
Heteroleptic Ru(II) cyclometalated complexes derived from benzimidazole-phenyl carbene ligands for dye-sensitized solar cells: an experimental and theoretical approach†
Tejaswi
Jella
ab,
Malladi
Srikanth
c,
Yarasi
Soujanya
c,
Surya Prakash
Singh
ab,
Lingamallu
Giribabu
*ab,
Ashraful
Islam
*d,
Liyuan
Han
d,
Idriss
Bedja
e and
Ravindra Kumar
Gupta
e
aInorganic & Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, and CSIR-Network Institutes for Solar Energy (CSIR-NISE), Tarnaka, and CSIR-Network Institutes for Solar Energy (CSIR-NISE), Hyderabad-500007, Telangana, India. E-mail: giribabu@iict.res.in; Fax: +91-40-27160921; Tel: +91-40-27191724
bAcademy of Scientific and Innovative Research (AcSIR), New Delhi, India
cMolecular Modelling Group, CSIR-Indian Institute of Chemical Technology, Hyderabad-500007, Telangana, India
dPhotovoltaic Materials Unit, National Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
eCRC, Optometry Department, College of Applied Medical Sciences, King Saudi University, Riyadh 11433, Saudi Arabia
First published on 14th December 2016
Abstract
We have designed and synthesized two new ligands based on N-heteroleptic/phenyl carbene (NH-phenyl C) i.e., 1-benzyl-2-(3,5-bis(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (L1) and 3-(1-benzyl-1H-benzol-2-yl)-10-hexyl-10-H-phenothiazene (L2), used as ancillary ligands to heteroleptic Ru(II) complexes for dye-sensitized solar cells. Both NCS groups of the N719 sensitizer are replaced with L1 (TC-1) and L2 (TC-3) to obtain cyclometalated Ru(II) complexes and one of 4,4′-dicarboxylato-2,2′-bipyridine (dcbpy) with L1 to obtain a heteroleptic Ru(II) complex (TC-2). The presence of two trifluoromethyl groups of the L1 ligand stabilizes the HOMO level of Ru(II) complexes and the presence of a phenothiazine moiety of the L2 ligand alters the absorption properties of the TC-3 complex. Both the ligands and the heteroleptic Ru(II) complexes are characterized by elemental analyses, ESI-MS, 1H NMR, absorption and emission spectroscopy as well as electrochemical methods. The absorption spectra of TC-1 and TC-3 are blue shifted, when compared to the standard N719 sensitizer. The assessment of these newly designed cyclometalated and heteroleptic Ru(II) complexes has revealed that TC-2 exhibits an efficiency of 7.63%, whereas TC-1 has an efficiency of 6.39% using an I−/I3− redox couple. DFT and nanosecond transient absorption kinetic studies have been adopted to understand the low efficiency of the TC-3 complex.
Introduction
Dye-sensitized solar cells (DSSCs) are being investigated extensively for their use in renewable energy technologies because of their great potential in terms of low fabrication cost, and flexible and high light-to-electrical energy conversion efficiency.1–8 DSSCs have achieved power conversion efficiencies of over 11%, which was demonstrated using a prototype device with bis(tetrabutylammonium)-cis-di(thiocyanato)-N,N′-bis(4-carboxylato-4′-carboxylic acid-2,2′-bipyridine)ruthenium(II) (the N719 dye) and trithiocyanato-4,4′4′′-tricarboxy-2,2′:6′,2′′-terpyridine ruthenium(II) (the black dye) sensitizers.9–12 In spite of this, these sensitizers show insufficient light-harvesting efficiencies in the near-IR region of the absorption spectra. The sensitizers are one of the key components in DSSCs as they absorb sunlight and induce intramolecular charge transfer from the ancillary ligand to the anchoring ligand with consequent electron injection into the conduction band of the TiO2 semiconductor. For this reason, numerous heteroleptic ruthenium complexes have been probed, where one of the two 4,4′-dicarboxylato-2,2′-bipyridine (dcbpy) anchoring ligands has been replaced by a functionalized 2,2′-bipyridine (bpy) ligand with either hydrophobic chains to improve the device stability or with extended π-conjugated systems that include thiophene, carbazole, triphenylamine, coumarine etc., to enhance the optical properties.13–22 Despite their stability and efficiency, modification of N∧N ancillary ligands involves tedious synthetic protocols that will affect the cost of the sensitizer. In contrast, Grätzel and co-workers reported cyclometalated ruthenium polypyridyl complexes by replacing the NCS ligand with an anionic carbon atom, C∧N as an alternative approach to structural modification.23 The synthesis of C∧N cyclometalated ligands involves simple synthetic protocols and inexpensive starting precursors. Herein, we report a set of substituted N-heterocyclic/phenyl carbene (NH-phenyl C) ruthenium complexes, in which one of the nitrogen atoms in the traditional bipyridine framework is replaced by a carbon atom, for use in DSSCs exhibiting good solar cell performances.The essential requirement of a dye suitable for DSSC applications is that the energy level of the lowest unoccupied molecular orbital (LUMO) of the sensitizer should be sufficiently high enough for efficient charge injection into the TiO2 conduction band, while the energy level of the highest occupied molecular orbital (HOMO) must be sufficiently low for efficient regeneration of the oxidized dye by the redox couple. Several methods have been adopted to stabilize the energy level of the HOMO through tuning of ancillary ligands and their substituents.22,24 The use of ruthenium complexes bearing ancillary ligands functionalized with C∧N cyclometalated ligands might be an alternative approach towards tuning the frontier orbitals of the dyes, which enhances their IPCE thresholds as well as device performance. A few examples of the preparation of metal-carbene complexes as sensitizers for DSSC applications have been reported in the literature. For example, Bessho et al. used fluorine substituted phenyl pyridine ligands to replace the NCS groups of the N719 sensitizer and reported a cyclometalated complex with an efficiency of 10.1%.23 Li and co-workers used the N-heterocyclic carbene/pyridine ancillary ligand and reported an efficiency of 9.69%.25 C∧N cyclometalated ligands reported in the literature till now, by using substituted benzimidazole as carbene and pyridine remainis as it is. Here, for the first time, to the best of our knowledge, we designed a couple of new C∧N cyclometalated ligands, 1-benzyl-2-(3,5-bis(trifluoromethyl)phenyl)-1H-benzo[d]imidazole (L1) and 3-(1-benzyl-1H-benzo[d]imidazol-2-yl)-10-hexyl-10H-phenothiazine (L2) by the modification of our previous work in which pyridine (benzimidazole-pyridine ligand) is replaced with a substituted phenyl carbene.13 For better stabilization of the HOMO level of the dyes, we introduced an electron withdrawing group CF3 in the phenyl ring of the NH-phenyl C ligand. Both NCS groups of the N719 dye were replaced with L1 to obtain the cyclometalated ruthenium complex (TC-1), one of the dcbpy ligands was replaced with the ancillary L1 ligand (TC-2), and to improve the absorption properties of TC-1, L1 was replaced with L2 having a phenothiazine moiety (TC-3). The structures of the ligands and ruthenium complexes are shown in Chart 1. Both the ligands and the sensitizers were characterized using various spectroscopic techniques, such as elemental analyses, mass spectrometry, 1H NMR, UV-Vis and emission spectroscopy and cyclic voltammetry. Finally, the photovoltaic performance of the newly designed sensitizers was studied by using a liquid redox couple.
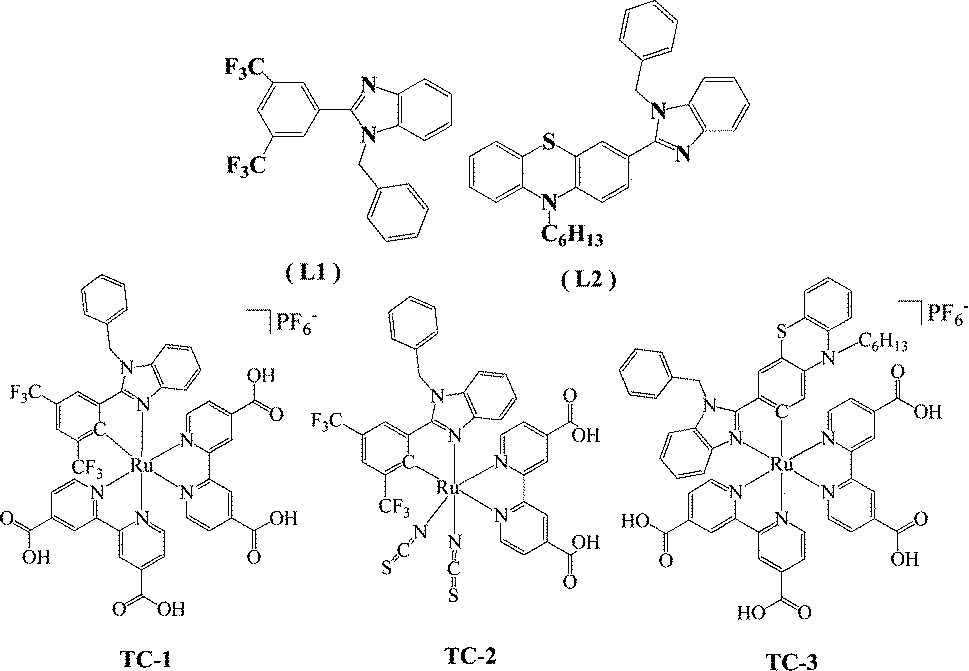 | ||
| Chart 1 Molecular structure of the ligands and ruthenium complexes. | ||
Experimental
Materials and methods
Commercially available reagents and chemicals were procured from Sigma-Aldrich. Analytical reagent (AR) grade solvents were used for the reactions while laboratory reagent (LR) grade solvents were used for purification and column chromatography. Dichloromethane, chloroform, acetonitrile and N,N-dimethyl formamide were dried in the presence of calcium hydride under a nitrogen atmosphere. Hexane was purified by Na metal with the addition of benzophenone and refluxed overnight, then distilled under vacuum and stored over 4 Å molecular sieves. Triethylamine was distilled over NaOH pellets. The ACME silica gel (100–200 mesh) was used for column chromatography. Thin-layer chromatography was performed on Merck-precoated silica gel 60-F254 plates. Either gravity or flash chromatography was performed for the purification of all compounds. All the reactions were carried out under a nitrogen or argon atmosphere using dry and degassed solvents.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethylacetate (8:2%, v/v) mixture as the eluent to obtain the desired compound 1. Yield (78%). Anal. calcd for C15H8F6N2O3 % (378.23): C, 47.63; H, 2.13; N, 7.41. Found: C, 47.65; H, 2.10; N, 7.45. ESI-MS: calculated (C15H8F6N2O3) 378. 1H NMR (500 MHz, CDCl3) (δ-ppm): 11.50 (1H, s), 8.95 (1H, dd, J = 5.1 Hz), 8.45 (2H, s), 8.33 (1H, dd, J = 5.0 Hz), 8.12 (1H, s), 7.8 (1H, m), 7.38 (1H, m).
:ethyl acetate (7:3%, v/v) as the eluent to obtain the desired product 2 in an 82% yield. Anal. calcd for C15H8F6N2 % (330.23): C, 54.56; H, 2.44; N, 8.48. Found: C, 54.60; H, 2.45; N, 8.45. ESI-MS calculated (C15H8F6N2) m/z: 330, found: 331 [M + H]. 1H NMR (500 MHz, CDCl3) (δ-ppm): 10.47 (1H, s), 8.50 (2H, s), 7.92 (1H, s), 7.72 (2H, s), 7.42 (2H, m).
: chloroform (7:3%, v/v) to obtain the desired compound L1 in an 85% yield. Anal. calcd for C22H14F6N2 % (563.33): C, 78.82; H, 8.04; N, 7.45. Found C, 78.85; H, 8.00; N, 7.47. ESI-MS: calcd m/z (C22H14F6N2): 420, found 421 [M + 1]+. 1H NMR (500 MHz, DMSO) (δ ppm): 8.30 (3H, m) 7.81 (1H, m), 7.65 (1H, m), 7.30 (5H, m), 7.02 (2H, m), 5.68 (2H, s).
:ethyl acetate (7:3 v/v) as the eluent to obtain the desired compound 4 in 88% yield. (Yellow solid, yield: 88%). Anal. calcd for C25H25N3S % (399.55): C, 75.15; H, 6.31; N, 10.52. Found C, 75.12; H, 6.30; N, 10.55. ESI-MS calculated (C25H25N3S) m/z: 399, found: 400 [M + H]. 1H NMR (500 MHz, CDCl3) (δ-ppm): 7.81 (1H, dd, J = 5.3 Hz), 7.70 (1H, d, J = 8.5 Hz), 7.53 (2H, m), 7.12 (3H, m), 7.02 (1H, dd, J = 5.0 Hz), 6.88 (1H, t, J = 7.5 Hz), 6.78 (1H, d, J = 8.1 Hz), 6.63 (1H, d, J = 8.6 Hz), 3.70 (2H, t, J = 7.2 Hz), 1.70 (2H, dt, J = 10.1 Hz), 1.36 (2H, m), 1.27 (4H, m), 0.85 (3H, t, J = 7.0 Hz).
:chloroform (7:3 v/v) to obtain compound L2 as a yellow colored powder in 85% yield. Anal. calcd for C32H31N3S % (489.22): C, 78.49; H, 6.38; N, 8.58. Found C, 78.52; H, 6.40; N, 8.55. ESI-MS: calcd m/z (C32H31N3S).489, found 490 [M + 1]+. 1H NMR (500 MHz, CDCl3) (δ ppm): 7.85 (1H, d, J = 8.0 Hz), 7.45 (2H, m), 7.36 (1H, m), 7.32 (5H, d, J = 5.5 Hz), 7.30 (2H, dd, J = 6.7 Hz), 7.20 (2H, m), 7.13 (4H, m), 6.92 (1H, td, J = 4.2 Hz), 6.85 (2H, d, J = 8.4 Hz), 5.45 (2H, s), 4.70 (1H, s), 3.86 (2H, m), 1.80 (2H, m), 1.43 (2H, m), 1.29 (4H, m), 0.87 (3H, t, J = 6.7 Hz).
:ethylacetate (8:2%, v/v) mixture as the eluent to obtain the desired compound 1. Yield (78%). Anal. calcd for C15H8F6N2O3 % (378.23): C, 47.63; H, 2.13; N, 7.41. Found: C, 47.65; H, 2.10; N, 7.45. ESI-MS: calculated (C15H8F6N2O3) 378. 1H NMR (500 MHz, CDCl3) (δ-ppm): 11.50 (1H, s), 8.95 (1H, dd, J = 5.1 Hz), 8.45 (2H, s), 8.33 (1H, dd, J = 5.0 Hz), 8.12 (1H, s), 7.8 (1H, m), 7.38 (1H, m).
:ethyl acetate (7:3%, v/v) as the eluent to obtain the desired product 2 in an 82% yield. Anal. calcd for C15H8F6N2 % (330.23): C, 54.56; H, 2.44; N, 8.48. Found: C, 54.60; H, 2.45; N, 8.45. ESI-MS calculated (C15H8F6N2) m/z: 330, found: 331 [M + H]. 1H NMR (500 MHz, CDCl3) (δ-ppm): 10.47 (1H, s), 8.50 (2H, s), 7.92 (1H, s), 7.72 (2H, s), 7.42 (2H, m).
: chloroform (7:3%, v/v) to obtain the desired compound L1 in an 85% yield. Anal. calcd for C22H14F6N2 % (563.33): C, 78.82; H, 8.04; N, 7.45. Found C, 78.85; H, 8.00; N, 7.47. ESI-MS: calcd m/z (C22H14F6N2): 420, found 421 [M + 1]+. 1H NMR (500 MHz, DMSO) (δ ppm): 8.30 (3H, m) 7.81 (1H, m), 7.65 (1H, m), 7.30 (5H, m), 7.02 (2H, m), 5.68 (2H, s).
:ethyl acetate (7:3 v/v) as the eluent to obtain the desired compound 4 in 88% yield. (Yellow solid, yield: 88%). Anal. calcd for C25H25N3S % (399.55): C, 75.15; H, 6.31; N, 10.52. Found C, 75.12; H, 6.30; N, 10.55. ESI-MS calculated (C25H25N3S) m/z: 399, found: 400 [M + H]. 1H NMR (500 MHz, CDCl3) (δ-ppm): 7.81 (1H, dd, J = 5.3 Hz), 7.70 (1H, d, J = 8.5 Hz), 7.53 (2H, m), 7.12 (3H, m), 7.02 (1H, dd, J = 5.0 Hz), 6.88 (1H, t, J = 7.5 Hz), 6.78 (1H, d, J = 8.1 Hz), 6.63 (1H, d, J = 8.6 Hz), 3.70 (2H, t, J = 7.2 Hz), 1.70 (2H, dt, J = 10.1 Hz), 1.36 (2H, m), 1.27 (4H, m), 0.85 (3H, t, J = 7.0 Hz).
:chloroform (7:3 v/v) to obtain compound L2 as a yellow colored powder in 85% yield. Anal. calcd for C32H31N3S % (489.22): C, 78.49; H, 6.38; N, 8.58. Found C, 78.52; H, 6.40; N, 8.55. ESI-MS: calcd m/z (C32H31N3S).489, found 490 [M + 1]+. 1H NMR (500 MHz, CDCl3) (δ ppm): 7.85 (1H, d, J = 8.0 Hz), 7.45 (2H, m), 7.36 (1H, m), 7.32 (5H, d, J = 5.5 Hz), 7.30 (2H, dd, J = 6.7 Hz), 7.20 (2H, m), 7.13 (4H, m), 6.92 (1H, td, J = 4.2 Hz), 6.85 (2H, d, J = 8.4 Hz), 5.45 (2H, s), 4.70 (1H, s), 3.86 (2H, m), 1.80 (2H, m), 1.43 (2H, m), 1.29 (4H, m), 0.87 (3H, t, J = 6.7 Hz).
Synthesis of ruthenium complexes
:acetonitrile (9:1%, v/v) as the eluent. The first yellow band was collected and evaporated to dryness, and then washed with dry hexane to obtain a pure yellow solid of either [Ru(CH3CN)4L1]+PF6− (C1) or [Ru(CH3CN)4L2]+PF6− (C2); this has been used for further steps.
The compound either C1 or C2 (0.204 mmol) and 2,2′-bipyridine-4,4′-dicarboxylic acid (dcbpy) (0.115 g, 0.470 mmol), and NaOH (0.033 g, 0.816 mmol) were dissolved in 10 ml of dry DMF under an inert atmosphere. The solution was heated to reflux for 16 h. Then the solvent was removed under reduced pressure to obtain a solid material. The obtained solid was dissolved in water and acidified with 0.2 M HNO3 until a dark red precipitate formed. The precipitate was filtered and purified using Sephadex column chromatography using the methanol:H2O (8:2 v/v) mixture as the eluent. The major reddish brown colour band was collected, concentrated and washed with diethyl ether to obtain the desired compound. [Ru(L1)(dcbpy)2]+PF6− (TC-1): red solid, 55% yield. Anal. calcd for C46H29F12N6O8PRu % (1154.06): C, 62.64; H, 3.32; N, 9.53. Found C, 62.60; H, 3.35; N, 9.50. ESI-MS: 1056 [M + 2Na]. ESI-MS: calcd m/z (C46H29F12N6O8PRu) 1154.06, found 1056 [M-PF6 + 1]+. 1H NMR (500 MHz, CD3OD) (δ ppm): 9.02 (1H, s), 8.92 (2H, s), 8.85 (1H, s), 8.78 (1H, s), 8.70 (1H, d), 8.56 (1H, s), 8.2 (1H, s), 8.08 (1H, d), 7.93 (1H, s), 7.84 (2H, dt), 7.78 (2H, dd), 7.65 (2H, m), 7.57 (4H, m), 7.37 (3H, m), 7.28 (1H, t), 7.10 (2H, d), 6.81 (1H, t), 6.12 (2H, s), 5.72 (1H, d).
[Ru(L1)(dcbpy)2]+PF6− (TC-1): red solid, 55% yield. Anal. calcd for C46H29F12N6O8PRu % (1154.06): C, 62.64; H, 3.32; N, 9.53. Found C, 62.60; H, 3.35; N, 9.50. ESI-MS: 1056 [M + 2Na]. ESI-MS: calcd m/z (C46H29F12N6O8PRu) 1154.06, found 1056 [M-PF6 + 1]+. 1H NMR (500 MHz, CD3OD) (δ ppm): 9.02 (1H, s), 8.92 (2H, s), 8.85 (1H, s), 8.78 (1H, s), 8.70 (1H, d), 8.56 (1H, s), 8.2 (1H, s), 8.08 (1H, d), 7.93 (1H, s), 7.84 (2H, dt), 7.78 (2H, dd), 7.65 (2H, m), 7.57 (4H, m), 7.37 (3H, m), 7.28 (1H, t), 7.10 (2H, d), 6.81 (1H, t), 6.12 (2H, s), 5.72 (1H, d).
[Ru(L2)(dcbpy)2]+PF6− (TC-3): red solid, 51% yield. Anal. calcd for C56H46F6N7O8PRuS % (1223.18): C, 70.68; H, 4.88; N, 10.31. Found C, 70.61; H, 4.85; N, 10.35. ESI-MS: calcd m/z (C56H46F6N7O8PRu) 1223.18, found 1077.8 [M-PF6 + 1]+.
Methods and instrumentation
1H NMR spectra were recorded on a 500 MHz INOVA spectrometer. Differential pulse voltammetric measurements were performed on a PC-controlled electrochemical analyser (CH instruments model CHI620C). All these experiments were conducted with a 1 mM concentration of compounds in DMF at a scan rate of 10 mV s−1 in which tetrabutylammonium hexafluorophosphate (TBAPF6) was used as the supporting electrolyte. Glassy carbon was used as the working electrode, a standard calomel electrode (SCE) was used as the reference electrode and the Pt wire was used as the auxiliary electrode. The optical absorption spectra were recorded on a Shimadzu (Model UV-3600) spectrophotometer. Steady-state fluorescence spectra were recorded on a Fluorolog-3 spectrofluorometer (Spex model, JobinYvon) for solutions with optical density at a wavelength of excitation of (λex) ≈ 0.05. Thermogravimetric measurements were carried out on a Mettler Toledo TGA/SDTA 851e instrument with a heating rate of 10 °C min−1 with 10 mg of the sample under a nitrogen atmosphere.Computational methodology
Ground state geometries optimized in the gas phase with the B3LYP functional with def2-TZVP with effective-core potential (ECP) was used for Ru, and for the rest of the elements def2-SVP was used.29 The TC-3 dye N-hexyl group on the phenothiazine was replaced by an ethyl group and the counter ion tetra(n-butyl)ammonium cation (TBA+) was modeled as the tetramethylammonium cation (TMA+) to reduce the computational cost. The polarizable continuum model (PCM) was used to optimize the geometries in methanol. TD-DFT (time-dependent density functional theory) calculations were performed on optimized geometries by using a modified CAM-B3LYP functional according to the method reported by Nakano et al. with μ = 0.15, α = 0.15, and β = 0.85.30 All the calculations were done in Gaussian 09.31 It has been previously reported by many researchers that the CAM-B3LYP functional gives reliable results for MLCT.31–34 Natural Bond Orbital (NBO) analysis was used to assess the metal to ligand back bonding or π acceptor strength of ligands L1 and L2 in the NBO Version 3.1 program.35 Electronic transitions analysis was carried out using the GaussSum software package.36Transient absorption spectroscopy
A laser flash photolysis spectrometer (model LP920, Edinburg) was used. It was associated with a Continuum Nd-YAG laser (Surelite; 10 Hz repetition rate; FWHM 5 ns). A Surelite optical parametric oscillator permits conversion of the second harmonic to a visible spectrum 400–820 nm. TiO2 samples (12 μm) sensitized with different dyes were subjected to 6 mJ cm−2 flash photolysis. The respective dyes were excited at 545 nm, 510 nm and 635 nm to assure homogeneous absorption profiles throughout the dye-sensitized TiO2 layers. The excited state decay of the dye and the recovery of its fundamental state were monitored by the change in the absorbed continuous wave light. A satisfactory signal-to-noise ratio was obtained by averaging the absorption spectra for 100–200 laser shots.Device fabrication and photovoltaic performance measurements
The TiO2 photoanode was prepared as reported previously.37 A fluorine-doped tin oxide (FTO) conducting glass substrate with a resistance of ∼10 ohm−2 was used. A screen printed double layer TiO2 film of (8 + 5) μm thickness (0.25 cm2 cell area) with a 8 μm transparent layer of TiO2 particles (approximately 20 nm in diameter) and a 5 μm scattering layer of TiO2 particles (approximately 400 nm in diameter) was prepared. The films were sintered at 500 °C for 1 h. The thickness of the films was measured using a Surfcom 1400A surface profiler (Tokyo Seimitsu Co. Ltd). A 0.2 mM solution of the dye in 1:1 (v/v) acetonitrile/tert-butyl alcohol was used to coat the TiO2 film. The TiO2 films were immersed in the dye solutions and then kept at 25 °C for 15 h. To assemble each cell, each dye-coated TiO2 film and a platinum-coated conducting glass were separated by a Surlyn spacer (40 μm thick) and sealed by heating the polymer frame at 100 °C. An electrolyte consisting of a mixture of 0.6 M dimethylpropyl-imidazolium iodide, 0.05 M I2, 0.1 M LiI, and 0.5 M tert-butylpyridine in acetonitrile was used in each cell. The current–voltage characteristics were measured using a black metal mask with an area of 0.25 cm2 under AM 1.5 sunlight (100 mW cm−2, WXS-155S-10: Wacom Denso Co. Japan). The IPCE spectra were measured using a monochromatic incident light of 1 × 1016 photons cm−2 in the direct current mode (CEP-2000BX, Bunko-Keiki).
Results and discussion
The details of the synthetic strategy adopted for the synthesis of both L1 and L2 and its ruthenium complexes TC-1, TC-2 and TC-3 are provided in Scheme 1. The L1 ligand was synthesized via the base catalyzed reaction of 3,5-bis(trifluoromethyl) benzoyl chloride and 2-nitroanaline followed by imidazole ring closure by using Fe/AcOH and was finally treated with benzyl chloride. Similarly, the L2 ligand was obtained by condensation of 10-hexyl-phenothiazine-3-carboxaldehyde with 2-nitroaniline and finally with N-alkyl arylation using benzyl chloride. Both the ligands were characterized using elemental analyses, 1H NMR, and ESI-MS spectroscopy (see the Experimental section in the ESI†). The mass spectra have molecular ion peaks at 421 and 400 corresponding to the presence of ligands L1 and L2, respectively. The synthesis of the Ru(II) complexes of TC-1, TC-2 and TC-3 was performed following standard procedures. The p-cymene complex was reacted with either L1 or L2 in acetonitrile to obtain an intermediate complex, followed by refluxing with the dcbpy ligand in DMF solvent to obtain cyclometalated Ru(II) complexes, TC-1 and TC-3, respectively. In contrast, the TC-2 complex was achieved by reacting L1 with p-cymene in DMF followed by treatment with the dcbpy ligand and excess KNCS at reflux temperature for 16 h. All new heteroleptic ruthenium sensitizers were purified using the sephadex (LH-20) column and a mixture of chloroform and methanol as the eluent. Preliminary characterization of these complexes was carried out via elemental analyses and mass spectroscopy. The mass spectra of all three ruthenium complexes consist of a molecular ion peak that corresponds to the proposed structure (see S14, S16 and S18, ESI†).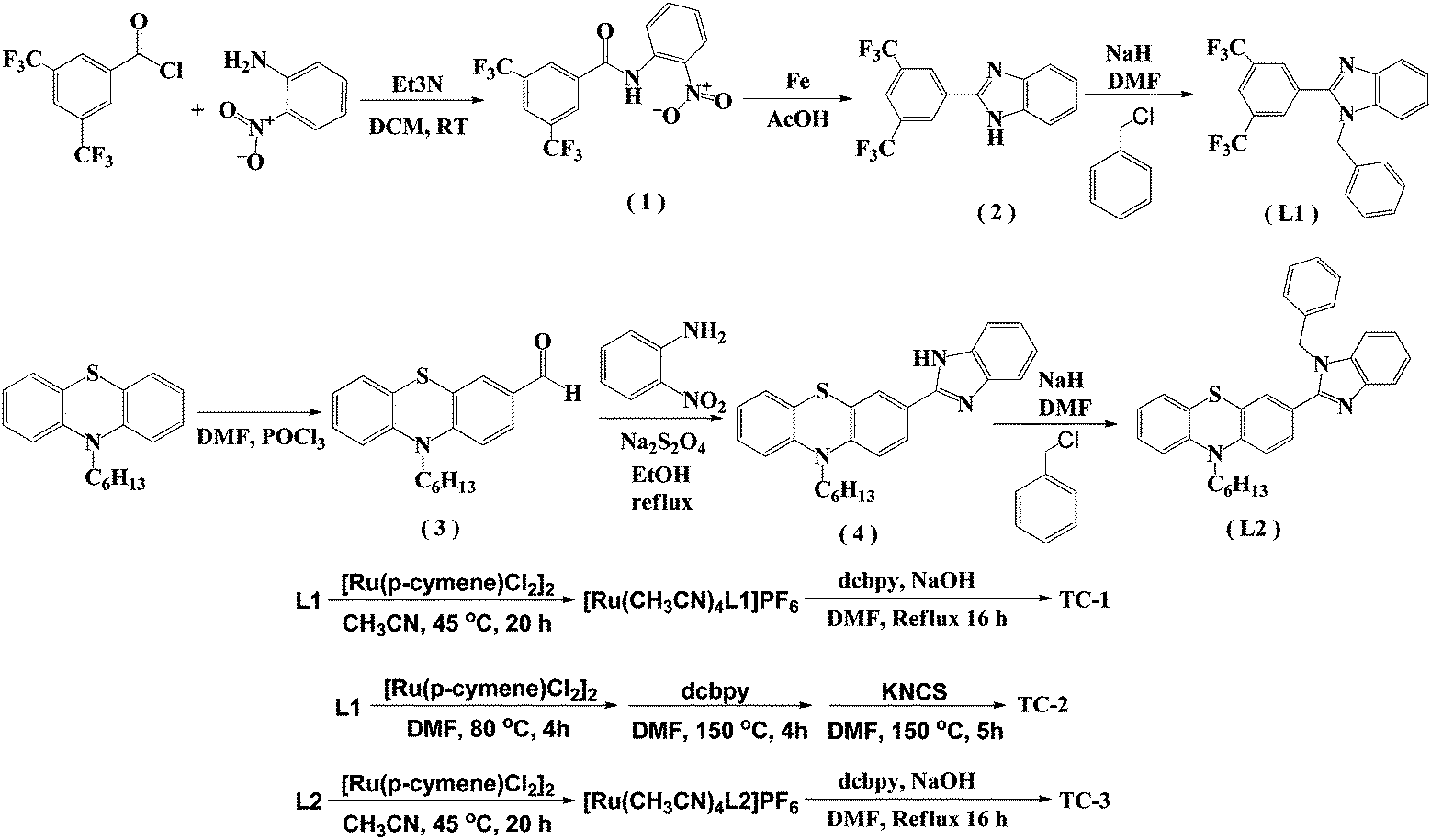 | ||
| Scheme 1 Synthetic route for the synthesis of NH-phenyl C ligands (L1 and L2) and ruthenium complexes (TC-1, TC-2, and TC-3). | ||
The proton NMR spectra of complexes TC-1, TC-2 and TC-3 are too complex for characterization because they contain diastereomers that cannot be separated using column chromatography (see S13, S15 and S17, ESI†).38 Consequently, the average effect of the inseparable diastereomers should be considered for the results discussed in the following.
Fig. 1 shows the electronic absorption spectra of all three heteroleptic ruthenium sensitizers along with standard N719 measured in methanol solvent at room temperature. The corresponding photophysical characterization data are summarized in Table 1. The absorption bands between the 400 and 600 nm regions can be ascribed to the metal to ligand charge transfer transitions in a singlet manifold (1MLCT). Fig. 1 and the data in Table 1 reveal that the absorption maximum of TC-1 is almost identical to that of the N719 sensitizer, but a new band appears at 477 nm with a high molar extinction coefficient (ε) compared to the standard N719 sensitizer whose spectrum shows a valley in this wavelength domain. On the other hand the absorption maximum of TC-2 is blue-shifted with low ε, as compared to the N719 sensitizer. In contrast, the absorption maxima of TC-3 are red-shifted and appear at 572 nm due to the presence of an N-hexyl phenothiazine moiety in its molecular structure. In both TC-1 and TC-2, the ε values are lower and they are comparable with that of the N719 sensitizer. However, absorption bands at 400 nm were observed with a large molar extinction coefficient due to the NH-phenyl C ligand. Fig. 1 also exemplifies the emission spectra of all the heteroleptic ruthenium complexes measured at RT in methanol solvent. The excitation of the lower energy MLCT transition of complexes TC-1, TC-2 and TC-3 resulted in the emission maxima at 730, 750 and 640 nm, respectively. Similar blue-shifted emission spectra were also observed for heteroleptic Ru(II) complexes having phenothiazine, as reported in the literature.39 Based on the optical properties, the E0–0 energy of complexes TC-1, TC-2, and TC-3 is found to be 1.92, 1.90, and 2.08 eV, respectively.
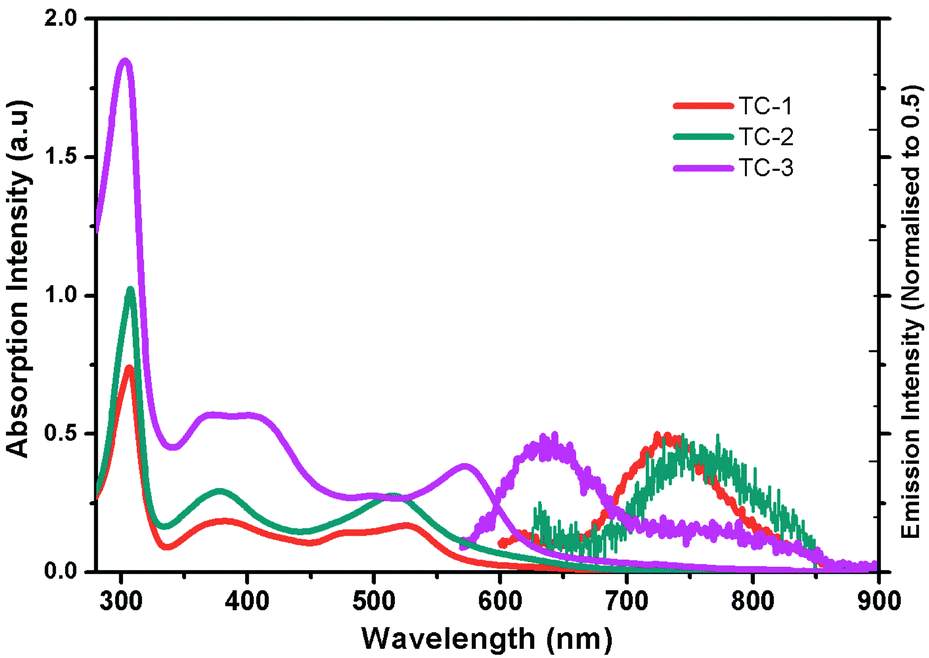 | ||
| Fig. 1 Absorption (left) and emission (right) spectra of ruthenium complexes TC-1, TC-2, and TC-3 in methanol. | ||
| Dye | ε π–π* (λmax) | ε π–π* (λmax) | ε MLCT (λmax) | λ em/nm | E 0–0 /eV | E HOMO /V | E LUMO /V |
|---|---|---|---|---|---|---|---|
| a Oxidation potentials EHOM (V vs. SCE) were measured for dyes in dry DMF in the presence of 0.1 M (n-C4H9)4NPF6. b E 0–0 was determined from the intersection of the absorption and emission spectra as shown in the figure. c E LUMO was determined as Eox − E0–0. | |||||||
| TC-1 | 18525 (306) | 6625 (381) | 6200 (525) | 730 | 1.92 | 0.82 | −1.10 |
| TC-2 | 25600 (307) | 7575 (381) | 7875 (514) | 750 | 1.90 | 1.02 | −0.88 |
| TC-3 | 46200 (303) | 7300 (378) | 9575 (572) | 645 | 2.08 | 0.63 | −1.45 |
| N719 | 35502 (308) | 10820 (381) | 9894 (526) | — | 1.85 | 0.97 | −0.88 |
The redox properties of all three heteroleptic ruthenium complexes along with standard N719 sensitizers are measured using a differential pulse voltammetric technique (DPV) in methanol solvent and 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6). The corresponding redox data are presented in Table 1. All three new sensitizers TC-1, TC-2 and TC-3 undergo one-electron reversible oxidation at 0.82, 1.02 and 0.63 V vs. NHE, respectively. The oxidation process can be assigned to Ru(II)/Ru(III) coupled with ligands L1 and L2. The second oxidation peak observed at 1.01 V in the case of TC-3 is probably due to the presence of the phenothiazine moiety. The potential level of the LUMO was determined from ELUMO = EHOMO − E0–0, the HOMO and LUMO values for comparison of the HOMO–LUMO levels of each sensitizer with respect to the TiO2 conduction band and the redox potential of the I−/I3− redox couple. The energy level diagram shows that the LUMO level of the cyclometalated TC-1 complex stabilized more than that of the cyclometalated TC-3 complex having electron withdrawing groups on the ancillary ligand. In all three heteroleptic Ru(II) complexes designed for this study, both electron injection and electron regeneration were found to be feasible when used as sensitizers for DSSCs (Fig. 2).
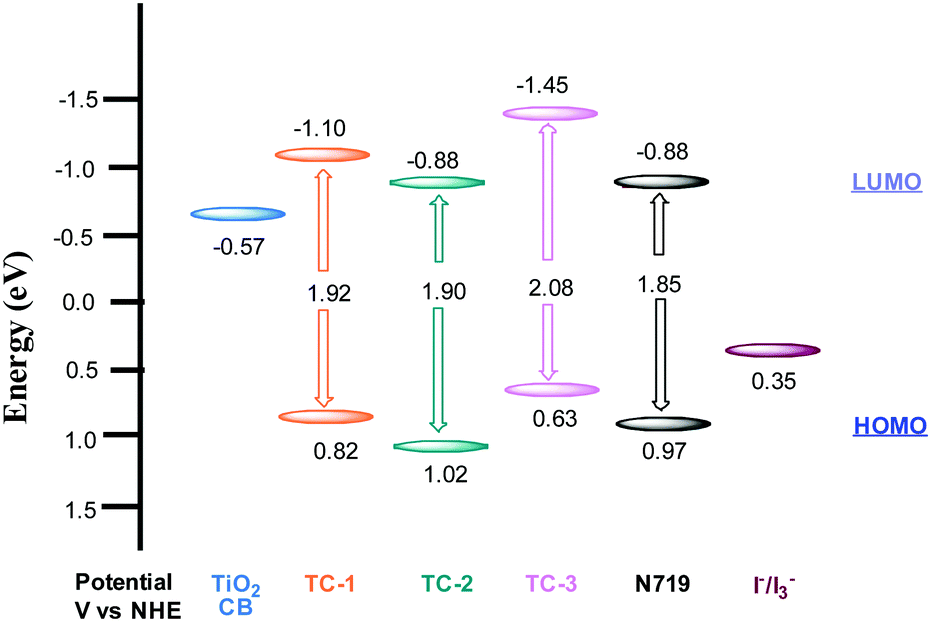 | ||
| Fig. 2 HOMO and LUMO levels obtained from electrochemical measurements. | ||
Device studies
The newly designed cyclometalated heteroleptic Ru(II) sensitizers were anchored onto a double layer nanocrystalline TiO2 film and their photovoltaic properties were determined using a liquid redox electrolyte (I−/I3−). Table 2 illustrates the photovoltaic performance characteristics of complexes TC-1, TC-2 and TC-3 along with the standard N719 sensitizer. A redox electrolyte consisting of a mixture of 0.6 M dimethylpropyl-imidazolium iodide, 0.05 M I2, 0.1 M LiI, with varying concentrations of tert-butylpyridine from 0.0 to 0.5 M in acetonitrile, was used in the device. Fig. 3a shows the IPCE spectra of all three sensitizers with a change in the concentration of TBP. We observed an IPCE of 80% and 86% at 540 nm for the TC-1 and TC-2 sensitizers by using 0.0 M TBP. In contrast, TC-3 showed an IPCE of 49% at 590 nm without using TBP. As the concentration of TBP increased, the IPCE value decreased in all the newly designed sensitizers. The photocurrent action spectrum resembles the absorption spectra except for a slight red shift of ca. 15 nm. The photoresponse of the thin films displays a broad spectral response covering the entire visible spectrum up to 800 nm using TC-1 and TC-2 dyes and up to 900 nm using the TC-3 dye.| Sensitizer | Concentration of TBP (mM) | IPCE (%) | J SC (mA/cm2) | V OC (mV) | ffb | η (%) |
|---|---|---|---|---|---|---|
| a Photoelectrode: TiO2 (8 + 4 μm and 0.158 cm2). b Error limits: JSC: ±0.20 mA cm−2, VOC = ±30 mV, ff = ±0.03, η = ±0.006. | ||||||
| TC-1 | 0.0 | 80 | 11.24 | 726 | 0.756 | 6.18 |
| TC-1 | 0.3 | 77 | 11.62 | 723 | 0.760 | 6.39 |
| TC-1 | 0.5 | 76 | 11.94 | 693 | 0.763 | 6.32 |
| TC-2 | 0.0 | 88 | 15.15 | 552 | 0.733 | 6.13 |
| TC-2 | 0.3 | 86 | 14.67 | 640 | 0.763 | 7.16 |
| TC-2 | 0.5 | 85 | 14.48 | 695 | 0.758 | 7.63 |
| TC-3 | 0.0 | 49 | 9.55 | 398 | 0.671 | 2.55 |
| TC-3 | 0.3 | 39 | 7.87 | 529 | 0.690 | 2.87 |
| TC-3 | 0.5 | 35 | 7.21 | 547 | 0.705 | 2.79 |
| N719 | 0.5 | 80 | 15.91 | 707 | 0.710 | 8.03 |
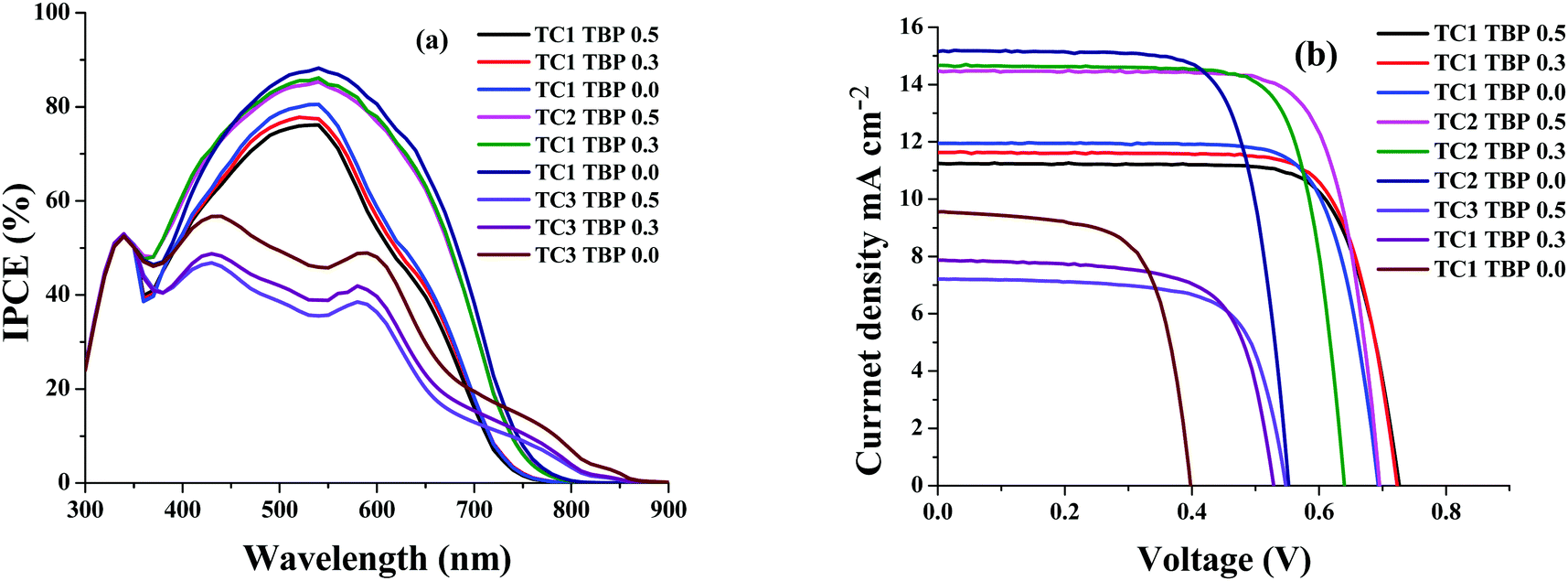 | ||
| Fig. 3 (a) Photocurrent action spectra and (b) current–voltage characteristics of complexes TC-1, TC-2, and TC-3. | ||
Fig. 3b shows the current–voltage characteristics of the device using all three heteroleptic ruthenium sensitizers with varying concentrations of TBP. It is reported in the literature that the addition of the TBP additive to the redox electrolyte has a large effect on device performance, as the surface charge of TiO2 is affected by decreasing the amount of adsorbed protons and/or Li+ ions.40 It also decreases the recombination of electrons in TiO2 with triiodide in the electrolyte by preventing triiodide access to the TiO2 surface and/or by complexation with iodine in the electrolyte.41 This is true in the case of the TC-2 and TC-3 sensitizers, as the concentration of TBP increases the open-circuit potential (VOC) and reaches a maximum value of 695 and 547 ± 30 mV, respectively. In contrast, VOC does not have much effect on the change in the concentration of TBP using the TC-1 sensitizer. Under standard global Air Mass (AM) 1.5 solar conditions, the complex TC-2 sensitizer based device gave a short-circuit photocurrent density (JSC) of 14.48 ± 0.20 mA cm−2, an open-circuit voltage (VOC) of 695 ± 30 mV and a fill factor (ff) of 0.758 ± 0.030, corresponding to an overall conversion efficiency η, derived from the equation: η = iSCVOC ff/light intensity, of 7.63%. Under similar test cell conditions, N719 showed an overall conversion efficiency of 8.03%. In contrast, TBP does not have much effect on the VOC of the device using TC-1 as the sensitizer and in fact, JSC marginally decreases with an overall η of 6.39%. The decrease in efficiency, when compared with the TC-1 complex, is probably due to the blue-shift of the absorption maxima. But, the efficiency of the device using TC-3 as the sensitizer showed a conversion η of ∼2.80%. We have adopted transient absorption studies to understand the effect of recombination on the efficiency of the devices.
Transient absorption study
Nanosecond transient absorption spectroscopy is an ideal tool for investigating DSSC kinetics between dye and photogenerated electrons in TiO2 as well as electrolyte redox mediators.41,42 These kinetics demonstrate how efficient DSSCs are.43,44Fig. 4 shows the transient absorption spectra of TC-1, TC-2 and TC-3 dye-sensitized TiO2 films in the presence and in the absence of the electrolyte. The decay profiles (here for the bleaching of the ground state of the dye) represent dye regeneration through the oxidation of I−. The fitting of the decays led to regeneration times. First, we can clearly keep faster regeneration times constant, measured in the presence of the electrolyte (5.0, 1.1 and 10 μs), which reflects the dynamics of regeneration of the dye by the electrolyte, compared to those in the absence of the latter (183, 51 and 96 μs), reflecting the dynamics of recombination of TiO2 photoinjected electrons with the oxidized dye. It is clear, here, that any dye regeneration due to the electrolyte is faster than that due to waste recombination with TiO2 photoinjected electrons and thus allows better collection of most of the photoinjected electrons into the external circuit.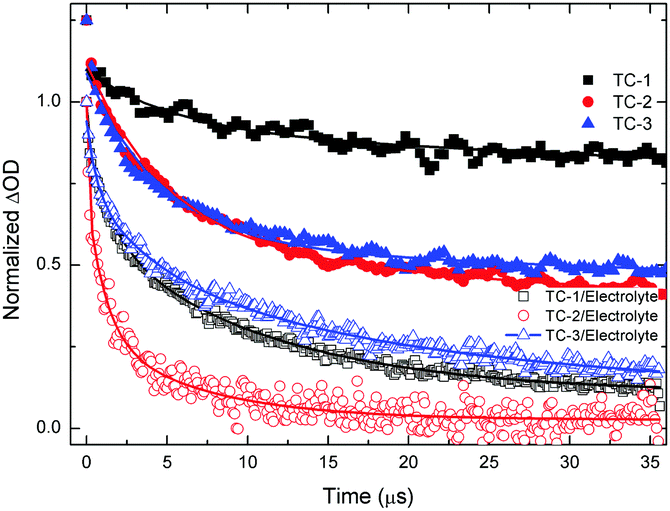 | ||
| Fig. 4 Transient absorbance decay profiles obtained upon nanosecond pulsed laser excitation (6 mJ, bandwidth 6 ns) on mesoporous TiO2 films sensitized, in the presence of air and electrolyte at laser excitation of 445 nm and monitored at λ = 550 nm with TC-1 dye and TC-2 dye and 580 nm with TC-3 dye. | ||
Regeneration times measured in the presence of redox mediators (1.1, 5.0 and 10 μs) for TC-2, TC-1 and TC-3 correlate well with the potential gradients between the respective oxidative dye potential and the LiI/I2 electrolyte redox potential (Fig. 2). Faster regeneration time correlates with a higher gradient potential. Another good corroboration was found regarding current density collected as (15.15, 11.24 and 9.55 mA cm−2), see Table 2, with respective regeneration times of 1.1, 5.0 and 10 μs, for TC-2, TC-1 and TC-3 dyes. Another interesting point regarding the electron transfer driving force (LUMO–TiO2CB) was observed. Above a certain threshold, having a higher driving force (like here for TC-3 dye, Fig. 2) does not necessarily allow a higher current density, on the contrary, it is the driving force for dye regeneration (LiI/I2 redox potential – HOMO) which mostly controls the current density collected.45
The driving force for electron transfer mostly controls the open circuit voltage (VOC = EF − I−/I3− potential) by filling the conduction band with photoelectrons thus increasing the Fermi energy level. We note here that the TC-3 dye does not seem to favor this concept. The TC-3 dye based device should present the highest VOC because of its highest LUMO level. The lower VOC measured might be due to a lower coverage of the largest TC-3 dye molecules to the TiO2 surface.
In order to understand the dye loading concentration on TiO2, we carried out absorption studies. For this reason, a 6 μm thick TiO2 layer was soaked in 0.2 mM dye solution (1:1 (v/v) acetonitrile/tert-butyl alcohol) at room temperature for 15 h and the photoanode was rinsed with fresh solvent to remove the unanchored sensitizer. Fig. 6 shows the absorption spectra of all three sensitizers adsorbed on TiO2. The absorption features of the complexes in solution as well as when anchored onto TiO2 are identical except for a slight red shift in absorption maxima due to interaction of the anchoring groups with the surface. From Fig. 5, it is clear that all three sensitizers, i.e., TC-1, TC-2 and TC-3, have identical absorption intensity (in the visible region) even though the molar extinction coefficient of TC-3 is higher than those of the remaining two sensitizers. This clearly suggests that the dye loading of TC-3 is lower compared to the other sensitizers.46
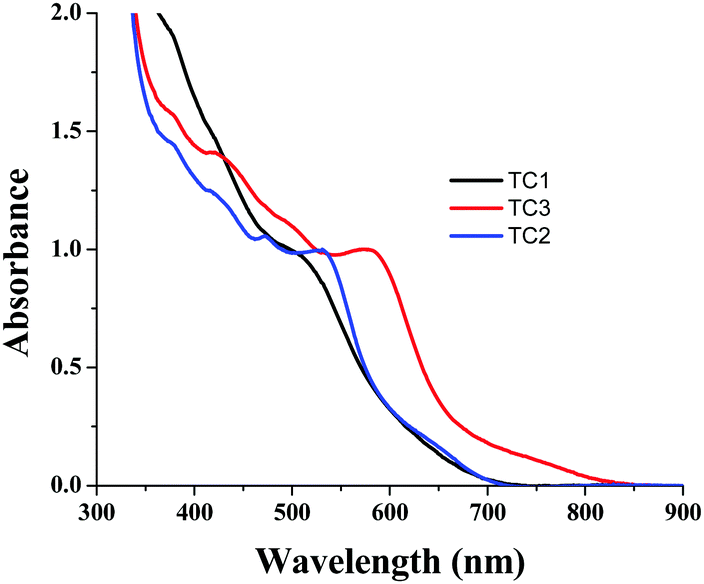 | ||
| Fig. 5 Absorption spectra of TC-1, TC-2 and TC-3 adsorbed on a 6 μm thick TiO2 layer. | ||
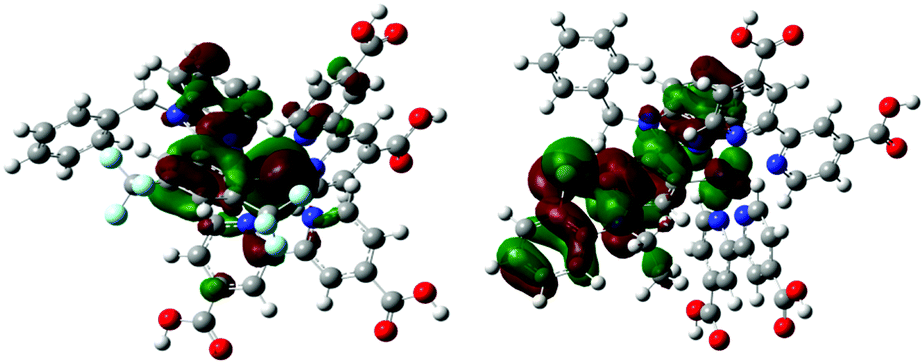 | ||
| Fig. 6 Bonding and anti-bonding interactions between the cyclometallating ligand with the ruthenium metal in the HOMO orbitals of the TC-1 and TC-2 complexes. | ||
Computational studies
Density-functional theory (DFT) calculations were carried out to analyse the electron withdrawing group and the annulation of the cyclometalating ligand (C^N), and their effects on the absorption spectra, energy levels, and efficiencies of these three complexes. Conjugated groups enhance molar absorption extinction coefficients, while the electron density at the metal center and the HOMO energy level are affected by the electron donating nature of the phenyl ring.47 A cyclometallating ligand with a more electron deficient π-accepting phenyl ring of the C^N ligand functionalized with –CF3 stabilizes the HOMO energy level, increases ΔH–L and also shows proper alignment with the I−/I3− redox couple leading to the efficient regeneration and higher VOC of the TC-11+ and TC-21− dyes (Table 3). Conversely, benzothiazine annulation to a phenyl carbene ligand destablizes the HOMO energy level of the TC-3 dye. In TC-31+ the Ru–C bond (2.0495) is shorter than all the studied complexes, and also the ∠C–CRu–C angle in phenyl carbene of the C^N ligand is 113.52° in TC-11+ and is 117.02° in TC-31+, which implies that TC-3 shows an increase of the p orbital character of the HOMO orbital and an increase of the σ-donor character.48| Dye | HOMO (eV) | LUMO (eV) | ΔE (eV) | NICS |
|---|---|---|---|---|
| TC-11+ | −5.7748 (−5.7098) | −3.0743 (−3.0234) | 2.7004 (2.6863) | −6.6 |
| TC-21− | −4.7993 (−4.9620) | −2.5132 (−2.5826) | 2.2860 (2.3793) | −6.9 |
| TC-22− | −4.5987 (−4.7843) | −2.2101 (−2.3306) | 2.3886 (2.4536) | −6.6 |
| TC-31+ | −5.0635 (−4.9720) | −3.0139 (−2.9342) | 2.0495 (2.0378) | −4.8 |
Counter ions (TBA+, PF6−) have an influence on the molecular energy levels and also the interaction between the dye and iodide ions increase via the S atom in the NCS ligand due to TBA+, which corresponds to the higher short-circuit photocurrent density of the TC-2 dye.49 In our present study, the counter ion tetra(n-butyl)ammonium cation (TBA+) is modeled as the tetramethylammonium cation (TMA+) to reduce the computational cost. Althougth the TC-2 complex HOMO energy level does not match the experimental trend, the energy gap (ΔH–L) trend corresponds to its absorption maxima trend. The energy levels of TC-11+ and TC-31+ are destabilized by the PF6− counter ion. In the TC-11+ complex, the HOMO orbital consists of bonding interactions between the metal and the L1 ligand. But the HOMO of the TC-3 complex consists of an anti-bonding interaction between the metal dx2−y2 orbital and the L2 ligand (see Fig. 6). Thus the π-accepting and donating abilities of the ligands at the Ru(II) center alter the bonding/antibonding overlap of the occupied molecular orbitals.50–52
The substitutional effects on the ring current of the phenyl π-electrons is assessed by calculation of the nucleus-independent chemical shift (NICS) at the centre of the ring (NICS(0)), and the increase in NICS for TC-11+ and for TC-21− can be attributed to delocalization of metal d electrons into π* orbitals of the C^N ligand (Table 3).
The second order perturbation energies reveal that TC-11+ and TC-21− have a higher back bonding energy due to greater π acceptor strength because of the electron withdrawing substituents (–CF3 substituted C ligand) that stabilize the π* orbital compared to TC-31+ (Table 4).
| Molecule | E(2) | ε(j) − ε(i) | F(i,j) |
|---|---|---|---|
| TC-1+ (LP*C*) | 9.34 | 0.14 | 0.038 |
| TC-2− (LP*C*) | 15.36 | 0.12 | 0.044 |
| TC-3+ (LP*C*) | 3.14 | 1.96 | 0.072 |
Fig. 7 shows the Mulliken spin-density analysis of one-electron oxidized complexes; for an electron deficient L1 ligand containing TC-1 and TC-2 complexes the spin density is mainly localized on the ruthenium metal atom, and for the TC-3 complex the spin density is delocalized throughout the L2 ligand. The Mulliken charges on the Ru metal atom for the parent complexes TC-11+, TC-21−, and TC-31+ are +0.353, +0.370 and +0.352 |e−|, respectively. Interestingly, for one-electron oxidized complexes the positive charge of fragmented spin densities (shown in Fig. 7) is localized on the ruthenium metal atom in the TC-1 and TC-2 complexes, but in TC-3 the positive charge is delocalized on an easily oxidizable phenothiazine ring. Oxidation of the cyclometallating ligand results in a less positive oxidation potential for TC-3 (+0.63 V). And this extensive delocalization of spin density on the organic ligand may be the reason for the longer regeneration time for TC-3 in the transient absorption studies.
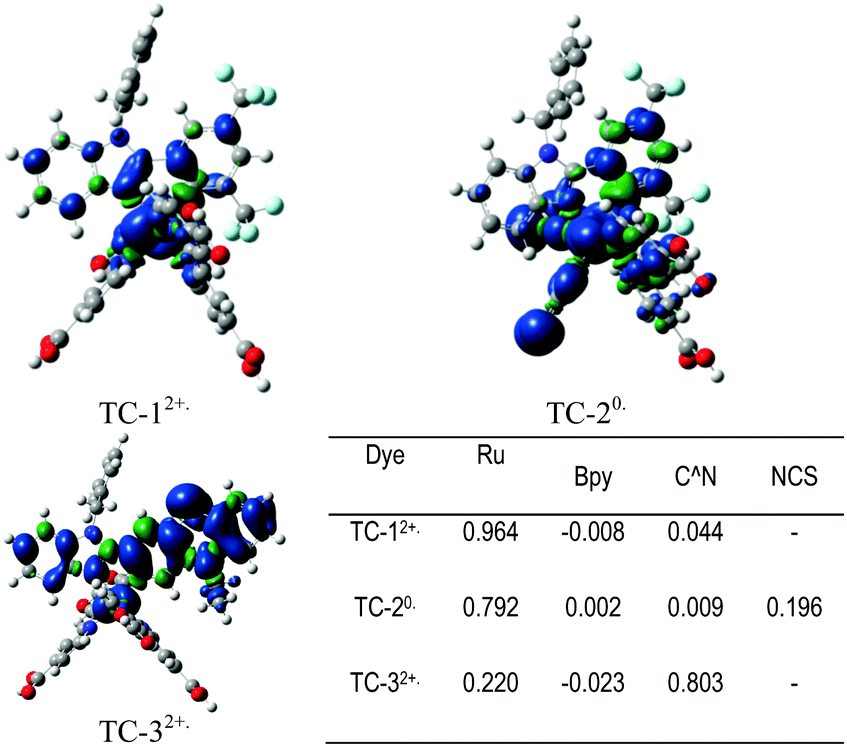 | ||
| Fig. 7 Calculated spin density distributions at UB3LYP/def2-SVP:def2-TZVP-ecp in methanol using the PCM solvation model. (Blue and green areas refer to regions of positive and negative spin density respectively.) | ||
A natural transition orbital (NTO) method was applied to obtain the optimal orbitals responsible for the electronic transitions. In this method, the occupied NTO (oNTO) can be seen as the “hole” orbital, and the virtual NTO (vNTO) is the “partical” orbital in which the electron is promoted to the excited state (Table 5 and S4, ESI†).53
| Dye | State | eV/nm | Major contribution MOs |
|---|---|---|---|
| TC-11+ | S5 | 2.542/487 (0.1932) | H−2 → L (36%), H−2 → L+1 (15%), H−1 → L (11%), H−1 → L+1 (28%) |
| S8 | 3.194/388 (0.0804) | H → L+2 (68%), H → L+4 (13%), H → L+5 (5%) | |
| S30 | 4.2928/288 (0.3508) | H−8 → L (11%), H−7 → L (12%), H−7 → L+1 (37%), H−1 → L+7 (6%) | |
| TC-21− | S1 | 1.7145/723 (0.0174) | H−2 → L (15%), H → L (75%) |
| S3 | 2.2730/545 (0.0857) | H−2 → L (64%), H → L (11%), H → L+1 (13%) | |
| S8 | 2.9375/422 (0.1169) | H−2 → L+1 (59%), H → L+3 (21%), H−1 → L+1 (5%) | |
| S26 | 4.2882/289 (0.2669) | H−10 → L (46%), H−5 → L+1 (18%), H → L+8 (16%) | |
| TC-31+ | S3 | 2.2408/553 (0.0188) | H−3 → L (36%), H−2 → L+1 (26%), H−1 → L (10%), H−1 → L+1 (9%), H → L (6%) |
| S5 | 2.3960/517 (0.1796) | H−3 → L (19%), H−2 → L+1 (48%), H−1 → L (7%), H−1 → L+1 (6%) | |
| S10 | 3.0215/410 (0.1294) | H → L+4 (10%), H → L+6 (42%), H−3 → L+1 (6%), H−1 → L+1 (3%), H−1 → L+4 (7%), H → L+1 (9%) | |
| S30 | 4.0428/306 (0.3981) | H → L+7 (20%), H → L+9 (10%), H−5 → L (8%), H−4 → L+1 (7%), H−3 → L+6 (7%), H−1 → L+6 (8%), H → L+13 (7%) | |
As shown in Table S4 (ESI†), the oNTO is localized either on the cyclometallating ligand or the ruthenium metal atom for TC-11+ and TC-21− complexes. In all the complexes the vNTO is localized on anchoring Bpy ligands, except the high energetic intraligand and interligand (ILCT/ILπ–π*) transitions (near ∼300 nm). For TC-11+ and TC-21− complexes MLCT transitions at 525 and 514 nm were observed. In the calculated absorption spectra of TC-21− instead of one, two MLCT bands are observed. And this low lying MLCT peak disappeared after simulating with two counter ions (calculated spectra with counterions are reported in Table S2, ESI†). The significant MLCT peak with high oscillator strength is slightly blue shifted when compared to the experimental absorption spectra. For TC-11+ the MLCT absorption band results majorly from ruthenium to one of the bypyridyl ligands. For TC-21− the MLCT absorption band appears due to the transition from both ruthenium and NCS groups to the bypyridyl ligand. For the TC-31+ complex, the transitions at 303 and 378 nm result mainly due to the metal to C^N ligand transition and intra-ligand transitions of the π-elongated cyclometallated ligand. The much red shifted absorption band centered at 572 nm for TC-31+ complex is found be to be due to both MLCT and LLCT.50 Based on the NTO transition analysis, the transition results from ruthenium and the cyclometallated ligand to two orthogonal bpy ligands. In particular, these two bpy ligands are bonded with the metal atom in the virtual orbitals of the LUMO and LUMO+1.
Conclusions
In conclusion, we have designed and synthesized cyclometalated and heteroleptic Ru(II) complexes based on the NH-phenyl C ligand. The absorption maxima of TC-1 and TC-2 are blue-shifted and the absorption maximum of TC-3 is red shifted, when compared to the standard N719 sensitizer. The molar extinction coefficient of all three complexes is lower than that of the N719 sensitizer. The electrochemical and theoretical properties indicate that the LUMO is above the TiO2 conduction band and the HOMO is below the redox potential of the redox couple. When applied to device studies, the heteroleptic TC-2 sensitizer showed an efficiency of 7.63%, whereas the cyclometalated complexes TC-1 and TC-3 showed efficiencies of only 6.39 and 2.87%, respectively, using a I−/I3− redox electrolyte. Nanosecond transient absorption studies indicated that the low performance of the TC-3 complex is due to the higher driving force required in order to inject the electrons into the TiO2 conduction band leading to a low-current density.Acknowledgements
The authors are thankful to CSIR-NISE for financial support to carry out this work. TJ thanks CSIR for financial support. A. Islam acknowledges the support from the JSPS KAKENHI grant no. 26288113. A. Islam and I. Bedja extend their appreciation to the International Scientific Partnership Program ISPP at King Saud University for funding this research work through ISPP #0019.References
- F. Bella, C. Gerbaldi, C. Barolo and M. Grätzel, Chem. Soc. Rev., 2015, 44, 3431–3473 RSC.
- C.-P. Lee, R. Y.-Y. Lin, L.-Y. Lin, C.-T. Li, T.-C. Chu, S.-S. Sun, J. T. Lin and K.-C. Ho, RSC Adv., 2015, 5, 23810–23825 RSC.
- L. Giribabu, R. Bolligarla and M. Panigrahi, Chem. Rec., 2015, 15, 760–788 CrossRef CAS PubMed.
- M. Ye, X. Wen, M. Wang, J. Locozzia, N. Zhang, C. Lin and Z. Lin, Mater. Today, 2015, 18, 155–162 CrossRef CAS.
- S. Zhang, X. Yang, Y. Numata and L. Han, Energy Environ. Sci., 2013, 6, 1443–1464 CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS PubMed.
- J. W. Bowers, H. M. Upadhyaya, S. Calnan, R. Hashimoto, T. Nakada and A. N. Tiwari, Prog. Photovoltaics, 2009, 17, 265–272 CAS.
- L. Han, A. Islam, H. Chen, M. Chandrasekharam, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057–6060 CAS.
- M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevery, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Muller, P. Liska, N. Valchopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- T. Jella, M. Srikanth, R. Bolligarla, Y. Soujanya, S. P. Singh and L. Giribabu, Dalton Trans., 2015, 44, 14697–14706 RSC.
- A. El-Shafei, M. Hussain, A. Islam and L. Han, Prog. Photovoltaics, 2013, 22, 958–969 Search PubMed.
- I. Stengel, A. Mishra, N. Pootrakulchote, S.-J. Moon, S. M. Zakeeruddin, M. Gratzel and P. Bäuerle, J. Mater. Chem. A, 2011, 21, 3726–3734 RSC.
- L. Giribabu, T. Bessho, M. Srinivasu, C. Vijaykumar, Y. Soujanya, V. G. Reddy, P. Y. Reddy, J.-H. Yum, M. Grätzel and M. K. Nazeeruddin, Dalton Trans., 2011, 40, 4497–4504 RSC.
- L. Giribabu, V. K. Singh, C. Vijaykumar, Y. Soujanya, V. G. Reddy and P. Y. Reddy, Adv. OptoElectron., 2011, 294353 Search PubMed.
- W.-C. Chang, H.-S. Chen, T.-Y. Li, N.-M. Hsu, Y. S. Tingare, C.-Y. Li, Y.-C. Liu, C. Su and W.-R. Li, Angew. Chem., Int. Ed., 2010, 49, 8161–8164 CrossRef CAS PubMed.
- L. Giribabu, C. Vijaykumar, C. S. Rao, V. G. Reddy, P. Y. Reddy, M. Chandrasekharam and Y. Soujanya, Energy Environ. Sci., 2009, 2, 770–773 CAS.
- F. Gao, Y. Wang, J. Zhang, D. Shi, M. Wang, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Gratzel, Chem. Commun., 2008, 2635–2637 RSC.
- C.-Y. Chen, S.-J. Wu, C.-G. Wu, J.-G. Chen and K.-C. Ho, Angew. Chem., Int. Ed., 2006, 45, 5822–5825 CrossRef CAS PubMed.
- P. Wang, C. Klein, R. Humphry-Baker, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2005, 127, 808–809 CrossRef CAS PubMed.
- T. Bessho, E. Yoneda, J.-H. Yum, M. Guglielmi, I. Tavernelli, H. Imai, U. Rothilsberger, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 5930–5934 CrossRef CAS PubMed.
- N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338–2345 CrossRef CAS PubMed.
- B. Zhou, B. Li, W. Yi and L. Ma, Bioorg. Med. Chem. Lett., 2013, 23, 3759–3763 CrossRef CAS PubMed.
- R. Schiffmann, A. Neugebauer and C. D. Klein, J. Med. Chem., 2006, 49, 511–522 CrossRef CAS PubMed.
- H. Tian, X. Yang, R. Chen, Y. Pan, L. Li, A. Hagfeldt and L. Sun, Chem. Commun., 2007, 3741–3743 RSC.
- C. Matijssen, M. C. Silva-Santisteban, I. M. Westwood, S. Siddiquie, V. Choi, P. Sheldrake, R. L. M. van Montfort and J. Blagg, Bioorg. Med. Chem. Lett., 2012, 20, 6630–6639 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- K. Okuno, Y. Shigeta, R. Kishi, H. Miyasaka and M. Nakano, J. Photochem. Photobiol., A, 2012, 235, 29–34 CrossRef CAS.
- E. W. Y. Wong and D. J. H. Emslie, Dalton Trans., 2015, 44, 11601–11612 RSC.
- M. J. Frisch, et al., Gaussian09 Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- M. J. G. Peach, P. Benfield, T. Helgaker and D. J. Tozer, J. Chem. Phys., 2008, 128, 044118 CrossRef PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- G. H. Rao, A. L. Rao, L. Giribabu and S. P. Singh, Photochem. Photobiol. Sci., 2016, 15, 287–296 CAS.
- W.-K. Huang, H.-P. Wu, P.-L. Lin, Y.-P. Lee and E. W.-G. Diu, J. Phys. Chem. Lett., 2012, 3, 1830–1835 CrossRef CAS PubMed.
- M. G. Murali, X. Wang, Q. Wang and S. Valiyaveettil, RSC Adv., 2016, 6, 57872–57879 RSC.
- S. E. Koops, B. O'Regarn, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- G. Beschloo, L. Häggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef PubMed.
- T. Swetha, S. Niveditha, K. Bhanuprakash, A. Islam, L. Han, I. Bedja, R. Fallahpour and S. P. Singh, Inorg. Chem. Commun., 2015, 51, 61–65 CrossRef CAS.
- R. K. Gupta and I. Bedja, Phys. Status Solidi A, 2014, 211, 1601–1604 CrossRef CAS.
- G. Boschloo and A. Hagfeldt, Chem. Phys. Lett., 2003, 370, 381–386 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J.-Y. Li, C. Lee, C.-Y. Chen, W.-L. Lee, R. Ma and C.-G. Wu, Inorg. Chem., 2015, 54, 10483–10489 CrossRef CAS PubMed.
- D. V. Pogozhev, M. J. Bezdek, P. A. Schauer and C. P. Berlinguette, Inorg. Chem., 2013, 52, 3001–3006 CrossRef CAS PubMed.
- A. Comas-Vives and J. N. Harvey, Eur. J. Inorg. Chem., 2011, 5025–5035 CrossRef CAS.
- H. Kusama, H. Sugihara and K. Sayama, J. Phys. Chem. C, 2011, 115, 2544–2552 CAS.
- S. Bellinger-Buckley, T.-C. Chang, S. Bag, D. Schweinfurth, W. Zhou, B. Torok, B. Sarkar, M.-K. Tsai and J. Rochford, Inorg. Chem., 2014, 53, 5556–5567 CrossRef CAS PubMed.
- I. M. Dixon, S. Khan, F. Alary, M. Boggio-Pasqua and J.-L. Heully, Dalton Trans., 2014, 43, 15898–15905 RSC.
- C.-H. Siu, C.-L. Ho, J. He, T. Chen, X. Cui, J. Zhao and W.-Y. Wong, J. Organomet. Chem., 2013, 748, 75–88 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS, 1H NMR spectral data of all compounds and tables of molecular orbital composition and calculated absorption spectral wavelength data. See DOI: 10.1039/c6qm00264a |
| This journal is © the Partner Organisations 2017 |