
β-Amyrin synthase from Euphorbia tirucalli L. functional analyses of the highly conserved aromatic residues Phe413, Tyr259 and Trp257 disclose the importance of the appropriate steric bulk, and cation–π and CH–π interactions for the efficient catalytic action of the polyolefin cyclization cascade†
Ryousuke
Ito
,
Chika
Nakada
and
Tsutomu
Hoshino
 *
*
Graduate School of Science and Technology and Department of Applied Biological Chemistry, Faculty of Agriculture, Niigata University, Ikarashi 2-8050, Nishi-ku, Niigata 950-2181, Japan. E-mail: hoshitsu@agr.niigata-u.ac.jp
First published on 2nd December 2016
Abstract
Many of the functions of the active site residues in β-amyrin synthase and its catalytic mechanism remain unclear. Herein, we examined the functions of the highly conserved Phe413, Tyr259, and Trp257 residues in the β-amyrin synthase of Euphorbia tirucalli. The site-specific mutants F416V and F416M showed nearly the same enzymatic activities as the wild type, indicating that π-electrons are not needed for the catalytic reaction. However, the F416A mutant yielded a large amount of the tetracyclic dammarane skeleton, with decreased production of β-amyrin. This indicates that the Phe416 residue is located near the D-ring formation site and works to position the oxidosqualene substrate correctly within the reaction cavity. On the other hand, the major catalysis-related function of the Tyr259 and Trp257 residues is to yield their π-electrons to the cationic intermediates. The Y259F variant showed nearly equivalent activity to that of the wild type, but aliphatic mutants such as the Ala, Val, and Leu variants showed significantly decreased the activity and yielded the tetracyclic dammarane scaffold, strongly demonstrating that the Tyr259 residue stabilizes the baccharenyl secondary cation via cation–π interaction. The aliphatic variants of Trp257 exhibited remarkably decreased enzymatic activity, and lupeol was produced in a high production ratio, indicating that Trp257 stabilizes the oleanyl cation via cation–π interaction. The aromatic Phe and Tyr mutants exhibited high activities owing to their more increased π-electron density relative to that of the aliphatic mutants, but lupeol was produced in a significantly high yield besides β-amyrin. The Trp residue is likely to be responsible for the robust binding of Me-30 through CH–π interaction. The decreased π-electron density of the Phe and Tyr mutants compared to that of Trp would have resulted in the high production of lupeol.
Introduction
Tetra- and pentacyclic triterpenoid scaffolds are formed by the enzymatic reactions of squalene and oxidosqualene cyclases. The cationic polyolefin cyclization cascade proceeds in a completely regio- and stereospecific fashion, yielding new C–C bonds and chiral centers, with four C–C bonds and seven chiral centers in the lanostane skeleton, five C–C bonds and nine chiral stereocenters in hopanoid, and five C–C bonds and eight chiral centers in β-amyrin.1 The structural diversity of triterpenoid scaffolds is remarkable, as about 150 different carbon frameworks have been found in nature.2 β-Amyrin 2 is widely distributed in plants and is a component of glycyrrhizin, which is a major bioactive compound with a wide range of pharmacological properties, and is used worldwide as a natural sweetener. As shown in Scheme 1, (3S)-2,3-oxidosqualene 1 is folded in a chair–chair–chair–boat–boat conformation.3 A proton attack on the epoxide ring initiates the polycyclization reaction. A 6,6-fused bicyclic ring skeleton (polypodatrienyl cation 3) is formed as the intermediate, which undergoes further cyclization to form the 6,6,5-fused tricyclic ring system (malabaricanyl cation 4). A ring expansion from the five- to the six-membered ring gives the 6,6,6-fused ring system, which undergoes further cyclization to generate the 6,6,6,5-fused tetracyclic dammarenyl cation 5. Subsequent ring enlargement of the five-membered D ring yields the 6,6,6,6-fused tetracyclic baccharenyl cation 6. Further cyclization yields the lupanyl cation 7 with the 6,6,6,6,5-fused ring system, and further ring expansion of 7 forms the final oleanyl cation 8 with a chair structure. Finally, 1,2-hydride shifts (i.e., H-18α to C-19 and H-13β to C-18) and the elimination of H-12α yield the product 2.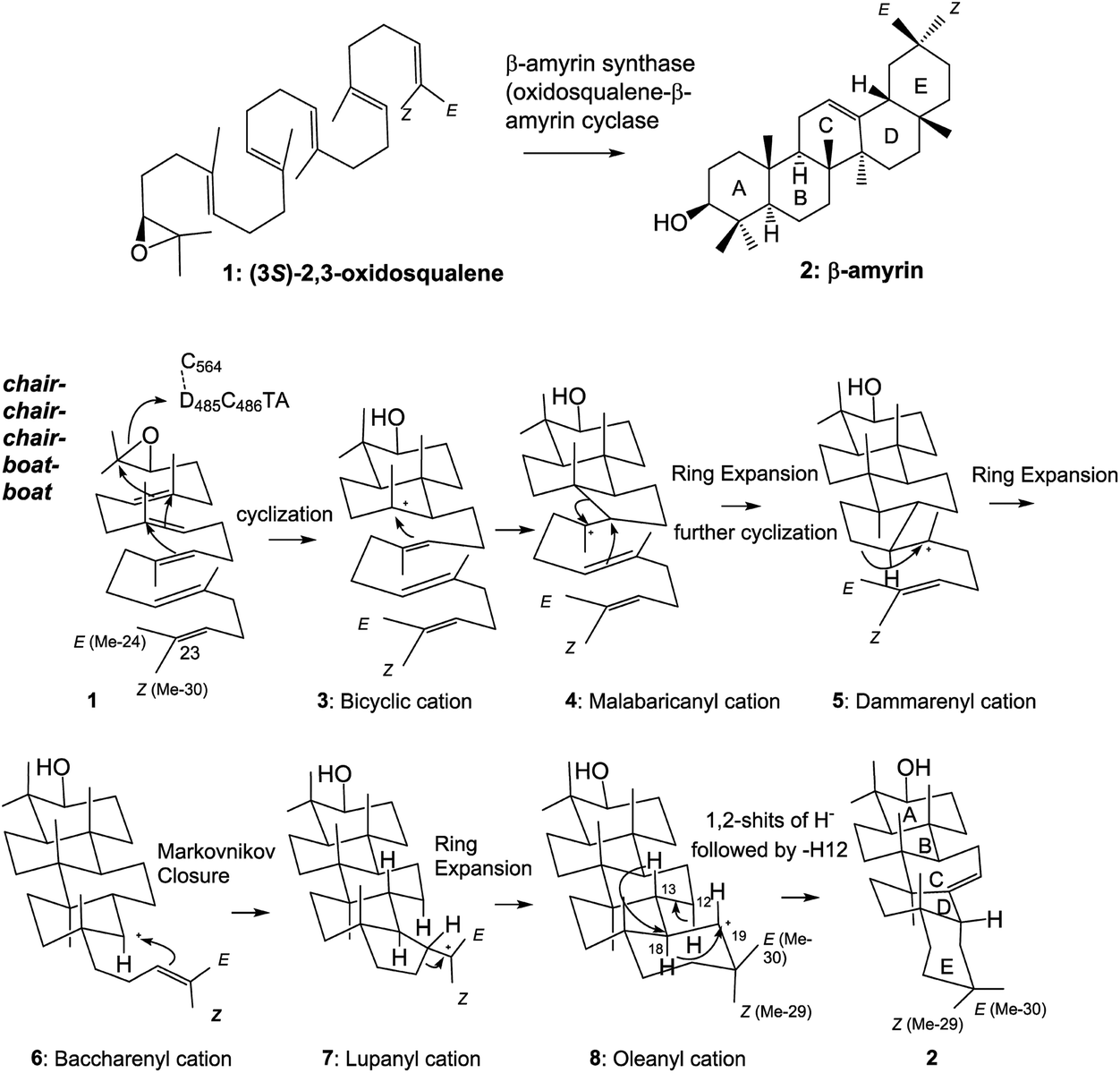 | ||
| Scheme 1 Cyclization pathway of (3S)-2,3-oxidosqualene 1 to generate β-amyrin triterpene 2. The substrate 1 is folded in a chair–chair–chair–boat–boat conformation in the enzyme cavity, and the proton released from the DCTA motif attacks the epoxide ring, leading to the sequential ring-forming reactions and construction of the 6,6,6,6,6-fused pentacyclic ring scaffold 8via the carbocationic intermediates 3–7. The oleanyl cation 8 undergoes the rearrangement reactions of 1,2-hydride shifts (H-18 → C-19 and H-13 → C-18) and deprotonation of the axial-oriented H-12 to yield β-amyrin 2. | ||
Functional studies on the active site residues of the lanosterol synthase of Saccharomyces cerevisiae4 and the hopene cyclase of Alicyclobacillus acidocaldarius5 have been carried out extensively, mainly by Wu and by our group, respectively. However, site-directed mutagenesis experiments on β-amyrin synthase are still limited. We have succeeded in completely purifying the β-amyrin synthase from Euphorbia tirucalli (EtAS),6 and reported the identification of some of its active sites. A proton attack on the oxirane ring is performed by the acidic Asp residue in the highly conserved D485C486TA motif.6 The Phe728 residue stabilizes the cationic intermediates 5–8 through cation–π interaction.7 As for Phe454, its steric size is more crucial than the π-electrons for the catalytic action.8 We have conducted site-directed mutagenesis experiments to further identify the active site residues. Here, we report the functional analyses of the Phe413, Trp257, and Tyr259 residues in EtAS, which are highly conserved in the family of β-amyrin synthases. Functional analysis of the conserved Phe413 residue has hitherto not been reported for any biological sources. This residue is also conserved in lupeol synthases (Fig. S1, ESI†). We analyzed the compound structures produced by various mutants and compared the enzymatic activities of each mutant. The Val and Met variants showed nearly the same enzymatic activities as the wild type, but the Tyr and Trp mutants exhibited significantly decreased activities. This finding strongly indicates that the steric bulk of Phe413, but not the π-electron density, is important for efficient catalysis. The Leu256 residue in the lupeol synthase of Olea europaea is highly conserved among various lupeol synthases, whereas Trp is conserved at the corresponding position in β-amyrin synthases (Fig. S1†). Kushiro et al. have succeeded in genetically engineering lupeol synthase into β-amyrin synthase.9 They reported that the site-specific mutagenesis of Leu256 → Trp provided β-amyrin, whereas that of Trp259 → Leu in the β-amyrin synthase of Panax ginseng (PNY) conversely generated lupeol.9 Based on the product distribution ratios of the single site-directed variants of PNY (W259L and Y261H), they have proposed the roles of Trp259 and Tyr in stabilizing the oleanyl and lupanyl cations through cation–π interaction, respectively, but they did not evaluate the enzyme activities of their respective mutants. To accurately verify this function of stabilizing the intermediate cation, the relative enzyme activities of various variants, including aliphatic and aromatic mutants must be estimated and compared with one another because aromatic active site residues have a crucial role in not only stabilizing cationic intermediates but also for placing the substrate at the correct position in the enzyme cavity as a result of appropriate steric bulk; this latter case was demonstrated by our mutational experiments of Phe454.8 The above-mentioned Trp259 and Tyr261 residues in PNY correspond to Trp257 and Tyr259 in EtAS (Fig. S1†). Therefore, to provide accurate insight into the functions of Trp257 and Tyr259, we have prepared various site-directed variants that are substituted with aliphatic and aromatic amino acids, and evaluated the product distribution ratios by each mutant. Furthermore, we compared the relative enzyme activities of each variant with that of the wild-type EtAS. In this paper, we report the detailed functions of Phe413, Tyr257, and Trp259 in EtAS. The function of Phe413 in providing appropriate steric volume, but not in furnishing π-electrons to the cationic intermediate, is addressed. Conversely, Tyr259 functions to provide the π-electrons to the baccharenyl cation 6, but not to the lupanyl cation 7 that was proposed by Kushiro et al.9 The Trp257 residue also works to stabilize the oleanyl cation 8 through cation–π interaction. Through our studies on the polycyclization cascade by substrate analogs10d–g and mutational experiments targeted for the amino acids around the E-ring formation site,10f we have reported that the Me-29 moiety of cation 8 is strongly captured through hydrophobic interaction, proposed as being CH–π interaction.10f Herein, we propose that the π-electrons of the Trp257 residue participate in the CH–π interaction to hold the Me-29 moiety of cation 8, leading to the ordered structure of the cation, and also function to stabilize 8via cation–π interaction.
Results and discussion
Analyses of the products of the F413X, Y259X, and W257X mutants
The following eight site-directed F413X mutants were prepared: F413A, F413V, F413M, F413S, F413T, F413Y, F413H, and F413W. These mutants were expressed in 100 mL cultures of S. cerevisiae GIL77, and the triterpene products that accumulated in the yeast cells were extracted with hexane after saponification with 15% KOH/MeOH. The hexane extracts were dried and acetylated with acetic anhydride–pyridine (Ac2O–Py). The hexane solution of the acetylated materials was subjected to gas chromatography (GC) analyses (Fig. 1). To isolate each of the products, 50 L of F413Y mutants was cultivated. After the yeast cells were saponified, the lipid materials were extracted with hexane, and then subjected to SiO2 column chromatography, eluting with hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc (100:5). The product-enriched fraction (220 mg) was acetylated with Ac2O–Py and subjected to normal high-performance liquid chromatography (HPLC) using hexane:tetrahydrofuran (hexane:THF = 100:0.02) as an eluent, yielding the acetates (Acs) of products 9–14 in a pure state in addition to the Ac of 2. The electron ionization mass spectrometry (EIMS) and proton nuclear magnetic resonance (1H-NMR) spectra (600 MHz, CDCl3) of 9-Ac revealed that this product was identical to that of polypoda-7,13,17,21-tetraen-3β-ol acetate (γ-polypodatetraene), which was previously isolated by us.8 The 1H- and 13C-NMR spectra of acetylated 10 (600 MHz, C6D6), as well as the two-dimensional (2D) NMR spectra, are shown in Fig. S2-2 through S2-8.† Two double bonds were found (δH 5.49, 1H, bs, H-16 and δH 5.41, 1H, t, J = 7.0 Hz, H-24; δC 121.2, d, C-16; δC 150.9, s, C-17; δC 125.4, d, C-24; and δC 131.1, s, C-25), indicating that the tetracyclic skeleton is assigned for 10. In the heteronuclear multiple-bond correlation (HMBC) spectrum, both Me-26 and Me-27 had clear cross-peaks with C-25 and C-24, confirming that the double bond is positioned at C-24–C-25. The doublet Me protons (δH 1.26, d, J = 6.8 Hz, 3H, Me-21) had HMBC correlations for C-17 and C-22 (δC 36.1, t). H-16 had a clear correlation spectroscopy cross-peak with H-15 (δH 1.81, 1H, m; 2.52, 1H, bd, J = 14.0 Hz). Me-30 (δH 1.241, 3H, s) showed a strong HMBC cross-peak with C-15 (δC 38.9, t). Thus, one of the two double bonds is situated at C-16–C-17. Analyses of all the NMR data (Fig. S2-9†) demonstrated that the structure of 10 is assigned as dammara-16(17),24-dien-3β-ol (see the structure in Scheme 2 and Fig. S2-9†). This compound is novel and has not been isolated from nature so far, although the stereochemical isomer protosta-16,24-dien-3β-ol (3β,8α,9β,13α,14β, see the structure shown in Fig. S2-9†) was isolated before.11a,b The EIMS spectrum and all the NMR spectra of 11-Ac were identical to those of dammara-20(21),24-dien-3β-ol, which was previously isolated from the F728W mutant.7 The NMR spectra of 12-Ac (400 MHz, C6D6), including the 2D NMR spectra, are shown in Fig. S3-2 through S3-9.† No olefinic proton was observed, but one double bond was found: δC 134.9, s, C-13; and δC 133.4, s, C-18. In the HMBC spectrum, clear cross-peaks were found between Me-27 (δH 1.33, 3H, s) and C-13, and between Me-28 (δH 1.33, 3H, s) and C-18 (Fig. S3-9†). This finding indicates that the double bond is positioned at C-13 and C-18. Me-29 (δH 0.959, 3H, s) had clear HMBC correlations with C-30 (δC 24.3, q) and C-20 (δC 33.4, s). Me-30 (δH 1.13, 3H, s) showed clear HMBC cross-peaks with C-29 (δC 32.5, q) and C-20. The HMBC data strongly indicated that the geminal methyl groups were located at C-20. The assignments of Me-29 and Me-30 were determined by the definitive nuclear Overhauser effect (NOE) of Me-28 and Me-30. Thus, a fully cyclized pentacycle was assigned for product 12. Detailed analyses of all the NMR spectra showed that the structure of 12 was δ-amyrin (olean-13(18)-3β-ol) (see structure 12 shown in Scheme 2 and Fig. S3-10†). This is the first case wherein δ-amyrin was produced by site-specific mutagenesis experiments. Products 13 and 14 were identified to be butyrospermol and tirucalla-7,24-dien-3β-ol, respectively, which were isolated in previous studies.6–8
:EtOAc (100:5). The product-enriched fraction (220 mg) was acetylated with Ac2O–Py and subjected to normal high-performance liquid chromatography (HPLC) using hexane:tetrahydrofuran (hexane:THF = 100:0.02) as an eluent, yielding the acetates (Acs) of products 9–14 in a pure state in addition to the Ac of 2. The electron ionization mass spectrometry (EIMS) and proton nuclear magnetic resonance (1H-NMR) spectra (600 MHz, CDCl3) of 9-Ac revealed that this product was identical to that of polypoda-7,13,17,21-tetraen-3β-ol acetate (γ-polypodatetraene), which was previously isolated by us.8 The 1H- and 13C-NMR spectra of acetylated 10 (600 MHz, C6D6), as well as the two-dimensional (2D) NMR spectra, are shown in Fig. S2-2 through S2-8.† Two double bonds were found (δH 5.49, 1H, bs, H-16 and δH 5.41, 1H, t, J = 7.0 Hz, H-24; δC 121.2, d, C-16; δC 150.9, s, C-17; δC 125.4, d, C-24; and δC 131.1, s, C-25), indicating that the tetracyclic skeleton is assigned for 10. In the heteronuclear multiple-bond correlation (HMBC) spectrum, both Me-26 and Me-27 had clear cross-peaks with C-25 and C-24, confirming that the double bond is positioned at C-24–C-25. The doublet Me protons (δH 1.26, d, J = 6.8 Hz, 3H, Me-21) had HMBC correlations for C-17 and C-22 (δC 36.1, t). H-16 had a clear correlation spectroscopy cross-peak with H-15 (δH 1.81, 1H, m; 2.52, 1H, bd, J = 14.0 Hz). Me-30 (δH 1.241, 3H, s) showed a strong HMBC cross-peak with C-15 (δC 38.9, t). Thus, one of the two double bonds is situated at C-16–C-17. Analyses of all the NMR data (Fig. S2-9†) demonstrated that the structure of 10 is assigned as dammara-16(17),24-dien-3β-ol (see the structure in Scheme 2 and Fig. S2-9†). This compound is novel and has not been isolated from nature so far, although the stereochemical isomer protosta-16,24-dien-3β-ol (3β,8α,9β,13α,14β, see the structure shown in Fig. S2-9†) was isolated before.11a,b The EIMS spectrum and all the NMR spectra of 11-Ac were identical to those of dammara-20(21),24-dien-3β-ol, which was previously isolated from the F728W mutant.7 The NMR spectra of 12-Ac (400 MHz, C6D6), including the 2D NMR spectra, are shown in Fig. S3-2 through S3-9.† No olefinic proton was observed, but one double bond was found: δC 134.9, s, C-13; and δC 133.4, s, C-18. In the HMBC spectrum, clear cross-peaks were found between Me-27 (δH 1.33, 3H, s) and C-13, and between Me-28 (δH 1.33, 3H, s) and C-18 (Fig. S3-9†). This finding indicates that the double bond is positioned at C-13 and C-18. Me-29 (δH 0.959, 3H, s) had clear HMBC correlations with C-30 (δC 24.3, q) and C-20 (δC 33.4, s). Me-30 (δH 1.13, 3H, s) showed clear HMBC cross-peaks with C-29 (δC 32.5, q) and C-20. The HMBC data strongly indicated that the geminal methyl groups were located at C-20. The assignments of Me-29 and Me-30 were determined by the definitive nuclear Overhauser effect (NOE) of Me-28 and Me-30. Thus, a fully cyclized pentacycle was assigned for product 12. Detailed analyses of all the NMR spectra showed that the structure of 12 was δ-amyrin (olean-13(18)-3β-ol) (see structure 12 shown in Scheme 2 and Fig. S3-10†). This is the first case wherein δ-amyrin was produced by site-specific mutagenesis experiments. Products 13 and 14 were identified to be butyrospermol and tirucalla-7,24-dien-3β-ol, respectively, which were isolated in previous studies.6–8
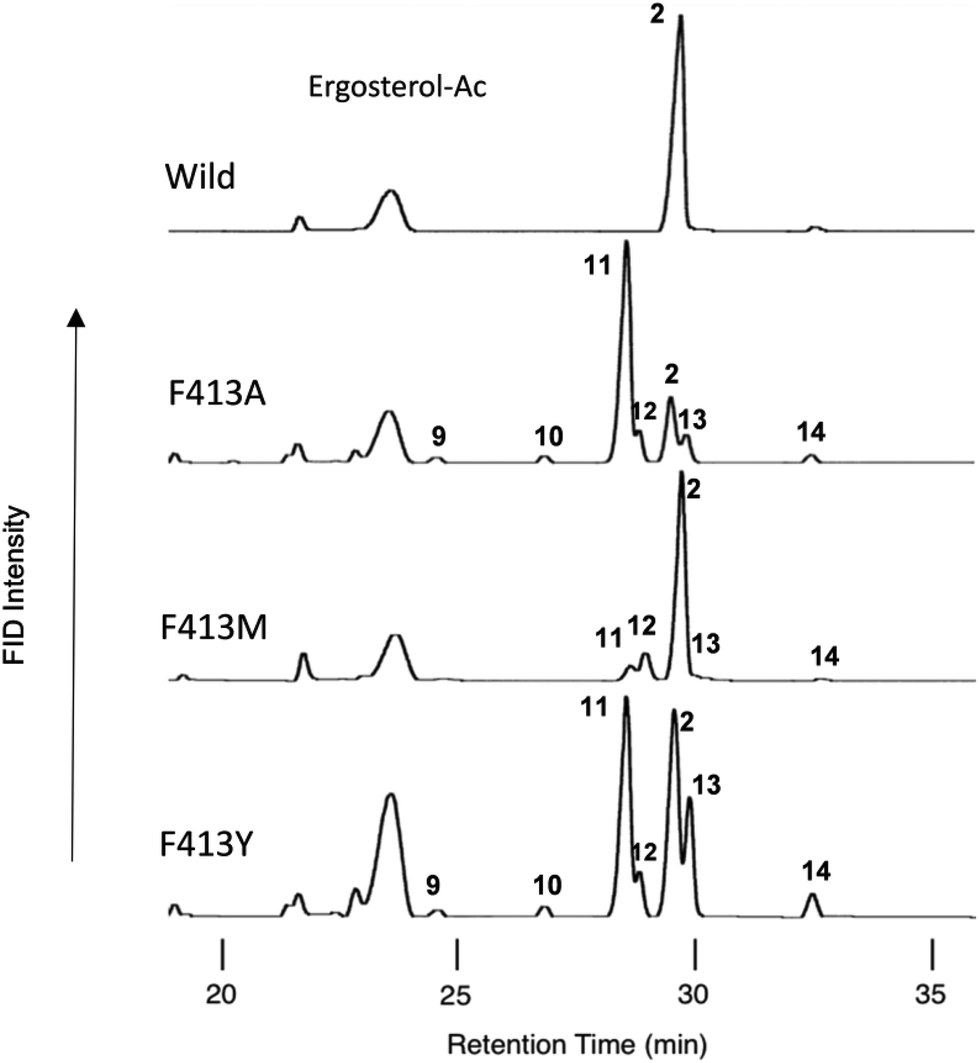 | ||
| Fig. 1 Gas chromatography (GC) profiles of the lipophilic acetylated materials produced by some mutants targeted for Phe413. Cultures (100 mL) of each mutant were centrifuged, and the cell pellets were subjected to saponification with 15% KOH/MeOH under reflux conditions, followed by extraction of the lipophilic materials with hexane extract (3 × 10 mL). The triterpene fraction including the products was obtained by partial purification on a SiO2 column to remove oxidosqualene, dioxidosqualene, and nontriterpene impurities (hexane:EtOAc = 100:1), followed by acetylation with Ac2O–Py. The acetate mixture was dissolved in 1.0 mL of hexane. A 0.5 μL aliquot of the hexane solution was injected into the GC apparatus. The GC conditions were as follows: capillary column, J & W DB-1 (30 m length, 0.32 mm I.D., 0.25 mm film thickness); injection temperature, 300 °C; column temperature, 245–270 °C (0.35 °C min−1). Product distribution patterns by the other variants are shown in Fig. S8.† | ||
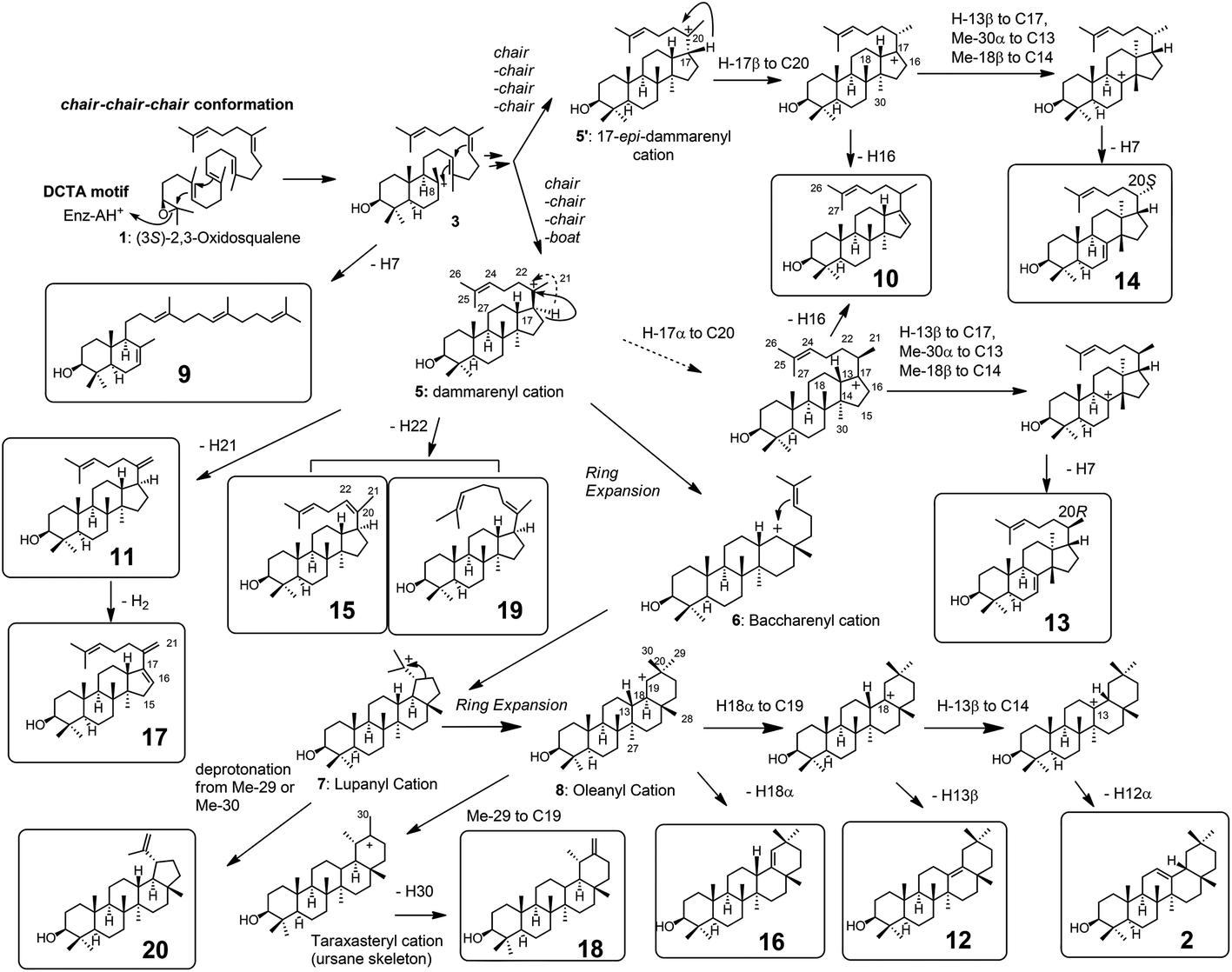 | ||
| Scheme 2 The cyclization pathway of oxidosqualene 1 to the cyclic products 9–20. The compound numberings are shown in Fig. 1 and 2. Compound names: 9: γ-polypodatetraen-3β-ol; 10: dammara-16,24-dien-3β-ol; 11: dammara-20(21),24-dien-3β-ol; 12: δ-amyrin (olean-13(18)-3β-ol); 13:butyrospermol; 14: tirucalla-7,24-dien-3β-ol; 15: dammara-(Z)-20(22)-24(25)-dien-3β-ol; 16: germanicol; 17: dammara-16(17),20(21),24-trien-3β-ol; 18: taraxasterol (urs-20(30)-en-3β-ol); 19: dammara-(E)-20(22)-24(25)-dien-3β-ol; and 20: lupeol. The carbon numbering systems are also shown here to aid the interpretation of the NMR data. | ||
Site-specific mutants targeted for the Tyr259 residue were constructed. Fig. 2A shows the GC traces of the lipophilic materials produced by the variants at position 259. The Y259H variant was cultivated (18 L) to isolate the triterpene products. The lipophilic materials were extracted with hexane after saponifying the yeast cells with KOH/MeOH. Partial purification was conducted via SiO2 column chromatography (hexane:EtOAc = 100:2.5). Next, the triterpene-enriched fraction including dioxidosqualene was acetylated with Ac2O–Py. A second SiO2 column chromatography allowed the successful isolation of the triterpene acetate fraction free of dioxidosqualene. A normal-phase HPLC (hexane:THF = 100:0.65) gave 15-Ac in a pure state. The EIMS spectrum of product 15-Ac showed a peak at m/z 468 (M+), as shown in Fig. S4-1.† Three olefinic methyl groups were found in the 1H-NMR spectrum of product 15 (400 MHz, CDCl3): δH 1.60 (3H, s, Me-21), δH 1.62 (3H, s, Me-26), and δH 1.68 (3H, s, Me-27). In addition, two double bonds were found: δH 5.15 (1H, t, J = 7.2 Hz, H-22) and δH 5.06 (t, J = 7.2 Hz, H-24); δC 136.9 (s, C-20), δC 124.8 (d, C-22), δC 123.8 (d, C-24), and δC 131.0 (s, C-25). In the HMBC spectrum, strong cross-peaks of Me-21 protons with C-17 (δC 41.2, d) and C-22 were evident. The protons of Me-26 and Me-27 had clear HMBC correlations with C-24 and C-25. Thus, these findings suggest that product 15-Ac consisted of a tetracyclic ring system with two double bonds present in the side chain. The double bond was determined to have Z geometry, because a definitive NOE of Me-21 and H-22 was observed (Fig. S4-6†). The detailed 2D NMR analyses (Fig. S4-9†) indicated that this product was dammara-20(22,Z),24-dien-3β-ol. Peak 16 was identified to be germanicol, by comparing 16-Ac with the authentic sample that was previously isolated from the F728 mutant.7 Product 17-Ac was isolated from the Y259L mutant. The purification method was essentially similar to that used for the His variant. EIMS (Fig. S5-1†) showed the peak at m/z 466 to be the highest ion; a value of 2 mass units is less than that for the triterpene acetate usually produced by oxidosqualene cyclization, suggesting that one more double bond is involved in 17-Ac. The 1D and 2D NMR spectra (400 MHz, C6D6) are shown in Fig. S5-2 through Fig. S5-8.† The 1H- and 13C-NMR spectra showed that three double bonds were involved in this molecule: δH 5.42 (very broad s, H-24), δH 5.23 (s, H-21), and δH 5.42 (s, H-21); δC 146.2 (s, C-17), δC 145.7 (s, C-20), δC 131.3 (s, C-25), δC 110.5 (t, C-21), δC 127.1 (d, C-16), and δC 124.9 (d, C-24). In the HMBC spectrum, the vinylidene protons (1H, 5.23, s; 1H, 5.14, s, H-21) had clear cross-peaks with C-17, C-20, and C-22 (δC 35.4, t), suggesting that 17-Ac is composed of a tetracyclic ring system; that is, dammara-20(21)-24-dien-3β-ol (product 11), containing one extra double bond at C-16–C-15. Deep insights into other NMR data further supported the proposed structure of 17, as shown in Scheme 2 and Fig. S5-9.† Product 17 is a novel compound (ACS SciFinder; https://scifinder.cas.org/scifinder/view/scifinder/scifinderExplore.jsf). Product 18-Ac was previously isolated from the F728W mutant.7 The production of 19 was very low, and thus the isolation of 19-Ac was unsuccessful. The EIMS spectrum of product 19-Ac was quite similar to that of 15-Ac (Fig. S6†), indicating that the fundamental cores of both products were identical. Thus, 19 is possibly assignable as dammara-(E)-20(22)-24-dien-3β-ol, which is further supported by the report that 19 was also produced from the Y261H mutant of PNY,9 which is equivalent to the Y259H mutant of EtAS. Compound 20 was identified as lupeol, which was previously isolated from the F728H variant.7
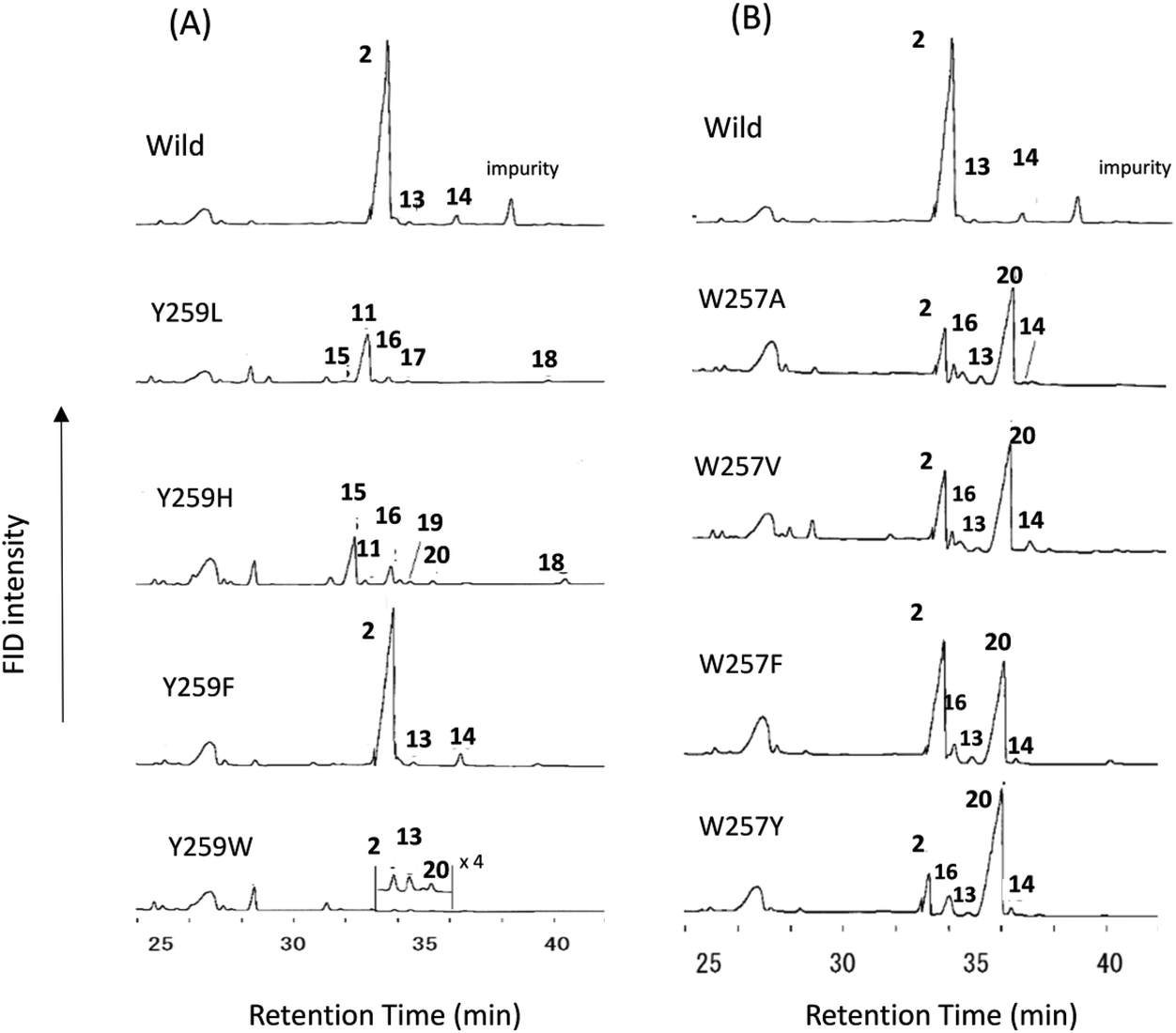 | ||
| Fig. 2 Gas chromatography (GC) traces of the lipophilic materials produced by the Y259X (A) and W257X mutants (B). The lipophilic materials were extracted with hexane after saponification of the yeast cells by refluxing with 15% KOH/MeOH. The hexane extract was evaporated to dryness. Then, 1.0 mL of hexane was added to the residue, and 0.5 μL of the mixture was injected into the GC apparatus. The gas chromatograms were obtained under the following conditions: capillary column, J & W DB-1 (30 m length, 0.32 mm I.D., 0.25 μm film thickness); injection temperature, 300 °C; column temperature, 190–250 °C (10 °C min−1), 250–268 (0.35 °C min−1), and 268–270 (3.0 °C min−1). The GC conditions were identical between (A) and (B). Of note, the hexane extracts were directly injected without acetylation treatment, which is different from the acetylated samples of Fig. 1. | ||
Cyclization pathway of 1 into products 9–20
Scheme 2 shows the biosynthetic pathway of 1 to form the enzymatic products 9–20, which were generated by the site-specific mutants targeted at the Phe413, Tyr259, and Trp257 residues. Substrate 1 was folded in a chair–chair conformation to give the bicyclic intermediate 3 with C-8 cation. Proton elimination of H-7 yielded product 9. Further cyclization of cation 3 could produce the tetracyclic cation 5 with H-17α via the tricyclic cationic intermediate 4. Cation 5 with H-17α was produced through the folding of 1 into a chair–chair–chair–boat conformation. In contrast, the folding of 1 into a chair–chair–chair–chair conformation gave the 17-epi-dammarenyl cation 5′ with H-17β. The H-17 of cations 5 and 5′ migrated to C-20 to give intermediates with a C-17 cation, which underwent the deprotonation of H-16 to yield 10. At the present time, the C-20 stereochemistry of 10 is not clear; thus, product 10 could be produced via either cations 5 or 5′. The C-17 cations thus produced via5 and 5′ underwent sequential 1,2-shift reactions as follows: H-13β to C-17, Me-30α to C-13, and Me-18β to C-14, to provide the C-8 cation. Next, the deprotonation reaction of H-7 could generate 13 and 14, which were produced via cations 5 and 5′, respectively, as shown in Scheme 2. The elimination of H-21 of 5 yielded product 11. The subsequent dehydrogenation (oxidation) reaction introduced the double bond at C-16 and C-17, yielding product 17. The elimination reaction of H-22 from cation 5 yielded products 15 and 19 at a production ratio of 30:1. The significantly higher yield of the Z-isomer 15 indicates that the side chain of cation 5 was tightly captured by this β-amyrin synthase. A ring enlargement of 5 gave the 6,6,6,6-fused baccharenyl cation 6. Further cyclization yielded the lupanyl cation 7 with a five-membered E-ring. The deprotonation reaction from the isopropyl group gave lupeol 20. Further ring expansion of the five-membered E-ring in cation 7 gave the 6,6,6,6,6-fused oleanyl cation 8. The shift of Me-29 to C-19 provided the taraxasteryl cation. The subsequent deprotonation from Me-30 gave product 18. The elimination of H-18α of cation 8 yielded 16. The 1,2-Shift of H-18α to the C-19 cation gave the oleanyl cation bearing the C-18 cation, and further deprotonation of H-13β led to δ-amyrin 12. The elimination of H-12α could furnish β-amyrin 2. Although the structures of 10 and 17 are quite similar, their biosynthetic pathways are significantly different. Compound 10 was produced only by the catalytic action of oxidosqualene cyclase. However, the production of 17 from 11 was not catalyzed by the cyclase, but a dehydrogenase (or oxidase) of S. cerevisiae GIL77 would have been involved in the biochemical formation of 17. Thus, 17 was produced by the two enzymes Y259L cyclase and dehydrogenase to introduce a double bond at C-16 and C-17.
Functional analysis of the Phe413 residue
Table 1 shows the product distribution ratio obtained by the various site-specific mutants. Notably, the Ala mutant yielded a significantly large amount of the tetracyclic products 10–14 (∼74%), but the amount of pentacyclic products (2 and 12, 24%) was lower than that produced by the wild type. A small amount of the bicyclic 9 (∼2%) was concomitantly produced. This finding implies that the π-electrons of Phe413 act to stabilize the intermediary dammarenyl cation through cation–π interaction. However, this idea is not correct, because the aliphatic Val and Met variants produced pentacycles in a high ratio (Table 1), despite the π-electrons being deficient. Thus, the steric size (van der Waals volume)12 at this position is important for efficient ring-forming reactions. The steric volume of the Ala residue is too small to complete the polycyclization cascade, leading to the production of premature cyclization products 9 and 10–14. Because the Val and Met residues are somewhat larger than the Ala moiety, this would have led to a high production of pentacycles (89.5–92.5%) in conjunction with a lower production of tetracycles (7.5–10.5%). In addition, the bicyclic 9 was produced by the Ala mutant, but not by the Val and the Met mutants; this would have occurred because Ala has a significantly smaller steric size than Val and Met. The product distribution ratio suggests that Phe413 is situated near the D-ring formation site in the reaction cavity. This suggestion coincides with the inference from the homology modeling of EtAS (Fig. S7.1†) constructed on the basis of the X-ray crystal structure of human lanosterol synthase. Fig. 3 shows the relative enzymatic activities of each of mutants against that of the native EtAS. The total amounts of the enzyme products that were estimated by GC analyses were divided by the enzyme amounts expressed in vivo that were determined by western blotting analyses (Fig. S9.1–S9.9†).6–8 The enzyme activities for the total amounts of triterpenes were not so much different among the wild type, and F413A, F413V, and F413M variants, although the amount of oleanane-type triterpene produced by the Ala mutant was much lower (as described above) than those of the Val and Met variants, indicating that the side chain of the Phe413 residue maintains crucial hydrophobic properties, but does not possess the electronic function of π-electrons. On the other hand, the Ser, Thr, Tyr, and His mutants yielded significantly lower activities for the production of total triterpenes, including tetracycles. This finding is not accountable in terms of the difference of the van der Waals volume of the side chain; the steric volume difference of Thr and Val, and that of Met and His is not so large (Fig. 3), but the variants bearing the OH group in the side chain (Ser and Thr) had significantly lower activities. As described below, our investigation showed that the Tyr259 residue could stabilize the baccharenyl cation 6via cation–π interaction. Hydrogen bonding may have occurred between the OH group of the polar amino acids and the phenolic OH group of Tyr259 because of the proximal distance between them (see Fig. S7.2†), which would have brought about the inappropriate placement of Tyr259. Otherwise, water molecule(s) may have inserted themselves in between the two groups through hydrogen bonding. The hydrogen bonds would have led somewhat to a failure by Tyr259 to stabilize intermediate 6, and thus the activities of the Ser, Thr, Tyr, and His mutants were lower than those of the Ala, Val, and Met variants, and the production of oleanane triterpenes (2 + 12) was also lower with the concomitant accumulation of tetracyclic products. This finding is further supported by the amino acid alignment of triterpene cyclases. Fig. S1† depicts the amino acid alignment of various oxidosqualene cyclases. Although the Phe413 residue is highly conserved in the β-amyrin and lupeol synthases that produce 6,6,6,6,6- and 6,6,6,6,5-fused pentacyclic skeletons, the residue is replaced by Tyr or Thr residues in cycloartenol and lanosterol synthases that yield the 6,6,6,5-fused tetracyclic scaffold. This amino acid alignment further validates our experimental results obtained from the product analyses of the polar Tyr, Thr, and Ser variants. Thus, Phe must be conserved at this position for the formation of oleanane and lupane scaffolds. The steric bulk of Trp is the largest among all of the natural amino acids, resulting in significantly lower activity (Fig. 3), due to the formation of the unfavorable (distorted) folding conformation of 1, which is far from the normal folding of the chair–chair–chair–boat–boat conformation, leading to looser binding to the enzyme.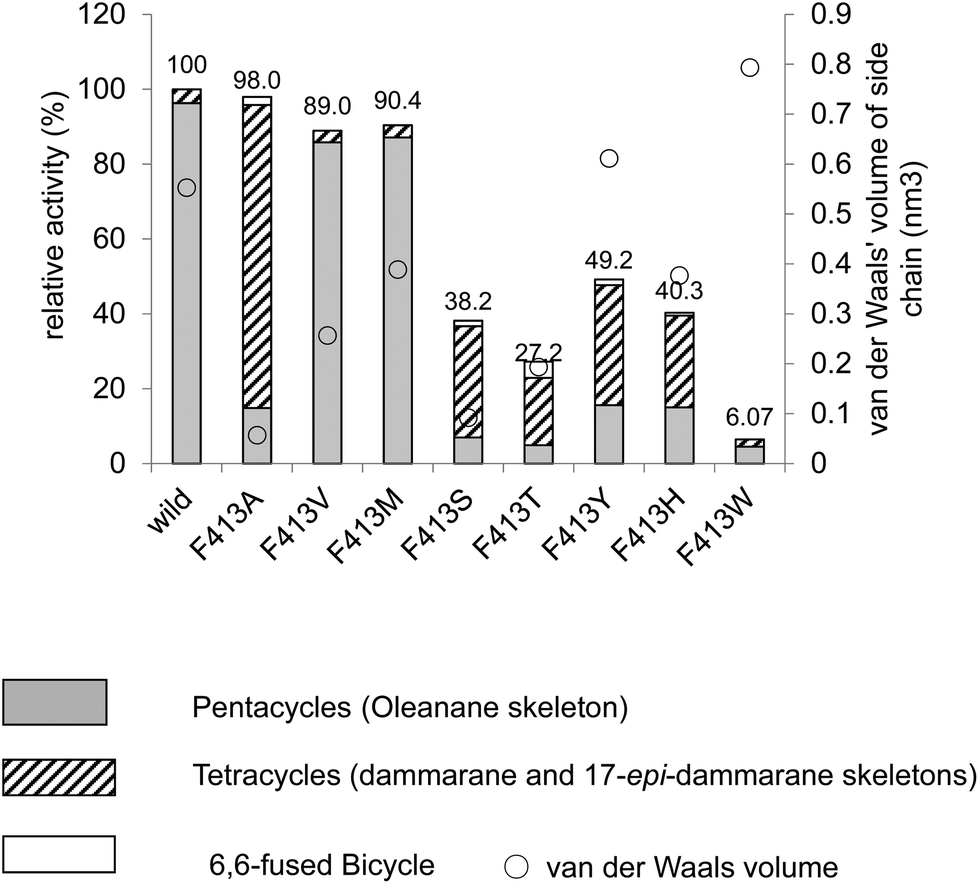 | ||
| Fig. 3 Enzymatic activities of the F413X mutants relative to that of the wild-type β-amyrin synthase from Euphorbia tirucalli (EtAS). The wild-type activity (100%) indicates the sum of the relative activities shown in Fig. S9.3 (production of 2) and Fig. S9.5† (production of 10, 11, 13 and 14). The activities for each mutant represent the total sum of the relative activities (%) estimated for the respective mutants, which are shown in Fig. S9.3, S9.5, S9.7, and S9.9 (ESI†). The total activities for the mutants thus obtained were evaluated against that of the wild-type EtAS (100%). Production of the pentacyclic oleanane skeleton derived from cation 8 roughly increased in proportion to the increase in the van der Waals volume (see the right-pointing arrow). The van der Waals volumes (nm3) for the side residues are as follows: Phe (wild type), 0.55298; Ala, 0.05702; Val, 0.25674; Leu, 0.37876; Met, 0.38872; Ser, 0.09204; Thr, 0.19341; Tyr, 0.6115; His, 0.37694; and Trp, 0.79351. The values are cited from the reference.12 | ||
| Product | Oleanane skeleton (pentacycle) | Dammarane skeleton (tetracycle) | Bicycle | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 12 | 2 + 12 | 10 | 11 | 13 (20R) | 14 (20S) | Total (10–14) | 9 | |
| Wild | 97.0 | 97.0 | 0.7 | 2.3 | 3.0 | ||||
| F413A | 17.9 | 6.1 | 24.0 | 2.0 | 62.9 | 6.8 | 2.2 | 73.8 | 2.1 |
| F413V | 88.4 | 1.1 | 89.5 | 8.1 | 0.2 | 2.2 | 10.5 | ||
| F413M | 81.0 | 11.5 | 92.5 | 5.8 | 0.6 | 1.1 | 7.5 | ||
| F413S | 10.8 | 6.7 | 17.5 | 4.8 | 69.3 | 5.0 | 0.8 | 79.9 | 2.6 |
| F413T | 22.0 | 6.0 | 28.0 | 3.9 | 54.0 | 5.6 | 1.7 | 65.2 | 6.7 |
| F413Y | 35.0 | 5.4 | 40.4 | 1.8 | 36.6 | 15.7 | 3.8 | 57.8 | 1.7 |
| F413H | 31.3 | 6.0 | 37.3 | 1.0 | 44.4 | 12.6 | 2.8 | 60.8 | 1.9 |
| F413W | 84.8 | 84.8 | 4.8 | 10.4 | 15.2 | ||||
Functional analysis of the Tyr259 moiety
Fig. 2A and Table 2 show the product distribution by each mutant that was targeted at position 259. The aliphatic amino acid mutants (Ala, Val, Ile, and Leu variants) yielded no production of 2 and the product ratio of tetracycles was significantly high. However, the aromatic Phe variant yielded 2 almost exclusively in a similar product distribution ratio as that of the wild type (Table 2). This strongly indicates that the π-electrons of Tyr259 stabilize the intermediary cation(s) through cation–π interaction. The aliphatic amino acid mutants generated the dammarenyl cation-derived product 11 in a high production ratio, definitively demonstrating that the Tyr259 residue works to stabilize the baccharenyl cation 6. Deficiency of the stabilization of cation 6 yielded the dammarenyl cation 5. This idea is in contrast to the proposal by Kushiro et al. that Tyr256 stabilizes lupanyl cation 7.9 No stabilization of cation 7 by the aliphatic mutants would have led to the production of cation 6-derived products, but no product from cation 6 was found (Table 2). It is noticeable that the His mutant produced tetracyclic 15 in the highest production ratio, indicating that the Tyr259 residue is located near the D-ring formation site and that the His variant could abstract the H-22 of the tetracyclic cation 5 because of its basic nature. This result is in good agreement with the finding that the His residue is conserved at this position among triterpene cyclases that produce tetracyclic scaffolds (Fig. S1†). The production ratio of the Z-isomer 15 and the E-isomer 19 was ∼30:1. The preferential formation of the Z-isomer indicated that the rotation of the side chain of the tetracyclic cation 5 was restricted, owing to the robust binding between the side chain of 5 and the EtAS enzyme. Therefore, it can be concluded that the function of the Tyr259 moiety is to stabilize the 6,6,6,6-fused secondary tetracyclic cation 6via cation–π interaction. Of further note is that the aliphatic mutants (A, V, I, L) also did not yield any amount of lupeol 20 (Table 2). Provided that Tyr259 is located in proximity to the oleanyl cation 8 to stabilize cation 8, the aliphatic mutants should have yielded lupeol 20via cation 7. This fact indicates that Tyr259 cannot stabilize cation 8, but exclusively stabilizes cation 6. The relative activity of the Trp mutant was significantly low (Fig. 4A). This would have occurred as a result of the incorporation of the significantly large steric bulk into the EtAS enzyme, leading to an unfavorable folding conformation of 1 and hence its looser binding to the altered enzyme.
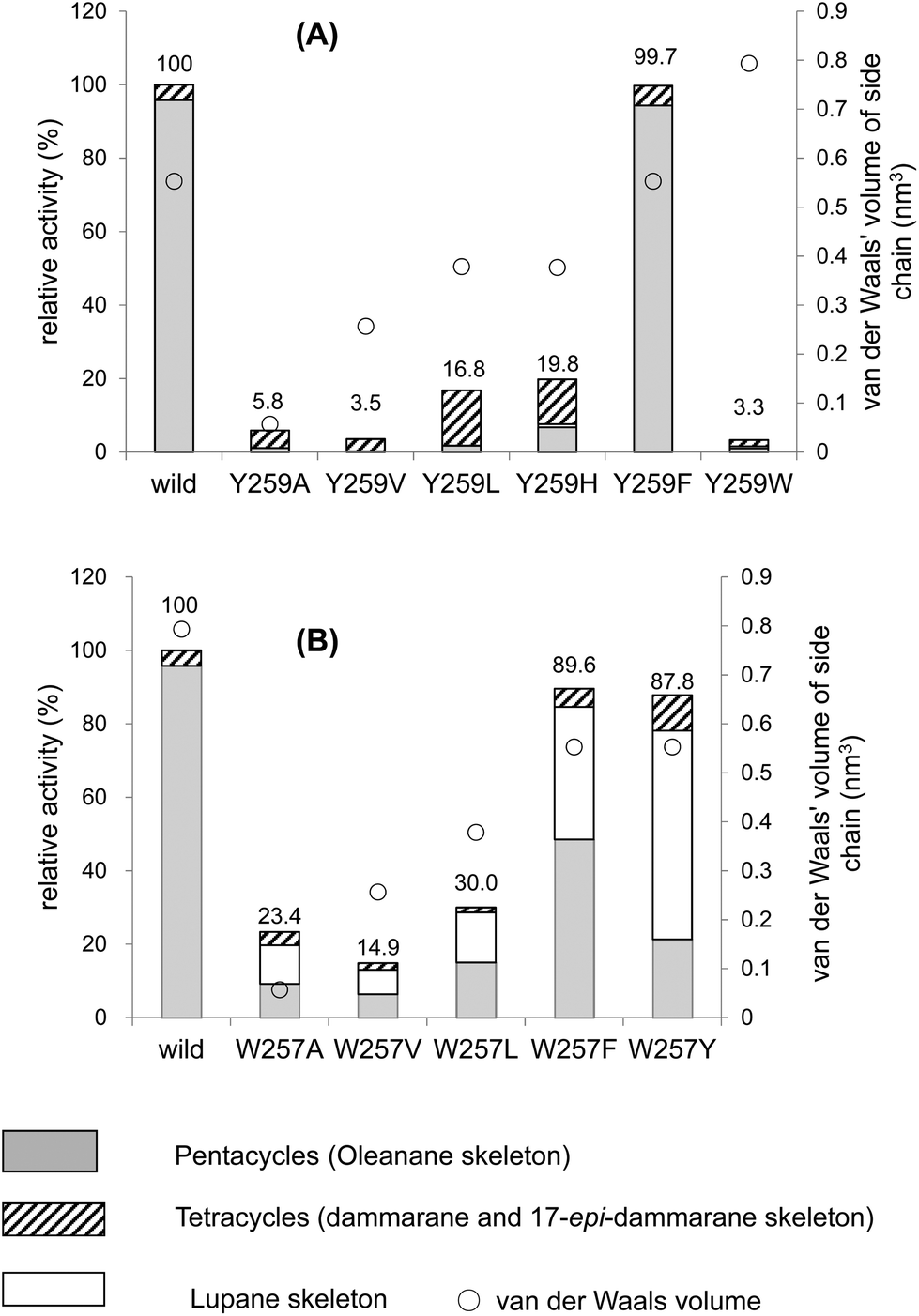 | ||
| Fig. 4 Enzymatic activities of the Y259X (A) and W257X mutants (B) relative to that of the wild-type β-amyrin synthase from Euphorbia tirucalli. | ||
| Products | Oleanyl skeleton | Lupanyl skeleton | Tetracyclic skeleton | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 16 | 18 | 2 + 16 + 18 | 20 | 15 | 19 | 15 + 19 | 11 | 17 | 11 + 17 | 13 | 14 | Tetracyclic total | |
| Wild | 97.0 | 97.0 | 0.7 | 2.3 | 3.0 | |||||||||
| Y259A | 19.0 | 1.8 | 20.8 | 1.9 | 1.9 | 51.6 | 18.1 | 69.7 | 7.6 | 79.2 | ||||
| Y259V | 4.8 | 4.8 | 95.2 | 95.2 | 95.2 | |||||||||
| Y259I | 0.7 | 0.7 | 99.3 | 99.3 | 99.3 | |||||||||
| Y259L | 4.6 | 2.1 | 6.7 | 8.7 | 8.7 | 77.4 | 7.2 | 84.6 | 93.3 | |||||
| Y259H | 16.6 | 3.7 | 20.3 | 3.7 | 69.6 | 2.4 | 72.0 | 2.0 | 2.0 | 76.0 | ||||
| Y259F | 94.7 | 94.7 | 1.5 | 3.8 | 5.3 | |||||||||
| Y259W | 40.9 | 40.9 | 9.5 | 49.6 | 49.6 | |||||||||
| W257A | 23.5 | 6.5 | 30.0 | 62.8 | 5.9 | 1.3 | 7.2 | |||||||
| W257V | 38.2 | 6.1 | 44.3 | 44.2 | 7.0 | 4.5 | 11.5 | |||||||
| W257L | 50.1 | 2.5 | 52.6 | 42.9 | 2.6 | 0.5 | 3.4 | |||||||
| W257F | 38.2 | 6.1 | 44.3 | 44.2 | 3.8 | 1.2 | 5.0 | |||||||
| W257Y | 21.2 | 0.7 | 21.9 | 64.5 | 10.9 | 2.7 | 13.6 | |||||||
Functional analysis of the Trp257 moiety
The W257A mutant yielded lupeol 20 as the major product (∼63%), but the production of pentacyclic compounds 2 and 16 was lower than that of the wild type (30%), as shown in Table 2. Whereas the wild type does not produce 20, the aliphatic mutants produced a large amount of this product. This finding indicates that the π-electrons of the Trp257 residue have a role in stabilizing the oleanyl cation 8via cation–π interaction. No stabilization of the secondary oleanyl cation 8 led to the production of the stable tertiary lupanyl cation 7, generating lupeol 20. The aromatic Phe and Tyr variants showed significantly higher activities than the aliphatic mutants, such as the Ala, Val, and Leu variants (Fig. 4B). This further indicates that the π-electrons at position 257 play a crucial role in stabilizing the intermediary cation 8 through cation–π interaction. The π-electron density of Trp in the native enzyme is more abundant than those of Phe and Tyr. Mecozzi et al. reported the cation–π-electron binding energies of the aromatic rings with Na+ as follows: 21.0 kcal mol−1 for His, 27.1 kcal mol−1 for Phe, 26.9 kcal mol−1 for Tyr, and 32.6 kcal mol−1 for Trp.13 Thus, the decreased π-electron density of the Phe and Tyr variants could not sufficiently stabilize cation 8, resulting in the high production of cation 7 and leading to the formation of 20. The Tyr mutant produced 20 in a higher yield than that of the Phe mutant, despite the binding energy being almost equivalent between them. The ionized phenoxide ion in the Tyr residue may have facilitated the deprotonation reaction from the isopropyl moiety in cation 7. Kushiro et al. reported that the mutation of W259 → Leu in PNY converted β-amyrin synthase into lupeol synthase.9 However, our results strongly indicate that the aliphatic Leu residue is not necessarily required at position 259 for the formation of lupeol, and that the aromatic Phe or Tyr variants can also generate lupeol 20 in a significantly high yield (Fig. 4B). Thus, this experimental result indicates that the conversion of β-amyrin synthase into lupeol synthase could also be attained by the incorporation of Phe and Tyr aromatic residues at this position. In a previous paper,10e,f we disclosed that the Me-30 moiety of 1 is essential for the formation of the oleanyl skeleton, and that Me-30, which corresponds to the Me-29 moiety of cation 8, is strongly captured by EtAS through hydrophobic interaction.10f We have proposed that CH–π interaction would occur between the Me-30 moiety of 1 and the π-electrons of aromatic amino acids.10f The π-electrons of the Trp257 residue are likely to play an important role in its tight binding with the Me-29 moiety of the ordered structure of oleanyl cation 8 (Scheme 3). The decreased CH–π interaction caused by the decreased π-electron density of Phe and Tyr could more or less produce a distortion to the ordered structure of 8 as a result of the decreased binding to Me-29 of 8, which could give rise to lupeol 20 in a significantly high yield (Fig. 4B).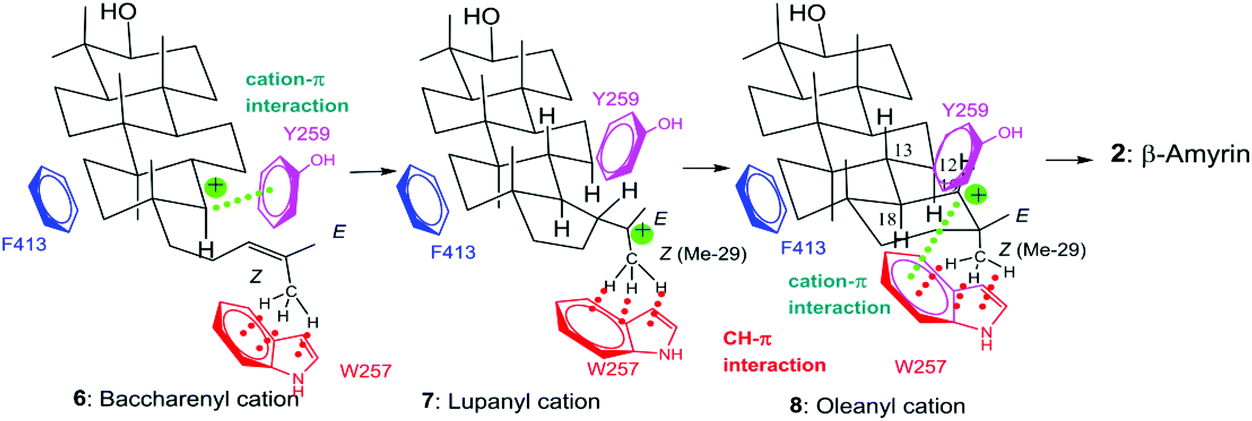 | ||
| Scheme 3 Illustration of the location of the aromatic residues of Phe413, Tyr259, and Trp257 in the reaction cavity and the catalytic functions. Phe413 binds to the D-ring formation site; Tyr259 stabilizes the baccharenyl cation 6 through cation–π interaction; Trp257 strongly captures the Z-Me of 8via CH–π interaction, leading to the formation of the final oleanyl cation, which is stabilized by Trp257 via cation–π interaction. Thus, the Trp257 residue possibly has dual functions. In previous studies,10e,f we had elucidated that 23Z-Me (Me-30) of oxidosqualene 1 binds more strongly to β-amyrin synthase than the 23E-Me (Me-24) group (see Scheme 1 for carbon numbering). The green dotted line denotes the cation–π interaction. The red dotted line represents the CH–π interaction between Trp257 and the Z-CH3 group of intermediates 6–8. Tyr259 and Phe7287 residues may also more or less have the additional function of CH–π interaction besides cation–π interaction. As shown in this scheme, Trp 257 is likely to participate in the binding of Me-29 (Z-Me) of cations 6 and 7. | ||
Conclusion
In conclusion, we have disclosed that aromatic active site residues perform critical functions not only to stabilize the cationic intermediates by π-electrons, but also to arrange the substrate at the correct position inside the reaction cavity, which is assisted by the appropriate steric bulk, leading to an efficient catalytic polycyclization reaction. As summarized in Scheme 3, the function of the Phe413 residue is to bind to the D-ring formation site through its appropriate bulkiness, while the Tyr259 residue stabilizes the intermediary cation 6via cation–π interaction. It is possible that the Trp257 residue has dual functions of stabilizing cation 8via cation–π interaction, and binding robustly with Me-29 of cations 7 and/or 8 through the CH–π complex. Therefore, this study on the function of the aromatic amino acid residues highlighted the following three contributions to the polycyclization reaction: (1) substrate binding via hydrophobic interaction with little involvement of π-electrons, which is attained by the appropriate steric bulkiness; (2) stabilization of intermediary cations via cation–π interactions; and (3) participation of the π-electrons in substrate binding through CH–π interactions, the concept of which has not been recognized hitherto in the field of triterpene cyclases. This is the first report to provide evidence for the occurrence of CH–π interaction between Trp and the Me-30 of 1. Finally, we succeeded in generating lupeol synthase from the β-amyrin synthase by altering Trp at position 257 into Tyr or Phe moieties. Notably, it is of interest that the Tyr variant produced a larger amount of lupeol than the Leu mutant, as shown in Fig. 4B.Experimental
Analytical methods
NMR spectra of the enzymatic products were recorded in CDCl3 on Bruker DMX 600 and DPX 400 spectrometers, the chemical shifts being given in ppm relative to the solvent peak δH = 7.26 and δC = 77.0 ppm as the internal reference for 1H- and 13C NMR spectra, respectively. In the C6D6 solution, the chemical shifts are given in ppm relative to the solvent peak (δH = 7.28 and δC = 128.0 ppm). The coupling constants J are given in Hz. GC analyses were done on a Shimadzu GC2014 chromatograph equipped with a flame ionization detector (a DB-1 capillary column, 30 m × 0.25 mm × 0.25 μm, J&W Scientific. Inc.). GC-MS spectra were on a JEOL SX 100 or a JEOL JMS-Q1000 GC K9 instrument equipped with a ZB-5 ms capillary column (30 m × 0.25 mm × 0.25 μm; Zebron) by using EI mode operated at 70 eV. High resolution-mass spectrometry (HR-MS) was performed on a JMS-T100GCV using electron ionization (EI) mode. HPLC was carried out with a Hitachi L-1700 (pump) and an L-7405 (UV detector), and the HPLC peaks were monitored at 210 or 214 nm. Specific rotation values were measured with a Horiba SEPA-300 polarimeter.Mutagenesis experiments
Mutagenesis experiments of Phe413, Tyr256 and Trp257 residues in wild-type pYES2-EtAS/CT were performed with the QuikChange site-directed mutagenesis method. The following oligonucleotide primers were used; substitutions are underlined. PCRs were conducted with a 16-cycle program: 98 °C for 0.5 min, 60 °C for 1 min, 68 °C for 9 min, and a final extension at 68 °C for 9 min. KOD-plus DNA polymerase (Toyobo) was used with dNTPs (0.2 mM), dimethylsulfoxide (5%) and MgSO4 (0.1 mM) in a final volume of 50 μL. Mutations were confirmed by DNA sequencing with the dideoxy chain-termination method and a Beckman Coulter CEQ8000 (Beckman Coulter).For the F413A variant:
sense primer, 5′-GGCATGAAAATGCAGAGC![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) GGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCC
GGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCC![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) GCTCTGCATTTTCATGCC-3′.
GCTCTGCATTTTCATGCC-3′.
For the F413V variant:
sense primer, 5′-GGCATGAAAATGCAGAGC>GGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the F413M variant:
sense primer, 5′-GGCATGAAAATGCAGAGCGGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the F413S variant:
sense primer, 5′-GGCATGAAAATGCAGAGCGGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the F413T variant:
sense primer, 5′-GGCATGAAAATGCAGAGCGGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the F413SY variant:
sense primer, 5′-GGCATGAAAATGCAGAGCGGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the F413H variant:
sense primer, 5′-GGCATGAAAATGCAGAGCGGGAGTCAGCAG-3′; and antisense primer, 5′-CTGCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the F413W variant:
sense primer, 5′-GGCATGAAAATGCAGAGCGGGAGTCAGC-3′; and antisense primer, 5′-GCTGACTCCCGCTCTGCATTTTCATGCC-3′.
For the Y259A variant:
sense primer, 5′-GCAAAAATGTGGTGTTGTCGGATGGTTTACATG-3′; and antisense primer, 5′-CATGTAAACCATCCGACAACACCACATTTTTGC.
For the Y259V variant:
sense primer, 5′-GCAAAAATGTGGTGTTGTCGGATGGTTTACATG-3′; and antisense primer, 5′-CATGTAAACCATCCGACAACACCACATTTTTGC-3′.
For the Y259I variant:
sense primer, 5′-GCAAAAATGTGGTGTTGTCGGATGGTTTACATGC-3′; and antisense primer, 5′-GCATGTAAACCATCCGACAACACCACATTTTTGC-3′.
For the Y259L variant:
sense primer, 5′-CCAGCAAAAATGTGGTGTTGTCGGATGGTTTAC-3′; and antisense primer, 5′-GTAAACCATCCGACAACACCACATTTTTGCTGG-3′.
For the Y259H variant:
sense primer, 5′-CAGCAAAAATGTGGTGTTGTCGGATGG-3′; and antisense primer, 5′-CCATCCGACAACACCACATTTTTGCTG-3′.
For the Y259F variant:
sense primer, 5′-CCAGCAAAAATGTGGTGTTGTCGGATGGTTTACATG-3′; and antisense primer, 5′-CATGTAAACCATCCGACAACACCACATTTTTGCTGG-3′.
For the Y259W variant:
sense primer, 5′-CAGCAAAAATGTGGTGTTGTCGGATGGTTTACATGC-3′; and antisense primer, 5′-GCATGTAAACCATCCGACAACACCACATTTTTGCTG-3′.
For the W257A variant:
sense primer, 5′-GCAAAAATGTGTTACTGTCGGATGGTTTACATG-3′; and antisense primer, 5′-CATGTAAACCATCCGACAGTAACACATTTTTGC-3′.
For the W257V variant:
sense primer, 5′-CCAGCAAAAATGTGTTACTGTCGGATGGTTTAC-3′; and antisense primer, 5′-CATGTAAACCATCCGACAGTAACACATTTTTGC-3′.
For the W257L variant:
sense primer, 5′-CCAGCAAAAATGTGTTACTGTCGGATGG-3′; and antisense primer, 5′-CCATCCGACAGTAACACATTTTTGCTGG-3′.
For the W257F variant:
sense primer, 5′-CCTATGCATCCAGCAAAAATGTGTTACTGTCGGATGG-3′; and antisense primer, 5′-CCATCCGACAGTAACACATTTTTGCTGGATGCATAGG-3′.
For the W257Y variant:
sense primer, 5′-CCTATGCATCCAGCAAAAATGTGTTACTGTCGGATGG-3′; and antisense primer, 5′-CCATCCGACAGTAACACATTTTTGCTGGATGCATAGG.
The cultural conditions of the mutants and measurements of in vivo protein expression levels
The detailed methods have been described in the previous papers.6–8Spectroscopic data of enzymatic products 10, 12, 15 and 17 acetates
![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) 3CO–), 1.89 (1H, m, H-12), 2.24 (2H, dt, J = 7.3, 7.3 Hz, H-23), 2.39 (1H, q-like, J = 6.6 Hz, H-20), 2.52 (1H, bd, J = 14.0 Hz, H-15), 2.87 (1H, bd, J = 10.9 Hz, H-13), 4.81 (1H, dd, J = 11.7, 4.6 Hz, H-3), 5.41 (1H, t, J = 7.0 Hz, H-24), 5.49 (1H, bs, H-16). 13C-NMR (150 MHz, C6D6) δ 16.3 (q, C-19), 16.8 (q, C-29), 17.7 (q, C-27), 18.0 (q, C-30), 18.3 (q, C-18), 18.5 (t, C-6), 19.0 (q, C-21), 20.8 (q,
3CO–), 1.89 (1H, m, H-12), 2.24 (2H, dt, J = 7.3, 7.3 Hz, H-23), 2.39 (1H, q-like, J = 6.6 Hz, H-20), 2.52 (1H, bd, J = 14.0 Hz, H-15), 2.87 (1H, bd, J = 10.9 Hz, H-13), 4.81 (1H, dd, J = 11.7, 4.6 Hz, H-3), 5.41 (1H, t, J = 7.0 Hz, H-24), 5.49 (1H, bs, H-16). 13C-NMR (150 MHz, C6D6) δ 16.3 (q, C-19), 16.8 (q, C-29), 17.7 (q, C-27), 18.0 (q, C-30), 18.3 (q, C-18), 18.5 (t, C-6), 19.0 (q, C-21), 20.8 (q, ![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H3CO–), 22.1 (t, C-11), 23.6 (t, C-12), 24.1 (t, C-2), 25.8 (q, C-26), 26.4 (t, C-23), 28.1 (q, C-28), 33.8 (d, C-20), 35.6 (t, C-7), 36.1 (t, C-22), 37.4 (s, C-10), 38.1 (s, C-4), 38.7 (t, C-1), 38.9 (t, C-15), 40.2 (s, C-8), 48.8 (d, C-13), 51.0 (d, C-9), 53.3 (s, C-14), 56.3 (d, C-5), 80.5 (d, C-3), 121.2 (d, C-16), 125.4 (d, C-24), 131.1 (s, C-25), 150.9 (s, C-17), 169.9 (s, CH3O–). EIMS (%): m/z 69 (100), 95 (63), 107 (60), 121 (45), 147 (43), 161 (57), 189 (58), 204 (47), 365 (28), 383 (25), 393 (28), 408 (30), 453 (40), 468 (M+, 21): see Fig. S2-1.† HRMS (EI): m/z: calcd for C32H52O2: 468.39673; found 468.39641. [α]25D = −32.5 (c = 0.02, CHCl3).
3CO–), 1.90 (1H, m, H-15), 1.95 (1H, m, H-12), 2.46 (1H, bd, J = 15.6 Hz, H-19), 2.78 (1H, m, H-12), 4.85 (1H, dd, J = 11.6, 4.8 Hz, H-3). 13C-NMR (100 MHz, C6D6) δ 16.6 (q, C-25), 16.9 (q, C-24), 17.9 (q, C-26), 18.6 (t, C-6), 20.9 (q, H3CO–), 21.6 (q, C-27), 22.0 (t, C-11), 23.8 (q, C-28), 24.1 (t, C-2), 24.3 (q, C-30), 25.4 (t, C-12), 26.9 (t, C-15), 28.2 (q, C-23), 32.5 (q, C-29), 33.4 (s, C-20), 34.9 (s, C-17), 35.1 (t, C-7), 35.7 (t, C-21), 37.06 (t, C-16), 37.32 (s, C-10), 37.9 (s, C-4), 38.5 (t, C-1), 39.0 (t, C-19), 39.8 (t, C-22), 41.3 (s, C-8), 45.0 (s, C-14), 50.8 (d, C-9), 55.6 (d, C-5), 80.5 (d, C-3), 133.4 (s, C-18), 134.9 (s, C-13), 169.9 (s, CH3O–). EIMS (%): m/z 95 (50), 109 (55), 189 (82), 205 (100), 218 (37), 393 (10), 408 (15), 453 (10), 468 (M+, 28): see Fig. S3-1.† HRMS (EI) m/z (M+): calcd for C32H52O2: calcd 468.39673; found. 468.39641. [α]20D = −17.1 (c = 0.035, CHCl3).
3CO–), 2.66 (2H, m, H-23), 2.73 (1H, m, H-17), 4.48 (1H, dd, J = 10.3, 6.4 Hz, H-3), 5.06 (1H, t, J = 7.2 Hz, H-24), 5.15 (1H, t, J = 7.2 Hz, H-22). 13C-NMR (100 MHz, CDCl3) δ 15.9 (q, C-30), 16.3 (q, C-19), 16.5 (q, C-29), 17.7 (q, C-27), 18.2 (t, C-6), 19.0 (q, C-21), 21.3 (q, H3CO–), 21.5 (t, C-11), 23.7 (t, C-2), 24.9 (t, C-12), 25.7 (q, C-26), 26.4 (t, C-23), 26.5 (t, C-16), 28.0 (q, C-28), 31.8 (t, C-15), 35.4 (t, C-7), 37.1 (s, C-10), 37.9 (s, C-4), 38.8 (t, C-1), 40.5 (s, C-8), 41.2 (d, C-17), 15.6 (q, C-18), 44.4 (d, C-13), 49.3 (s, C-14), 50.9 (d, C-9), 56.0 (d, C-5), 80.9 (d, C-3), 123.8 (d, C-24), 124.8 (d, C-22), 131.0 (s, C-25), 136.9 (s, C-20), 171.0 (s, CH3O–). EIMS (%): m/z 69 (41), 81 (38), 93 (100), 109 (45), 121 (32), 136 (48), 189 (55), 468 (M+, 18): see Fig. S4-1.† HRMS (EI) m/z (M+): calcd for C32H52O2: 468.39673; found. 468.39780. [α]20D = 63.3 (c = 0.035, CHCl3).
3CO-), 2.21 (1H, bd, J = 12.5 Hz, H-12), 2.45 (2H, m, H-23), 2.47 (1H, m, H-22), 2.49 (1H, m, H-15), 2.57 (1H, m, H-22), 3.05 (1H, dd, J = 13.0, 3.2 Hz, H-13), 4.80 (1H, dd, J = 11.6, 4.8 Hz, H-3), 5.14 (1H, s, H-21), 5.23 (1H, s, H-21), 5.42 (1H, very broad s, H-24), 5.84 (1H, bs, H-16). 13C-NMR (100 MHz, C6D6) δ 16.1 (q, C-30), 16.3 (q, C-19), 16.8 (q, C-29), 17.8 (q, C-27), 18.3 (q, C-18), 18.5 (t, C-6), 20.9 (q, H3CO–), 22.0 (t, C-11), 24.0 (t, C-12), 24.1 (t, C-2), 25.8 (q, C-26), 28.0 (t, C-23), 28.1 (q, C-28), 35.4 (t, C-22), 35.5 (t, C-7), 37.2 (s, C-10), 38.1 (s, C-4), 38.6 (t, C-1), 39.0 (t, C-15), 40.1 (s, C-8), 48.4 (d, C-13), 50.6 (d, C-9), 53.4 (s, C-14), 56.2 (d, C-5), 80.4 (d, C-3), 110.5 (t, C-21), 124.9 (d, C-24), 127.1 (d, C-16), 131.3 (s, C-25), 145.7 (s, C-20), 146.2 (s, C-17), 170.0 (s, CH3O–). The assignments may be exchangeable between C-2 and C-12, and between C-18 and C-29. EIMS (%): m/z 69 (97), 159 (100), 189 (78), 363 (40), 423 (63), 466 (M+, 11): see Fig. S5-1.† HRMS (EI) m/z (M+): calcd for C32H50O2: 466.38108; found. 466.38168. Optical rotation was not determined due to the significant loss of the sample during handling.
H3CO–), 22.1 (t, C-11), 23.6 (t, C-12), 24.1 (t, C-2), 25.8 (q, C-26), 26.4 (t, C-23), 28.1 (q, C-28), 33.8 (d, C-20), 35.6 (t, C-7), 36.1 (t, C-22), 37.4 (s, C-10), 38.1 (s, C-4), 38.7 (t, C-1), 38.9 (t, C-15), 40.2 (s, C-8), 48.8 (d, C-13), 51.0 (d, C-9), 53.3 (s, C-14), 56.3 (d, C-5), 80.5 (d, C-3), 121.2 (d, C-16), 125.4 (d, C-24), 131.1 (s, C-25), 150.9 (s, C-17), 169.9 (s, CH3O–). EIMS (%): m/z 69 (100), 95 (63), 107 (60), 121 (45), 147 (43), 161 (57), 189 (58), 204 (47), 365 (28), 383 (25), 393 (28), 408 (30), 453 (40), 468 (M+, 21): see Fig. S2-1.† HRMS (EI): m/z: calcd for C32H52O2: 468.39673; found 468.39641. [α]25D = −32.5 (c = 0.02, CHCl3).
3CO–), 1.90 (1H, m, H-15), 1.95 (1H, m, H-12), 2.46 (1H, bd, J = 15.6 Hz, H-19), 2.78 (1H, m, H-12), 4.85 (1H, dd, J = 11.6, 4.8 Hz, H-3). 13C-NMR (100 MHz, C6D6) δ 16.6 (q, C-25), 16.9 (q, C-24), 17.9 (q, C-26), 18.6 (t, C-6), 20.9 (q, H3CO–), 21.6 (q, C-27), 22.0 (t, C-11), 23.8 (q, C-28), 24.1 (t, C-2), 24.3 (q, C-30), 25.4 (t, C-12), 26.9 (t, C-15), 28.2 (q, C-23), 32.5 (q, C-29), 33.4 (s, C-20), 34.9 (s, C-17), 35.1 (t, C-7), 35.7 (t, C-21), 37.06 (t, C-16), 37.32 (s, C-10), 37.9 (s, C-4), 38.5 (t, C-1), 39.0 (t, C-19), 39.8 (t, C-22), 41.3 (s, C-8), 45.0 (s, C-14), 50.8 (d, C-9), 55.6 (d, C-5), 80.5 (d, C-3), 133.4 (s, C-18), 134.9 (s, C-13), 169.9 (s, CH3O–). EIMS (%): m/z 95 (50), 109 (55), 189 (82), 205 (100), 218 (37), 393 (10), 408 (15), 453 (10), 468 (M+, 28): see Fig. S3-1.† HRMS (EI) m/z (M+): calcd for C32H52O2: calcd 468.39673; found. 468.39641. [α]20D = −17.1 (c = 0.035, CHCl3).
3CO–), 2.66 (2H, m, H-23), 2.73 (1H, m, H-17), 4.48 (1H, dd, J = 10.3, 6.4 Hz, H-3), 5.06 (1H, t, J = 7.2 Hz, H-24), 5.15 (1H, t, J = 7.2 Hz, H-22). 13C-NMR (100 MHz, CDCl3) δ 15.9 (q, C-30), 16.3 (q, C-19), 16.5 (q, C-29), 17.7 (q, C-27), 18.2 (t, C-6), 19.0 (q, C-21), 21.3 (q, H3CO–), 21.5 (t, C-11), 23.7 (t, C-2), 24.9 (t, C-12), 25.7 (q, C-26), 26.4 (t, C-23), 26.5 (t, C-16), 28.0 (q, C-28), 31.8 (t, C-15), 35.4 (t, C-7), 37.1 (s, C-10), 37.9 (s, C-4), 38.8 (t, C-1), 40.5 (s, C-8), 41.2 (d, C-17), 15.6 (q, C-18), 44.4 (d, C-13), 49.3 (s, C-14), 50.9 (d, C-9), 56.0 (d, C-5), 80.9 (d, C-3), 123.8 (d, C-24), 124.8 (d, C-22), 131.0 (s, C-25), 136.9 (s, C-20), 171.0 (s, CH3O–). EIMS (%): m/z 69 (41), 81 (38), 93 (100), 109 (45), 121 (32), 136 (48), 189 (55), 468 (M+, 18): see Fig. S4-1.† HRMS (EI) m/z (M+): calcd for C32H52O2: 468.39673; found. 468.39780. [α]20D = 63.3 (c = 0.035, CHCl3).
3CO-), 2.21 (1H, bd, J = 12.5 Hz, H-12), 2.45 (2H, m, H-23), 2.47 (1H, m, H-22), 2.49 (1H, m, H-15), 2.57 (1H, m, H-22), 3.05 (1H, dd, J = 13.0, 3.2 Hz, H-13), 4.80 (1H, dd, J = 11.6, 4.8 Hz, H-3), 5.14 (1H, s, H-21), 5.23 (1H, s, H-21), 5.42 (1H, very broad s, H-24), 5.84 (1H, bs, H-16). 13C-NMR (100 MHz, C6D6) δ 16.1 (q, C-30), 16.3 (q, C-19), 16.8 (q, C-29), 17.8 (q, C-27), 18.3 (q, C-18), 18.5 (t, C-6), 20.9 (q, H3CO–), 22.0 (t, C-11), 24.0 (t, C-12), 24.1 (t, C-2), 25.8 (q, C-26), 28.0 (t, C-23), 28.1 (q, C-28), 35.4 (t, C-22), 35.5 (t, C-7), 37.2 (s, C-10), 38.1 (s, C-4), 38.6 (t, C-1), 39.0 (t, C-15), 40.1 (s, C-8), 48.4 (d, C-13), 50.6 (d, C-9), 53.4 (s, C-14), 56.2 (d, C-5), 80.4 (d, C-3), 110.5 (t, C-21), 124.9 (d, C-24), 127.1 (d, C-16), 131.3 (s, C-25), 145.7 (s, C-20), 146.2 (s, C-17), 170.0 (s, CH3O–). The assignments may be exchangeable between C-2 and C-12, and between C-18 and C-29. EIMS (%): m/z 69 (97), 159 (100), 189 (78), 363 (40), 423 (63), 466 (M+, 11): see Fig. S5-1.† HRMS (EI) m/z (M+): calcd for C32H50O2: 466.38108; found. 466.38168. Optical rotation was not determined due to the significant loss of the sample during handling.
Acknowledgements
This work was supported in part by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, no. 25450150 (C) and 18380001(B).References
- Review.
(a) K. U. Wendt, G. E. Schulz, E. J. Corey and D. R. Liu, Angew. Chem., Int. Ed., 2000, 39, 2812–2833 CrossRef CAS
; (b) T. Hoshino and T. Sato, Chem. Commun., 2002, 291–301 RSC
- R. Xu, G. C. Fazio and S. P. T. Matsuda, Phytochemistry, 2004, 65, 261–291 CrossRef CAS PubMed
-
(a) A. Eschenmoser, L. Ruzicka, O. Jeger and D. Arigoni, Helv. Chim. Acta, 1955, 38, 1890–1940 CrossRef CAS
-
(a) C. H. Chang, H. Y. Wen, W. S. Shie, C. T. Liu, M. E. Li, Y. T. Liu, W. H. Li and T. K. Wu, Org. Biomol. Chem., 2013, 11, 4214–4219 RSC
-
(a) N. Morikubo, Y. Fukuda, K. Ohtake, N. Shinya, D. Kiga, K. Sakamoto, M. Asanuma, H. Hirota, S. Yokoyama and T. Hoshino, J. Am. Chem. Soc., 2006, 128, 13184–13194 CrossRef CAS PubMed
- R. Ito, Y. Masukawa and T. Hoshino, FEBS J., 2013, 280, 1267–1280 CrossRef CAS PubMed
- R. Ito, I. Hashimoto, Y. Masukawa and T. Hoshino, Chem. – Eur. J., 2013, 19, 17150–17158 CrossRef CAS PubMed
- R. Ito, Y. Masukawa, C. Nakada, K. Amari, C. Nakano and T. Hoshino, Org. Biomol. Chem., 2014, 12, 3836–3846 CAS
- T. Kushiro, M. Shibuya, K. Masuda and Y. Ebizuka, J. Am. Chem. Soc., 2000, 122, 6816–6824 CrossRef CAS
- Substrate analog experiments for β-amyrin synthase.
(a) I. Abe, Y. Sakano, H. Tanaka, W. Lou, H. Noguchi, M. Shibuya and Y. Ebizuka, J. Am. Chem. Soc., 2004, 126, 3426–3427 CrossRef CAS PubMed
-
(a) T. K. Wu, Y. C. Chang, Y. T. Liu, C. H. Chang, H.-Y. Wen, W.-H. Li and W. S. Shie, Org. Biomol. Chem., 2011, 9, 1092–1097 RSC
- Z.-h. Lin, H.-x. Long, Z. Bo, Y.-q. Wang and Y.-z. Wu, Peptides, 2008, 29, 1798–1805 CrossRef CAS PubMed
- S. Mecozzi, A. P. West Jr. and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10566–10571 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Amino acid alignment of oxidosqualene cyclases, spectroscopic data of enzymatic products, homology modeling, GC profiles of enzymatic products produced by F413X mutants, and the relative enzymatic activities of various mutants. See DOI: 10.1039/c6ob02539k |
| This journal is © The Royal Society of Chemistry 2017 |