
Second-generation total synthesis of aplyronine A featuring Ni/Cr-mediated coupling reactions†
Ichiro
Hayakawa
*a,
Keita
Saito
b,
Sachiko
Matsumoto
b,
Shinichi
Kobayashi
b,
Ayaka
Taniguchi
b,
Kenichi
Kobayashi
b,
Yusuke
Fujii
b,
Takahiro
Kaneko
b and
Hideo
Kigoshi
*b
aDivision of Applied Chemistry, Graduate School of Natural Science and Technology, Okayama University, 3-1-1 Tsushima-naka, Kita-ku, Okayama 700-8530, Japan. E-mail: ichiro.hayakawa@okayama-u.ac.jp
bDepartment of Chemistry, Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba 305-8571, Japan. E-mail: kigoshi@chem.tsukuba.ac.jp
First published on 31st October 2016
Abstract
Second-generation total synthesis of aplyronine A, a potent antitumor marine macrolide, was achieved using Ni/Cr-mediated coupling reactions as key steps. The overall yield of the second-generation synthetic pathway of aplyronine A was 1.4%, obtained in 38 steps based on the longest linear sequence. Compared to our first-generation synthetic pathway of aplyronine A, the second-generation synthesis greatly improved both the yield and number of steps. In particular, we improved the stereoselectivity in the construction of the C13 stereogenic center and the C14–C15 (E)-trisubstituted double bond using the asymmetric Ni/Cr-mediated coupling reaction. Furthermore, we established efficient reaction conditions for the asymmetric Ni/Cr-mediated coupling reaction between the C21–C28 segment and C29–C34 segment. Thus, this coupling reaction proceeded with an equimolar ratio of each segment.
Introduction
Aplyronine A (1) is a marine macrolide isolated from the Japanese sea hare Aplysia kurodai by Yamada and co-workers (Fig. 1).1 Aplyronine A (1) exhibited strong cytotoxicity against HeLa S3 cells in vitro, and potent antitumor activities in vivo against P388 leukemia, Lewis lung carcinoma, and Ehrlich carcinoma.2 Previously, we have revealed structure–activity relationships,3 target proteins,4 and mechanisms of action of aplyronine A (1)5 based on organic synthesis. Aplyronine A (1) possesses unique mechanisms of action. Thus, aplyronine A (1) forms a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 heterotrimeric complex with actin and tubulin, and inhibits tubulin polymerization. Like aplyronine A (1), FK-506 and rapamycin are known to induce protein–protein interactions (PPIs) in natural products.6 In addition, inhibitors of tubulin polymerization, such as vinblastine, have been clinically important drugs, especially for breast cancer.7 Therefore, aplyronine A (1) can be expected to become a lead compound in novel-type anticancer drugs.
:1:1 heterotrimeric complex with actin and tubulin, and inhibits tubulin polymerization. Like aplyronine A (1), FK-506 and rapamycin are known to induce protein–protein interactions (PPIs) in natural products.6 In addition, inhibitors of tubulin polymerization, such as vinblastine, have been clinically important drugs, especially for breast cancer.7 Therefore, aplyronine A (1) can be expected to become a lead compound in novel-type anticancer drugs.
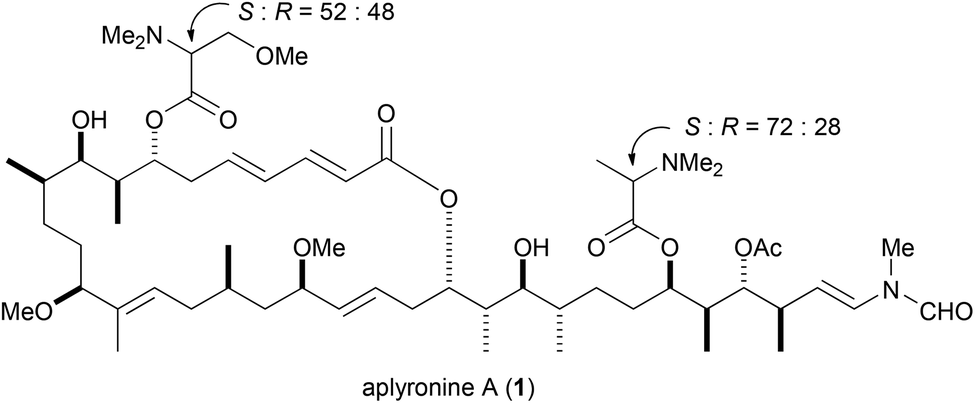 | ||
| Fig. 1 Structure of aplyronine A (1). | ||
The potent and unique biological activities of aplyronine A (1) have made it an attractive synthetic target.8 Several groups have reported approaches for synthesizing aplyronine A (1) and its related derivatives. In 2013, Paterson and co-workers achieved total synthesis of aplyronine C,9 which is a natural analogue of aplyronine A (1). Prior to that report, we described the first total synthesis of aplyronine A (1).10 This synthesis made it possible to evaluate in vivo antitumor activities, and to prepare chemical probes to elucidate the mechanisms of action of aplyronine A (1). However, our previous synthetic route included a few steps with low yield and poor stereoselectivity. We therefore sought an efficient second-generation route to aplyronine A (1) and its derivatives that could provide a practical supply for further biological studies. Here, we describe the efficient second-generation total synthesis of aplyronine A (1), featuring Ni/Cr-mediated coupling reactions as key steps.
In our first-generation total synthesis of aplyronine A (1),10 we constructed the C14–C15 (E)-trisubstituted double bond using Julia coupling because of the advantage that recovering the unreacted sulfone segment was much easier than using Wittig phosphonium salts.11 However, Julia coupling between ketone 2 and sulfone 3, and the subsequent reduction of the hydroxy sulfone, produced the undesired (Z)-olefin 5 (19%) and C14 tertiary alcohol 6 (25%), along with the desired (E)-olefin 4 (44%) (Scheme 1A). In addition, when macrolactonization of seco-acid 7, which has two hydroxy groups at C23 and C25 as possible reaction sites, was conducted under the modified Yamaguchi conditions,12 the desired 24-membered lactone 8 and the undesired 26-membered lactone 9 were produced in a ratio of ca. 4:3. Therefore, the undesired 26-membered lactone 9 had to be isomerized to 24-membered lactone 8via an ester exchange reaction with Ti(OiPr)4 (Scheme 1B). We consider that these problems of poor stereoselectivity and/or regioselectivity could be improved by using the Ni/Cr-mediated coupling reaction.13 In 2012, we reported the synthesis of the aplyronine A–mycalolide B hybrid molecule consisting of the macrolactone portion in aplyronine A (1) and the side chain portion in mycalolide B, featuring Ni/Cr-mediated coupling reactions as key steps.14 This synthesis improved the problems outlined above. Thus, we planned the second-generation total synthesis of aplyronine A (1) based on the synthetic strategy of the aplyronine A–mycalolide B hybrid molecule.
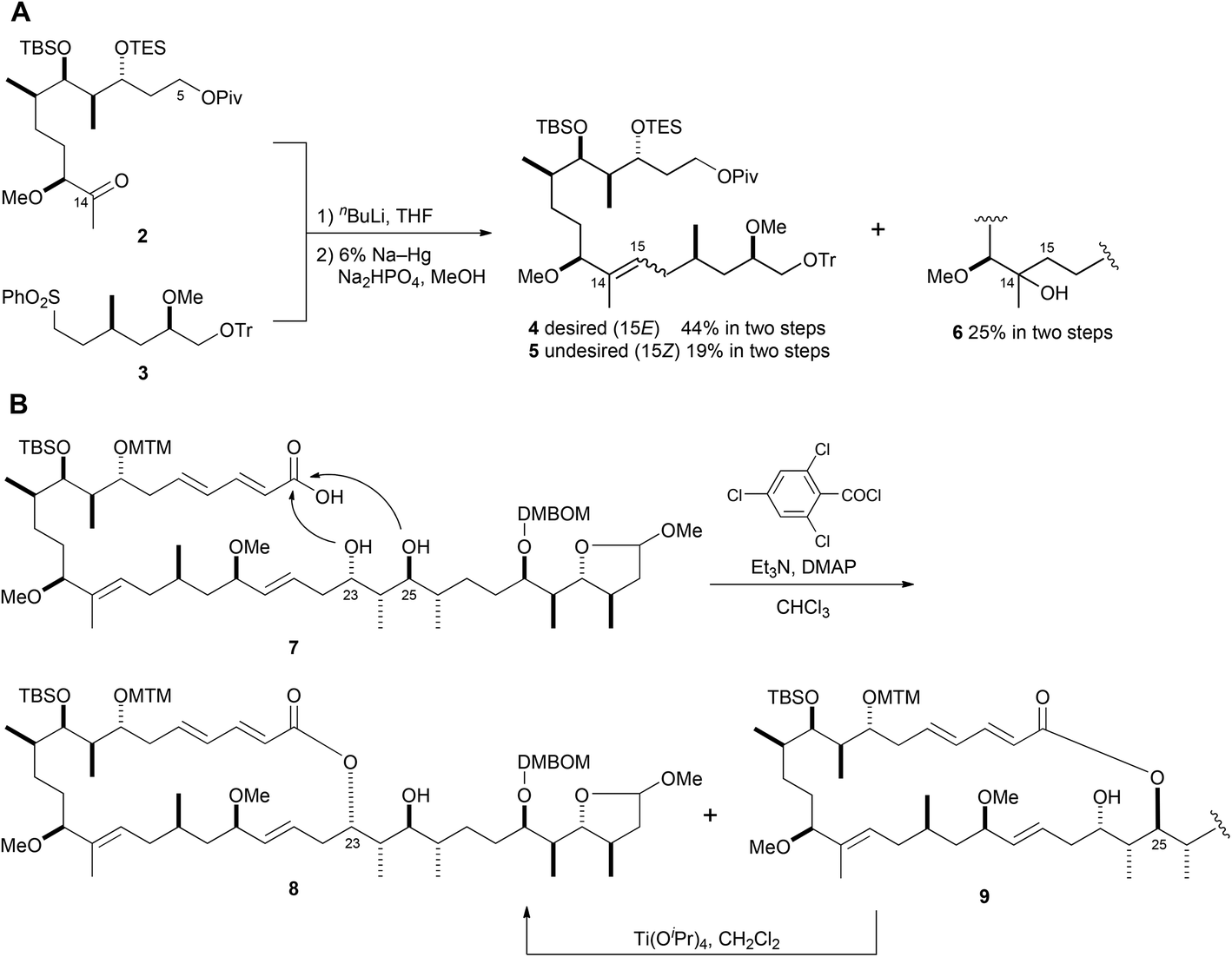 | ||
| Scheme 1 First-generation synthetic route; A: Julia coupling; B: modified Yamaguchi lactonization and isomerization. | ||
Results and discussion
The second-generation retrosynthetic pathway of aplyronine A (1) is shown in Scheme 2. Aplyronine A (1) would be obtained from 10, the same intermediate as in our first-generation synthesis.10 The macrolactone portion in 10 might be assembled from the cyclization precursor 11 using the intramolecular Ni/Cr-mediated coupling (Nozaki–Hiyama–Takai–Kishi coupling) reaction.13 The cyclization precursor 11 can be constructed by intermolecular esterification between carboxylic acid 12 and alcohol 13. In our previous work,15 carboxylic acid 12, which is the C1–C19 segment, was prepared from aldehyde 14 and iodoolefin 15 by an asymmetric Ni/Cr-mediated coupling reaction.16 This strategy has the benefit of forming the C14–C15 (E)-trisubstituted double bond and the C13 stereogenic center simultaneously. In addition, the C20–C34 segment 13 might also be obtained from iodoolefin 16 and aldehyde 1714 by an asymmetric Ni/Cr-mediated coupling reaction followed by hydrogenation.
 | ||
| Scheme 2 Retrosynthetic pathway of aplyronine A (1). | ||
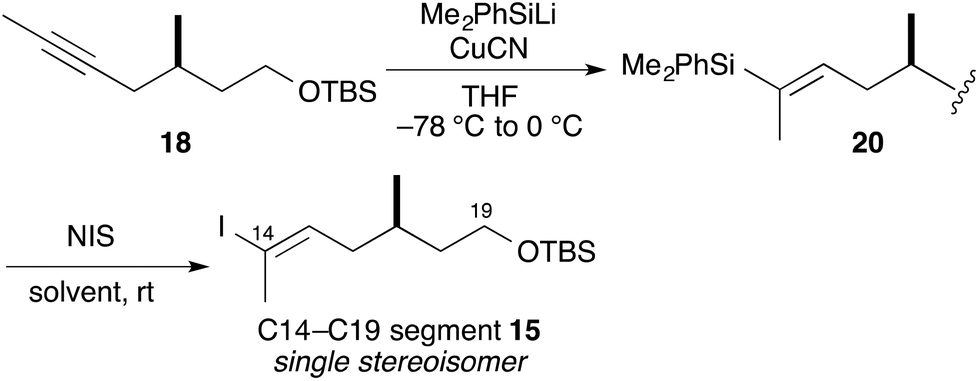
In our preliminary report,15 iodoolefin 15 was stereoselectively constructed from compound 18 by regioselective hydrozirconation with Schwartz's reagent (Scheme 3).17 However, this reaction was not reproducible (0–70%) and afforded reduced compounds, such as 19, as by-products in some cases. Thus, preparation of iodoolefin 15 required optimization.
 | ||
| Scheme 3 Previous synthetic route: hydrozirconation of 18. | ||
We attempted silylcupration of the internal alkyne 18, with subsequent iododesilylation (Table 1).18 Thus, treatment of 18 with CuCN and PhMe2SiLi19 gave vinylsilane 20 as a single isomer with quantitative yield. We next tried iododesilylation of 20 with NIS. The Holton and Zakarian groups reported stereoselective iododesilylation with NIS in hexafluoroisopropanol.20 We attempted to reproduce the Zakarian-reported conditions20b to obtain the desired iodoolefin 15 in moderate yields (entries 1 and 2). Treatment of vinylsilane 20 with NIS in MeCN–THF (3:1) afforded the desired iodoolefin 15 as a single isomer (entry 3).21 This modification made the reaction reproducible and increased the yield of the desired iodoolefin 15 to 96% in two steps.
|
|
||
|---|---|---|
| Entry | Solvent | 15 in two steps (from 18) |
| 1 | (CF3)2CHOH | 39% |
| 2 | (CF3)2CHOH–CH2Cl2 (4:1) |
58% |
| 3 | MeCN–THF (3:1) |
96% |
We next investigated the Ni/Cr-mediated coupling reaction between the C5–C13 segment 1415 and C14–C19 segment 15 (Table 2). The constructions of trisubstituted olefins via the asymmetric Ni/Cr-mediated coupling reaction have been few and far between, and investigation of this reaction was a synthetic challenge.22 We first tried the Ni/Cr-mediated coupling reaction under standard conditions (0.1 w/w% NiCl2/CrCl2, DMSO)13 without a chiral ligand. We found that the reaction was completed to give the desired coupling compound 22 in 96% yield as a 1:1 diastereomixture at C13. This result suggested that the stereochemistry of C13 could be controlled by the Ni/Cr-mediated coupling reaction with a chiral ligand. Thus, we screened several known chiral sulfonamide ligands for asymmetric Ni/Cr-mediated coupling reactions,15 but known sulfonamide ligands with high yield and stereoselectivity were not found in this screening.15b Results of the screening, however, led to the following conclusions: (1) a large substituent on the sulfonamide group prevented the transmetalation of the vinyl-Ni(II) species to Cr(III) ones and thereby decreased the yield of the coupling product; (2) a large substituent on the oxazoline ring induced high stereoselectivity because of its steric hindrance; and (3) electron-donating substituents on the benzene ring improved the stereoselectivity due to enhanced affinity to Cr, which reduced the ligand-free Ni/Cr-mediated coupling reaction.15 Therefore, in consideration of all the above, we designed and synthesized the chiral ligand 21, which has a large tBu group on the oxazoline ring, a small methyl group on the sulfonamide group, and three methoxy groups on the benzene ring, based on the chiral sulfonamide ligands developed by Kishi (Fig. 2).15
 | ||
| Fig. 2 Modified chiral sulfonamide ligand 21. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | NiCl2 (dppp) (equiv.) | CrCl2 (equiv.) | Ligand 21 (equiv.) | Proton-sponge® (equiv.) | Yield (%) 22a+22b | d.r. |
| a Calculated by 1H NMR. | ||||||
| 1 | 0.60 | 8.0 | 8.0 | 8.0 | Quant | 3.7:1 |
| 2 | 0.15 | 3.1 | 3.5 | 3.0 | 88 | 7.3:1 |

We used our modified ligand 21 to carry out an asymmetric Ni/Cr-mediated coupling reaction between the C5–C13 segment 14 and C14–C19 segment 15 (Table 2). In entry 1, the coupling reaction with ligand 21 (8.0 equiv.) and CrCl2 (8.0 equiv.) yielded the coupling compound 22 as a diastereomeric mixture (22a/22b = 3.7:1, quantitative yield).15,23 We speculated that one reason for the low diastereoselectivity was that there was insufficient formation of the Cr–ligand complex. Thus, we next tried a coupling reaction with CrCl2 (3.1 equiv.) and a small excess (against CrCl2) of ligand 21 (3.5 equiv.), and found that the diastereoselectivity was greatly improved (22a/22b = 7.3:1, 88% yield) (entry 2). The C13 isomers 22a and 22b were separated by silica gel column chromatography, and the desired isomer 22a was converted into the C1–C19 segment 12 by our previously established synthetic route (63% in eight steps).15 The undesired isomer 22b could be inverted into the former by the Mitsunobu reaction24 or the oxidation–asymmetric reduction sequence.
We next examined the synthesis of the C20–C34 segment 13. The C21–C28 segment 16 and C29–C34 segment 17 were synthesized using the Mukaiyama Sn(II)-promoted aldol reaction,25 Takai olefination,26 and Roush crotylboration,27 respectively, as key steps (see the ESI†). Thus, we attempted the Ni/Cr-mediated coupling reaction between the C21–C28 segment 16 and C29–C34 segment 17 (Table 3). We first checked the substrate controllability of the chiral methyl group at the α-position of the aldehyde group in 17. The ligand-free Ni/Cr-mediated coupling reaction (1 w/w% NiCl2/CrCl2, DMSO)13 proceeded through the Felkin–Anh model to give priority to the desired diastereomer 23. However, the yield and diastereoselectivity were moderate (50% yield, 4.6:1). Thus, we next examined the Ni/Cr-mediated coupling reaction with our modified chiral sulfonamide ligand 21.15 In entry 1, the asymmetric Ni/Cr-mediated coupling reaction proceeded smoothly to afford the coupling compound 23 in 80% yield as a single diastereomer.23 In this case, the coupling reaction was performed with 2.0 equiv. of vinyl iodide 16. Generally, ca. 1.5–2.0 equiv. of a vinyl iodide against an aldehyde is used for the Ni/Cr-mediated coupling reaction. However, because the preparation of vinyl iodide 16 required more steps than that of aldehyde 17 in this case, we investigated the molar ratio of vinyl iodide 16 and aldehyde 17 in this asymmetric Ni/Cr-mediated coupling reaction.28 In entry 2, the asymmetric Ni/Cr-mediated coupling reaction with an equimolar mixture of vinyl iodide 16 and aldehyde 17 gave the desired allylic alcohol 23 in 56% yield. Also, this coupling reaction proceeded slowly, and led to the decomposition of aldehyde 17. In this result, the coupling yield was decreased from entry 1. However, the yield in entry 2 was acceptable when the consumption of vinyl iodide 16 was considered. Thus, we next optimized the coupling conditions using an equimolar mixture of vinyl iodide 16 and aldehyde 17. For the enhanced reaction rate, we attempted this coupling reaction at a higher concentration, but the yield was slightly decreased (entry 3). We assumed that the cause of this decreased yield was that the reaction system was rendered acidic by the sulfonamide proton in ligand 21. In entry 4, the reduction of each reagent to half the original amount was found to give the best yield (64%). Therefore, we improved the condition of the asymmetric Ni/Cr-mediated coupling reaction with an equimolar mixture of vinyl iodide 16 and aldehyde 17.

Next, we attempted to synthesize the C20–C34 segment 13 from the desired coupling compound 23 (Scheme 4). Reduction of the olefin in 23 and subsequent protection of the secondary hydroxy group gave DMBOM [{(3,4-dimethoxybenzyl)oxy}methyl] ether,10 which was converted into diol 24 by selective desilylation of the silylene acetal group. Silylation of the diol group in 24 with TES groups gave di-TES ether, which was transformed into aldehyde 25 by selective removal of the primary TES group and oxidation of the resultant primary hydroxy group. Aldehyde 25 was converted into the C20–C34 segment 13 by Takai olefination26 and selective removal of the TES group. Although the C20–C34 segment 13 was obtained as an inseparable mixture of olefin stereoisomers (E/Z = 4.5:1), the stereoisomers could be chromatographically separated after several steps.
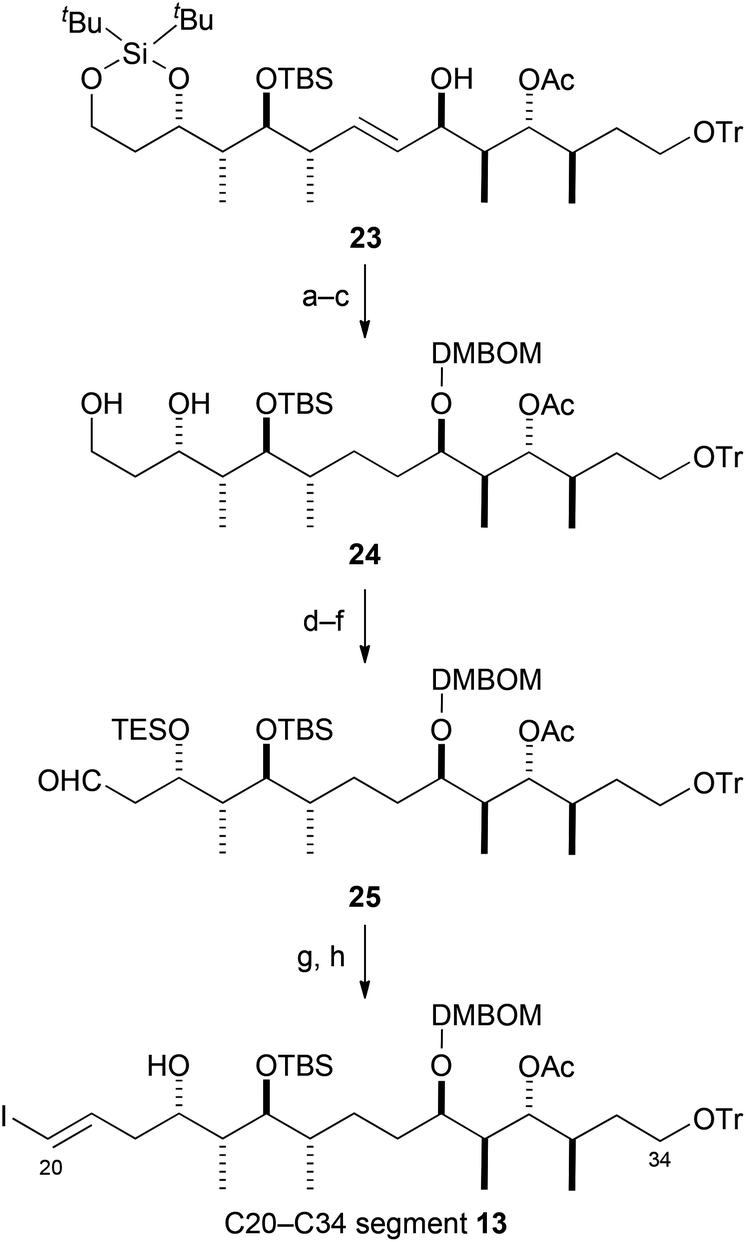 | ||
| Scheme 4 Synthesis of C20–C34 segment 13. Reagents and conditions: (a) H2, Pd(OH)2/C, NaHCO3, EtOH, rt, 92%; (b) DMBOMCl, iPr2NEt, CH2Cl2, 30 °C, 94%; (c) nBu4NF, AcOH, THF, −5 °C, 91%; (d) TESCl, imidazole, DMF, rt, quant; (e) NH4F, MeOH, rt, quant; (f) Dess–Martin periodinane, py, CH2Cl2, rt, 93%; (g) CrCl2, CHI3, THF, rt, 91%, E/Z = 4.5:1; (h) AcOH, H2O, THF, rt, 86%. | ||
With both fragments C1–C19 segment 12 and C20–C34 segment 13 in hand, synthesis of a precursor for the intramolecular Ni/Cr-mediated coupling reaction was examined next. In preparation for this, we followed our synthetic route for the aplyronine A–mycalolide B hybrid molecule (Scheme 5).14 Thus, the C1–C19 segment 12 and C20–C34 segment 13 were esterified under Yamaguchi conditions29 to give ester 26, the O19 TBS group of which was selectively removed to provide alcohol 27. The resultant primary hydroxy group of 27 was oxidized to afford aldehyde 11 as a cyclization precursor.
 | ||
| Scheme 5 Synthesis of a precursor for the intramolecular Ni/Cr-mediated coupling reaction. Reagents and conditions: (a) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, toluene, rt, 96%; (b) AcOH, H2O, THF, rt, 93%; (c) Dess–Martin periodinane, CH2Cl2, rt, 90%. | ||
The intramolecular Ni/Cr-mediated coupling reaction was examined next (Table 4). Treatment of aldehyde 11 with 2.0 w/w% NiCl2/CrCl2 in DMSO (c = 10 mM)13 gave the desired cyclization compound 28 (54%) and its diastereomer 29 (35%) (entry 1).23 This intramolecular Ni/Cr-mediated coupling reaction proceeded smoothly at a 26-times higher concentration than that of the modified Yamaguchi macrolactonization12 in our first-generation total synthesis of aplyronine A (1) (c = 0.39 mM).30 The stereoisomer at the C20–C21 double bond could be separated after the intramolecular Ni/Cr-mediated coupling reaction of 11. Then, we examined the asymmetric intramolecular Ni/Cr-mediated coupling reaction of 11 using our modified chiral ligand 30 (ent-21) (entry 2). Unexpectedly, the yield and diastereoselectivity of this cyclization were not improved (28: 41%, 29: 18%). We thought that steric hindrance of the vinyl–Cr(III)/ligand complex of 11 interfered with the intramolecular addition to the aldehyde group. The undesired diastereomer 29 could be converted into the desired allylic alcohol 28 by using a sequence of Dess–Martin oxidation and CBS reduction.31 To convert the undesired diastereomer 29 into the desired allylic alcohol 28, we followed the procedure reported by Paterson.9b
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Solvent | Yield (%) | |
| 28 | 29 | |||
| 1 | 2.0 w/w% NiCl2/CrCl2 (NiCl2: 0.1 equiv.; CrCl2: 5.0 equiv.) | DMSO (10 mM) | 54 | 35 |
| 2 | NiCl2 (dppp) (0.05 equiv.) | MeCN (2 mM) | 41 | 18 |
| CrCl2 (12 equiv.) | ||||
| Ligand 30 (12 equiv.) | ||||
| Proton-sponge® (11 equiv.) | ||||

|
||||

Methylation of the hydroxy group at C19 in allylic alcohol 28 afforded methyl ether 10, which was our intermediate in the first-generation total synthesis of aplyronine A (1) (Scheme 6).10 The methyl ether 10 gave spectral data (1H NMR and 13C NMR spectroscopy, HRMS, and optical rotation) that were in full agreement with those of our authentic intermediate.3a To convert methyl ether 10 into aplyronine A (1), we followed our first-generation total synthesis (see the ESI†). The 1H NMR data of synthetically obtained aplyronine A (1) were in good agreement with those of natural aplyronine A (1) (see the ESI†).
 | ||
| Scheme 6 Second-generation total synthesis of aplyronine A (1). Reagents and conditions: (a) MeI, NaH, THF, 35 °C, 90%. | ||
Conclusions
In conclusion, we achieved second-generation total synthesis of aplyronine A (1). The overall yield of the second-generation synthetic pathway of aplyronine A (1), based on the longest linear sequence, was 1.4% in 38 steps (the total number of steps was 80 from the commercial materials), a considerable improvement over our first-generation synthetic pathway of aplyronine A (1), which gave 0.39% overall yield in 47 steps in the longest linear sequence. We especially improved the stereoselectivity of the construction of the C13 stereogenic center and the C14–C15 (E)-trisubstituted double bond using the asymmetric Ni/Cr-mediated coupling reaction. Moreover, we established efficient reaction conditions for the asymmetric Ni/Cr-mediated coupling reaction between the C21–C28 segment 16 and the C29–C34 segment 17. This coupling reaction proceeded well with an equimolar ratio of each segment. We consider that this synthetic strategy, which uses Ni/Cr-mediated coupling reactions as key steps, could be useful for the preparation of structurally diverse derivatives of aplyronine A (1). In fact, we have prepared aplyronine A (1) and an aplyronine A–mycalolide B hybrid molecule using this strategy. The strategy is now being applied to the synthesis of other derivatives based on aplyronine A (1) for the development of lead compounds of novel-type anticancer agents.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas ‘Chemical Biology of Natural Products’ (Grant Number JP23102014) and for Scientific Research (Grant Number JP26242073) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)/Japanese Society for the Promotion of Science (JSPS). I. H. thanks the Okayama Foundation for Science and Technology, the Naito Foundation, the Kurata Grant awarded by the Kurata Memorial Hitachi Science and Technology Foundation, the NOVARTIS Foundation (Japan) for the Promotion of Science, and the Suzuken Memorial Foundation for their financial support.Notes and references
- (a) K. Yamada, M. Ojika, T. Ishigaki, Y. Yoshida, H. Ekimoto and M. Arakawa, J. Am. Chem. Soc., 1993, 115, 11020 CrossRef CAS; (b) M. Ojika, H. Kigoshi, T. Ishigaki and K. Yamada, Tetrahedron Lett., 1993, 34, 8501 CrossRef CAS; (c) M. Ojika, H. Kigoshi, T. Ishigaki, M. Nisiwaki, I. Tsukada, K. Mizuta and K. Yamada, Tetrahedron Lett., 1993, 34, 8505 CrossRef CAS; (d) M. Ojika, H. Kigoshi, T. Ishigaki, I. Tsukada, T. Tsuboi, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7441 CrossRef CAS.
- M. Ojika, H. Kigoshi, Y. Yoshida, T. Ishigaki, M. Nisiwaki, I. Tsukada, M. Arakawa, H. Ekimoto and K. Yamada, Tetrahedron, 2007, 63, 3138 CrossRef CAS.
- (a) H. Kigoshi, K. Suenaga, T. Mutou, T. Ishigaki, T. Atsumi, H. Ishikawa, A. Sakakura, T. Ogawa, M. Ojika and K. Yamada, J. Org. Chem., 1996, 61, 5326 CrossRef CAS; (b) K. Suenaga, N. Kamei, Y. Okugawa, M. Takagi, A. Akao, H. Kigoshi and K. Yamada, Bioorg. Med. Chem. Lett., 1997, 7, 269 CrossRef CAS; (c) H. Kigoshi, K. Suenaga, M. Takagi, A. Akao, K. Kanematsu, N. Kamei, Y. Okugawa and K. Yamada, Tetrahedron, 2002, 58, 1075 CrossRef CAS; (d) O. Ohno, M. Morita, T. Kitamura, T. Teruya, K. Yoneda, M. Kita, H. Kigoshi and K. Suenaga, Bioorg. Med. Chem. Lett., 2013, 23, 1467 CrossRef CAS PubMed.
- (a) S. Saito, S. Watabe, H. Ozaki, H. Kigoshi, K. Yamada, N. Fusetani and H. Karaki, J. Biochem., 1996, 120, 552 CrossRef CAS PubMed; (b) T. Kuroda, K. Suenaga, A. Sakakura, T. Handa, K. Okamoto and H. Kigoshi, Bioconjugate Chem., 2006, 17, 524 CrossRef CAS PubMed; (c) K. Hirata, S. Muraoka, K. Suenaga, T. Kuroda, K. Kato, H. Tanaka, M. Yamamoto, M. Takata, K. Yamada and H. Kigoshi, J. Mol. Biol., 2006, 356, 945 CrossRef CAS PubMed; (d) M. Kita, Y. Hirayama, M. Sugiyama and H. Kigoshi, Angew. Chem., Int. Ed., 2011, 50, 9871 CrossRef CAS PubMed; (e) M. Kita, Y. Hirayama, K. Yamagishi, K. Yoneda, R. Fujisawa and H. Kigoshi, J. Am. Chem. Soc., 2012, 134, 20314 CrossRef CAS PubMed; (f) M. Kita, K. Yoneda, Y. Hirayama, K. Yamagishi, Y. Saito, Y. Sugiyama, Y. Miwa, O. Ohno, M. Morita, K. Suenaga and H. Kigoshi, ChemBioChem, 2012, 13, 1754 CrossRef CAS PubMed.
- M. Kita, Y. Hirayama, K. Yoneda, K. Yamagishi, T. Chinen, T. Usui, E. Sumiya, M. Uesugi and H. Kigoshi, J. Am. Chem. Soc., 2013, 135, 18089 CrossRef CAS PubMed.
- FK506: J. Liu, J. D. Farmer Jr., W. S. Lane, J. Friedman, I. Weissman and S. L. Schreiber, Cell, 1991, 66, 807 CrossRef CAS PubMed ; rapamycin: (a) C. J. Kuo, J. Chung, D. F. Fiorentino, W. M. Flanagan, J. Blenis and G. R. Crabtree, Nature, 1992, 358, 70 CrossRef CAS PubMed; (b) E. J. Brown, M. W. Albers, T. B. Shin, K. Ichikawa, C. T. Keith, W. S. Lane and S. L. Schreiber, Nature, 1994, 369, 756 CrossRef CAS PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790 CrossRef CAS PubMed.
- Review: (a) K. Yamada, M. Ojika, H. Kigoshi and K. Suenaga, Nat. Prod. Rep., 2009, 26, 27 RSC; (b) I. Hayakawa and H. Kigoshi, Heterocycles, 2015, 91, 1137 CrossRef CAS.
- (a) I. Paterson, S. J. Fink, L. Y. W. Lee, S. J. Atkinson and S. B. Blakey, Org. Lett., 2013, 15, 3118 CrossRef CAS PubMed; (b) I. Paterson, C. J. Cowden and M. D. Woodrow, Tetrahedron Lett., 1998, 39, 6037 CrossRef CAS; (c) I. Paterson, M. D. Woodrow and C. J. Cowden, Tetrahedron Lett., 1998, 39, 6041 CrossRef CAS; (d) I. Paterson, S. B. Blakey and C. J. Cowden, Tetrahedron Lett., 2002, 43, 6005 CrossRef CAS.
- H. Kigoshi, M. Ojika, K. Suenaga, T. Mutou, J. Hirano, A. Sakakura, T. Ogawa, M. Nisiwaki and K. Yamada, Tetrahedron Lett., 1994, 35, 1247 CrossRef CAS; H. Kigoshi, M. Ojika, T. Ishigaki, K. Suenaga, T. Mutou, A. Sakakura, T. Ogawa and K. Yamada, J. Am. Chem. Soc., 1994, 116, 7443 CrossRef.
- M. Julia and J.-M. Paris, Tetrahedron Lett., 1973, 14, 4833 CrossRef.
- M. Hikota, Y. Sakurai, K. Horita and O. Yonemitsu, Tetrahedron Lett., 1990, 31, 6367 CrossRef CAS.
- (a) H. Jin, J. Uenishi, W. J. Christ and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 5644 CrossRef CAS; (b) K. Takai, M. Tagashira, T. Kuroda, K. Oshima, K. Utimoto and H. Nozaki, J. Am. Chem. Soc., 1986, 108, 6048 CrossRef CAS PubMed.
- K. Kobayashi, Y. Fujii, Y. Hirayama, S. Kobayashi, I. Hayakawa and H. Kigoshi, Org. Lett., 2012, 14, 1290 CrossRef CAS PubMed.
- (a) K. Kobayashi, Y. Fujii, I. Hayakawa and H. Kigoshi, Org. Lett., 2011, 13, 900 CrossRef CAS PubMed; (b) We screened various sulfonamide ligands, including benzenesulfonamides and isopropyloxazolines, for this asymmetric reaction and found that ligand 21 showed the best performance. See ref. 15a.
- (a) W. Yan, Z. Li and Y. Kishi, J. Am. Chem. Soc., 2015, 137, 6219 CrossRef CAS PubMed; (b) J. Li, W. Yan and Y. Kishi, J. Am. Chem. Soc., 2015, 137, 6236 Search PubMed and references cited therein.
- J. Schwartz and J. A. Labinger, Angew. Chem., Int. Ed. Engl., 1976, 15, 333 CrossRef.
- D. P. Stamos, A. G. Taylor and Y. Kishi, Tetrahedron Lett., 1996, 37, 8647 CrossRef CAS.
- I. Fleming, T. W. Newton and F. Roessler, J. Chem. Soc., Perkin Trans. 1, 1981, 2527 RSC.
- (a) A. Zakarian, A. Batch and R. A. Holton, J. Am. Chem. Soc., 2003, 125, 7822 CrossRef CAS PubMed; (b) E. A. Ilardi, C. E. Stivala and A. Zakarian, Org. Lett., 2008, 10, 1727 CrossRef CAS PubMed.
- H. Fuwa, M. Ebine and M. Sasaki, J. Am. Chem. Soc., 2006, 128, 9648 CrossRef CAS PubMed.
- (a) Kishi reported stereoselective construction of a trisubstituted secondary allylic alcohol group by means of a catalytic Ni/Cr-mediated coupling reaction;22b; (b) X. Liu, X. Li, Y. Chen, Y. Hu and Y. Kishi, J. Am. Chem. Soc., 2012, 134, 6136 CrossRef CAS PubMed.
- (a) The stereochemistry was confirmed by modified Mosher's method (see the ESI†); ; (b) I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- O. Mitshunobu, Synthesis, 1981, 1 CrossRef.
- (a) T. Mukaiyama, R. W. Stevens and N. Iwasawa, Chem. Lett., 1982, 11, 353 CrossRef; (b) I. Paterson and R. D. Tillyer, Tetrahedron Lett., 1992, 33, 4223 Search PubMed.
- K. Takai, K. Nitta and K. Utimoto, J. Am. Chem. Soc., 1986, 108, 7408 CrossRef CAS.
- W. R. Roush, A. D. Palkowitz and K. Ando, J. Am. Chem. Soc., 1990, 112, 6348 CrossRef CAS.
- (a) Kishi et al. developed a heterobimetallic (Ni/Cr) catalyst for the asymmetric Ni/Cr-mediated coupling reaction. This heterobimetallic catalyst promoted the reaction with high yield and diastereoselectivity under conditions with a low catalytic amount (2–3 mol%) and an equimolar ratio of coupling partners;28b; (b) X. Liu, J. A. Henderson, T. Sasaki and Y. Kishi, J. Am. Chem. Soc., 2009, 131, 16678 CrossRef CAS PubMed.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CrossRef CAS.
- (a) Namba and Kishi reported a catalytic intramolecular Ni/Cr-mediated coupling reaction without the use of high-dilution techniques;30b; (b) K. Namba and Y. Kishi, J. Am. Chem. Soc., 2005, 127, 15382 CrossRef CAS PubMed.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, characterization of data and NMR spectra of all new compounds. See DOI: 10.1039/c6ob02241c |
| This journal is © The Royal Society of Chemistry 2017 |