
Pseudopeptidic compounds for the generation of dynamic combinatorial libraries of chemically diverse macrocycles in aqueous media†
Joan
Atcher
,
Jordi
Solà
 * and
Ignacio
Alfonso
*
* and
Ignacio
Alfonso
*
Department of Biological Chemistry and Molecular Modelling, IQAC-CSIC, Jordi Girona, 18-26, 08034, Barcelona, Spain. E-mail: jordi.sola@iqac.csic.es; ignacio.alfonso@iqac.csic.es
First published on 22nd November 2016
Abstract
A straightforward four-step synthesis leads to the preparation of C2-symmetric dithiols containing a central aromatic core and amino acid side chains. These building blocks allow the preparation of dynamic covalent libraries of pseudopeptidic macrocycles in aqueous media that cover a broad range of polarities, functional groups and bulkiness mirroring the diversity found in natural peptides. The versatility of the generated dynamic libraries has been illustrated by the amplification of two different members from the same library upon the action of two biologically relevant templates.
Introduction
The generation of new, dynamic systems has attracted the attention of researchers and has shown a great potential in a plethora of areas.1 Since the early reports by the groups of Lehn and Sanders, Dynamic Covalent Chemistry (DCvC) has demonstrated an enormous utility to synthesize structurally complex compounds, especially interlocked and multicomponent assemblies, usually under thermodynamic control.2 Thus, fascinating structures such as helicates3 were initially reported by template-assisted synthesis and, more importantly, the new methodology paved the way to the generation of more complex and more beautiful structures such as catenanes4 and knots.5 On the other hand, the combination of building blocks (BBs) by reversible covalent bonds allows the generation of libraries of products, usually (but not exclusively) cyclic structures with different levels of complexity. Playing with different BBs in a combinatorial approach led to the new research area of Dynamic Combinatorial Chemistry (DCC). Thanks to the dynamic nature of their bonds, these mixtures of exchanging compounds, the Dynamic Covalent Libraries (DCLs), can respond to external stimuli altering the different proportions of final structures.6 Apart from being a methodology to synthesize molecular receptors,7 the study of dynamic systems has evolved as an area of research itself within the sub-discipline of Systems Chemistry and it has important implications in diverse research fields, from chemical biology to the chemistry of materials.8 More recently, a special interest has been focused on the study of out-of-equilibrium assemblies which can allow reaching different states of the system.9 Importantly, life operates far from equilibrium and its extraordinary complicated molecular machinery operates in a kinetically controlled regime and, therefore, the study of such non-thermodynamically controlled systems has obvious implications in pre-biotic chemistry.One of the many possibilities that DCC offers is to deliver materials that can interact with biologically relevant compounds. DCC methods have been used to synthesize molecular receptors,7 catalysts10 or sensors.11 Among different reversible bonds imine,12 hydrazone13 or disulphide14 bond formation and exchange have been widely used in DCC. However, when addressing challenges related to biological chemistry, organic chemists usually face the difficulty to work in aqueous media. Sanders and Otto,15 Ravoo,16 our group17 and others18 have employed the formation and metathesis of disulphide bonds, since they take place spontaneously in water at slightly basic pH. The addition of an oxidant like DMSO as co-solvent allows working at even slightly acidic pHs and significantly accelerates the disulphide formation and exchange reactions.19 However, the solubility of organic compounds in water is usually very limited and narrows the toolkit of compounds available for chemists. This limitation conditions the development of even minimalistic new experiments and, therefore, there is a need for the synthesis of compounds which are soluble in aqueous conditions and present the characteristics necessary for the formation of reversible covalent bonds in aqueous media.
The synthesis of molecules containing amino acids allows taking advantage of the vast diversity of functionalities present in these building blocks.20 Pseudopeptides are molecules incorporating natural amino acids and non-natural structural parts21 and they have been widely used for the development of compounds with many important applications. Examples are reported in which pseudopeptidic backbones are used for preparing structured and stimuli-responsive materials,22 peptide nucleic acids (PNAs),23 catalysts,24 and bio-active compounds.25 Additionally, pseudopeptides have also been used for the development of molecular receptors for a variety of templates including anions, inorganic cations and peptides.26
We previously reported the use of pseudopeptidic dithiols in dynamic covalent libraries.17 In this article we report the synthetic procedure for the formation of twelve pseudopeptidic dithiols (4a–l in Schemes 1–3) that were designed in order to be used as bipodal building blocks (BBs) for the generation of disulphide-based dynamic combinatorial libraries with minimalistic peptide-like structural information. The structure of the BBs is based on a common C2-symmetric scaffold consisting of a central m-phenylenediamine that joins two identical arms, each formed by an amino acid residue. At the N-terminus of the two amino acids, dithiols 4a–k incorporate a mercaptoacetyl moiety. Our BBs suppose a new addition to the quantity and variety of molecular Lego pieces available for chemists working in the field. All of them were conceived with a common chemical backbone, with the only exception of 4l where the use of cysteine allows a simpler, more compact, structure.
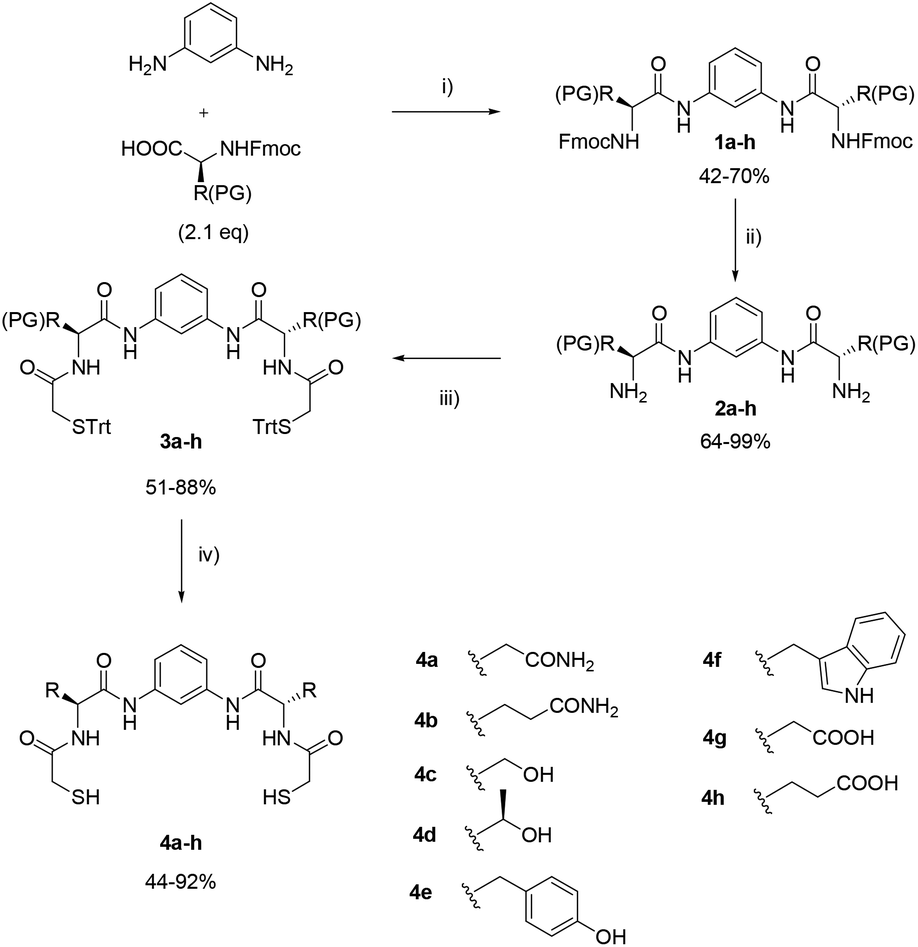 | ||
| Scheme 1 Synthetic route to dithiols 4a–h. Reagents and conditions: (i) dry DMF, DCCD (2.4 eq.), HOBt (2.4 eq.), DIPEA (4.7 eq.), 60 h; (ii) 20% piperidine in DMF; (iii) dry DMF, tritylsulfanyl acetic acid (2.1 eq.), EDC (2.4 eq.), HOBt (2.4 eq.), DIPEA (4.7 eq.), 24–48 h; (iv) CH2Cl2: TFA, TIS (8 eq.), EDT (12 eq.). | ||
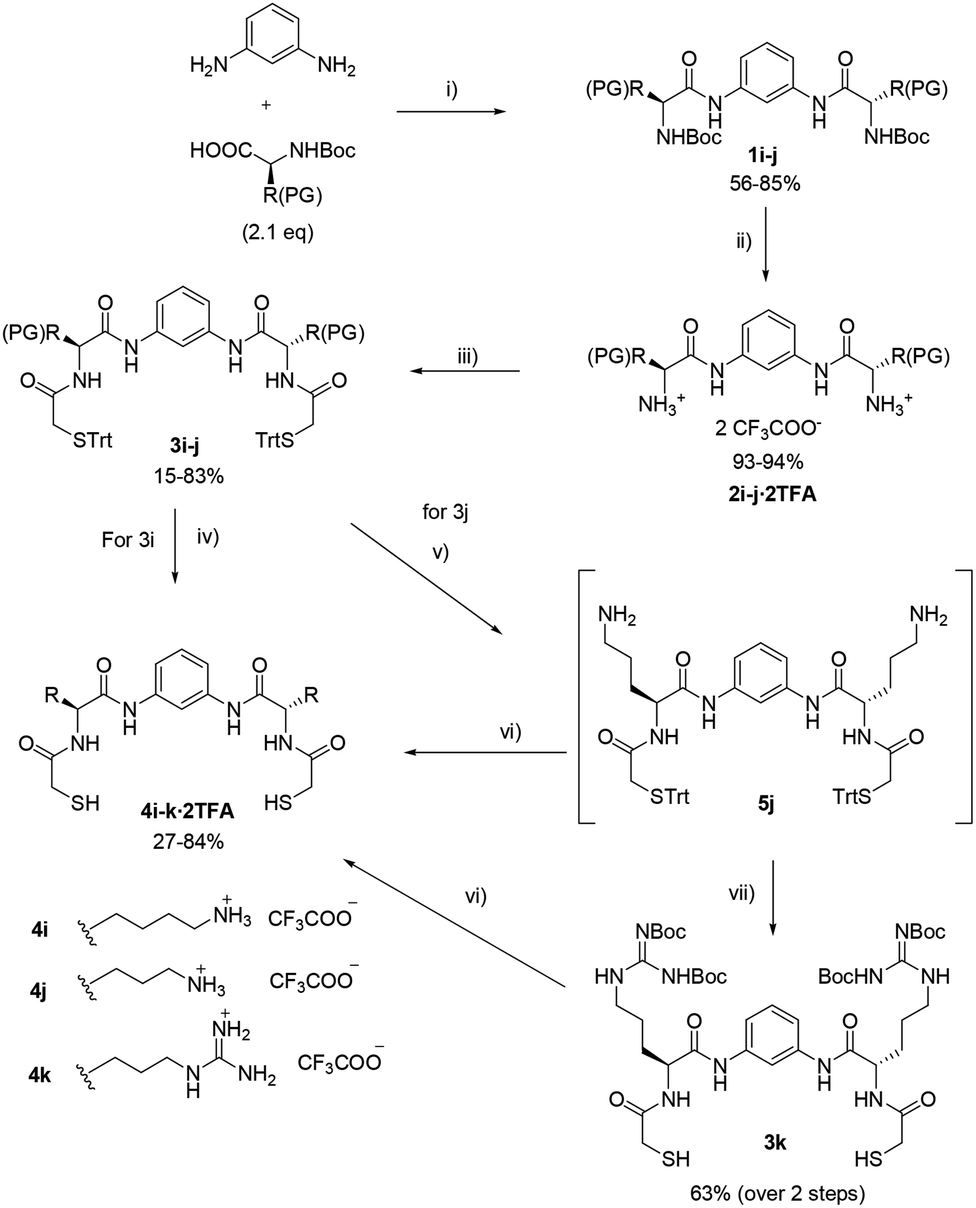 | ||
| Scheme 2 Synthetic route to dithiols 4i–k. Reagents and conditions: (i) dry DMF, HBTU (2.4 eq.), DIPEA (4.7 eq.); (ii) TFA, CH2Cl2; (iii) dry DMF, tritylsulfanyl acetic acid (2.1 eq.), EDC (2.4 eq.), HOBt (2.4 eq.), DIPEA (4.7 eq.), 24–48 h; (iv) HBr/CH3COOH, CH2Cl2, TIS (8 eq.); (v) CH2Cl2, PhSiH3 (24 eq.), Pd(PPh3)4 (0.13 eq.); (vi) CH2Cl2: TFA, TIS (8 eq.), EDT (12 eq.); (vii) CH2Cl2, triethylamine (3.2 eq.), 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine (3.0 eq.). | ||
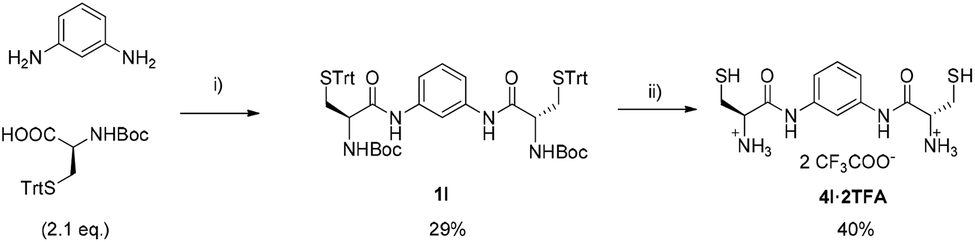 | ||
| Scheme 3 Synthetic route to dithiol 4l: (i) dry DCM, DCCD (2.4 eq.), HOBt (2.4 eq.), 60 h. (ii) CH2Cl2: TFA, TIS (8 eq.), EDT (12 eq.). | ||
Results and discussion
Synthesis of the building blocks
The C2-symmetric dithiols described herein are easily accessible by standard organic synthesis procedures and present good solubility in water or water-organic solvent mixtures. The BBs cover a broad range of polarities, incorporating negative, positive or neutral charges at physiological pH. Besides, different functional groups, such as alcohol, amine, guanidine, carboxylic acid, amide, phenol and indol can be included in the structures thanks to the chemical diversity of the natural amino acids.The rigid geometry of the m-phenylenediamine predisposes the components to the formation of macrocycles (mostly dimers), preventing the formation of the monomeric disulphides, i.e. the products of the intramolecular cyclization. The synthesis of the dithiol building blocks was carried out using two different protecting group strategies. For most of the compounds (4a–h) an Fmoc-protected amino acid was used with acid-cleavable protecting groups on the side chains (Scheme 1).
Thus, after diacylation of m-phenylenediamine with the corresponding amino acids using dicyclohexylcarbodiimide and hydroxybenzotriazole (DCCD/HOBt) as coupling reagents and dry DMF as solvent, compounds 1a–h were obtained. Fmoc-removal was carried out with 20% (v/v) piperidine in dry DMF yielding amines 2a–h. The thiol groups were incorporated as trityl-protected thioglycolic acid with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and HOBt as coupling reagents in dry DMF, to obtain intermediates 3a–h. Final deprotection using trifluoroacetic acid (TFA) in dichloromethane and triisobutylsilane/ethylenedithiol (TIS/EDT) as scavengers afforded final compounds 4a–h. The final products were purified by reverse phase chromatography in acidic water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile mixtures when necessary. The identity of the protecting groups used in this sequence was chosen according to orthogonality requirements and it is disclosed in Table 1.
:acetonitrile mixtures when necessary. The identity of the protecting groups used in this sequence was chosen according to orthogonality requirements and it is disclosed in Table 1.
| Entry | Compound | PG | Amino acid |
|---|---|---|---|
| 1 | 1a, 2a, 3a | Trt | Asn |
| 2 | 1b, 2b, 3b | Trt | Gln |
| 3 | 1c, 2c, 3c | t Bu | Ser |
| 4 | 1d, 2d, 3d | t Bu | Thr |
| 5 | 1e, 2e, 3e | t Bu | Tyr |
| 6 | 1f, 2f, 3f | Boc | Trp |
| 7 | 1g, 2g, 3g | t Bu | Asp |
| 8 | 1h, 2h, 3h | t Bu | Glu |
For building blocks 4i–k different strategies were employed depending on the nature of the corresponding amino acid functionalities. All their syntheses began with the coupling of m-phenylenediamine to an N-Boc protected amino acid (Scheme 2). Removal of the Boc protecting group conducted to diamines 2i–j that were diacylated with trityl protected thioglycolic acid to yield intermediates 3i–j. For final deprotection, an acidic cleavage using scavengers TIS/EDT was used. For lysine derivative with the side chain protected with a Cbz-group an HBr/CH3COOH solution was used together with TIS. Compound 4i was isolated as TFA salt after reverse phase chromatography (eluent: mixtures of H2O and MeCN containing 0.1% and 0.07% TFA respectively).
The ornithine derivative 4j required deprotection in two steps, firstly side-chain allyl group was removed by using phenylsilane and Pd(PPh3)4 catalyst to yield compound 5j (which was not purified), followed by acidic removal of the trityl group yielded the Orn-derivative 4j as TFA salt. This route also allowed the synthesis of the Arg-derivative using 5j as starting material which was reacted with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine to furnish Boc-protected guanidine 3k. Conventional deprotection employing TFA and TIS/EDT yielded 4k as TFA salt. Preparation of compound 4k by alternative routes proved troublesome. Again, the choice of protecting groups was realised according to orthogonality taking into account the corresponding amino acid side chains. Table 2 shows the combination of protecting groups employed for the synthesis of compounds bearing basic side chains.
| Entry | Compound | PG | Amino acid |
|---|---|---|---|
| 1 | 1i, 2i, 3i | Cbz | Lys |
| 2 | 1j, 2j, 3j | Alloc | Orn |
| 3 | 3k | Boc | Arg |
Finally, cysteine derivative 4l was synthesised in only two steps as the amino acid already contains the thiol functionality and so the incorporation of the thioglycolic moiety is not necessary. Thus, after diacylation of m-phenylenediamine with Boc-protected cysteine bearing a trityl group on the side chain, an acidic cleavage of the protecting groups directly furnished 4l (also as TFA salt, Scheme 3) with a slightly different scaffold than other thiols.
All the synthesised dithiol building blocks 4a–l were fully characterised by NMR and mass spectrometry. Hence, we have set up a robust synthetic methodology that gives access to C2-symmetric pseudopeptide-dithiols with a broad range of functionalities thanks to amino acid side chain diversity. Thus, very polar compounds at neutral pH such as protonated amines and guanidines or anionic carboxylates are available. Other compounds of intermediate polarity like amide or alcohol are also accessible. Finally, hydrophobic side chains like those derived from aromatic amino acids (Trp and Tyr) can also be easily prepared.
Oxidation of the building blocks in aqueous mixtures
To demonstrate this broad range of functional groups that results in a vast variability in molecular diversity, we oxidised each of the BBs individually from 1 mM samples. To ensure complete solubility we tested different solvent mixtures containing 20% of DMSO and buffered water (bis-Tris pH 6.5 to have a final concentration of 50 mM) and acetonitrile as co-solvents. Most building blocks investigated were soluble in mixtures containing 20–60% buffer and 60–20% acetonitrile. However, Trp-derivate 4f precipitated at mixtures containing more than 40% water. For this reason we established 40% buffer, 40% acetonitrile and 20% DMSO as ideal conditions to work with all BBs available. Under these conditions, after 8 days of reaction at room temperature, complete oxidation of the BBs was observed (HPLC monitoring) that rendered the corresponding macrocyclic dimers (as inferred by UPLC-MS analysis). The minor formation of the corresponding trimers could be also detected in some cases (Scheme 4).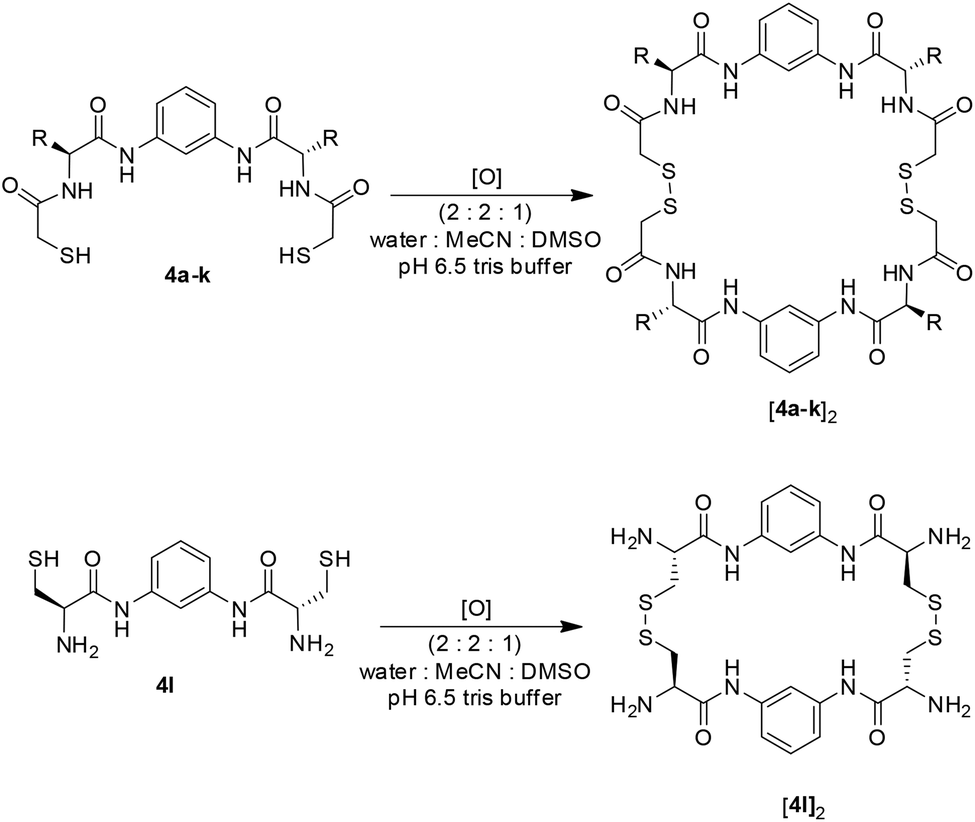 | ||
| Scheme 4 Oxidation of BBs 4a–l to form the corresponding cyclic dimers. | ||
In Fig. 1 we show the overlapped HPLC traces of the individual dimeric species using the same solvent gradient to illustrate the variety in functionalities of the amino acid derivatives. As can be easily appreciated from the traces, a broad range of polarities were observed. At short retention times positively charged species are eluted (the HPLC analysis is performed in acidic media). The most polar component is [4l]2 which presents a smaller and more compact structure, with a higher positive charge density. It is followed by the components having Orn, Lys and Arg side chains ([4j]2, [4i]2 and [4k]2 respectively). In the middle of the HPLC traces the polar and neutral compounds are eluted (those having Gln, Asn or Ser residues) followed by the carboxylic acid-side chain species (Glu, Asp) that are neutral under the HPLC analysis conditions, but anionic in the reaction media. The more hydrophobic Thr-based compound [4d]2 appears between the two carboxylic acid derivatives. Finally, at high percentage of acetonitrile the highly hydrophobic Tyr- and Trp-based compounds are eluted (the latter nearly at 100% MeCN).
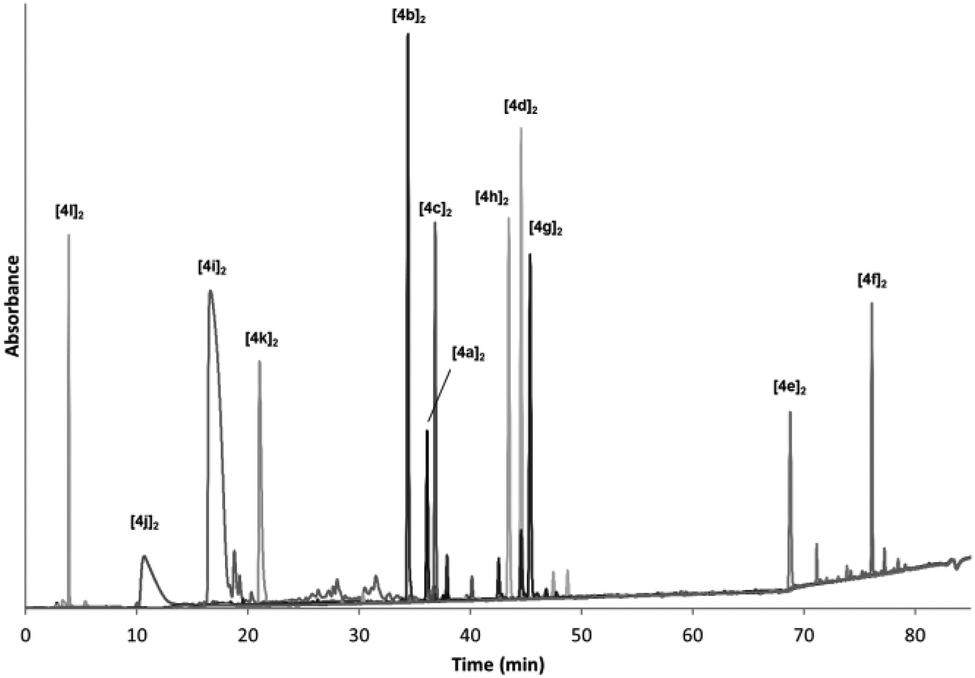 | ||
| Fig. 1 Overlapped RP-HPLC (H2O:MeCN, 20 mM formic acid) traces of individual BBs after oxidation to form the respective dimers (C18, 8 min at 2% MeCN, then linear gradient from 2% to 40% over 57 min and then linear gradient to 100% over 15 min). Vertical axis shows absorbance in arbitrary units. | ||
Finally, to illustrate the utility of the described building blocks as components for the generation of complex DCLs, different sets of mixtures of BBs were prepared that show the ability to generate molecular and functional diversity. In a first mixture, a positively charged derivative 4j, a neutral compound 4d and the two hydrophobic components 4e and 4f were included at 0.5 mM concentration of each BB in the reaction conditions described above (40% buffer, 40%, MeCN, 20% DMSO). Under these conditions all of the 10 expected dimers can be observed (as evidenced by UPLC-MS analysis) plus two of the possible trimers. Other very minor peaks could not be unambiguously identified by UPLC-MS (Fig. 2).
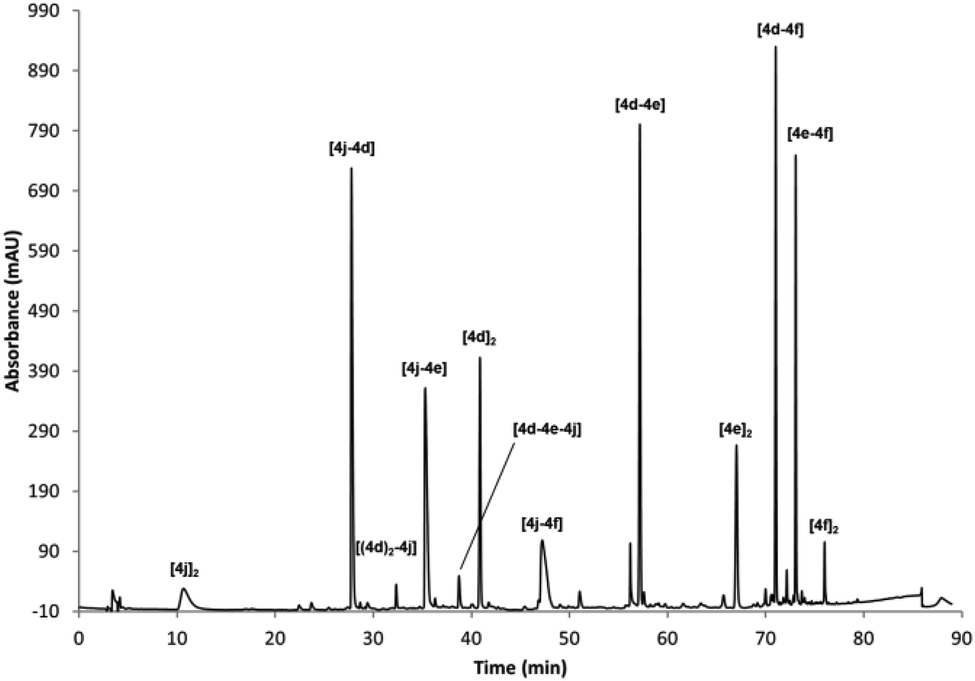 | ||
| Fig. 2 RP-HPLC traces of the oxidised mixture of 4d, 4e, 4f and 4j (0.5 mM each) in 40% buffer (pH 6.5), 40% MeCN, 20% DMSO. | ||
A different DCL was generated using the same conditions by mixing BBs 4b, 4g, 4e and 4l (Fig. 3). This reaction rendered more complex HPLC traces, where only nine dimers could be identified. The homodimer of the Cys-based compound [4l]2 was not formed under these reaction conditions, probably because it is strongly disfavoured as it has a higher charge density. All the other possible dimers were identified, but in this case no trimers were unambiguously assigned although some minor compounds could not be identified by UPLC-MS. According to our own experience Asp residues proved troublesome in some cases forming undesired compounds due to side reactions (i.e. formation of aspartimides).
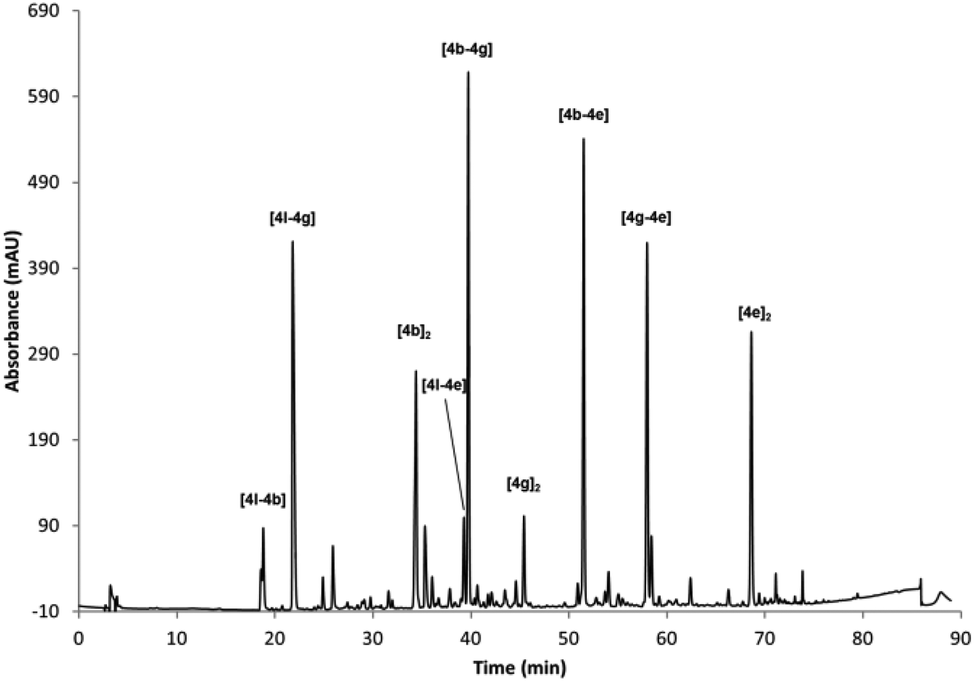 | ||
| Fig. 3 RP-HPLC traces of the oxidised mixture of 4b, 4g, 4e and 4l (0.5 mM each) in 40% buffer (pH 6.5), 40%, MeCN, 20% DMSO. | ||
Response to biologically relevant stimuli
Once the conditions for the efficient generation of dynamic libraries of pseudopeptidic macrocycles were established, we turned our attention to the effect of biologically relevant stimuli. To this aim we designed a library capable to respond to the presence of compounds with very different structures and charge (Fig. 4). We chose the linear polycationic spermine alkaloid and a cyclic polyanionic sugar-like molecule such as phytic acid, two relevant and structurally dissimilar compounds as benchmarks to test the potential of our systems. In order to produce a library that could respond to these stimuli, we chose a positively charged side chain (4i; Lys derivative), a negatively charged compound (4h; Glu), a hydrophobic functionality (4e; Tyr) and a neutral polar group capable of establishing hydrogen bond interactions (4d; Thr). Using the reaction conditions described earlier a new library was formed (Fig. 4a). In this new library the main species were the neutral dimers (hetero or homodimers) and the heterodimer combining Glu and Lys ([4h–4i]) side chains, which compensate opposite charges. Once characterised the outcome of the new library, we analysed the effect of the chosen stimuli to this dynamic mixture. Starting from the same stock solution different amounts of either spermine or phytic acid were used in the reaction mixtures (from 0.125 mM to 10 mM of the templates, Fig. S80–S81 in ESI†) and the response was analysed by HPLC. In the case of spermine, non-surprisingly, the homodimer of glutamic acid [4h]2 was found to be amplified from the mixture by a 3.5 factor (10 mM of spermine in Fig. 4b, also see Fig. S80 in the ESI†), which suggests an electrostatic interaction between the tetranionic macrocycle and the tetracationic spermine (see below). However, a different response was obtained when phytic acid was added to the same library: increasing amounts of phytate induced an amplification of the threonine homodimer [4d]2 (1.6-fold for 10 mM phytate in Fig. 4c, also see Fig. S81 in the ESI†). This observation is consistent with other responses found in dynamic mixtures of Thr-containing peptides, where the presence of anionic carbohydrates amplified this structural motif.16 In both cases the identity of the amplified compounds was unequivocally confirmed by ESI-MS and by adding true samples of the corresponding dimers to the equilibrated library, and analysing the outcome by HPLC. Thus, different species were amplified from the same library upon the action of different templates.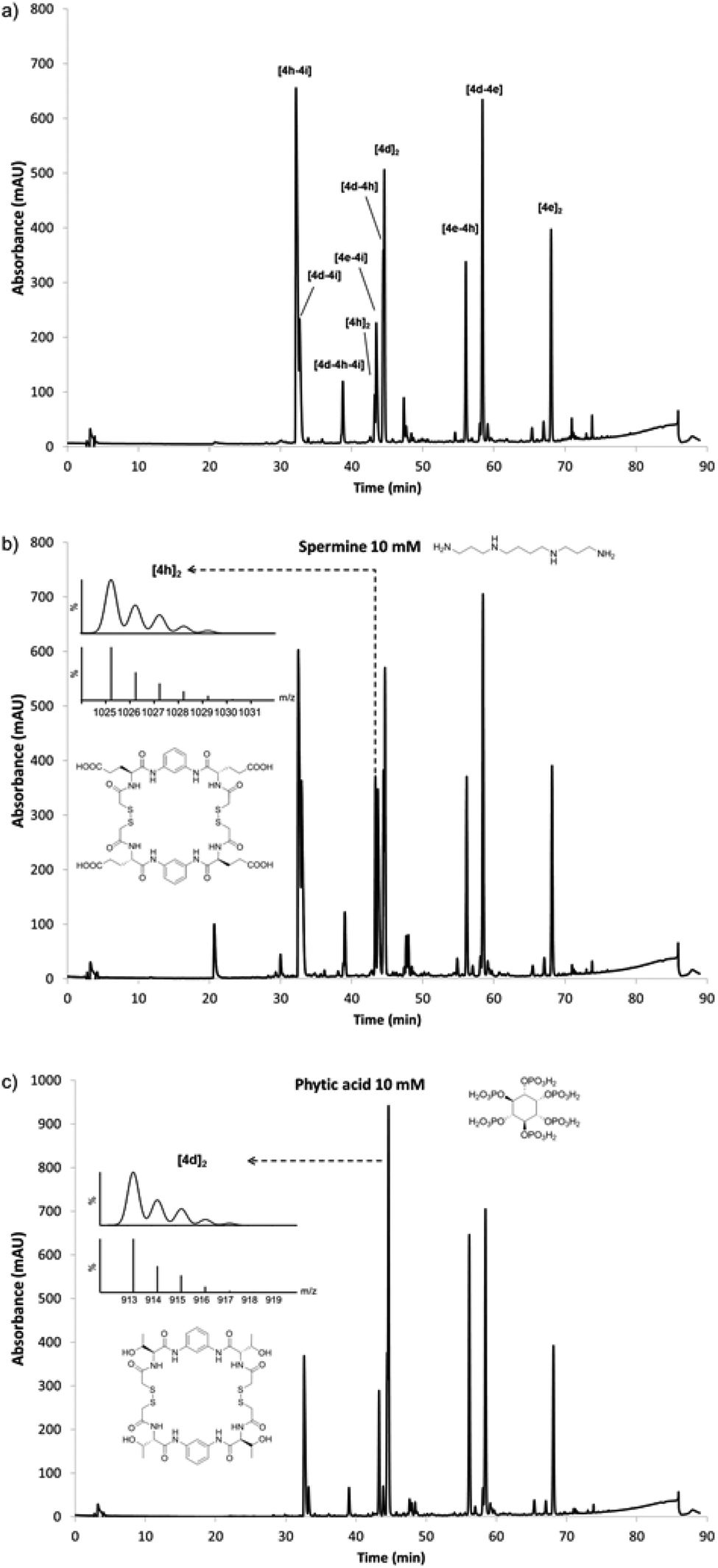 | ||
| Fig. 4 Response of a library composed of 4d, 4e, 4h and 4i to the presence of different compounds. (a) Library at pH 6.5 in the absence of stimuli. (b) Library distribution in the presence of 10 mM of spermine showing the amplified species. (c) Library distribution in the presence of 10 mM of phytic acid highlighting the amplified species. The insets show the experimental and simulated ESI-MS spectra of the corresponding peaks. | ||
The interaction between the [4h]2 dimer and spermine was qualitatively supported by 1H NMR (500 MHz, 4:4:2 H2O:CD3CN:DMSO-d6, d11-Tris buffer pH 7.0, 298 K).27 When a sample containing the Glu-derived macrocycle was treated with an excess of spermine several proton signals were perturbed (Fig. 5 and ESI† for details). Thus, the two amide NH proton signals (NHa and NHb) were downfield shifted, while the methylene vicinal to the carboxylate of the side chains (Hγ) moved upfield. Regarding the aromatic protons, the singlet H1 was deshielded while the other signals (H2,3) were slightly shielded. These changes confirmed the host–guest interaction between the [4h]2 macrocycle and the polyamine.
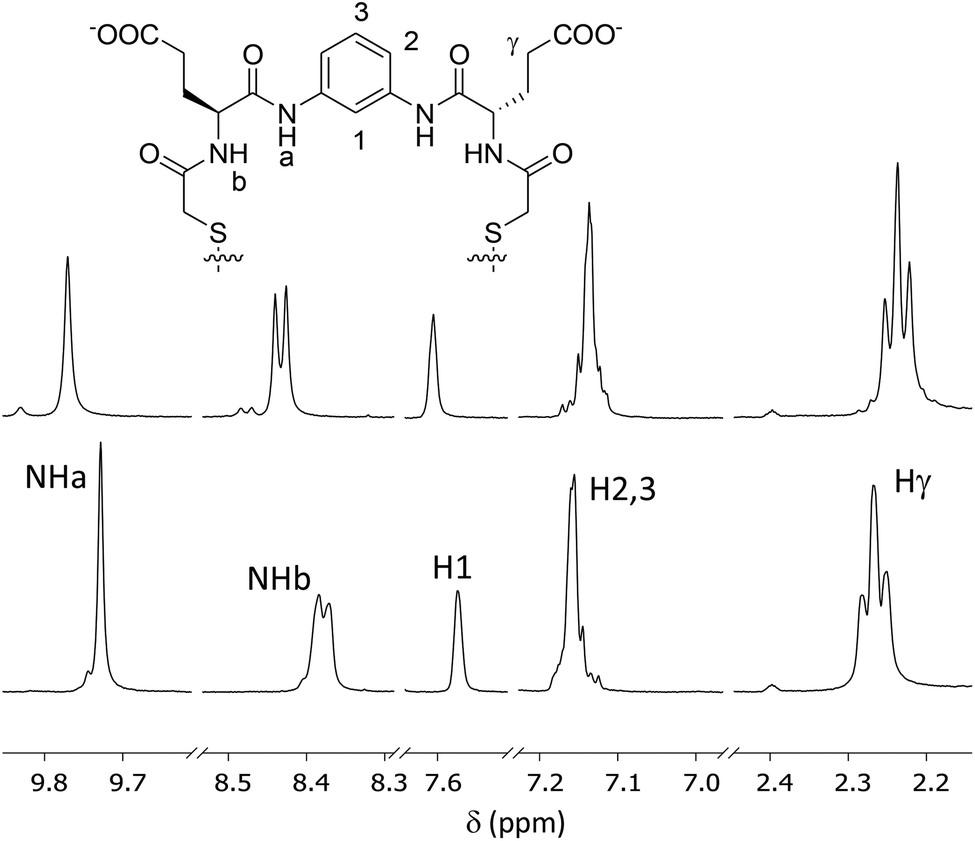 | ||
| Fig. 5 Partial 1H NMR spectra (500 MHz, 4:4:2 H2O:CD3CN:DMSO-d6, d11-Tris buffer pH 7.0, 298 K) of [4h]2 alone and upon the addition of 20 equivalent of spermine (upper trace). Proton assignment of arbitrarily numbered proton signals is also shown. | ||
Conclusions
We have described the efficient synthesis of a set of pseudopeptidic dithiols that present C2 symmetry and peptide-like information, and are suitable for the generation of Dynamic Combinatorial (Covalent) Libraries (DCLs) in aqueous media. The synthesis involves an easy four-step procedure using standard solution-phase peptide chemistry. The compounds described herein contain a rigid aromatic scaffold that predispose to the formation of macrocyclic (mainly dimeric) compounds upon disulphide formation and allows monitoring by HPLC-UV traces. The disulphide bond is an important structural moiety in natural peptide-like macrocycles with biological implications.28 On the other hand the amino acid side chains provide a diversity of functionalities that cover a broad range of polarities as demonstrated by the comparison of the individual HPLC analysis of the oxidised compounds. Despite this broad range of behaviour water/organic solvent mixtures allow for the generation of DCLs even in the presence of the most hydrophobic components. Two different DCLs have been prepared to show the potential of the synthesised building blocks and confirmed the formation of almost all possible dimers in those libraries. Our experiments underscore the capability of the described BBs to generate chemical diversity in aqueous media, which is a useful tool in the fields of systems chemistry and bio-molecular recognition. In fact, as a proof of concept, we have designed a representative dynamic library of macrocycles that responded to the presence of structurally different substrates. Thus, we observed the amplification of different macrocyclic receptors upon the action of either spermine or phytic acid. Overall, the molecular diversity of the building blocks and the versatility in the generation of the corresponding DCLs by disulphide formation will open the way for the future applications of these dynamic systems in biomolecular recognition.Acknowledgements
This work was supported by the Spanish Ministry of Economy and Competitiveness (MINECO/FEDER, CTQ2012-38543-C03-03, CTQ2015-70117-R and CSIC13-4E-2076), EU (FP7-PEOPLE-2012-CIG-321659) and Generalitat de Catalunya (2014 SGR 231). Personal financial support for J. S. (MINECO, Ramón y Cajal contract) and J. A. (CSIC and European Social Fund, JAE-predoc fellowship) are gratefully acknowledged. We also thank Dr Yolanda Pérez for the helpful assistance in the acquisition of the water-suppression NMR experiments.Notes and references
- (a) J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC; (b) J.-M. Lehn, in Constitutional Dynamic Chemistry, ed. M. Barboiu, 2012, vol. 322, pp. 1–32 Search PubMed; (c) J.-M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 2836–2850 CrossRef CAS PubMed.
- (a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed; (b) R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101–108 RSC; (c) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed.
- (a) B. Hasenknopf, J.-M. Lehn, N. Boumediene, A. Dupont-Gervais, A. Van Dorsselaer, B. Kneisel and D. Fenske, J. Am. Chem. Soc., 1997, 119, 10956–10962 CrossRef CAS; (b) B. Hasenknopf, J.-M. Lehn, N. Boumediene, E. Leize and A. Van Dorsselaer, Angew. Chem., Int. Ed., 1998, 37, 3265–3268 CrossRef CAS; (c) V. E. Campbell, X. de Hatten, N. Delsuc, B. Kauffmann, I. Huc and J. R. Nitschke, Nat. Chem., 2010, 2, 684–687 CrossRef CAS PubMed.
- (a) H. Y. Au-Yeung, G. D. Pantos and J. K. M. Sanders, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10466–10470 CrossRef CAS PubMed; (b) F. B. L. Cougnon, N. A. Jenkins, G. D. Pantos and J. K. M. Sanders, Angew. Chem., Int. Ed., 2012, 51, 1443–1447 CrossRef CAS PubMed; (c) F. B. L. Cougnon, N. Ponnuswamy, N. A. Jenkins, G. D. Pantos and J. K. M. Sanders, J. Am. Chem. Soc., 2012, 134, 19129–19135 CrossRef CAS PubMed.
- (a) C. D. Meyer, R. S. Forgan, K. S. Chichak, A. J. Peters, N. Tangchaivang, G. W. V. Cave, S. I. Khan, S. J. Cantrill and J. F. Stoddart, Chem. – Eur. J., 2010, 16, 12570–12581 CrossRef CAS PubMed; (b) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed; (c) N. Ponnuswamy, F. B. L. Cougnon, J. M. Clough, G. D. Pantoş and J. K. M. Sanders, Science, 2012, 338, 783–785 CrossRef CAS PubMed; (d) T. Prakasam, M. Lusi, M. Elhabiri, C. Platas-Iglesias, J.-C. Olsen, Z. Asfari, S. Cianferani-Sanglier, F. Debaene, L. J. Charbonniere and A. Trabolsi, Angew. Chem., Int. Ed., 2013, 52, 9956–9960 CrossRef CAS PubMed.
- (a) R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS PubMed; (b) N. Hafezi and J.-M. Lehn, J. Am. Chem. Soc., 2012, 134, 12861–12868 CrossRef CAS PubMed; (c) P. Nowak, M. Colomb-Delsuc, S. Otto and J. Li, J. Am. Chem. Soc., 2015, 137, 10965–10969 CrossRef CAS PubMed.
- (a) W. Li and B. Yan, Front. Biotechnol. Pharm., 2002, 3, 85–95 CAS; (b) S. Otto, R. L. E. Furlan and J. K. M. Sanders, Science, 2002, 297, 590–593 CrossRef CAS PubMed; (c) S. Otto, Curr. Opin. Drug Discovery Dev., 2003, 6, 509–520 CAS.
- (a) J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS PubMed; R. A. R. Hunt and S. Otto, Chem. Commun., 2011, 47, 847–858 Search PubMed; (b) N. Giuseppone, Acc. Chem. Res., 2012, 45, 2178–2188 CrossRef CAS PubMed; (c) S. Otto, Acc. Chem. Res., 2012, 45, 2200–2210 CrossRef CAS PubMed.
- (a) J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed; (b) M. Colomb-Delsuc, E. Mattia, J. W. Sadownik and S. Otto, Nat. Commun., 2015, 6 Search PubMed; (c) J. W. Sadownik, E. Mattia, P. Nowak and S. Otto, Nat. Chem., 2016, 8, 264–269 CrossRef CAS PubMed.
- P. Dydio, P. A. R. Breuil and J. N. H. Reek, Isr. J. Chem., 2013, 53, 61–74 CrossRef CAS.
- (a) A. Buryak and K. Severin, J. Comb. Chem., 2006, 8, 540–543 CrossRef CAS PubMed; (b) A. Herrmann, Chem. – Eur. J., 2012, 18, 8568–8577 CrossRef CAS PubMed; (c) X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981 RSC.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- R. Nguyen and I. Huc, Chem. Commun., 2003, 942–943 RSC.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- (a) S. Otto, R. L. E. Furlan and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 12063–12064 CrossRef CAS; (b) K. R. West, K. D. Bake and S. Otto, Org. Lett., 2005, 7, 2615–2618 CrossRef CAS PubMed; (c) A. R. Stefankiewicz, M. R. Sambrook and J. K. M. Sanders, Chem. Sci., 2012, 3, 2326–2329 RSC.
- M. Rauschenberg, S. Bomke, U. Karst and B. J. Ravoo, Angew. Chem., Int. Ed., 2010, 49, 7340–7345 CrossRef CAS PubMed.
- (a) J. Atcher, A. Moure and I. Alfonso, Chem. Commun., 2013, 49, 487–489 RSC; (b) J. Solà, M. Lafuente, J. Atcher and I. Alfonso, Chem. Commun., 2014, 50, 4564–4566 RSC; (c) J. Atcher, A. Moure, J. Bujons and I. Alfonso, Chem. – Eur. J., 2015, 21, 6869–6878 CrossRef CAS PubMed; (d) M. Lafuente, J. Atcher, J. Solà and I. Alfonso, Chem. – Eur. J., 2015, 21, 17002–17009 CrossRef CAS PubMed.
- (a) S. Ladame, A. M. Whitney and S. Balasubramanian, Angew. Chem., Int. Ed., 2005, 44, 5736–5739 CrossRef CAS PubMed; (b) M. T. Cancilla, M. M. He, N. Viswanathan, R. L. Simmons, M. Taylor, A. D. Fung, K. Cao and D. A. Erlanson, Bioorg. Med. Chem. Lett., 2008, 18, 3978–3981 CrossRef CAS PubMed; (c) K. B. Jadhav, R. J. Lichtenecker, A. Bullach, B. Mandal and H.-D. Arndt, Chem. – Eur. J., 2015, 21, 5898–5908 CrossRef CAS PubMed.
- J. Atcher and I. Alfonso, RSC Adv., 2013, 3, 25605–25608 RSC.
- G. C. Barrett and D. T. Elmore, Amino Acids and Peptides, Cambridge University Press, 1998 Search PubMed.
- Y.-D. Wu and S. Gellman, Acc. Chem. Res., 2008, 41, 1231–1232 CrossRef CAS PubMed.
- (a) I. Alfonso, M. Bru, M. I. Burguete, E. García-Verdugo and S. V. Luis, Chem. – Eur. J., 2010, 16, 1246–1255 CrossRef CAS PubMed; (b) J. Rubio, I. Alfonso, M. I. Burguete and S. V. Luis, Soft Matter, 2011, 7, 10737–10748 RSC; (c) W. Edwards and D. K. Smith, Chem. Commun., 2012, 48, 2767–2769 RSC.
- (a) P. Wittung, P. E. Nielsen, O. Buchardt, M. Egholm and B. Norden, Nature, 1994, 368, 561–563 CrossRef CAS PubMed; (b) P. E. Nielsen and G. Haaima, Chem. Soc. Rev., 1997, 26, 73–78 RSC.
- F. Rodríguez-Llansola, B. Escuder and J. F. Miravet, J. Am. Chem. Soc., 2009, 131, 11478–11484 CrossRef PubMed.
- (a) P. Kokkonen, M. Rahnasto-Rilla, P. H. Kiviranta, T. Huhtiniemi, T. Laitinen, A. Poso, E. Jarho and M. Lahtela-Kakkonen, ACS Med. Chem. Lett., 2012, 3, 969–974 CrossRef CAS PubMed; (b) E. Zervoudi, E. Saridakis, J. R. Birtley, S. S. Seregin, E. Reeves, P. Kokkala, Y. A. Aldhamen, A. Amalfitano, I. M. Mavridis, E. James, D. Georgiadis and E. Stratikos, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19890–19895 CrossRef CAS PubMed; (c) N. Micale, K. Scarbaci, V. Troiano, R. Ettari, S. Grasso and M. Zappalà, Med. Res. Rev., 2014, 34, 1001–1069 CrossRef CAS PubMed.
- (a) E. Faggi, A. Moure, M. Bolte, C. Vicent, S. V. Luis and I. Alfonso, J. Org. Chem., 2014, 79, 4590–4601 CrossRef CAS PubMed; (b) S. V. Luis and I. Alfonso, Acc. Chem. Res., 2014, 47, 112–124 CrossRef CAS PubMed; (c) E. Faggi, C. Vicent, S. V. Luis and I. Alfonso, Org. Biomol. Chem., 2015, 13, 11721–11731 RSC; (d) I. Alfonso, Chem. Commun., 2016, 52, 239–250 RSC; (e) E. Faggi, Y. Pérez, S. V. Luis and I. Alfonso, Chem. Commun., 2016, 52, 8142–8145 RSC.
- The spectra were recorded using water suppression: M. Liu, X. Mao, C. He, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125–129 CrossRef CAS.
- B. K. W. Chung and A. K. Yudin, Org. Biomol. Chem., 2015, 13, 8768–8779 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and characterization details, full NMR and ESI-MS spectra of 4a–l, high resolution ESI-MS spectra of all the described macrocycles, DCL generation procedure and titration experiments. See DOI: 10.1039/c6ob02441f |
| This journal is © The Royal Society of Chemistry 2017 |