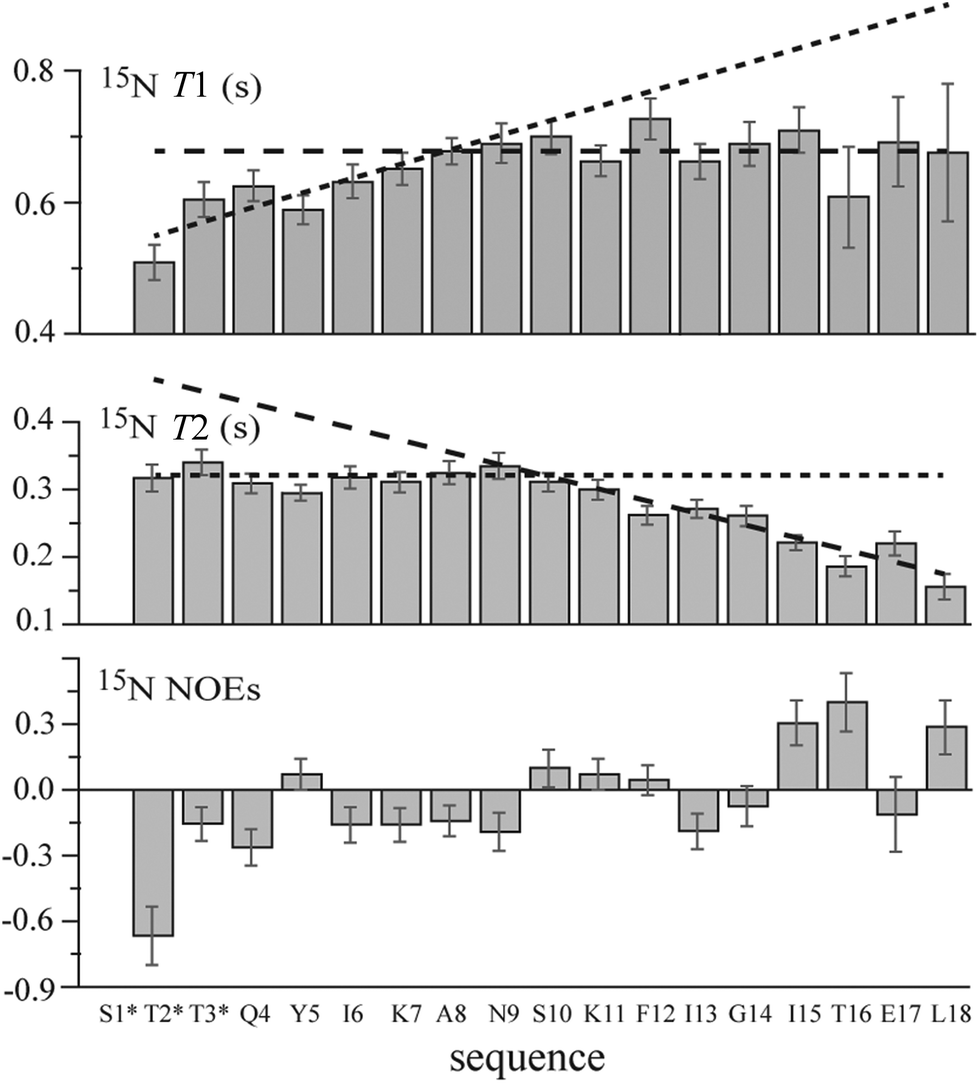 | ||
| Fig. 5 15N relaxation parameters for MAG-Tn3. The dotted lines highlight the different dynamic behavior of MAG-Tn3 along the sequence, notably the reduced N-ter end-fraying from Thr2* to approximately Ala8. For residues T16, E17 and L18, the mean value calculated from the differentiated chain values is reported. | ||
For the GalNAc NH group, the 15N T1 and T2 values are larger for GalNAc 1 than for GalNAc 2 and 3 (T1 = 0.88, 0.72 and 0.71 ± 0.03 s and T2 = 0.49, 0.41 and 0.34 ± 0.03 s for GalNAc1, 2 and 3, respectively). This suggests greater freedom in the motions of the N-acetyl group in GalNAc-Ser1 than in GalNAc-Thr2 and -Thr3. In fact, it was shown that hydrogen bonding between the N-acetyl group of GalNAc residues and the carbonyl group of the glycosylated AA was always detected in the case of threonine but not in the case of serine.40,43
Despite the large errors due to the low concentration of the 15N natural abundance sample, an increase in the NOEs from negative to positive values was observed from Thr2* to Leu18 (Fig. 5), which is in agreement with a progressive increase in flexibility when going from the lysine core bonded residues to the free N-ter residues.
In conclusion, all of the NMR results indicate that the four peptidic chains of the MAG-Tn3 molecule behave in an approximately similar fashion. Their flexibility increases starting from their lysine core attachment point up to the N-ter. However, the glycosylation of the three N-ter residues induces a preferred extended conformation in the Tn3 antigen motif and reduces the N-ter end-fraying.
The structural arrangement of the N-ter Tn3, and most likely the one of the overall MAG, plays a role in its immunological properties. A number of innovative studies have demonstrated that both the Tn display and environment within glycopeptides (e.g. antigen density, peptide backbone) are critical to modulate their molecular recognition by Tn-specific antibodies.39,44–47 Moreover the impact of the proper design of Tn-based vaccines on their immunogenicity has been highlighted. In particular the carrier and the spacer arm attaching the Tn to the carrier have a profound effect on the level and on the diversity of antibody responses.48,49
Because no conformational difference was observed between the MAG-Tn3 glycopeptide chains and the linear glycopeptide, the improved immunogenicity of the MAG-Tn3 tetramer compared to that of the monomer14 is not due to particular conformational glycopeptidic epitopes. It rather may account for the Tn density effect for its recognition by macrophage galactose-type lectin (MGL) on antigen-presenting cells.50 MGL is expressed on dendritic cells and carries a single carbohydrate recognition domain for Gal/GalNAc residues.51 Thus, we have shown that antigen processing and presentation of the T-cell peptide are increased by the Tn density, most likely by clustering MGL on the dendritic cell surface that enhances receptor-mediated uptake of MAG-Tn3 in the dendritic cell endocytic processing compartment.
As reported earlier for the MAP construct by Esposito and co-workers,20 the dendrimeric structure appears to be optimized for interactions, compared to the peptide monomer. The MAG-Tn3 allows the four-peptide chains to sweep the space with larger-amplitude motions at the N-ter, to present the antigenic motif at the surface as an outstretched hand ready for interaction with its partners. Thus, the MAG-Tn3 offers a multiplicity of interactions including with the galactose-specific lectins and the immunoglobulins which both contribute to its immunogenic properties. In addition, through its four flexible peptidic arms, MAG-Tn3 can display a large variety of Tn discrete conformational epitopes that may better mimic the diversity of cancer-associated Tn clusters.
Conclusions
Despite their enormous potential as medicines and/or immunotherapeutics, carbohydrate-based drugs remain underused, in part because of their challenging synthesis. Herein, we report a new process allowing multigram preparation of a highly pure glycopeptide as an anti-cancer therapeutic vaccine, and we demonstrate its potency in humanized mice. We also provide new insights into its molecular structure in solution, which may aid in the future design of such complex molecules. These results led to a phase I clinical trial, ongoing in breast cancer patients. In contrast to many others, this vaccine candidate is chemically-defined, which is a key advantage for consistent studies and safe clinical evaluation. The importance of carbohydrate- and peptide-based drugs is being increasingly recognized. The current study demonstrates the feasibility of the large-scale synthesis of a complex glycopeptide and it opens new avenues for the use of synthetic glycopeptides as drugs in humans.Experimental section
General synthesis methods
The synthesis of 5 was performed by a stepwise solid-phase method using Fmoc chemistry. The AA side-chain protective groups used were Trt on Gln and Asn, Boc on Lys7 and Lys11, Fmoc on Lys19 and Lys20, tBu on Tyr, Ser, Thr, and Glu. The synthesis of 5via route I was performed according to a previously described protocol.15The net peptide content was determined by nitrogen analysis or quantitative AAA using a Beckman 6300 analyzer after hydrolysis of the compounds with 6 N HCl at 110 °C for 20 h. The HPLC/MS analyses were performed on an Alliance model 2695 system coupled to a model 2487 UV detector (220 nm) and to a Q-Tofmicro™ spectrometer (Micromass) with an electrospray ionization (positive mode) source (Waters, France). The samples were cooled to 4 °C on the autosampler. A linear gradient was applied with acetonitrile + 0.025% formic acid (A)/water + 0.04% TFA + 0.05% formic acid (B) over a period of 20 min. Either a Zorbax 300SB C18 column (3.5 μm, 3 × 150 mm) (Agilent, France) (gradient 13–53% A) or an XBridge™ BEH130 C18 column (3.5 μm, 2.1 × 150 mm) (Waters, France) (gradient 15–40% A) was used. The source temperature was maintained at 120 °C and the desolvation temperature at 400 °C. The cone voltage was 40 V. The ESMS analyses were recorded in the positive mode by direct infusion in the same spectrometer with a source temperature and a desolvation temperature maintained at 80 °C and 250 °C, respectively. The samples were dissolved at a concentration of 5 μM in water/acetonitrile (1/1) containing 0.1% formic acid. MaxEnt 1 Software (Waters, France) was used for the deconvolution of mass spectra. The purity of 5 was analyzed by RP-HPLC using an Agilent 1200 pump system with a UV detector at 220 nm. A Zorbax 300SB C18 column (3.5 μm, 3 × 150 mm) (Agilent, France) was used, and a gradient of acetonitrile + 0.1% TFA (A)/water + 0.1% TFA (B) was applied over a period of 40 min, from 13 to 53% A (0.8 mL min−1, retention time 20.5 min).
The molar equivalents of all of the reagents are indicated relative to the reactive amino groups. The molar amounts of crude protected intermediates 3 and 4 were calculated on the basis of the starting Fmoc-β-Ala-resin 1 substitution. The overall yields include all the synthetic steps from 1. These yields were calculated on the net peptide content of the isolated product 5 based on the first β-Ala residue loading on the resin.
Syntheses
The AA couplings (1.75 equiv. for Lys20 and 2 equiv. for the other AA) were performed in DMF (111 mL) at room temperature with DIC/HOBt (1.5 to 2 equiv. each). The AA in positions 15–16 and 9–10 were incorporated as Fmoc-Ile-Thr(ΨMe,Mepro)-OH and Fmoc-Asn(Trt)-Ser(ΨMe,Mepro)-OH (2 equiv.), respectively. After 30 min, a fresh portion of DIC (1.5 to 2 equiv.) was added to the reaction mixture. The coupling steps were monitored using the Kaiser test. From Leu18 to Ser1, after 1 h of coupling with DIC/HOBt, the PyBOP reagent was added (0.5 equiv. for Lys20 and dipeptides, 1 equiv. for the other AA) and the pH was adjusted to 7 by dropwise addition of diisopropylethylamine (DIPEA). After 30 min, the resin was washed with DMF (240 mL) (5 times, 2 min per cycle) and an acetylation step was carried out from Leu18 to Thr2. The acetylation was performed at room temperature with acetic anhydride (1 equiv.) and pyridine (1 equiv.) in DMF (111 mL). After 20 min, the resin was washed with DMF (240 mL) (6 times, 2 min per cycle). After the incorporation of Tyr5, the resin was extensively washed with DMF (240 mL) (8 times, 2 min per cycle) and CH2Cl2 (240 mL) (8 times, 2 min per cycle) prior to drying.
After the incorporation of Tyr5, the assembly was reiterated on a 0.15 and 4.65 mmol scale using the same protocol to afford the peptide-resin 2 for 3 and 4, respectively.
ESMS: 12409.589 (C553H855N107O213 calcd 12410.465).
ESMS: 14141.433 (C733H999N107O177 calcd 14141.610).
From 4, the TfOH method: compound 4 (200 mg, 0.014 mmol) was dissolved in TFA (2.96 mL), DMS (1.78 mL) (Sigma-Aldrich, France) and m-cresol (587 μL) (Sigma-Aldrich, France) at room temperature. The solution was cooled to −10 °C and TfOH (587 μL) (Fluka, France) was added. The mixture was stirred for 1 h 15 min at −10 °C (TfOH/TFA/DMS/m-cresol 1/5/3/1 v/v/v/v). The product was precipitated with diethyl ether and, after centrifugation, the pellet was dissolved in water and lyophilized to yield 372 mg of the crude glycopeptide. The product was dissolved in 0.05 M ammonium acetate buffer (7.7 mL) and the pH was adjusted to 7 with 1 M ammonia. After 1 h at room temperature, the solution was lyophilized to yield 412 mg of the crude product. The product was purified with a gradient from 27/73 to 40/60, as previously described. The purification afforded 3.9 mg (net peptide content) of 5 in 95.9% purity. The overall yield was 1.6%.
From 4, the H2 method: compound 4 (10 g, 1.1 mmol) was dissolved in N-methylpyrrolidone (NMP)/H2O (87.5/12.5, 800 mL v/v). After 10% Pd/C type 39 (4 g) (Johnson Matthey, UK) was added, the reaction was stirred at 37 °C under a pressure of 5 bars for 170 h. Two additional portions of the catalyst were added after 72 h (2 g) and 120 h (2 g). At the end of the reaction, the catalyst was filtered on Celite and washed with NMP/H2O (87.5/12.5 v/v). The resulting filtrate (including that of similar reactions, 1.35 mmol in total) was diluted with H2O (until NMP/H2O 10/90 v/v) and purified by RP-HPLC in two steps. The primary purification was carried out on a Vydac C18 column (300 Å, 10–15 μm, 50 mL min−1) with TFA/H2O/CH3CN (0.1/94.9/5.0 v/v/v) (A) and TFA/H2O/CH3CN (0.1/49.9/50.0 v/v/v) (B) as the eluents. The gradient was 0% B for 15 min, 0–40% B over 5 min, and 40–80% B over 60 min. A secondary purification with AcOH/H2O/CH3CN (0.5/94.5/5.0 v/v/v) (A) and AcOH/H2O/CH3CN (0.5/49.5/50.0 v/v/v) (B) was carried out on the same column. The gradient was 0% B for 15 min, 0–20% B over 5 min, and 20–60% B over 60 min. After concentration by RP-HPLC on a Daisogel SP-300-10-ODS-AP column (20 mL min−1, isocratic TFA/H2O/CH3CN 0.1/49.9/50.0 v/v/v), the solution was evaporated on a rotary evaporator and lyophilized to afford 225 mg (net peptide content) of 5 in 96.3% purity. The overall yield was 1.5%.
AAA: Ala 4 (4), Asn 3.9 (4), Glu + Gln 7.8 (8), Gly 4.1 (4), Ile 11.0 (12), Leu 4.1 (4), Lys 11.0 (11), Phe 3.8 (4), Ser 7.0 (8), Thr 10.5 (12), Tyr 3.7 (4). ESMS: 10897.387 (C481H783N107O177 calcd 10897.123).
NMR experiments
NMR spectra were acquired at 303 K on a Varian NMR System 600 spectrometer equipped with a cryogenically-cooled triple resonance 1H{13C/15N} PFG probe (Agilent Technologies). The samples were dissolved in a 15% v/v D2O buffered solution (50 mM deuterated sodium acetate at pH 3.9 prepared with acetic acid D4 100% D, D2O 99.97% D and sodium deuteroxide 40% w/w solution in D2O 99.5% D, Euriso-top, Saint-Aubin, France) and transferred into a 4 mm NMR tube (Shigemi Inc., Alison Park, USA). The final sample concentration ranged from 0.3 to 0.8 mM, except for that used in the 15N relaxation study (1.2 mM). The 1H chemical shifts were referenced to external DSS (0 ppm), and the 13C and 15N chemical shifts were referenced indirectly to DSS.52Resonance assignments were determined using the following 2D experiments: 1H, 1H COSY,531H, 1H TOCSY (mixing times of 40, 60 and 80 ms),541H,1H NOESY (mixing times of 50, 100 and 200 ms),551H–13C edited HSQC and 1H–13C HSQC-TOCSY (mixing time of 30 and 60 ms).56 In addition, a 1H–15N HSQC was employed to elucidate the overlapped HN, αH region in the NOESY experiments.57
The conformational study of MAG-Tn3 5 was carried out using the following NMR measurements: (i) chemical shifts, (ii) 3JNH,Hα and 3JNH,H2 coupling constants extracted from 1D or 2D spectra with a 1H resolution of 0.1 Hz and 0.8 Hz, respectively, (iii) 3JHα,Hβ and 3JHα,Hβ′ coupling constants measured in a 2D DQF-COSY experiment performed with a resolution of 0.6 Hz on a sample dissolved in 100% D2O buffered solution, (iv) 1H–1H NOE cross-peak intensities measured for a mixing time of 200 ms (detailed in the ESI†) and (v) T1 and T2 15N relaxation times and steady-state {1H}–15N NOEs at natural abundance at 14.1 Tesla.58,59
Analysis of immune responses
Mice were kept in the Institut Pasteur animal house with water and food supplied ad libitum. The study was performed in compliance with French guidelines (project CETEA no. 2013-126).The immunogenicity of the MAG-Tn3 glycopeptide was assessed in DR1 or DR1*A2 mice which are transgenic for human HLA-DR1 or HLA-DR1 and HLA-A2.60 For this purpose, the mice were i.p. immunized every three weeks with 10 μg of MAG-Tn3 in alum (1 mg) supplemented with 10 μg of CpG1826 oligonucleotide (Sigma). One week after each boost, sera were collected and tested for anti-Tn antibodies by ELISA using the Tn3-G7KG biotinylated glycopeptide.14 Serial dilutions of sera were performed and bound antibodies were revealed using the goat anti-mouse IgG peroxidase conjugate (Sigma) and an o-phenyldiamine/H2O2 substrate. The plates were read photometrically at 492 nm. The results are expressed as the mean antibody titer of individual sera ± SD.
Sera were also tested at 1/50 dilution by flow cytometry for recognition of Tn-expressing human Jurkat tumor cells.15 First the cells were incubated for 30 min with sera at 4 °C in PBS containing 5% fetal calf serum and 0.05% sodium azide. Then, the cells were incubated for 30 min with anti-mouse IgG conjugated to phycoerythrin (Caltag). The cells were analyzed on an FACS Calibur flow cytometer (BD), and the analysis was performed using Flowjo software (Tree Star Inc.). The data are shown as histograms corresponding to the fluorescence of cells incubated with the secondary reagent alone or with sera and reported as the mean of the fluorescence intensities.
The biological activity of the sera was evaluated for their ability to induce death of Tn-positive Jurkat cells. Jurkat cells were incubated with sera diluted 50-fold for 15 min at 4 °C and then with goat anti-IgG (Southern Biotech) for 45 min at RT. Cell death was measured by FACS using propidium iodide (PI). The cytotoxicity was calculated as the percent of PI-positive cells. The results are expressed as the mean cytotoxicity of individual test sera ± SD.
Author contributions
C. A., M. D., C. L., R. L-M., and S. B. designed and coordinated research; C. G., C. S., E. E., T. C., Y.-M. C., E. D., and F. B. performed research; C. S., M. D., C. L., R. L-M., and S. B. analyzed data; and C. S., R. L-M., and S. B. wrote the paper.Conflict of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful to F. Groh for the AAA, to Dr Y. C. Lone for providing DR1 and DR1*A2 transgenic mice, and to Dr L. Mulard for critical review of the manuscript. We also wish to thank Dr J.-M. Poudrel, Dr J.-M. Cauvin, Dr C. Coindet, and Dr D. Monnaie (Lonza Braine SA) for their contribution to the process development. We gratefully acknowledge financial support from the Institut Pasteur, and we wish to thank the donators of the MAG-Tn3 program. This work was also supported by grants from the Ligue Nationale Contre le Cancer (Equipe Labellisée 2014) and Banque Privée Européenne to CL.References
- G. F. Springer, Science, 1984, 224, 1198–1206 CAS.
- T. Buskas, P. Thompson and G.-J. Boons, Chem. Commun., 2009, 5335–5349 RSC.
- D. Feng, A. S. Shaikh and F. Wang, ACS Chem. Biol., 2016, 11, 850–863 CrossRef CAS PubMed.
- M. Glaffig, B. Palitzsch, N. Stergiou, C. Schull, D. Strassburger, E. Schmitt, H. Frey and H. Kunz, Org. Biomol. Chem., 2015, 13, 10150–10154 CAS.
- T. Ju, V. I. Otto and R. D. Cummings, Angew. Chem., Int. Ed., 2011, 50, 1770–1791 CrossRef CAS PubMed.
- V. Lakshminarayanan, P. Thompson, M. A. Wolfert, T. Buskas, J. M. Bradley, L. B. Pathangey, C. S. Madsen, P. A. Cohen, S. J. Gendler and G.-J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 261–266 CrossRef CAS PubMed.
- P. Niederhafner, M. Reinis, J. Sebestik and J. Jezek, J. Pept. Sci., 2008, 14, 556–587 CrossRef CAS PubMed.
- O. Renaudet, G. Dasgupta, I. Bettahi, A. Shi, A. B. Nesburn, P. Dumy and L. BenMohamed, PLoS One, 2010, 5, e11216 Search PubMed.
- T. C. Shiao and R. Roy, New J. Chem., 2012, 36, 324–339 RSC.
- S. F. Slovin, S. J. Keding and G. Ragupathi, Immunol. Cell Biol., 2005, 83, 418–428 CrossRef CAS PubMed.
- R. M. Wilson and S. J. Danishefsky, J. Am. Chem. Soc., 2013, 135, 14462–14472 CrossRef CAS PubMed.
- R. Roy, Drug Discovery Today, 2004, 1, 327–336 CrossRef CAS PubMed.
- S. Bay, R. Lo-Man, E. Osinaga, H. Nakada, C. Leclerc and D. Cantacuzene, J. Pept. Res., 1997, 49, 620–625 CrossRef CAS PubMed.
- R. Lo-Man, S. Vichier-Guerre, S. Bay, E. Dériaud, D. Cantacuzène and C. Leclerc, J. Immunol., 2001, 166, 2849–2854 CrossRef CAS.
- R. Lo-Man, S. Vichier-Guerre, R. Perraut, E. Dériaud, V. Huteau, L. BenMohamed, O. M. Diop, P. O. Livingston, S. Bay and C. Leclerc, Cancer Res., 2004, 64, 4987–4994 CrossRef CAS PubMed.
- J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 5409–5413 CrossRef CAS.
- P. Niederhafner, J. Sebestik and J. Jezek, J. Pept. Sci., 2005, 11, 757–788 CrossRef CAS PubMed.
- H. Cai, Z.-Y. Sun, M.-S. Chen, Y.-F. Zhao, H. Kunz and Y.-M. Li, Angew. Chem., Int. Ed., 2014, 53, 1699–1703 CrossRef CAS PubMed.
- S. Keil, A. Kaiser, F. Syed and H. Kunz, Synthesis, 2009, 1355–1369 CAS.
- G. Esposito, F. Fogolari, P. Viglino, S. Cattarinussi, M. T. Demagistris, L. Chiappinelli and A. Pessi, Eur. J. Biochem., 1993, 217, 171–187 CrossRef CAS PubMed.
- W. Kowalczyk, M. Monso, B. G. de la Torre and D. Andreu, J. Pept. Sci., 2011, 17, 247–251 CrossRef CAS PubMed.
- E. H. Nardin, J. M. Calvo-Calle, G. A. Oliveira, P. Clavijo, R. Nussenzweig, R. Simon, W. Zeng and K. Rose, Vaccine, 1998, 16, 590–600 CrossRef CAS PubMed.
- P. Niederhafner, L. Bednarova, M. Budesinsky, M. Safarik, S. Ehala, J. Jezek, L. Borovickova, V. Fucik, V. Cerovsky and J. Slaninova, Amino Acids, 2010, 39, 1553–1561 CrossRef CAS PubMed.
- D. Laubreton, S. Bay, C. Sedlik, C. Artaud, C. Ganneau, E. Dériaud, S. Viel, A. L. Puaux, S. Amigorena, C. Gérard, R. Lo-Man and C. Leclerc, Cancer Immunol. Immunother., 2016, 65, 315–325 CrossRef CAS PubMed.
- J. P. Tam and Y. A. Lu, J. Am. Chem. Soc., 1995, 117, 12058–12063 CrossRef CAS.
- J. Sebestik, P. Niederhafner and J. Jezek, Amino Acids, 2011, 40, 301–370 CrossRef CAS PubMed.
- M. Monso, W. Kowalczyk, D. Andreu and B. G. de la Torre, Org. Biomol. Chem., 2012, 10, 3116–3121 CAS.
- J. C. Spetzler and J. P. Tam, Int. J. Pept. Protein Res., 1995, 45, 78–85 CrossRef CAS PubMed.
- V. Cavallaro, P. Thompson and M. Hearn, J. Pept. Sci., 2001, 7, 262–269 CrossRef CAS PubMed.
- M. Monso, J. Tarradas, B. G. de la Torre, F. Sobrino, L. Ganges and D. Andreu, J. Pept. Sci., 2011, 17, 24–31 CrossRef CAS PubMed.
- R. A. Boykins, M. Joshi, C. Syin, S. Dhawan and H. Nakhasi, Peptides, 2000, 21, 9–17 CrossRef CAS.
- T. Wohr, F. Wahl, A. Nefzi, B. Rohwedder, T. Sato, X. C. Sun and M. Mutter, J. Am. Chem. Soc., 1996, 118, 9218–9227 CrossRef.
- J. P. Tam, W. F. Heath and R. B. Merrifield, J. Am. Chem. Soc., 1986, 108, 5242–5251 CrossRef CAS.
- N. Gaidzik, U. Westerlind and H. Kunz, Chem. Soc. Rev., 2013, 42, 4421–4442 RSC.
- M. C. Galan, D. Benito-Alifonso and G. M. Watt, Org. Biomol. Chem., 2011, 9, 3598–3610 CAS.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS PubMed.
- C. Nativi and O. Renaudet, ACS Med. Chem. Lett., 2014, 5, 1176–1178 CrossRef CAS PubMed.
- D. Valmori, A. Sabbatini, A. Lanzavecchia, G. Corradin and P. M. Matricardi, J. Immunol., 1994, 152, 2921–2929 CAS.
- A. Borgert, J. Heimburg-Molinaro, X. Song, Y. Lasanajak, T. Ju, M. Liu, P. Thompson, G. Ragupathi, G. Barany, D. F. Smith, R. D. Cummings and D. Live, ACS Chem. Biol., 2012, 7, 1031–1039 CrossRef CAS PubMed.
- F. Corzana, J. H. Busto, G. Jimenez-Oses, M. Garcia de Luis, J. L. Asensio, J. Jimenez-Barbero, J. M. Peregrina and A. Avenoza, J. Am. Chem. Soc., 2007, 129, 9458–9467 CrossRef CAS PubMed.
- J. Schuman, D. Qiu, R. R. Koganty, B. M. Longenecker and A. P. Campbell, Glycoconjugate J., 2000, 17, 835–848 CrossRef CAS PubMed.
- D. M. Coltart, A. K. Royyuru, L. J. Williams, P. W. Glunz, D. Sames, S. D. Kuduk, J. B. Schwarz, X. T. Chen, S. J. Danishefsky and D. H. Live, J. Am. Chem. Soc., 2002, 124, 9833–9844 CrossRef CAS PubMed.
- F. Corzana, J. H. Busto, M. Garcia de Luis, J. Jimenez-Barbero, A. Avenoza and J. M. Peregrina, Chemistry, 2009, 15, 3863–3874 CrossRef CAS PubMed.
- O. Blixt, E. Clo, A. S. Nudelman, K. K. Sorensen, T. Clausen, H. H. Wandall, P. O. Livingston, H. Clausen and K. J. Jensen, J. Proteome Res., 2010, 9, 5250–5261 CrossRef CAS PubMed.
- H. Coelho, T. Matsushita, G. Artigas, H. Hinou, F. J. Canada, R. Lo-Man, C. Leclerc, E. J. Cabrita, J. Jimenez-Barbero, S. Nishimura, F. Garcia-Martin and F. Marcelo, J. Am. Chem. Soc., 2015, 137, 12438–12441 CrossRef CAS PubMed.
- N. Martinez-Saez, J. Castro-Lopez, J. Valero-Gonzalez, D. Madariaga, I. Companon, V. J. Somovilla, M. Salvado, J. L. Asensio, J. Jimenez-Barbero, A. Avenoza, J. H. Busto, G. J. Bernardes, J. M. Peregrina, R. Hurtado-Guerrero and F. Corzana, Angew. Chem., Int. Ed., 2015, 54, 9830–9834 CrossRef CAS PubMed.
- D. Mazal, R. Lo-Man, S. Bay, O. Pritsch, E. Deriaud, C. Ganneau, A. Medeiros, L. Ubillos, G. Obal, N. Berois, M. Bollati-Fogolin, C. Leclerc and E. Osinaga, Cancer Immunol. Immunother., 2013, 62, 1107–1122 CrossRef CAS PubMed.
- E. Kagan, G. Ragupathi, S. S. Yi, C. A. Reis, J. Gildersleeve, D. Kahne, H. Clausen, S. J. Danishefsky and P. O. Livingston, Cancer Immunol. Immunother., 2005, 54, 424–430 CrossRef CAS PubMed.
- Z. Yin, S. Chowdhury, C. McKay, C. Baniel, W. S. Wright, P. Bentley, K. Kaczanowska, J. C. Gildersleeve, M. G. Finn, L. BenMohamed and X. Huang, ACS Chem. Biol., 2015, 10, 2364–2372 CrossRef CAS PubMed.
- T. Freire, X. Zhang, E. Deriaud, C. Ganneau, S. Vichier-Guerre, E. Azria, O. Launay, R. Lo-Man, S. Bay and C. Leclerc, Blood, 2010, 116, 3526–3536 CrossRef CAS PubMed.
- M. Tsuiji, M. Fujimori, Y. Ohashi, N. Higashi, T. M. Onami, S. M. Hedrick and T. Irimura, J. Biol. Chem., 2002, 277, 28892–28901 CrossRef CAS PubMed.
- D. S. Wishart, C. G. Bigam, J. Yao, F. Abildgaard, H. J. Dyson, E. Oldfield, J. L. Markley and B. D. Sykes, J. Biomol. NMR, 1995, 6, 135–140 CrossRef CAS PubMed.
- M. Rance, O. W. Sorensen, G. Bodenhausen, G. Wagner, R. R. Ernst and K. Wuthrich, Biochem. Biophys. Res. Commun., 1983, 117, 479–485 CrossRef CAS PubMed.
- C. Griesinger, G. Otting, K. Wuthrich and R. R. Ernst, J. Am. Chem. Soc., 1988, 110, 7870–7872 CrossRef CAS.
- S. Macura, Y. Huang, D. Suter and R. R. Ernst, J. Magn. Reson., 1981, 43, 259–281 CAS.
- W. Willker, D. Leibfritz, R. Kerssebaum and W. Bermel, Magn. Reson. Chem., 1993, 31, 287–292 CrossRef CAS.
- L. E. Kay, P. Keifer and T. Saarinen, J. Am. Chem. Soc., 1992, 114, 10663–10665 CrossRef CAS.
- N. A. Farrow, R. Muhandiram, A. U. Singer, S. M. Pascal, C. M. Kay, G. Gish, S. E. Shoelson, T. Pawson, J. D. Formankay and L. E. Kay, Biochemistry, 1994, 33, 5984–6003 CrossRef CAS PubMed.
- L. E. Kay, L. K. Nicholson, F. Delaglio, A. Bax and D. A. Torchia, J. Magn. Reson., 1992, 97, 359–375 CAS.
- A. Pajot, M. L. Michel, N. Fazilleau, V. Pancre, C. Auriault, D. M. Ojcius, F. A. Lemonnier and Y. C. Lone, Eur. J. Immunol., 2004, 34, 3060–3069 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Four figures, three tables, detailed NMR analyses (chemical shift, coupling constants, 1H, 1H NOEs). See DOI: 10.1039/c6ob01931e |
| ‡ These authors contributed equally to this work. |
| § Present address: Institut Pasteur, Unité d'Histopathologie Humaine et Modèles Animaux, Paris, France. |
| This journal is © The Royal Society of Chemistry 2017 |