
The 5-chlorouracil:7-deazaadenine base pair as an alternative to the dT:dA base pair†
E.
Eremeeva
a,
M.
Abramov
a,
P.
Marlière
b and
P.
Herdewijn
*ab
aKU Leuven, Rega Institute for Medical Research, Medicinal Chemistry, Minderbroedersstraat 10, BE-3000 Leuven, Belgium. E-mail: piet.herdewijn@kuleuven.be
biSSB, Genopole, CNRS, UEVE, Université Paris-Saclay, 5 rue Henri Desbruères, 91030 Evry Cedex, France
First published on 5th December 2016
Abstract
5-Chloro-2′-deoxyuridine as a possible component of a chemically modified genome has been discussed in terms of its influence on duplex stability and DNA polymerase incorporation properties. The search for its counterpart among different deoxyadenosine analogs (7-deaza-, 8-aza- and 8-aza-7-deaza-2′-deoxyadenosines) showed that the stable duplex formation as well as the synthesis of long constructs, more than 2 kb, were successful with the 5-chloro-2′-deoxyuridine and 7-deaza-2′-deoxyadenosine combination and with Taq DNA polymerase.
Introduction
The coding genome of every species on earth is composed of four bases: adenine paired with thymine and guanine paired with cytosine. Only one exception to this rule is known to date since the report by Kirnos et al.,1 where it was shown that the adenine base was completely substituted with a modified 2-aminoadenine in S-2L cyanophage DNA. It is generally accepted that the selection of the canonical four bases by nature relies on chemical contingency and their potential to sustain evolution. However, there is no reason to accept that no alternative chemistry would be able to sustain life in the same way. Investigations on the development of ‘artificial’ genomes would serve the aim of a better understanding of present-day life, and the goal to be able to substitute the ‘natural’ four bases by chemicals that are not naturally available. Implementation of this fully modified information system in vivo would provide us with organisms that, preferentially, should be used as new bio-safe tools in synthetic biology.2 Moreover, the stable synthetic sequences can be the target for the development of advanced non-immunogenic therapeutics.3–6A first criterion to select the bases for a synthetic genome is that these compounds are not naturally occurring so that these organisms cannot survive in a natural ecosystem. However, this is the necessary criterion for nucleobases as alternative genetic molecules, but it is certainly not a sufficient criterion for selection. The criteria for a chemically modified genome (xenome) are further based on (physico)chemical, biochemical, metabolic and genetic considerations. The first step toward the development of such a genome has been realized by the full substitution of the thymine base with a 5-chlorouracil base,7 as well as partial replacement of thymine (75%) with a non-canonical 5-hydroxymethyluracil base,8 in the genomic DNA of E. coli. Furthermore, in our previous work,9 we demonstrated that the bacterial machinery could successfully accept a synthetic gene with all four substituted nucleobases as an information template.
Chemically speaking, the selection of new base pairs capable of sustaining life is initially based on their chemical stability. Parameters to be considered are their pKa, which should be preferentially more than four magnitudes higher than the pKa of its purine counterpart,10 their ability to undergo stacking interactions, and hydrogen bond formation. These properties were analyzed for the 5-chlorouracil base. A chlorine atom has a similar van der Waals radius (1.80 Å) as a methyl group (2.0 Å), as occurring in thymine. In contrast, a chlorine atom has a negative inductive effect situated between the inductive effects of a fluorine and a bromine atom. A methyl group shows a positive inductive effect situated between the effects of a hydrogen atom and an ethyl group. The properties of 5-chlorouracil and thymine are therefore similar, but not identical, which is an important prerequisite to start the in vivo evolution process. 5-Chloro-2′-deoxyuridine (5-Cl-dU or T2 in Fig. 1) is a chemically stable compound.11 Also, structurally, a B-form DNA containing an A:T2 or an A:T base pair shows nearly identical geometries.12 Due to the better leaving group properties of bromine versus chlorine, 5-bromo-2′-deoxyuridine reacts with cysteine to produce 2′-deoxyuridine at pH 7.2, which is not the case with 5-Cl-dU.13 An additional advantage of 5-chlorouracil is that it can function as a substrate for transglycosylase, making the parent nucleotide biochemically available.14 5-Chlorouracil is metabolically stable; no or minimal dehalogenation may occur in vivo, as would be the case with the bromine and iodine counterparts.15,16 No organic chlorine-containing compounds are present in regular cellular biochemistry. However, one percent of the characterized natural compounds contain chlorine,17 while chloride is the most abundant halide in ocean water. Drinking water disinfected using chlorination is a source of chlorinated organic compounds in nature,18 and 5-chlorouracil and 5-chlorouridine occur in the chlorinated effluent coming from sewage treatment plants.19 Chloride can be oxidized by hydrogen peroxide (Cl2/Cl−: −1.36 eV; H2O2/H2O: −1.8 eV) using haloperoxidases, which makes Cl+ accessible for electrophilic halogenations in vivo.20 Chlorinase is an example of an enzyme that may introduce a carbon–chlorine bond. Human neutrophils may use myeloperoxidase and H2O2 to chlorinate uracil to 5-chlorouracil.21 Chlorinated biochemicals are therefore potentially metabolically accessible in different ways. The possibility that 5-chlorouracil is recognized as a nucleobase and incorporated into DNA has been demonstrated by the observation that 5-chlorouracil causes a decrease in the growth rate of E. coli due to its incorporation into bacterial DNA.22 5-Chloro-2′-deoxyuridine was therefore considered to be an ideal substitute for thymidine in an evolution process leading to the semi-synthetic genome.7 The genetic toxicity of 5-chlorouracil may have contributed to this chemical genome evolution.23
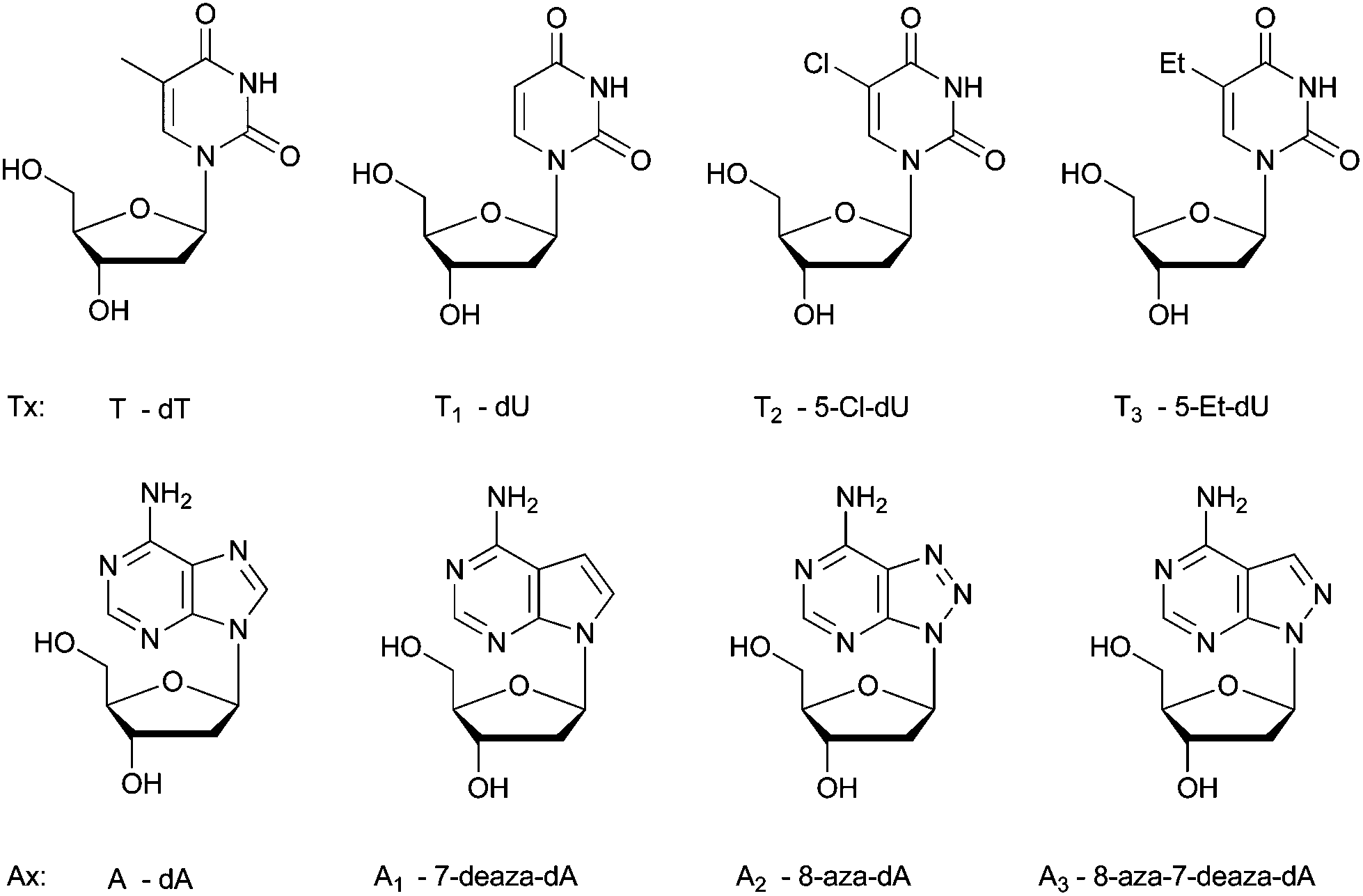 | ||
| Fig. 1 The chemical structures of the investigated nucleotides (Tx and Ax). | ||
Herein, we further evaluated the possibility of using 5-chlorouracil as a component of the synthetic genome relying on physicochemical and biochemical considerations. We also investigated the alternatives for its counterpart among adenosine analogs.
Results and discussion
Physicochemical studies
When selecting modified nucleosides as a constituent of DNA in vivo, the modified bases should give rise to a thermodynamically stable genome, which is more difficult to predict since many parameters are involved in hybridization processes in cells. Also, the modifications should not result in an extremely stable genome, and the artificial genome should be able to exert its biological functions, involving protein–nucleic acid interactions, for example, with polymerases. The modified bases should not hamper these interactions. Here, we have experimentally analyzed the potential of 5-Cl-dU (T2, pKa 7.9, Table S1 in the ESI†) to form a thermodynamically stable base pairing system with purine nucleosides, in comparison with thymidine (T, pKa 9.7) and other potential 2′-deoxyuridine analogues (Tx) that could have been used for this purpose, 2′-deoxyuridine (T1, pKa 9.3) and 5-ethyl-2′-deoxyuridine (T3, pKa 9.6).24 The structures of the investigated modified nucleosides are shown in Fig. 1 and the syntheses are described in the ESI.† The model is based on simple Tm measurements. An important motivation to perform this analysis is the search for a synthetic purine–pyrimidine base pair that may replace the canonical A:T base pair in the genome of E. coli. Therefore, the stability is evaluated using adenine (A, pKa 3.5) as a purine counterpart, and, likewise, using other purine bases that might be candidates as a substitute for adenine in a synthetic genome (Ax analogs, Fig. 1). These purine nucleosides are 7-deaza-2′-deoxyadenosine (A1, pKa 5.2),25 8-aza-2′-deoxyadenosine (A2, pKa 2.4),26 and 8-aza-7-deaza-2′-deoxyadenosine (A3, pKa 4.2, Seela F., personal communication). Based on pKa values, we would not expect that 5-Cl-dU would give rise to very stable base pairing,10 as the difference between the pKa value of 5-Cl-dU and the pH of an aqueous physiological medium is much smaller than that in the case of the other pyrimidine nucleosides (T, T1, T3). Based on the ΔpKa rule between the hydrogen bond donor and the hydrogen bond acceptor in an aqueous medium, 5-Cl-dU should form base pairs with A1 (ΔpKa = 2.7) and A3 (ΔpKa = 3.7), which are less stable than those with A (ΔpKa = 4.4). The ΔpKa value of, for example, the A3:T2 pair is similar to the ΔpKa value of the weakly pairing 2,4-diamino-5-aminopyrimidine:thymidine (ΔpKa = 3.8) system.27 However, chlorine with its larger dipole moment (MeCl 1.87 D, which is similar to water 1.85 D) could influence stacking/hydrophobic interactions considerably. Moreover, the hydrogen bond strength is also dependent on the geometry of the hydrogen bonding network between the two bases, and the ease with which solvating waters are eliminated.The Tm values (Table 1) observed for the hybridization of the different purine modified oligonucleotides, with the thymidine containing oligonucleotides, are in agreement with previous literature data. In general, 7-deaza-dA gives less stable duplexes than dA pairing with thymidine (T)28–32 or deoxyuridine (T1),33 while the introduction of 8-aza-dA or 8-aza-7deaza-dA just slightly changes the Tm of duplexes.34–36 The differences in stability of the A:T, A2:T, and A3:T base pairs are minor in the case of one Ax substitution, although when incorporating three modified bases in line, the A2 nucleosides seem to give more destabilizing duplexes (Table 1). Invariably, the ranking order (stability of duplexes using different pyrimidine nucleosides) is T2 > T > T1 > T3. When incorporating one, two or three T2 residues, the obtained duplexes (with A, A1 and A3 as a partner) are invariably more stable than those with other Tx pyrimidines. The stabilities of duplexes containing T2 and these three purine partners are very similar. The exception is duplexes containing A2:T2 pairs, which are in general somewhat less stable than the others, although this represents a base pair with the largest ΔpKa value (ΔpKa = 5.5).
| A | A1 | A2 | A3 | G | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A: DNA duplexes with one Ax/Tx modification: 5′-CCG TAxA ATG ACC-3′ and 3′-GGC ATxT TAC TGG-5′. B: DNA duplexes with two Ax/Tx modifications: 5′-CCG TAxA ATG AxCC-3′ and 3′-GGC ATxT TAC TxGG-5′. C: DNA duplexes with three Ax/Tx modifications: 5′-CCG TAxAx AxTG ACC-3′ and 3′-GGC ATxTx TxAC TGG-5′. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| A | T | 48.6 | 48.0 | 48.1 | 48.4 | 42.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1 | 48.5 | 47.9 | 48.0 | 48.2 | 41.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T2 | 49.0 | 48.9 | 47.9 | 48.9 | 41.0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T3 | 47.1 | 46.2 | 46.7 | 47.0 | 40.9 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| B | T | 48.6 | 46.5 | 48.7 | 48.5 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1 | 47.8 | 45.8 | 47.9 | 48.1 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T2 | 49.9 | 49.1 | 49.2 | 49.9 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T3 | 45.4 | 43.4 | 45.9 | 45.7 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| C | T | 48.5 | 45.4 | 47.6 | 48.2 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T1 | 46.6 | 43.5 | 45.8 | 46.3 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T2 | 49.6 | 49.4 | 47.3 | 49.3 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| T3 | 44.7 | 40.8 | 44.0 | 44.0 | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The ΔpKa values of the four different purines with the three other pyrimidines (T, T1, T3, Table S1 in the ESI†) are larger than those with T2. The hydrogen bonding in water–phosphate buffer might be stronger for these three pyrimidines than those for T2 (paired with the different purines), but the stability of the duplexes is weaker than that with 5-Cl-dU. The exception of halogen-substituted bases to the ΔpKa rule has been noted before,10 and may be attributed to the strong C–Cl dipole moment, influencing stacking/hydrophobic interactions. The Tm values of the duplexes with 5-Cl-dU as pyrimidine analogs do not decrease when one, two or three modifications are introduced in the oligonucleotides. 5-Cl-dU gives, thermodynamically, potentially the most stable genome (of these four pyrimidine nucleosides), and hydrophobic stacking interactions are more important than hydrogen bonding for explaining these differences. Previously, the lack of difference between the Tm values of A:T2 and A:T containing oligonucleotides was described.37 This stabilizing effect may have contributed to the success of the T → 5-Cl-dU genome substitution.7 Preferential pairing partners for 5-Cl-dU in a chemically modified base-pair would be 8-aza-7-deaza-dA and 7-deaza-dA, although 8-aza-dA cannot be excluded, and further selection depends largely on metabolic considerations. It should also be remembered that this study on the thermal stability of duplexes has been performed in vitro and that the thermodynamic stability of duplexes may be significantly different in an in vivo situation.
Another simple analysis that can be carried out (using Tm measurements) is mismatch discrimination. The lower the difference in stability between an A:Tx base pair and a G:Tx base pair, the more the transition mutations, A:T to G:C, that would be expected to occur during evolution. A:Tx forms a classical Watson–Crick base pair, while G:Tx forms a wobble base pair. Therefore, Tm values were also measured of oligonucleotide duplexes incorporating one G:Tx mismatch and compared with the stability of the same oligonucleotide with the canonical A:T base pair (Table 1A). From the table, it can be observed that the stability of the duplexes considerably decreases in all cases when introducing the mismatch and that the ΔTm is the highest when using chlorouracil as a pyrimidine partner (ΔTm = 8.0 between A:T and A:T2 containing duplexes). This effect may be sequence selective (which was not investigated), but it seems that also in view of the mismatch discrimination, 5-Cl-dU may be a better choice as a pyrimidine partner than uracil and 5-ethyluracil in a synthetic genome.
Primer extension studies
Further, the feasibility of using 7-deaza-dA, 8-aza-dA and 8-aza-7-deaza-dA as modified nucleosides in vitro (aptamers) and in vivo (in artificial genomes) was evaluated by incorporation studies of their corresponding triphosphates (dAxTP, Fig. 2). The model primer–template duplex had 7 overhanging T residues. We verified as catalysts thermophilic Taq and Vent exo- DNA polymerases (Taq and Vent exo-, respectively), as well as mesophilic DNA polymerases, Klenow fragment exo- and α subunit of E. coli DNA polymerase III (KF exo- and PolIIIα, respectively).38 We performed the extension reactions in the presence or absence of Mn2+ ions, which is known to improve the efficiency of incorporation of modified nucleotides, by decreasing the fidelity of polymerases.39 In all cases, P + 7 compounds (sometimes an extended non-templated product, up to P + 9) were obtained, which means that dAxTP are well accepted by a diverse group of DNA polymerases, but with different efficiencies. In general, triphosphate of A1 showed uniformly good incorporation abilities among different dAxTP (with or without Mn2+, with all the studied polymerases) with the yield of full-sized product formation compatible with standard dNTP incorporation. The extension reactions with two other dA-analogs, A2 and A3, were also successful, although they showed more DNA polymerase and Mn2+-ion dependency. In general, Vent exo- scored better than Taq polymerase and there was no significant difference between E. coli enzymes in the case of A2 and A3 (Fig. S1 and Table S2 in the ESI†). The presence of manganese was essential for the successful incorporation of A3 in reactions with Vent exo- and in reactions with A2 catalyzed by KF exo-.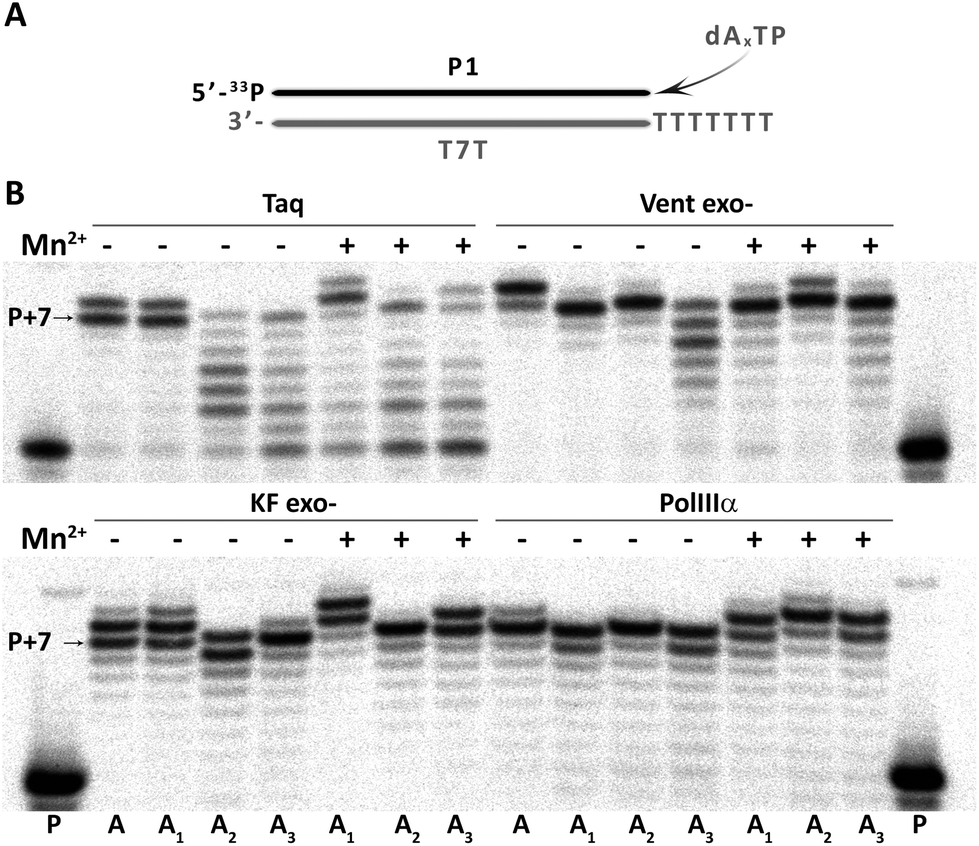 | ||
| Fig. 2 Enzymatic incorporation of dAxTP by different DNA polymerases. (A) Schematic representation of the experiment. (B) Phosphorimages of the extension reaction after 30 min of reaction in the absence or presence of 1 mM MnCl2. P – primer only. P + 7 – full length product. | ||
Encouraged by these results, we further evaluated the potential of using these modified adenosine nucleotides in combination with the T2 analog in a primer extension reaction with a longer template (with 37 diverse nucleotides to be extended, Fig. S2 in the ESI†). We evaluated various times of extension reaction catalyzed by different concentrations of the thermophilic and mesophilic DNA polymerases. All dAxTP showed good incorporation efficiency (>50% after already 3 min of reaction), although dA3TP incorporation was somewhat slower than dA1TP and dA2TP. In general, the incorporation efficiency of thermophilic polymerases decreased in the order: A1 ≥ A2 > A3, and Vent exo- catalyzed extension with modified substrates faster than the Taq polymerase. Among mesophilic DNA polymerases, KF exo- was more efficient than PolIIIα in the incorporation of dAxTP alone or together with T2 modified triphosphates (Fig. S2 in the ESI†).
PCR amplification studies
Next, we performed PCR amplification with triphosphate mixtures containing different dAxTP and dTTP or dT2TP as counterparts together with natural dCTP and dGTP. In the first PCR experiments, we used Cy3 and Cy5 fluorescently labeled primers and a natural 57mer template (Fig. 3). Cy3-containing PCR product can be formed using Cy3-reverse primer and either an initial DNA template or a newly synthesized A–T-substituted sequence, while Cy5-labeled PCR product synthesis with Cy5-forward primer can proceed only using an A–T modified sequence as the template (Fig. S3 in the ESI†). Therefore, we can compare the incorporation efficiencies of different modified triphosphates as well as the recognition of A–T substituted templates by DNA polymerases using yields of Cy3- and Cy5-containing PCR products.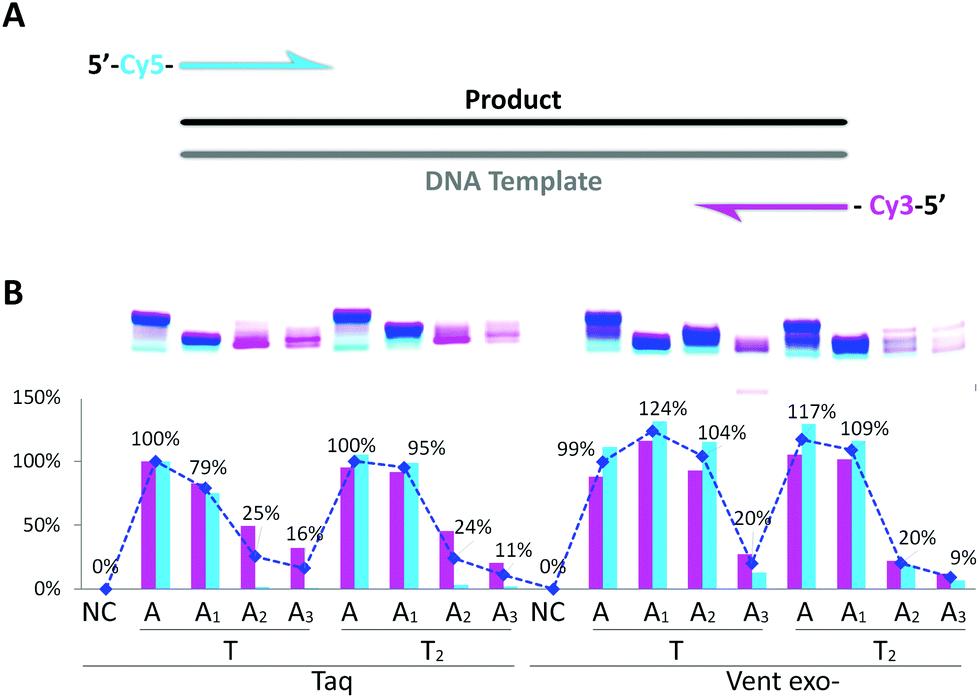 | ||
| Fig. 3 PCR amplification of 57mer DNA template in the presence of natural dNTP or dAxTP, dT2TP together with dCTP and dGTP triphosphates. (A) Schematic representation of primers and template. (B) Image of 15% denaturing PAGE with relative yields shown below. Cy3- or Cy5-labeled PCR products are shown in pink or in light blue, respectively. Total double stranded PCR products are shown as dark blue line with average yields in percentage. Yield of natural PCR product formed by Taq polymerase was taken as 100%. PCR reactions were performed with 25 U ml−1 Taq or Vent exo- DNA polymerases. NC – negative control, PCR without dNTP. Full images of PAGE can be found in Fig. S3 in the ESI.† | ||
From Fig. 3, we can see that Taq DNA polymerase is not capable of using A2- and A3-containing sequences as templates, only Cy3-labeled PCR product was forming (pink bands). In these cases, Vent exo- was more successful, although still providing the low yields with A2:T2 and A3:T2 combinations. On the other hand, both DNA polymerases were very efficient in incorporating dA1TP together with dT2TP as well as in recognition of A1 and T2 containing templates, providing yields of the PCR products as 95% or 110% compared to natural product formation for Taq or Vent exo- DNA polymerases, respectively. Moreover, in PCR reactions catalyzed by Taq DNA polymerase, accumulation of A1:T2 containing PCR product can be observed with increasing the number of cycles. This further demonstrates that this pair of analogs can be successfully used in the synthesis of double-stranded fragments with completely substituted A:T content (Fig. S4 in the ESI†). These data are consistent with our previous results9 where we demonstrated that Taq DNA polymerases could replicate in vitro the 57mer DNA template containing 7-deaza-dA and 5-Cl-dU in the presence of the corresponding triphosphates with natural dGTP and dCTP.
Further, we examined PCR amplification of A–T modified fragments with lengths of 149 bp or 360 bp. For this purpose, we performed a PCR experiment similar to Seela et al.40 We used different plasmids as templates with M13 fluorescently labeled primers and either natural dNTP or modified (dAxTP, dT2TP, dGTP and dCTP = dNxTP) sets of triphosphates (Fig. 4). The results proved again that A2 and A3 triphosphates are very poor substrates for both the tested DNA polymerases, which were not able to produce the full length PCR products in reactions containing the corresponding triphosphates together with dT2TP. In contrast, dA1TP in combination with dT2TP gave an abundant amount of the full length dsDNA products catalyzed by Taq polymerase, with the yields of 113% or 94% for 149 bp or 360 bp fragments respectively. It is interesting that Vent exo- DNA polymerase demonstrated less efficiency than Taq in PCR amplification of the longer sequences with both A and T substitutions. However, Vent exo- was still more efficient in incorporating 8-aza-dA pairing with thymidine leading to the yield of 28% with 149 bp fragment, but 5% with 360 bp. Invariably, 8-aza-7-deaza-dA was the least effective adenosine analog giving less than 2% yield with both polymerases and templates.
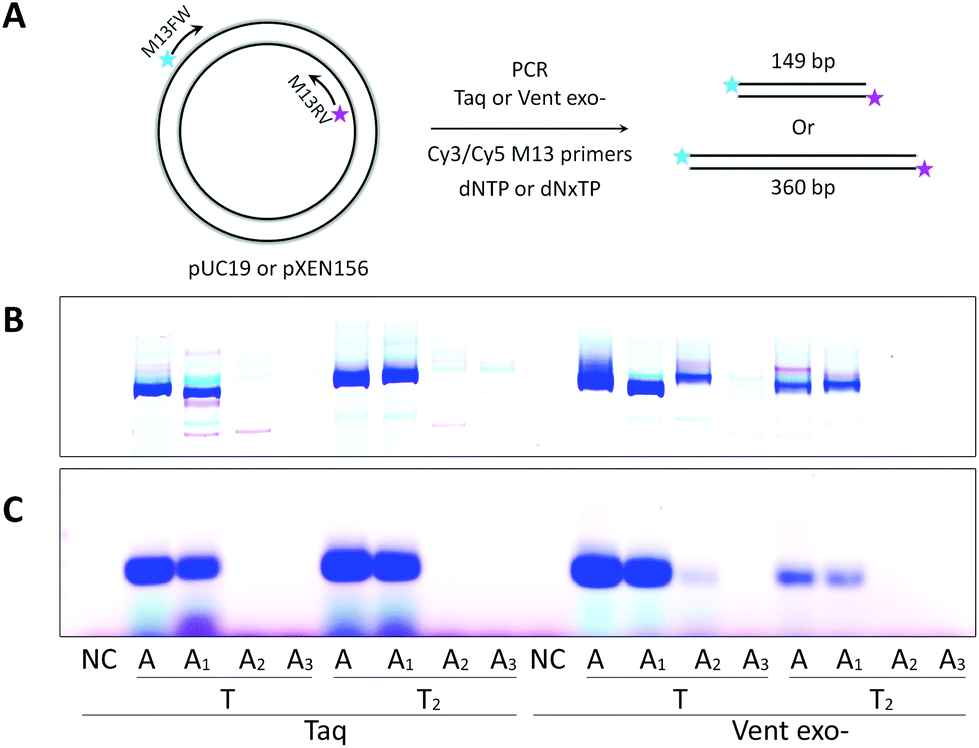 | ||
| Fig. 4 PCR amplification of the plasmids in the presence of natural (dNTP) or dAxTP, dT2TP together with dGTP and dCTP (dNxTP) triphosphates. (A) Schematic representation of natural or modified fragment synthesis with Cy3-M13RV and Cy5-M13FW primers and plasmids, pUC19 is a template for the 149 bp fragment and pXEN156 is for 360 bp fragments. 25 U ml−1 Taq or Vent exo- DNA polymerases were used. (B) Image of 15% denaturing PAGE gel with 149 bp product. (C) Image of 2% agarose gel with 360 bp product. NC – negative controls, PCR without dNTP. | ||
In order to further prove the in vitro replication of longer templates in the absence of the initial DNA template, we performed PCR amplification of plasmids as described above for 149 bp and 360 bp products with the best combination of triphosphates, dA1TP and dT2TP together with natural dCTP and dGTP, M13 primers without fluorescent dyes, and Taq DNA polymerase as a catalyst. The PCR samples were digested by Dpn I restriction enzyme and agarose gel purified to eliminate the initial DNA plasmid from the samples (Fig. 5). The resulting DNA or fully A1–T2 substituted (XNA) PCR products were used as templates for the next amplification step already with fluorescently labeled primers to visualize product formation. The results demonstrate that both, 149 and 360 bp, modified fragments with completely substituted A:T content by 7-deaza-dA and 5-Cl-dU were excellent templates for in vitro replication. Product formation was even better in PCR with the XNA template and dNxTP than in the control reaction with the natural template and dNTP (Fig. 5) for both model fragments (103% or 124% compared to the control for 149 bp and 360 bp fragments, respectively).
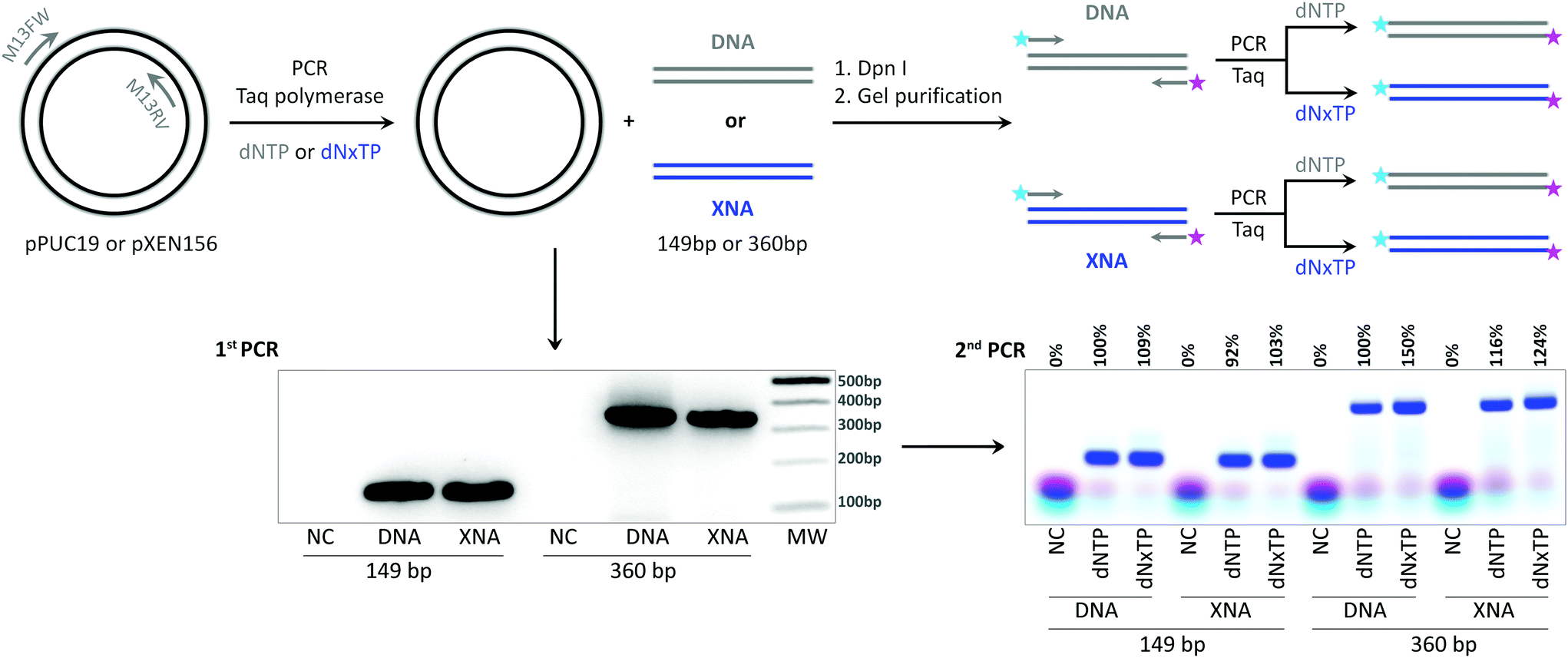 | ||
| Fig. 5 In vitro replication of A1 and T2 containing sequences by PCR with Taq DNA polymerase. Schematic representation of the synthesis of natural (DNA) and A1–T2 substituted (XNA) fragments using DNA plasmid as template and primers without labels, followed by Dpn I cleavage and agarose gel purification. The second PCR was performed with the resulting purified 149 bp or 360 bp, DNA (as positive control) or XNA fragments, and Cy3/Cy5-labeled primers. 2% agarose gel images represent the resulting PCR product after the 1st PCR and after the 2nd PCR with relative yields to the natural PCR product. NC – negative control, PCR without dNTP. dNxTP is a triphosphate set containing dA1TP, dT2TP, dGTP and dCTP. MW – molecular weight ladder. | ||
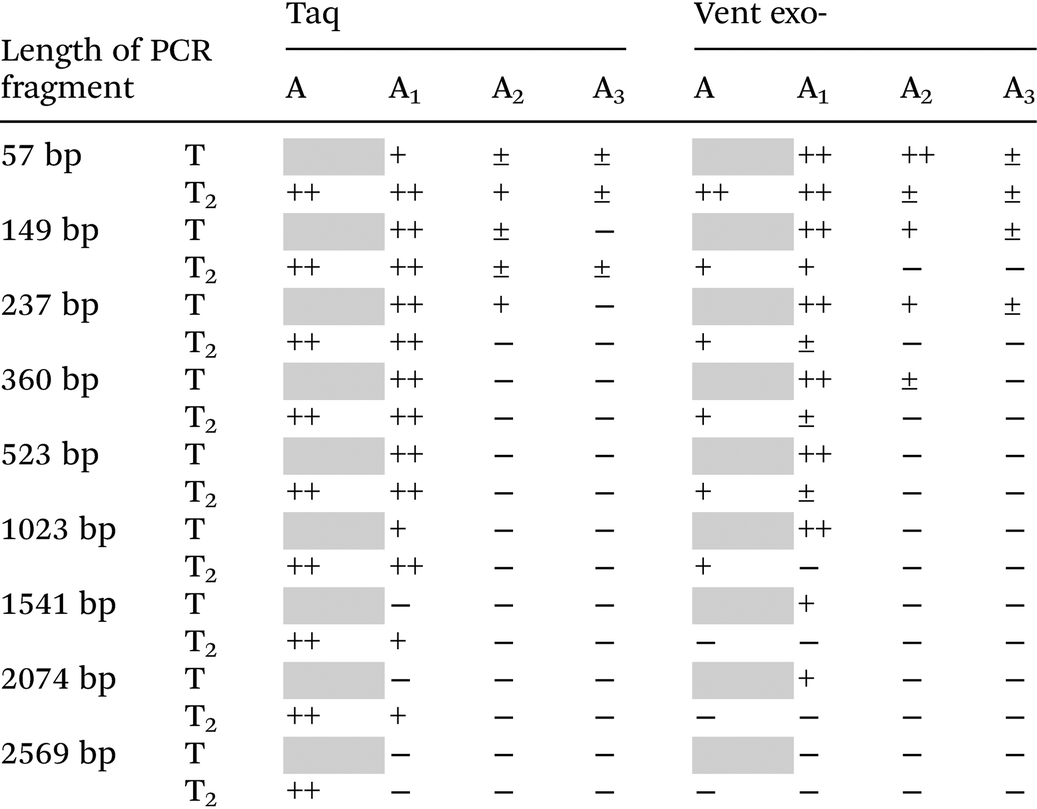
Lastly, we verify the possibility of using different dAxTP together with dT2TP in PCR amplification of various templates with different lengths, as long as 2569 bp (Table 2 and Fig. S5, ESI†). Some examples of successful PCR amplification of DNA sequences with completely substituted nucleobase content by 7-deaza-modified purines together with 5-substituted pyrimidines have been shown. In these studies, the simultaneous incorporation of two,41–43 three44,45 or all four46–48 different base-modified triphosphates proceeded during PCR with the lengths of DNA template of 62–300 bp. In our previous work, we have shown that the fully substituted sequence as long as 525 bp can be replicated by Taq DNA polymerase with all four base modifications (denoted “DZA”: A1:T2 together with 7-deaza-dG:5-fluoro-dC).9
| a The average data from several independent experiments. 30 cycles of PCR were performed with natural dCTP and dGTP and 25 U ml−1 Taq or Vent exo- DNA polymerases. The relative yield of PCR reactions is represented as follows: ‘−’, no PCR product formation (<5%); ‘±’, low yield of PCR product formation (5–25%); ‘+’, moderate yield of PCR product (25–65%); ‘++’, high yield of PCR product (>65%); grey squares are 100% natural PCR product. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Here we further examined the limits of synthesis of functional sequences with altered A:T content. This can be interesting for the simple and efficient production of long chemically redesigned DNA constructs, for example, partial or entire artificial genes, or even complex plasmids, which is not possible to synthesize by traditional chemical synthesis. These modified DNA structures can possess increased thermal and endonuclease stability.9
As shown in Table 2 only reactions with A1 triphosphate pairing with T2 are able to produce long PCR products, as long as 2074 bp (±46%) catalyzed by Taq DNA polymerase. In contrast to incorporation studies, Vent exo- demonstrated limited abilities in PCR amplification compared to Taq DNA polymerase (the maximum length of A1–T2 substituted product is 523 bp with a yield of ±13%). In PCR amplification reactions catalyzed by Taq DNA polymerase, it seems that the T2 base stabilizes the A1, since the yield of the A1–T substituted product was always lower than A1:T2, and the full length PCR product formation with A1:T stopped after 1541 bp (<5% with A1:T compared to ±33% of A1:T2 containing product, Fig. S5, ESI†). Therefore, in long amplicon synthesis, we can observe a preference for base pair formation between 7-deaza-dA and 5-Cl-dU to 7-deaza-dA with natural dT (the mechanism has not yet been studied).
Conclusions
The present study provides the (bio)chemical motivation to select 5-Cl-dU as a pyrimidine partner in a synthetic genome, together with an analysis of its potential modified purine counterpart. The preferable combination is 5-Cl-dU:7-deaza-dA, which showed reliable yield in replication in vitro of the long templates (up to 2074 bp). This is the first example of the synthesis of an A–T-substituted, very long DNA construct, which can be utilized for the straightforward production of artificial genetic templates, >2 kb, bearing the desirable functions into a cell.3,9Experimental
Chemical synthesis of modified oligonucleotides and deoxyribonucleoside 5′-triphosphates
The phosphoramidites were incorporated into the DNA sequences through solid-phase DNA synthesis49 on an automated RNA synthesizer. The A1, A3, and T1 containing modified oligonucleotides were obtained using commercially available amidites (Glen Research). The chemical synthesis of A2 modified phosphoramidites was previously described.26,36 The T2 and T3 phosphoramidites were synthesized as O5′-DMTr derivatives starting from 5-Cl-2′-deoxyuridine50 and commercial 5-ethyl-2′-deoxyuridine, respectively. A1–A3 and T2 2′-deoxyribonucleosides were converted to their 5′-triphosphates in a one-pot reaction by the Ludwig method.29,37,51,52 See the ESI† for more details.Oligodeoxyribonucleotides
All unmodified oligonucleotides (primers and templates, Table S4†) used in the incorporation and PCR studies were purchased from Integrated DNA Technologies (IDT, Leuven), then they were purified by 15 or 20% denaturing polyacrylamide gel electrophoresis (PAGE) according to a standard procedure and quantified by NanoDrop (ThermoScientific). Some primers were 5′-labeled with [γ-33P]ATP (PerkinElmer) using T4 polynucleotide kinase (New England Biolabs) according to standard procedures. Labeled oligonucleotides were further purified on Illustra MicroSpin G-25 columns (GE Healthcare).Incorporation of dAxTP into short DNA duplex with 7T-base overhang by different DNA polymerases
50 nM radioactively-labeled primer P1 was annealed to 125 nM template T7T containing 7 overhanging T residues at the 5′-end (Table S4†) and the mixture was heated at 95 °C for 5 min followed by slow cooling to 4 °C and equilibration to room temperature. The enzymes used in the study were thermostable DNA polymerases Taq and Vent exo- DNA polymerases (New England Biolabs), as well as mesophilic DNA polymerases: Klenow fragment exo- of DNA polymerase I (New England Biolabs) and the α subunit of E. coli DNA polymerase III (PolIIIα).38 Enzymatic reactions were performed in total volume 20 μl of 1× ThermoPol buffer (New England Biolabs) for reactions with thermophilic polymerases (Taq and Vent exo-), or 1× NEBuffer 2 (New England Biolabs) for KF exo- or 1× PolIII buffer (20 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 10 mM DTT, 20 mg L−1 BSA, 4% glycerol) for reactions with PolIIIα. The final reaction mixtures contained 50 nM radiolabeled primer–template duplex (P1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :T7T), 25 U ml−1 Taq, Vent exo-, KF exo- or 1200 U ml−1 of PolIIIα, 100 μM dAxTP and with or without 1 mM MnCl2. Mixtures were incubated at 72 °C (for thermostable enzymes Vent exo- and Taq) or 37 °C for KF exo- or 30 °C for PolIIIα, either for 30 or 60 min. Reactions were quenched by addition of 20 μL of 2× loading buffer (95% formamide, 0.05% bromophenol blue, and 50 mM ethylenediaminetetraacetic acid), heated at 90 °C for 5 min before analysis by 20% PAGE. Products were visualized by phosphorimaging. The amounts of radioactivity in the bands corresponding to the products of enzymatic reactions were determined with the Cyclone imaging device and the Optiquant image analysis software (PerkinElmer).
:T7T), 25 U ml−1 Taq, Vent exo-, KF exo- or 1200 U ml−1 of PolIIIα, 100 μM dAxTP and with or without 1 mM MnCl2. Mixtures were incubated at 72 °C (for thermostable enzymes Vent exo- and Taq) or 37 °C for KF exo- or 30 °C for PolIIIα, either for 30 or 60 min. Reactions were quenched by addition of 20 μL of 2× loading buffer (95% formamide, 0.05% bromophenol blue, and 50 mM ethylenediaminetetraacetic acid), heated at 90 °C for 5 min before analysis by 20% PAGE. Products were visualized by phosphorimaging. The amounts of radioactivity in the bands corresponding to the products of enzymatic reactions were determined with the Cyclone imaging device and the Optiquant image analysis software (PerkinElmer).
Incorporation of dAxTP and 5-Cl-dUTP using a 57mer DNA template and different DNA polymerases
Incorporation of dAxTP and dT2TP was performed as described before, but 50 nM radioactively-labeled primer P2 was annealed to 125 nM T37 template (Table S4†). Reactions were mixed in 50 μl solutions of 1× ThermoPol buffer (New England Biolabs) for thermostable polymerases, Taq and Vent exo-, or in 1× NEBuffer 2 (New England Biolabs) for KF exo- polymerase, or 1× PolIII buffer for PolIIIα polymerase. The final reaction mixtures included 200 μM dNTP (natural dNTP or modified dAxTP, dT2TP), 50 or 100 U ml−1 of Taq or Vent exo- polymerases, 25 or 50 U ml−1 KF exo-, or 2400 U ml−1 PolIIIα. Reactions with thermostable polymerases were incubated at 72 °C for 3–60 min, and at 37 °C for 1–120 min with mesophilic polymerases. 5 μl aliquots were quenched by adding 5 μl of 2× loading buffer and analyzed by 15% denaturing PAGE as described above.PCR amplification of 57mer template with dAxTP and 5-Cl-dUTP using thermophilic DNA polymerases
PCR amplification of 50 nM 57mer template (T57, Table S4†) with 0.5 μM primers (PrFW and PrRV or Cy5-PrFW and Cy3-PrRV) and 200 μM natural or modified dNTP was performed in 20 μl of 1× reaction buffer (ThermoPol buffer) with 25 U ml−1 Taq or Vent exo- DNA polymerases (both from New England Biolabs). The samples were initially denatured at 95 °C for 30 s, followed by 30 cycles of repetitive denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s and extension at 68 °C for 30 s, and the final extension at 68 °C for 10 min. The resulting PCR samples were either mixed with 5 μl of 6× Purple gel loading dye (New England Biolabs), loaded into 3% agarose gel with ethidium bromide, and visualized using a UV transilluminator (VWR), or 5 μl of samples were mixed with 5 μl of 2× loading buffer and analyzed by 15% PAGE. Fluorescently labeled samples were scanned using a Typhoon 9500 imaging system (GE Healthcare Life Sciences), and analyzed using ImageQuant TL v8.1 software (GE Healthcare Life Sciences).PCR amplification of 149 bp amplicons with dAxTP and 5-Cl-dUTP using thermophilic DNA polymerases
PCR amplification of 149 bp amplicons was performed using 10 ng of the pUC19 plasmid with 0.5 μM M13 primers ((Cy5-)M13FW and (Cy3-)M13RV). The reactions were mixed in 20 μl of 1× reaction buffer (ThermoPol buffer, New England Biolabs) with 200 μM natural or modified dNTP and 25 U ml−1 Taq or Vent exo- DNA polymerases. The PCR procedure included an initial denaturation at 95 °C for 30 s, followed by 30 cycles of repetitive denaturation at 95 °C for 30 s, annealing at 60 °C for 30 s and extension at 68 °C for 30 s, and the final extension at 68 °C for 10 min. The resulting 20 μl PCR samples were added to 5 μl of 6× Purple gel loading dye and loaded into 3% agarose gel (with or without ethidium bromide for Cy3/Cy5-labeled samples). Gels were visualized using a UV transilluminator (VWR) or a Typhoon 9500 imaging system (GE Healthcare Life Sciences).PCR amplification of 237 bp amplicons with dAxTP and 5-Cl-dUTP using thermophilic DNA polymerases
PCR amplification of 237 bp amplicon was performed using 10 ng of pXEN156 plasmid with 0.5 μM R67 primers (R67FW and R67RV). The reactions were mixed in 20 μl of 1× reaction buffer (ThermoPol buffer, New England Biolabs) with 200 μM natural or modified dNTP, and 25 U ml−1 Taq or Vent exo- DNA polymerases. The PCR procedure included an initial denaturation at 95 °C for 30 s, followed by 30 cycles of repetitive denaturation at 95 °C for 30 s, annealing at 54 °C for 30 s and extension at 68 °C for 30 s, and the final extension at 68 °C for 10 min. The resulting 20 μl PCR samples were added to 5 μl of 6× Purple gel loading dye and loaded into 2% agarose gel with ethidium bromide. The gel was visualized using a UV transilluminator (VWR).PCR amplification of 360 bp amplicons with dAxTP and 5-Cl-dUTP using thermophilic DNA polymerases
PCR amplification of 237 bp amplicons was performed using 10 ng of pXEN156 plasmid with 0.5 μM M13 primers ((Cy5-)M13FW and (Cy3-)M13RV). The reactions were mixed with 20 μl of 1× reaction buffer (ThermoPol buffer, New England Biolabs) with 200 μM natural or modified dNTP, and 25 U ml−1 Taq or Vent exo- DNA polymerases. The PCR procedure included an initial denaturation at 95 °C for 30 s, followed by 30 cycles of repetitive denaturation at 95 °C for 30 s, annealing at 54 °C for 30 s and extension at 68 °C for 30 s, and the final extension at 68 °C for 10 min. The resulting 20 μl PCR samples were added to 5 μl of 6× Purple gel loading dye and loaded into 2% agarose gel (with or without ethidium bromide for Cy3/Cy5-labeled samples). The gel was visualized using a UV transilluminator (VWR) or a Typhoon 9500 imaging system (GE Healthcare Life Sciences).PCR amplification of 532–2569 bp amplicons with dAxTP and 5-Cl-dUTP using thermophilic DNA polymerases
For PCR amplification of long amplicons (523, 1023, 1541, 2074 and 2569 bp), 25 ng pXEN156 vector was used as a DNA template. Reactions were performed in 20 μl of 1× ThermoPol buffer (New England Biolabs) with 0.5 μM primers (LongFW1 and LongFW2, LongRV1–LongRV4, see Table S4† for details), 25 U ml−1 Taq or Vent exo- DNA polymerases (New England Biolabs) and the 200 μM triphosphates. The samples were initially denatured at 95 °C for 30 s, followed by 30 cycles of repetitive denaturation at 95 °C for 30 s, annealing at 58 °C for 30 s (for 2569 bp at 62 °C) and extension at 68 °C for 30 s (for 2569 bp for 1 min) and the final extension at 68 °C for 10 min. The resulting 20 μl PCR samples were mixed with 5 μl of 6× Purple gel loading dye and loaded into 1% agarose gel with ethidium bromide. The gel was visualized using a UV transilluminator (VWR).In vitro replication experiment with 149 bp and 360 bp amplicons
PCR amplifications of 149 bp and 360 bp amplicons were performed by the same method as that described above, but in 50 μl of 1× ThermoPol buffer with 0.5 μM M13RV and M13FW primers without Cy3 and Cy5 fluorescent dyes, with or without dNTP (DNA – positive control or NC – negative control), or with dA1TP, dT2TP, dCTP and dGTP triphosphate (dNxTP) set at a concentration 200 μM each. 40 μl of reactions were digested by 100 U DpnI restriction enzyme (New England Biolabs) in 50 μl of 1× CutSmart buffer (New England Biolabs) for 1 hour at 37 °C, followed by enzyme heat inactivation at 80 °C for 20 min and 2% agarose gel purification with a NucleoSpin Gel and PCR Purification Clean-up kit (Macherey-Nagel) according to the manufacturer's instructions. Samples were eluted two times with 25 μl volume of the elution buffer (NE, Macherey-Nagel) and quantified using a CLARIOstar plate reader with a VIS plate (BMG Labtech). 5 nM of the resulting samples (149 bp or 360 bp, DNA and XNA) were subjected to a new PCR reaction with 0.5 μM Cy3-M13RV and Cy5-M13FW primers, with or without dNTP (DNA or NC), or XNA triphosphate sets at a concentration of 200 μM each, 25 U ml−1 Taq DNA polymerase in the final volume 20 μl of 1× ThermoPol buffer. The PCR programmes were as described above for 149 bp and 360 bp. The resulting 20 μl PCR products were loaded on 2% agarose gel (without ethidium bromide) with 5 μl of 6× loading buffer (without the bromophenol blue dye), scanned using a Typhoon 9500 imaging system (GE Healthcare Life Sciences), and analyzed using ImageQuant TL v8.1 software (GE Healthcare Life Sciences).Acknowledgements
The authors are indebted to Guy Schepers for oligonucleotide synthesis and Dr J. Rozenski for mass spectrometry analysis. This work was supported by FWO (Vlaanderen) (G.078014N) and Research Fund, KU Leuven (OT/14/128). The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013)/ERC Grant agreement no. ERC-2012-ADG_20120216/320683. The authors also thank Chantal Biernaux for editorial help.Notes and references
- M. D. Kirnos, I. Y. Khudyakov, N. I. Alexandrushkina and B. F. Vanyushin, Nature, 1977, 270, 369–370 CrossRef CAS PubMed.
- P. Herdewijn and P. Marlière, Chem. Biodiversity, 2009, 6, 791–808 CAS.
- N. Tarashima, H. Ando, T. Kojima, N. Kinjo, Y. Hashimoto, K. Furukawa, T. Ishida and N. Minakawa, Mol. Ther. Nucleic Acids, 2016, 5, e274 CrossRef CAS PubMed.
- A. I. Taylor, S. Arangundy-Franklin and P. Holliger, Curr. Opin. Chem. Biol., 2014, 22, 79–84 CrossRef CAS PubMed.
- G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef CAS PubMed.
- J. Zhou and J. Rossi, Nat. Rev. Drug Discovery, 2016 DOI:10.1038/nrd.2016.199.
- P. Marlière, J. Patrouix, V. Döring, P. Herdewijn, S. Tricot, S. Cruveiller, M. Bouzon and R. Mutzel, Angew. Chem., Int. Ed., 2011, 50, 7109–7114 CrossRef PubMed.
- A. P. Mehta, H. Li, S. A. Reed, L. Supekova, T. Javahishvili and P. G. Schultz, J. Am. Chem. Soc., 2016, 138, 7272–7275 CrossRef CAS PubMed.
- E. Eremeeva, M. Abramov, L. Margamuljana, J. Rozenski, V. Pezo, P. Marlière and P. Herdewijn, Angew. Chem., Int. Ed., 2016, 55, 7515–7519 CrossRef CAS PubMed.
- X. Zhang and R. Krishnamurthy, Angew. Chem., Int. Ed., 2009, 48, 8124–8128 CrossRef CAS PubMed.
- A. Van Schepdael, N. Ossembe, P. Herdewijn, E. Roets and J. Hoogmartens, J. Pharm. Biomed. Anal., 1993, 11, 345–351 CrossRef CAS PubMed.
- A. Patra, J. Harp, P. S. Pallan, L. Zhao, M. Abramov, P. Herdewijn and M. Egli, Nucleic Acids Res., 2013, 41, 2689–2697 CrossRef CAS PubMed.
- B. C. Pal, J. Am. Chem. Soc., 1978, 100, 5170–5174 CrossRef CAS.
- A. Munch-Petersen, Metabolism of nucleotides, nucleosides, and nucleobases in microorganisms, Academic Press, 1983 Search PubMed.
- D. W. Visser, D. M. Frisch and B. Huang, Biochem. Pharmacol., 1960, 5, 157–164 CrossRef CAS PubMed.
- C. Garrett, Y. Wataya and D. V. Santi, Biochemistry, 1979, 18, 2798–2804 CrossRef CAS PubMed.
- N. Winterton, Green Chem., 2000, 2, 173–225 RSC.
- N. R. Council, Drinking Water and Health, National Academies Press, Washington, DC, 1987, vol. 7 Search PubMed.
- R. L. Jolley, J. Water Pollut. Control Fed., 1975, 47, 601–618 CAS.
- M. Morrison and G. R. Schonbaum, Annu. Rev. Biochem., 1976, 45, 861–888 CrossRef CAS PubMed.
- J. P. Henderson, J. Byun, J. Takeshita and J. W. Heinecke, J. Biol. Chem., 2003, 278, 23522–23528 CrossRef CAS PubMed.
- D. B. Dunn and J. D. Smith, Biochem. J., 1957, 67, 494–506 CrossRef CAS PubMed.
- S. M. Morris, Mutat. Res., Genet. Toxicol., 1993, 297, 39–51 CrossRef CAS.
- E. Privat, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1996, 354, 151–156 CrossRef PubMed.
- H. Rosemeyer and F. Seela, Helv. Chim. Acta, 1988, 71, 1573–1585 CrossRef CAS.
- Z. Kazimierczuk, U. Binding and F. Seela, Helv. Chim. Acta, 1989, 72, 1527–1536 CrossRef CAS.
- R. Krishnamurthy, Acc. Chem. Res., 2012, 45, 2035–2044 CrossRef CAS PubMed.
- F. Seela and H. Thomas, Helv. Chim. Acta, 1995, 78, 94–108 CrossRef CAS.
- F. Seela, H. Driller, A. Kehne and K. Kaiser, Chem. Scr., 1986, 26, 173–178 CAS.
- L. J. Latimer and J. S. Lee, J. Biol. Chem., 1991, 266, 13849–13851 CAS.
- F. Seela and A. Kehne, Biochemistry, 1987, 26, 2232–2238 CrossRef CAS PubMed.
- F. Seela, H. Berg and H. Rosemeyer, Biochemistry, 1989, 28, 6193–6198 CrossRef CAS PubMed.
- F. Seela, J. Ott and D. Franzen, Nucleic Acids Res., 1982, 10, 1389–1397 CrossRef CAS PubMed.
- F. Seela and K. Kaiser, Helv. Chim. Acta, 1988, 71, 1813–1823 CrossRef CAS.
- F. Seela, C. Wei, G. Becher, M. Zulauf and P. Leonard, Bioorg. Med. Chem. Lett., 2000, 10, 289–292 CrossRef CAS PubMed.
- F. Seela, I. Munster, U. Luchner and H. Rosemeyer, Helv. Chim. Acta, 1998, 81, 1139–1155 CrossRef CAS.
- J. D. Hoheisel and H. Lehrach, FEBS Lett., 1990, 274, 103–106 CrossRef CAS PubMed.
- A. Giraut, R. Abu El-Asrar, P. Marlière, M. Delarue and P. Herdewijn, ChemBioChem, 2012, 13, 2439–2444 CrossRef CAS PubMed.
- S. Tabor and C. C. Richardson, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 4076–4080 CrossRef CAS.
- F. Seela, A. Röling, A. Roling, B. Chemie, F. B. Chemie, U. Osnabrock and D. Osnabrock, Nucleic Acids Res., 1992, 20, 55–61 CrossRef CAS PubMed.
- T. Gourlain, A. Sidorov, N. Mignet, S. J. Thorpe, S. E. Lee, J. a. Grasby and D. M. Williams, Nucleic Acids Res., 2001, 29, 1898–1905 CrossRef CAS PubMed.
- P. Čapek, H. Cahová, R. Pohl, M. Hocek, C. Gloeckner and A. Marx, Chem. Eur. J., 2007, 13, 6196–6203 CrossRef PubMed.
- I. V. Kutyavin, Biochemistry, 2008, 47, 13666–13673 CrossRef CAS PubMed.
- M. Kuwahara, S. Hososhima, Y. Takahata, R. Kitagata, A. Shoji, K. Hanawa, A. N. Ozaki, H. Ozaki and H. Sawai, Nucleic Acids Res. Suppl., 2003, 37–38 CrossRef CAS.
- M. Hollenstein, Org. Biomol. Chem., 2013, 11, 5162–5172 CAS.
- S. Jäger and M. Famulok, Angew. Chem., Int. Ed., 2004, 43, 3337–3340 CrossRef PubMed.
- S. Jäger, G. Rasched, H. Kornreich-Leshem, M. Engeser, O. Thum and M. Famulok, J. Am. Chem. Soc., 2005, 127, 15071–15082 CrossRef PubMed.
- T. Tasara, B. Angerer, M. Damond, H. Winter, S. Dörhöfer, U. Hübscher and M. Amacker, Nucleic Acids Res., 2003, 31, 2636–2646 CrossRef CAS PubMed.
- S. L. Beaucage and M. H. Caruthers, in Current Protocols in Nucleic Acid Chemistry, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2001, ch. 3, unit 3.3 Search PubMed.
- E. K. Ryu and J. N. Kim, Nucleosides Nucleotides, 1989, 8, 43–48 CAS.
- J. Ludwig, Acta Biochim. Biophys. Acad. Sci. Hung., 1981, 16, 131–133 CAS.
- J. D. Rose, W. B. Parker, H. Someya, S. C. Shaddix, J. A. Montgomery and J. A. Secrist, J. Med. Chem., 2002, 45, 4505–4512 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob02274j |
| This journal is © The Royal Society of Chemistry 2017 |