DOI:
10.1039/C7QM00058H
(Research Article)
Mater. Chem. Front., 2017,
1, 1413-1421
Investigation of luminescence and structural properties of ZnO nanoparticles, synthesized with different precursors
Received
7th February 2017
, Accepted 18th February 2017
First published on 21st February 2017
Abstract
Zinc oxide nanoparticles (ZnO NPs) were synthesized by chemical methods using different precursors (zinc chloride, zinc nitrate hexahydrate and zinc acetate) and media (water, ethanol, and methanol). An effort has been made to precisely control the particle size and optical band gap by various methods, while maintaining the crystal structure. The particle sizes of the synthesized ZnO nanoparticles were found to be in the range of 5 nm to 20 nm, by Transmission Electron Microscopy (TEM). X-ray diffraction (XRD) confirmed the hexagonal crystal structure of all the samples, with a variation in the average crystalline size, in accordance with the TEM studies. Absorption spectra of these nanoparticles exhibit broad peaks in the range of 274 to 376 nm. Optical band gap values of the synthesized ZnO NPs were found to be in the range of 3.2 eV to 3.32 eV. The photoluminescence (PL) spectra show two emission peaks; one at 393–420 nm which corresponds to band gap excitonic emission, and another located at 520–550 nm, due to the presence of defects. Variations in peak positions of the emission spectra are due to changes in the defect densities on the surfaces of the nanoparticles which were synthesized with different precursors. The number of sub peaks obtained from Gaussian peak function fitting of PL spectra shows the possible energy levels, the types of defects present in the samples and also their influence on the optical properties.
1 Introduction
Semiconductors on the nanometer scale have gained remarkable interest because of their optical, electrical and chemical properties which make them desirable for various applications.1 The optical properties of these materials are specifically being investigated for their use in optoelectronics, photovoltaics and sensing.2 Zinc oxide (ZnO) is a direct band gap (3.37 eV) semiconductor at room temperature and has a large exciton binding energy of 60 meV.3 Moreover, its relatively high thermal and mechanical stabilities make it an ideal candidate for device applications.4–7 It is superior to conventional materials like phosphorus and sulfur because it is more resistant to UV rays, has a higher electrical conductivity and also has strong ferromagnetic properties at room temperature.8 The size of the nanoparticles, the temperature and the defects in the crystalline structure affect the photoluminescence properties of the ZnO NPs. Because of their photoluminescence properties they are used in field emission display equipment, such as televisions, LCDs, LEDs and OLEDs.9 ZnO nanorods as gas sensors are highly sensitive to NO2 gas with a very low detection limit of 10 ppb at 250 °C, and thus provide short response and recovery times.10 Zinc oxide nanostructures have large specific surface areas, high pore volumes, low toxicities and long life-spans11 and are being used as promising materials for antibacterial applications,12 chemical absorbents,13 polymer additives14 and photocatalysts.15,16 Interestingly ZnO NPs have tunable optical and electrical properties due to their large band gaps,17 thus are suitable for a wide range of applications like Ultra Violet (UV) detectors and UV emitting devices,18,19 solar cells,20 light emitting diodes,21,22 and gas sensing materials.23 In addition, zinc oxide is also suitable for generating UV light24 and for biosensing.25 In the last few decades researchers have reported various methods for synthesizing ZnO nanoparticles, such as the sol gel process,26 chemical vapor deposition,27 microwave synthesis,28 direct precipitation,29 homogeneous precipitation,30 micro emulsion synthesis,31 spray pyrolysis,32 plasma synthesis,33 ball milling,34 wet chemical synthesis,35 solvo-thermal and hydro thermal synthesis,36 the pulsed laser ablation technique37 and the flame combustion method.38 Kumar et al. synthesized rod like ZnO nanoparticles with an average particle size between 10.0 and 12.0 nm, by the microemulsion route.39 Virendra Chandore et al. used a microwave assisted combustion method for the synthesis of ZnO NPs with particle sizes around 13 nm,40 using dc thermal plasma. Hsiu-Fen Lin et al. synthesized ZnO NPs with an average particle size of 31 nm and optical band gap of about 2.5 eV.33 PK Giri et al. used the ball milling method for synthesizing ZnO NPs and they found a particle size in the range of 7 to 35 nm. The optical absorption spectra of these NPs showed an absorption peak at 368 nm.41 Ghaffarian et al. synthesized ZnO NPs by the spray pyrolysis method and found the particle size to be in the range of 10–25 nm.42 From the above methods, the chance of impurities in the ball milling method is higher, while the others are either more expensive or require complex equipment, so it is necessary to search for a simple route which needs less equipment, yet giving a high yield. There are several reports on the size and morphology dependent green luminescence intensity of ZnO nanoparticles but in their synthesis methods size variation or morphology variation occurs by varying only particular parameters like solvent, solvent concentration, temperature etc. but in our synthesis methods we take an arbitrary solvent, precursor and temperature.
In the present work, we have synthesized ZnO nanoparticles using different precursors and solvents via a chemical route. The possible energy levels and defects have been investigated in detail by the optical characterization of the ZnO nanoparticles. In addition, the detailed structural investigation of different sized ZnO nanoparticles has been also done.
2 Materials and methods
Zinc chloride (Rankem, purity 98.0%), zinc nitrate hexahydrate (Merck, purity ≥96%), zinc acetate dihydrate (Merck, purity ≥98%), polyvinyl pyrrolidone (Lobal chemie), potassium hydroxide (Merck, purity ≥84%), sodium hydroxide (Fisher scientific purity, 97.0%), ammonia solution (Rankem, purity 25.0%), dimethyl sulphoxide (Himedia, purity 99.5%), thioglycerol (Otto, purity 98%), ethanol (Changshu angyuan chemical), methanol (Rankem, purity 99.0%), tea leaf (dry form) and de-ionized (DI) water were used as received.
The X-Ray diffraction patterns of the ZnO nanoparticles that were synthesized using different precursors and solvents were recorded by a PANalytical X-ray diffractometer. The step size and scan step time of the X-ray diffractometer were 0.02° and 0.5 seconds, respectively, in reflection mode between 20°–80°. TEM analysis was carried out by using a Technai T20 FEI to confirm the particle size and shape of the ZnO nanoparticles. Diffuse reflectance spectra were taken by a Cary 4000 UV-vis spectrometer for evaluation of the band gap of ZnO nanoparticles. In order to investigate the luminescence properties and possible defects, room temperature photoluminescence spectra were recorded by LABHR-UV-EVO, using a He–Cd laser with 30 mW of power, at an excitation wavelength (λexc) of 325 nm.
3 Synthesis of ZnO nanoparticles
3.1 ZnO by ZnCl2 in water medium
1.1 g of ZnCl2 was dissolved in 20 mL of DI water under constant stirring at 90 °C (solution A). Another solution was prepared by dissolving 4 g of NaOH in 20 mL of DI water (solution B). 4 mL of solution B was added drop wise into solution A and the resultant milky white solution without any precipitation was left to stir for 2 hours. This solution was then kept for sufficient time for the NPs to settle down. The supernatant was removed, washed 5 times with DI water and dried at 100 °C for 30 minutes. ZnO nanoparticles were obtained in white powder form.43
3.2 ZnO by ZnCl2 in methanol medium
1.090 g of ZnCl2 was dissolved in 20 mL of methanol and 0.96 g of NaOH was dissolved in 30 mL of methanol. Both solutions were kept under constant stirring for 40 minutes. NaOH solution was added drop wise into the zinc chloride solution for 20 minutes which was stirred for 2 hours. The solution was then kept in a dark chamber overnight. The obtained precipitate was centrifuged for 5 minutes then washed 5 times with DI water and annealed in an air atmosphere at 300 °C. A white powder of ZnO nanoparticles was obtained.44
3.3 ZnO by ZnCl2 in ethanol medium
The ZnO nanoparticles were synthesized by the same procedure as discussed in Section 3.2 but using ethanol in place of methanol as the medium. All the experimental parameters and conditions were the same. The ZnO NPs were obtained in a light brown colored powder form.44
3.4 ZnO by Zn(NO3)2·6H2O in water medium
The wet chemical method was used to synthesize the ZnO nanoparticles by dissolving 2.9747 g of Zn(NO3)2·6H2O in 100 mL of DI water (solution A) and 0.8924 g of polyvinyl pyrrolidone in 50 mL of DI water (solution B). Both solutions were stirred for 30 minutes at 60 °C then NH4OH was added drop wise into solution A until the pH reached 7.5 (solution C) and then it was stirred for 15 minutes. Solution B was then added to solution C and the resultant solution was kept at 60 °C for 1 hour. The mixture was then cooled to room temperature and kept overnight in a dark chamber. The precipitate was filtered with a 0.2 μm filter and washed 2–3 times with DI water and then dried at 60 °C for 12 hours.35
3.5 ZnO by green synthesis method
Zinc acetate dehydrate was dissolved in 70 mL of distilled water to make a 0.2 M solution and stirred for 30–40 minutes. Another solution of 5 g of dried green tea leaf powder in 100 mL of distilled water was prepared and stirred for 2 hours at 80 °C, then cooled to room temperature and filtered through Whatman filter paper. 30 mL of this green tea extract was mixed homogeneously with the zinc acetate solution. The solution was filtered and dried at room temperature. The obtained powder was annealed at 300 °C for 1 hour.45
3.6 ZnO by zinc acetate in DMSO medium
Zinc acetate (0.4 M) and potassium hydroxide (2.4 M) were stirred separately in 40 mL dimethyl sulfoxide and 20 mL ethanol, respectively. The potassium hydroxide solution was added drop wise into the zinc acetate solution. Thioglycerol (0.25 mL) was added to the above solution which then turned milky. The white filtrate was washed three times with methanol and DI water and then dried at room temperature.46 The schematic for the syntheses of the ZnO nanoparticles using different precursors and solvents is shown in Fig. 1.
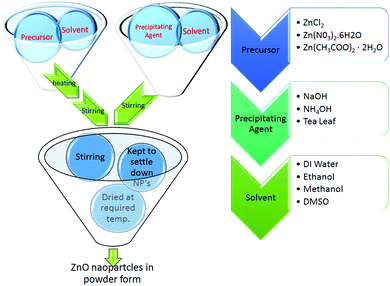 |
| | Fig. 1 Schematic for synthesis of all ZnO NPs. | |
4 Results and discussion
4.1 X-ray diffraction (XRD)
The XRD patterns of the ZnO nanoparticles which were prepared by Methods 3.1 to 3.6 are shown in Fig. 2. These XRD patterns of the nanoparticles show a single phase crystalline nature, corresponding to a wurtzite hexagonal crystal structure. Morphological studies by TEM (Fig. 3 and 4) confirm a single phase. Spurious low intensity peaks in the range 20–35 degrees in the XRD patterns (Fig. 2, Method 3.2) are observed and can be attributed to the multiphase product or impurity in the sample. However, the occurrence of a multiphase induces a change in the morphology of nanoparticles,47,48 which is not observed in our case (Fig. 3c). Thus, the extra peaks can be assigned to impurity in the sample. The ZnO nanoparticles synthesized by Methods 3.1–3.6 all have the characteristic peaks of ZnO. It can be concluded from these characteristic peaks that ZnO has a hexagonal crystal structure and matches the space group of P63mc. The relative intensity of the XRD peaks corresponding to a particular plane depends on the morphology49 but the TEM images confirm that the morphologies of all the ZnO samples are similar which indicate the variation in intensity is not due to the morphology but it may be due to agglomeration. In Method 3.6 the capping agent DMSO was used to control agglomeration and the particle size was reduced, which was indicated by the broadening of peaks in the XRD pattern. The lower intensity of the peaks suggests a decrease in crystalline quality of the nanoparticles when using a capping agent. The average crystalline size of these nanoparticles was determined by the Debye Scherrer equation for each orientation| | 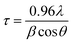 | (1) |
where τ is average crystalline size, β is the full width at half maximum (FWHM) of the peak, λ is the wavelength of the X-ray (for Cu 1.54 Å) and θ is the angle of diffraction. The broadening of peaks indicates that the synthesized nanoparticles are in the nanometer range. The calculated average crystalline size found for the particles from Method 3.1 was 21 nm which is similar to a previous result.43 For Method 3.2 the size was 26 nm which is higher than a previous result (∼5.36 nm).63 The size reduced to ∼16 nm when methanol was replaced by ethanol (Method 3.3). For Method 3.4 the average crystalline size was ∼17 nm, which is nearly half of that from a previous result.35 For Method 3.5 the crystalline size was 9 nm which is smaller than a previous result45 and for Method 3.6 the size was found to be ∼7 nm. The lattice parameters of the ZnO nanoparticles are as shown in Table 1. It can be clearly seen from Fig. 2 that the broadening of peaks in Method 3.6 is greater compared to the other methods, indicating that the particles synthesized by Method 3.6 have a smaller particle size. It is also clear that the average crystalline size of the ZnO NPs synthesized using zinc acetate is smaller than those synthesized with a zinc chloride precursor. The size was found to be the smallest from the above methods when ethylene glycol was used as the capping agent (Method 3.6).
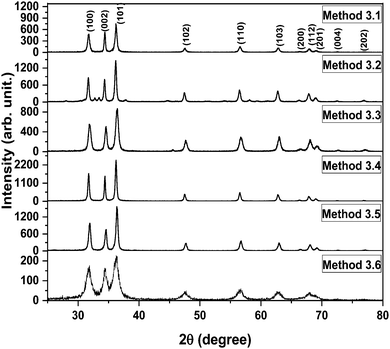 |
| | Fig. 2 XRD spectra of all the ZnO nanoparticles. | |
 |
| | Fig. 3 TEM images and diffraction patterns of ZnO synthesized by Methods 3.1 (a and b) and 3.2 (c and d). | |
 |
| | Fig. 4 TEM images and diffraction patterns of ZnO synthesized by Methods 3.3 (a and b), 3.4 (c and d), 3.6 (e and f) and 3.5 (g and h). | |
Table 1 Lattice parameters for all the samples and corresponding JCPDS card numbers
| Method |
Precursor |
Solvent |
Lattice parameter |
JCPDS card no. |
| 3.1 |
Zinc chloride |
DI water |
a = b = 3.2490 Å, c = 5.2060 Å |
01-079-0206 |
| 3.2 |
Zinc chloride |
Methanol |
a = b = 3.2170 Å, c = 5.2130 Å |
01-076-0704 |
| 3.3 |
Zinc chloride |
Ethanol |
a = b = 3.2427 Å, c = 5.1948 Å |
01-075-0576 |
| 3.4 |
Zinc nitrate |
DI water |
a = b = 3.2501 Å, c = 5.2070 Å |
01-079-2205 |
| 3.5 |
Zinc acetate |
DI water |
a = b = 3.2427 Å, c = 5.1948 Å |
01-079-0576 |
| 3.6 |
Zinc acetate |
DMSO |
a = b = 3.2417 Å, c = 5.1876 Å |
01-079-0205 |
4.2 Transmission electron microscopy (TEM)
Fig. 3a, c and 4a show the TEM images of the ZnO nanoparticles synthesized using a ZnCl2 precursor with water, methanol and ethanol, respectively (Methods 3.1, 3.2 and 3.3). It can be observed that the particles have an almost spherical shape in both ethanol (Method 3.3) and methanol media (Method 3.2) (Fig. 1b and c) and the selected area diffraction patterns (SADP) (Fig. 3b, d and 4b) show the polycrystalline nature of the NPs, which is in good agreement with the XRD results. The average particle size of ZnO in ethanol (3.3) was found to be 14 ± 2 nm, which also agrees with the XRD spectrum. When ethanol was replaced by methanol, keeping the same procedure, the average size of particles increased to 18 ± 2 nm, while the shape and size distribution was similar. In the case of water as the solvent (3.1), a well dispersed rod like structure was observed with an average diameter of 18 ± 2 nm. Fig. 4c shows the TEM image of the ZnO nanoparticles prepared by zinc nitrate in DI water; the nanoparticles are almost spherical in shape with an average size of 16 ± 2 nm, which is almost twice that of the previous result.35 The TEM images and selected area diffraction patterns of the ZnO nanoparticles prepared by Methods 3.5 and 3.6 are shown in Fig. 4g and h and Fig. 4e and f, respectively. Selected area diffraction patterns of both of these samples show the polycrystalline nature of the nanoparticles which also agree with XRD results (Fig. 1e and f). The TEM images show that the shapes of the nanoparticles are almost spherical with small agglomeration. The particle sizes were been found to be 5 ± 2 nm and 13 ± 2 nm, respectively, which also agree with the XRD results. It is clear from the TEM study that the average diameters of the ZnO NPs are almost the same if we use zinc chloride as the precursor (Methods 3.1, 3.2 and 3.3). It is also clear that average diameters of nanoparticles are smaller if we use precursor zinc nitrate and zinc acetate compared to zinc chloride. The average diameter of the ZnO NPs was found to be the smallest among the above methods when the capping agent ethylene glycol was used (Method 3.6).
4.3 Diffuse reflectance spectroscopy (DRS)
The diffuse reflectance spectra of all of the ZnO samples are shown in Fig. 5. Strong reflections were found above 400 nm for all samples. The absorbance has been calculated using the Kubelka–Munk equation in the limiting case of an infinitely thick sample at any wavelength:| | 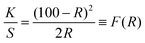 | (2) |
F(R) is the remission or Kubelka–Munk function. When the material scatters in a perfectly diffuse manner, the K–M absorption coefficient K becomes equal to 2α (K = 2α). The corresponding absorption spectra for the ZnO NPs are shown in Fig. 6. Absorption peaks were observed at 368 nm, 374 nm, 372 nm, 370 nm, 368 nm and 260 nm, respectively, indicating the presence of a blue shift with a decrease in particle size with respect to bulk ZnO (= 376 nm; 3.3 eV). The observed blue shift could be attributed to confinement effects. In the parabolic band structure, the band gap Eg, and coefficient α of a direct band gap semiconductor are related through the equation50where α is the linear absorption coefficient of the material, hν is the photon energy and B is a proportionality constant. In this case, considering the K–M scattering coefficient S as a constant with respect to the wavelength, and using the remission function in eqn (2), we obtain the expression:| | | [F(R)hν]2 = B(hν − Eg) | (4) |
The band gap energies of the samples are measured by extrapolation of the linear portion of the graph between the modified Kubelka–Munk (K–M) function [F(R)hν]2versus photon energy (hν), as shown in Fig. 7. The extrapolation of the straight lines in Fig. 7 gives the value of the band gap energies. The optical band gap is found to be size dependent and there is an increase in the band gap of the semiconductor with a decrease in particle size. The optical band gap values obtained for the ZnO nanoparticles prepared by the above Methods 3.1–3.6 are 3.25, 3.21, 3.23, 3.23, 3.2 and 3.32 eV, respectively. There is no significant change in the optical band gaps of ZnO prepared by Methods 3.1 to 3.5 because the particle sizes are almost the same. A remarkable change in the band gap of 0.12 eV was observed when the particle size decreased from 12 nm to 5 nm. The smaller band gap value in the as-synthesized ZnO nanoparticles compared to those of bulk materials (∼3.3 eV) may be attributed to planar defects, like stacking faults, twin boundaries and intrinsic defects, for example oxygen vacancies, zinc vacancies and Zn and O interstitials (as shown in the energy level diagram in Fig. 9) found in ZnO materials. It is clear from the UV-Vis spectra (Fig. 6) that there were no sharp changes observed in the peak positions in comparison to bulk ZnO. A large blue shift is observed when zinc acetate was used as precursor. The observed results are summarized in Table 2.
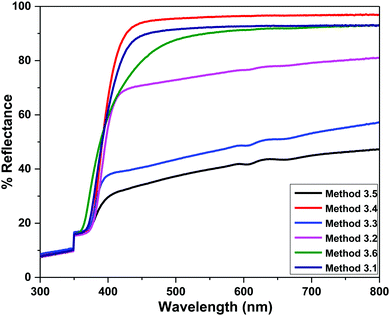 |
| | Fig. 5 Room temperature diffuse reflectance spectra of the ZnO NPs prepared by Methods 3.1 to 3.6. | |
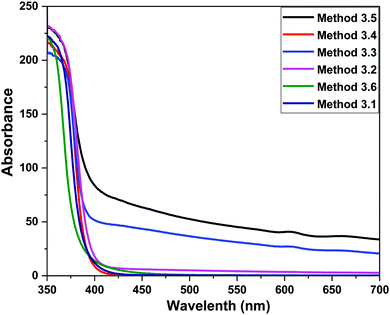 |
| | Fig. 6 UV Vis absorption spectra of the ZnO NPs prepared by Methods 3.1 to 3.6. | |
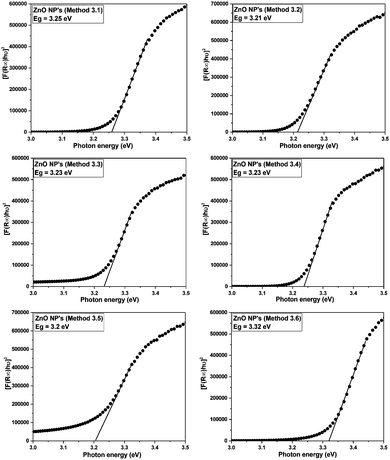 |
| | Fig. 7 Kubelka–Munk function versus energy plots of all the ZnO samples. | |
Table 2 Summary of present work
| Method |
Precursor |
Solvent |
d
XRD (nm) |
d
TEM (nm) |
E
g (eV) |
|
d
XRD is average crystallite size, dTEM is average diameter and Eg is optical band gap of ZnO NPs. |
| 3.1 |
Zinc chloride |
DI water |
21 |
18 ± 2 |
3.25 |
| 3.2 |
Zinc chloride |
Methanol |
26 |
17 ± 2 |
3.21 |
| 3.3 |
Zinc chloride |
Ethanol |
16 |
14 ± 2 |
3.23 |
| 3.4 |
Zinc nitrate |
DI water |
17 |
13 ± 2 |
3.23 |
| 3.5 |
Zinc acetate |
DI water |
9 |
12 ± 2 |
3.20 |
| 3.6 |
Zinc acetate |
DMSO |
7 |
5 ± 2 |
3.32 |
It can observed from Table 2 that by using DI water as the solvent and the precursors zinc chloride, zinc nitrate and zinc acetate both the average crystalline size and the average diameter decrease. It can also be seen from Table 2 that by keeping the precursor the same and changing the solvent to ethanol, DI water or methanol, the average diameter and the optical band gaps were almost same but the average crystalline size increased. In the case of zinc acetate as the precursor, when either DI water or DMSO were used as the solvent the average crystalline size remained almost the same but the average diameter changed. It is also clear that the average crystalline size and average diameter of the ZnO NPs synthesized using Method 3.6 were found to be smaller than for all the other used methods (Methods 3.1 to 3.5) but the optical band gap was larger due to the quantum confinement effect. Because of the capping agent (EG in this case) the average particle size decreased and a blue shift occurred in the absorption spectra.51
4.4 Photoluminescence (PL) spectroscopy
In order to study the luminescence properties and possible defects in the samples, photoluminescence spectra were studied at room temperature (Fig. 8) with an excitation wavelength (λexc) of 325 nm, over a wavelength range of 350 nm to 650 nm, for the ZnO nanopowders prepared by the different methods. The PL spectra of the ZnO NPs exhibit two emission peaks, one in the UV region (between 370–420 nm) which corresponds to the near band gap excitonic emission (band to band transition).52 The second emission peak is located in the range of 420–650 nm which is attributed to the presence of singly ionized oxygen vacancies (surface defects).53 The reason for the second emission might be the radiative recombination of a photo generated hole with an electron occupying the oxygen vacancy.54 The visible band observed in all of the ZnO samples is due to the existence of structural imperfections and IR emission from the n-type ZnO.
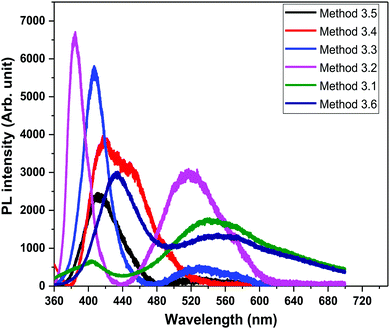 |
| | Fig. 8 PL spectra of all the ZnO samples (λexc = 325 nm). | |
The emission bands of all of the ZnO samples were fitted by multiple Gaussian peak functions with optimal full width at half maxima (FWHM). The number of sub-peaks in all of the ZnO PL spectra reveals the presence of defects and their influence on the optical properties. Fig. 9 shows the PL spectra along with the corresponding energy band diagrams of the ZnO samples and the corresponding parameters from multi-peak fitting are tabulated in Table 3. The presence of multiple peaks in the visible region of the electromagnetic spectrum is typical for nanocrystalline ZnO.55 The ZnO NPs prepared by Methods 3.1 to 3.6 have band edge peaks at 432, 382, 407, 412, 408 and 374 nm, respectively. The ZnO NPs prepared by Methods 3.2 and 3.3 have their emission peaks in the violet and green region while the NPs prepared by Method 3.4 have an additional blue emission peak. The NPs synthesized by Methods 3.5 and 3.6 have emission in the violet, green and orange-red regions and Method 3.1 have emission in the green and orange regions. The violet emission usually originates from the zinc interstitial (Zni) defects, where an electronic transition occurs from the Zni level to the valence band.56 The green emission in all of the samples is due to transitions from Zni to the valence bands of oxygen (VO).57 The presence of more than one green emission may be due to the different shallow energy levels in close vicinity inside the band gap (Fig. 9, Methods 3.2 and 3.3) which is associated with interface trap in the grain boundaries and dislocations.58,59 The other reason may be the coexistence of interstitial sites with different energy configurations such as isolated or split interstitials. The blue emission observed in the ZnO NPs prepared by Method 3.4 may be due to the transition from the Zni level to VZn. However, it could also arise from the surface deep trap states.56 The orange-red emission occurring in the ZnO NPs prepared by Methods 3.5 and 3.6 is due to a Zni to Oi transition.60 In the case of Methods 3.2 and 3.3, the higher intensities show the large population of low lying energy levels in the range of 434–485 nm.
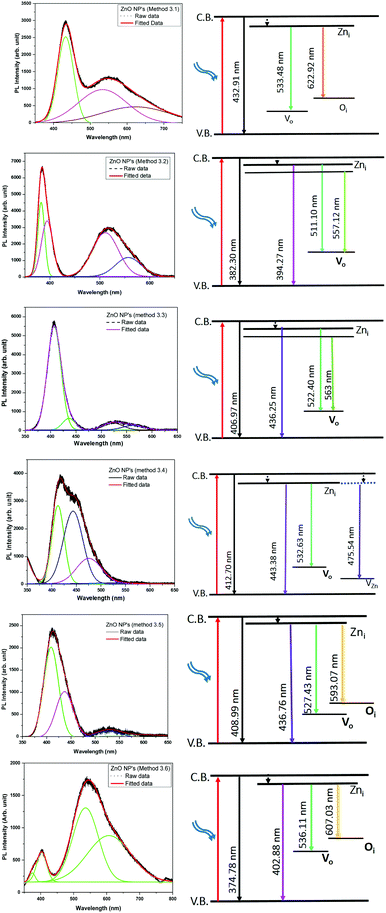 |
| | Fig. 9 Gaussian fitted photoluminescence spectra and corresponding possible energy level diagrams for all of the ZnO samples (λexc = 325 nm). | |
Table 3 Band positions, relative intensities and bandwidths in individual bands after Gaussian fitting of the PL of the ZnO NPs
| Method |
Peak position (nm) |
Peak intensity (counts) |
Band width (nm) |
| 3.1 |
432.91 |
2538 |
50.78 |
| 533.48 |
988.58 |
156.35 |
| 622.92 |
483.29 |
218.81 |
|
|
| 3.2 |
382.30 |
67.6 |
16.30 |
| 394.27 |
50.6 |
27.10 |
| 511.10 |
39.9 |
61.29 |
| 557.12 |
17.4 |
60.96 |
|
|
| 3.3 |
406.97 |
98.4 |
31.81 |
| 436.25 |
11.1 |
29.84 |
| 522.40 |
6.8 |
46.11 |
| 563.00 |
4.4 |
60.96 |
|
|
| 3.4 |
412.70 |
73.9 |
29.61 |
| 443.38 |
68.7 |
46.78 |
| 475.54 |
24.03 |
60.10 |
| 532.63 |
0.8 |
92.42 |
|
|
| 3.5 |
408.99 |
82.2 |
36.26 |
| 436.76 |
43.3 |
42.11 |
| 527.43 |
6.02 |
57.23 |
| 593.07 |
2.5 |
147.64 |
|
|
| 3.6 |
374.78 |
139.73 |
19.85 |
| 402.88 |
457.2 |
35.64 |
| 536.11 |
1143.50 |
92.02 |
| 607.03 |
718 |
155.13 |
Theoretically, the Zni, VZn and oxygen interstitial states (Oi energy levels) are located nearly 0.22, 3.06 and 2.28 eV below the conduction band, respectively, whereas the Vo level is 0.9 eV above the valence band.58 The deep level emissions occur in the green (2.5 eV) and orange-red (1.95 eV) regions, which are in good agreement with our experimental results. As observed in Fig. 9, the higher intensities of the violet emissions in Methods 3.2 and 3.3, as compared to the other methods, may due to a higher concentration of grain boundaries, dislocations and surface traps. The nanopowders show an intense violet emission along with emission in the blue and green bands. The position of the most intense peak lies in the range of 380–410 nm, which corresponds to the violet band. The blue and green emission bands which are in the region of 435–593 nm are similar to other reports.57,61 The relative intensities (Methods 3.2, 3.3, 3.4 and 3.5) in the visible region of 530–593 nm are very low compared to the ZnO NPs prepared by Methods 3.1 and 3.6. This is because of the presence of the defects at the surface of the nanoparticles. The defect density changes with the surface to volume ratio and according to the changes in the morphology and size of the NPs. Furthermore, variations in peak positions in the emission spectra are attributed to the changes in the defect densities on the surfaces of the NPs.62
5 Conclusion
ZnO nanoparticles were synthesized successfully by simple chemical routes using different precursors and solvents. The synthesized NPs were characterized by XRD, TEM, DRS and PL. The XRD and TEM results confirm the crystalline nature and single phase formation of the ZnO NPs. The DRS results revealed that there are no drastic changes in absorption edges and the optical band gaps of the ZnO NPs prepared by Methods 3.1 to 3.5, while a large blue shift was observed for the NPs synthesized by Method 3.6 with an optical band gap of 3.32 eV. It can be concluded that Method 3.6 can be used for smaller size and large band gap ZnO NPs. The zinc interstitial defects were present in all of the ZnO samples prepared with different precursors and solvents. Oxygen interstitial defects were present in the ZnO NPs prepared by Methods 3.1, 3.5 and 3.6.
Acknowledgements
The authors are thankful to DST New Delhi. Authors KA and MK acknowledge the financial support from INSPIRE Faculty award. Thanks to MRC, MNIT Jaipur for providing research facilities.
References
- V. Y. Davydov, V. Lundin, A. Smirnov, N. Sobolev, A. Usikov and A. Emel'yanov, Semiconductors, 1999, 33, 1 CrossRef CAS.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- V. Srikant and D. R. Clarke, J. Appl. Phys., 1998, 83, 5447–5451 CrossRef CAS.
- Y. Huang, X. Bai and Y. Zhang, J. Phys.: Condens. Matter, 2006, 18, L179 CrossRef CAS.
- D. King and R. Nix, J. Catal., 1996, 160, 76–83 CrossRef CAS.
- Y. K. Mishra, S. Kaps, A. Schuchardt, I. Paulowicz, X. Jin, D. Gedamu, S. Freitag, M. Claus, S. Wille and A. Kovalev,
et al.
, Part. Part. Syst. Charact., 2013, 30, 775–783 CrossRef CAS.
- N. Faraji, C. Ulrich, N. Wolff, L. Kienle, R. Adelung, Y. K. Mishra and J. Seidel, Adv. Electron. Mater., 2016, 2, 201600138 Search PubMed.
- Z. Li, W. Zhong, X. Li, H. Zeng, G. Wang, W. Wang, Z. Yang and Y. Zhang, J. Mater. Chem. C, 2013, 1, 6807–6812 RSC.
- A. Kołodziejczak-Radzimska and T. Jesionowski, Materials, 2014, 7, 2833–2881 CrossRef.
- R. Kumar, O. Al-Dossary, G. Kumar and A. Umar, Nano-Micro Lett., 2015, 7, 97–120 CrossRef.
- D. Yu, R. Cai and Z. Liu, Spectrochim. Acta, Part A, 2004, 60, 1617–1624 CrossRef PubMed.
- T. Baird, P. J. Denny, R. Hoyle, F. McMonagle, D. Stirling and J. Tweedy, J. Chem. Soc., Faraday Trans., 1992, 88, 3375–3382 RSC.
- R. P. Singh, V. K. Shukla, R. S. Yadav, P. K. Sharma, P. K. Singh and A. C. Pandey, Adv. Mater. Lett., 2011, 2, 313–317 CrossRef CAS.
- Y. Peng, A.-W. Xu, B. Deng, M. Antonietti and H. Cölfen, J. Phys. Chem. B, 2006, 110, 2988–2993 CrossRef CAS PubMed.
- G. Chinchen, P. Denny, D. Parker, M. Spencer and D. Whan, Appl. Catal., 1987, 30, 333–338 CrossRef CAS.
- T. Reimer, I. Paulowicz, R. Röder, S. Kaps, O. Lupan, S. Chemnitz, W. Benecke, C. Ronning, R. Adelung and Y. K. Mishra, ACS Appl. Mater. Interfaces, 2014, 6, 7806–7815 CAS.
- K. H. Kim, K. C. Park and D. Y. Ma, J. Appl. Phys., 1997, 81, 7764–7772 CrossRef CAS.
- Z. L. Wang, J. Phys.: Condens. Matter, 2004, 16, R829 CrossRef CAS.
- X. Li, J. Song, Y. Liu and H. Zeng, Curr. Appl. Phys., 2014, 14, 521–527 CrossRef.
- K. Keis, C. Bauer, G. Boschloo, A. Hagfeldt, K. Westermark, H. Rensmo and H. Siegbahn, J. Photochem. Photobiol., A, 2002, 148, 57–64 CrossRef CAS.
- T. K. Gupta, J. Am. Ceram. Soc., 1990, 73, 1817–1840 CrossRef CAS.
- D. Costenaro, F. Carniato, G. Gatti, L. Marchese and C. Bisio, New J. Chem., 2013, 37, 2103–2109 RSC.
- E. Comini, Anal. Chim. Acta, 2006, 568, 28–40 CrossRef CAS PubMed.
- J. Wang, X. Sun, A. Wei, Y. Lei, X. Cai, C. M. Li and Z. Dong, Appl. Phys. Lett., 2006, 88, 233106 CrossRef.
- T. Pauporté and D. Lincot, Electrochim. Acta, 2000, 45, 3345–3353 CrossRef.
- S. Reda, Mater. Sci. Semicond. Process., 2010, 13, 417–425 CrossRef CAS.
- B. Zhang, N. Binh, Y. Segawa, K. Wakatsuki and N. Usami, Appl. Phys. Lett., 2003, 83, 1635–1637 CrossRef CAS.
- L. Nehru, V. Swaminathan and C. Sanjeeviraja, Powder Technol., 2012, 226, 29–33 CrossRef CAS.
- H. Wang, C. Li, H. Zhao and J. Liu, Adv. Powder Technol., 2013, 24, 599–604 CrossRef CAS.
- R. K. Sharma and R. Ghose, Ceram. Int., 2015, 41, 967–975 CrossRef CAS.
- S. K. Lim, S.-H. Hwang, S. Kim and H. Park, Sens. Actuators, B, 2011, 160, 94–98 CrossRef CAS.
- T. Tharsika, A. Haseeb, S. Akbar and M. Thanihaichelvan, Ceram. Int., 2015, 41, 5205–5211 CrossRef CAS.
- H.-F. Lin, S.-C. Liao and S.-W. Hung, J. Photochem. Photobiol., A, 2005, 174, 82–87 CrossRef CAS.
- M. Zdujić, O. Milošević and L. Ć. Karanović, Mater. Lett., 1992, 13, 125–129 CrossRef.
- M. Debanath and S. Karmakar, Mater. Lett., 2013, 111, 116–119 CrossRef CAS.
- Y.-X. Wang, J. Sun, X. Fan and X. Yu, Ceram. Int., 2011, 37, 3431–3436 CrossRef CAS.
- E. Fazio, A. Mezzasalma, G. Mondio, F. Neri and R. Saija, Appl. Surf. Sci., 2013, 272, 30–35 CrossRef CAS.
- J. R. Jensen, T. Johannessen, S. Wedel and H. Livbjerg, J. Catal., 2003, 218, 67–77 CrossRef CAS.
- H. Kumar and R. Rani, Int. Lett. Chem., Phys. Astron., 2013, 14, 26–36 CrossRef.
- V. Chandore, G. Carpenter, R. Sen and N. Gupta, Int. J. Environ. Sci.: Dev. Monit., 2013, 4, 45–47 Search PubMed.
- P. Giri, S. Bhattacharyya, D. K. Singh, R. Kesavamoorthy, B. Panigrahi and K. Nair, J. Appl. Phys., 2007, 102, 093515 CrossRef.
- H. R. Ghaffarian, M. Saiedi, M. A. Sayyadnejad and A. M. Rashidi, Iran. J. Chem. Chem. Eng., 2011, 30, 1–6 CAS.
- V. Parthasarathi and G. Thilagavathi, Int. J. Pharm. Pharm. Sci., 2011, 3, 392–398 CAS.
- S. S. Ashtaputre, A. Deshpande, S. Marathe, M. Wankhede, J. Chimanpure, R. Pasricha, J. Urban, S. Haram, S. Gosavi and S. Kulkarni, Pramana, 2005, 65, 615–620 CrossRef CAS.
- S. Senthilkumar and T. Sivakumar, Int. J. Pharm. Pharm. Sci., 2014, 6, 461–465 Search PubMed.
-
S. K. Kulkarni, Nanotechnology: Principles and Practices, Springer, Mahaveer Street, Darayagang, New Delhi, 1st edn, 2009 Search PubMed.
- F. Qian, P. C. Lan, T. Olson, C. Zhu, E. B. Duoss, C. M. Spadaccini and T. Y.-J. Han, Chem. Commun., 2016, 52, 11627–11630 RSC.
- A. Kumar, L. K. Jangir, Y. Kumari, M. Kumar, V. Kumar and K. Awasthi, J. Appl. Polym. Sci., 2016, 133, 44091 CrossRef.
- M. Inoue and I. Hirasawa, J. Cryst. Growth, 2013, 380, 169–175 CrossRef CAS.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- A. Singh, V. Viswanath and V. Janu, J. Lumin., 2009, 129, 874–878 CrossRef CAS.
- R. Cross, M. De Souza and E. S. Narayanan, Nanotechnology, 2005, 16, 2188 CrossRef CAS PubMed.
- J. Ye, S. Gu, F. Qin, S. Zhu, S. Liu, X. Zhou, W. Liu, L. Hu, R. Zhang and Y. Shi,
et al.
, Appl. Phys. A: Mater. Sci. Process., 2005, 81, 759–762 CrossRef CAS.
- X. Wu, G. Siu, C. Fu and H. Ong, Appl. Phys. Lett., 2001, 78, 2285–2287 CrossRef CAS.
- S. Bhattacharyya and A. Gedanken, J. Phys. Chem. C, 2008, 112, 659–665 CAS.
- B. Debnath, G. Halder and S. Bhattacharyya, Sci. Adv. Mater., 2014, 6, 1160–1169 CrossRef CAS.
- Y. Gong, T. Andelman, G. F. Neumark, S. O'Brienand and I. L. Kuskovsky, Nanoscale Res. Lett., 2007, 2, 297 CrossRef CAS.
- C. H. Ahn, Y. Y. Kim, D. C. Kim, S. K. Mohanta and H. K. Cho, J. Appl. Phys., 2009, 105, 013502 CrossRef.
- S. Studenikin, N. Golego and M. Cocivera, J. Appl. Phys., 1998, 84, 2287–2294 CrossRef CAS.
- A. Djurišić, Y. Leung, K. Tam, L. Ding, W. Ge, H. Chen and S. Gwo, Appl. Phys. Lett., 2006, 88, 103107 CrossRef.
- T. K. Kundu, N. Karak, P. Barik and S. Saha, Int. J. Soft Comput. Eng., 2011, 1, 19–24 Search PubMed.
- A. Oudhia, A. Choudhary, S. Sharma, S. Aggrawal and S. Dhoble, J. Lumin., 2014, 154, 211–217 CrossRef CAS.
- A. O. Awodugba and A.-M. O. Ilyas, AJSC, 2013, 2, 41–44 Search PubMed.
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a










