
Insights into the origin of aggregation enhanced emission of 9,10-distyrylanthracene derivatives†
Jibo
Zhang‡
a,
Suqian
Ma‡
a,
Honghua
Fang
b,
Bin
Xu
*a,
Hongbo
Sun
b,
Im
Chan
 cd and
Wenjing
Tian
*a
cd and
Wenjing
Tian
*a
aState Key Laboratory of Supramolecular Structure and Materials, Jilin University Qianjin Street No. 2699, Changchun 130012, P. R. China. E-mail: xubin@jlu.edu.cn; wjtian@jlu.edu.cn
bState Key Laboratory on Integrated Optoelectronics, Jilin University, Qianjin Street No. 2699, Changchun 130012, P. R. China
cKonkuk University – Fraunhofer Institute for Solar Energy Systems Next Generation Solar Cell Research Center(KFnSC), Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 143-701, Republic of Korea
dDepartment of Chemistry, Konkuk University, 120 Neungdong-ro, Gwangjin-gu, Seoul 143-701, Republic of Korea
First published on 1st March 2017
Abstract
9,10-Distyrylanthracene (DSA) and its four derivatives are investigated by both steady state and ultrafast spectroscopy to reveal the intrinsic photophysical process upon excitation. Intramolecular rotation around the vinyl moiety plays an important role in the whole photophysical process in addition to the electronic properties of the peripheral substituents. In dilute solutions, DSA derivatives possess a twisted structure in the ground state that eventually relaxes to a planar structure within picoseconds. The fluorescence process is dominated by the relaxed excited state, and the quantum yield is affected by competition between the nonradiative and radiative deactivations. The enhanced fluorescence of the molecular aggregates originates from the optically allowed S1–S0 transition together with the suppressed nonradiative deactivation via molecular stacking. These findings provide an in-depth understanding of the origin of the aggregation enhanced emission process, and may be applicable for the fine design of DSA based molecules with enhanced fluorescence and novel structures beyond DSA.
Introduction
The development of organic molecules exhibiting efficient solid-state luminescence is an extremely challenging task due to the aggregation effect of molecules which usually causes the concentration quenching of luminescence in molecular aggregates.1–3 Great efforts have been made to deal with the quenching4–7 among which aggregation-induced/enhanced emission (AIE/AEE) stands out from the competition.8–11 In such a process the non-emissive fluorophores turn out to be highly luminescent in the aggregated state favoring optoelectronic applications.10–15 The family of AIE molecules was initiated by the study on hexaphenylsilole16 and tetraphenylethene17 derivatives, and is continuously growing with precisely designed fluorophores18 and the corresponding mechanisms proposed, such as the restriction of intramolecular motions (RIM),19 the formation of J-aggregates8 and the elimination of a charge transfer state.20Among the AIE molecules, 9,10-distyrylanthracene (DSA) and its derivatives have been widely utilized in applications for solid state light-emission,21,22 bioimaging,23 and chemical and biosensing24,25 for their high fluorescence quantum yield in molecular aggregates. However, there is still a lack of a general understanding of the photophysical process regarding the photoluminescence of DSA derivatives, specifically in the excited state. Herein, we present a mechanistic study using five DSA-based molecules with both steady state and ultrafast spectroscopy to reveal the intrinsic photophysical process upon excitation as well as the properties of the subsequent excited states from which the fluorescence originates. The unique structure of DSA derivatives prefers the ultrafast intramolecular rotation around the vinyl moiety, and therefore the molecular configuration is sensitively affected. It is indicated that the large Stokes shift in dilute solutions comes from the relaxed state with an extended conjugation upon photoexcitation competing with the fast nonradiative deactivation process that leads to the formation of a low quantum yield. In addition to the photophysical behavior of the molecule, the effects of a supramolecular structure on the properties of molecular aggregates are also investigated. Single crystal structures are included to investigate the enhanced fluorescence in molecular aggregates. The optically allowed S1–S0 transition combined with the suppressed nonradiative deactivation via molecular stacking leads to a significant enhancement in fluorescence of the crystals. These results provide an insight into the photophysical process of DSA derivatives and may contribute to a further design of DSA based molecules with efficient fluorescence.
Experimental
The derivatives share a similar divinylanthracene (DVA) structure in spite of the different aryl groups attached. The synthesis routes can be found elsewhere.21,22,27,28 Among the five molecules, DSA acts as the prototype. BDPVA has a twisted structure arising from the huge steric hindrance around the biphenyl moiety, so that it is as an example with a forced twisted structure. BP2VA, BMOSA and BTVA are then selected to demonstrate the effect of electron-withdrawing/donating on fluorescence. Experimental details are summarized in the ESI.†Results and discussion
Basic photophysical properties
The absorption and photoluminescence (PL) spectra of the derivatives in tetrahydrofuran are shown in Fig. 1 and Table 1. The concentration is kept at 10−5 M to eliminate the effect of aggregation. A characteristic absorption band at ∼410 nm is found among all the derivatives, which accounts for the π–π* transition of DVA.22 The substitution of phenyl has a mere effect on the vibronic transition since the π–π* band is only shifted by about 10 nm. In contrast, the fluorescence spectra are highly dependent on the substitutions. For instance, DSA exhibits a broad band peaking at 615 nm, while the emission peak of BDPVA is at 524 nm, and the emission peak is further shifted to 697 nm for BTVA. The Stokes shift of these derivatives is as high as ∼7000 cm−1. Generally, the large Stokes shift is caused by the significant decrease in the energy of the excited state after photoexcitation. Since the molecules are excited at the lowest absorption band, the transitions with higher energy (e.g. S0–S2) are not involved. Therefore, the energy decrease in the S1 state could be caused by several processes, such as a charge transfer state,29 excimer30 and large geometrical conversion between a DVA unit and the neighboring segments.31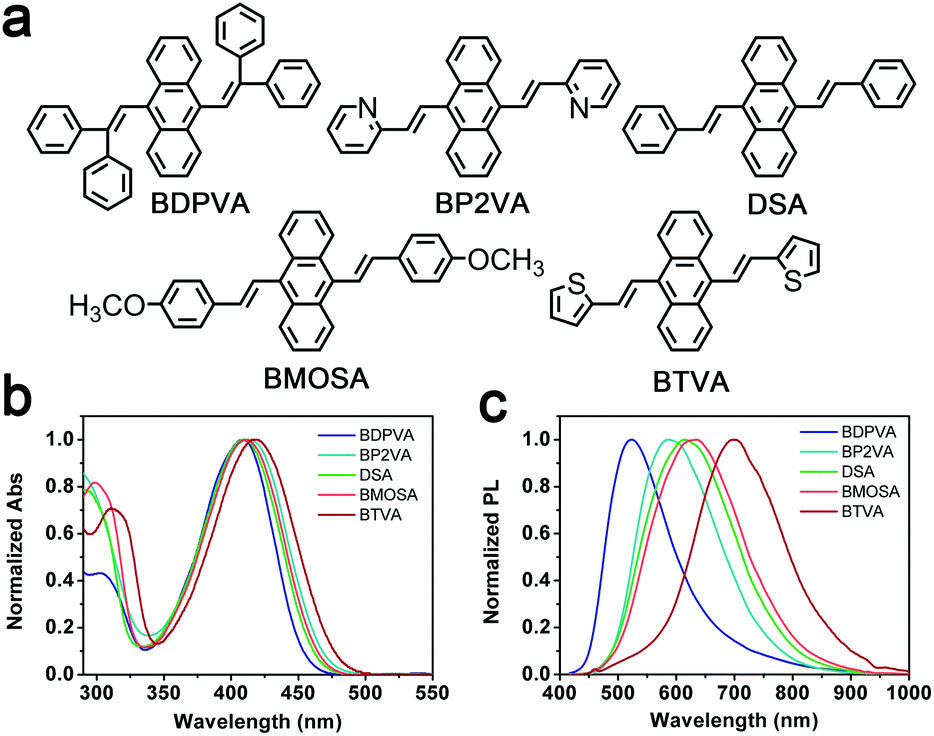 | ||
| Fig. 1 (a) Molecular structure of the DSA derivatives. (b) Normalized absorption and (c) fluorescence spectra of the derivatives in tetrahydrofuran at 10−5 M. | ||
| Derivative | λ abs (nm) | λ PL (nm) | Stokes shift (cm−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TOLa | CFa | THFa | TOL | CF | THF | TOL | CF | THF | |
| a Abbreviations: TOL: toluene; CF: chloroform; THF: tetrahydrofuran. | |||||||||
| BDPVA | 410 | 410 | 409 | 525 | 533 | 518 | 5343 | 5629 | 5144 |
| BP2VA | 415 | 416 | 412 | 575 | 587 | 586 | 6705 | 7003 | 7207 |
| DSA | 409 | 409 | 410 | 586 | 614 | 615 | 7326 | 8104 | 8130 |
| BMOSA | 414 | 414 | 415 | 616 | 650 | 649 | 7863 | 8712 | 8688 |
| BTVA | 417 | 418 | 418 | 679 | 697 | 698 | 9253 | 9636 | 9597 |
Our previous study has shown that DSA derivatives (AnPHZ2 and AnPHZ4, molecular structures shown in Scheme S1, ESI†) with strong electron donating groups prefer to form the intramolecular charge transfer (ICT) state to lower the excited state energy especially in a polar medium.29 Nevertheless, the derivatives involved here do not show significant solvatochromic effects as summarized in Table 1. Toluene, chloroform and tetrahydrofuran are chosen due to their gradually increased orientational polarizability (Δf) of 0.014, 0.149 and 0.211 according to the following equation.32
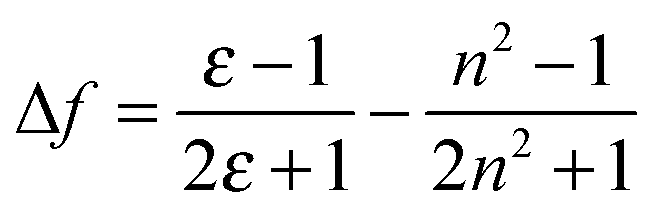
Both the electron-withdrawing and donating groups may cause the redistribution of electron density with a subsequent ICT in the excited state. And the ICT state could be characterized by the greatly increased Stokes shift in polar solvents.32 BP2VA possesses the electron-withdrawing pyridyl moieties and exhibits a small increase in the Stokes shift from 6705 cm−1 in toluene to 7207 cm−1 in tetrahydrofuran. Similarly, BTVA has a nearly unchanged Stokes shift in the three solvents. The fluorescence Stokes shift as a function of the orientational polarizability Δf for the derivatives in different solvents is also summarized in Fig. S1 (ESI†). The mere increase in the Stokes shift should be attributed to the slightly increased dipole moment of a locally excited (LE) state.32 So, none of the derivatives have shown typical evidence for the formation of the ICT state. Although the further characterization in high polar solvents is limited by the poor solubility, clues could be found from the comparison between the derivatives and AnPHZ2.29 For instance, the PL peaks of AnPHZ2 and BTVA are at 633 and 680 nm, respectively, in toluene where the twist intramolecular charge transfer (TICT) is suppressed by the low polarity and the emission is from the LE state. Phenothiazine is a stronger electron donating group than thiophene, so it should contribute more to increasing the highest occupied molecular orbital (HOMO) and end up with an emission with lower energy. However, our observation proves conversely. In consideration of the previous results from AnPHZ2, it is likely that BTVA has a more planar structure to lower the excited energy owing to the extended conjugation. And more importantly, it can be concluded that the large Stokes shift is not brought about by the ICT state. Further investigation was carried out to confirm the hypothesis, such as ultrafast spectroscopy and crystal structure analysis.
The molecular configuration in a rigid matrix
The configuration of a molecule is somewhat confined by its intrinsic steric hindrance.21 As for DSA derivatives, the steric hindrance induced by the hydrogen atoms on anthracene and vinyl moieties usually causes the DVA to deviate from a strictly planar configuration (see Fig. S2, ESI†). In contrast, there is no significant steric hindrance existing among the vinyl and peripheral aromatic substitutions. Hence the dihedral angle between anthracene and vinyl (α1) is much higher than that between vinyl and peripheral aryl moieties (α2).21 It should be noted that the biphenyl group in BDPVA leads to the formation of a twisted structure of the biphenyl-vinyl due to the steric hindrance arising from the adjacent phenyls. Therefore, the aryl vinyl moiety is partially involved in the conjugation of anthracene which is highly dependent on the dihedral angle.22The absorption spectra of the derivatives shown in Fig. 1b demonstrate their similar conjugation extent in the ground state, in which the structures are highly twisted around the vinyl-anthracene bond to minimize the ground state energy. In order to eliminate the potential effect of intramolecular rotation on the conjugation extent upon excitation, the derivatives are dispersed in rigid matrices, for instance the transparent polymer and frozen solvent, to maintain their configurations. As shown in Fig. 2, all the derivatives exhibit similar absorption when dispersed in poly(methyl methacrylate) (PMMA) at 2 wt% to eliminate aggregation, in spite of the low signal-to noise ratio from the mere concentration. The PL spectra are significantly different from those in solutions. All the derivatives have shown green emission centering at ∼510 nm with lowered FWHM (peak width half-height) indicating the suppressed intramolecular rotation and vibration. BDPVA has the absorption and emission bands with the highest energy indicating the lowest conjugation extent, which is reasonable according to its twisted structure.22 The spectra of BP2VA, BMOSA and BTVA are slightly different from that of DSA, as can be ascribed to the electronic effects of the substitutions through the conjugated vinyl. Similar PL spectra were observed from the dispersion of the derivatives in 2-methyltetrahydrofuran at 77 K (Fig. S3, ESI†), where the intramolecular rotation and vibration were further restricted by both the rigid matrix and the low temperature. The emission peaks are ∼20 nm blue-shifted compared to those in PMMA, with vibration coupled sub peaks becoming obvious. The phenomena in a rigid matrix clearly demonstrate the effect of intramolecular rotation on the emission spectra. BDPVA has a forced twisted structure so that the Stokes shift is about the same in solution and a rigid matrix because of the lower contribution from the biphenylvinyl moiety (high α2).22 While for the other four derivatives, in which the arylvinyl is planar (low α2), even a small decrease in α1 (intramolecular rotation) upon excitation may help to extend the conjugation of anthracene so as to lower the excited state energy.31 Therefore, the Stokes shift is much smaller when dispersed in a rigid matrix. Besides, it should be noted that the mere rotation and vibration can still take place in PMMA due to thermal activation as indicated by the slight blue-shifts in frozen 2-methyltetrahydrofuran solution, let alone in room temperature solutions. So far, clear evidence has been provided by steady state spectroscopy to investigate the configurational change in DSA derivatives upon photoexcitation. Ultrafast spectroscopy was then carried out to obtain a direct view of the evolution of molecular configurations.
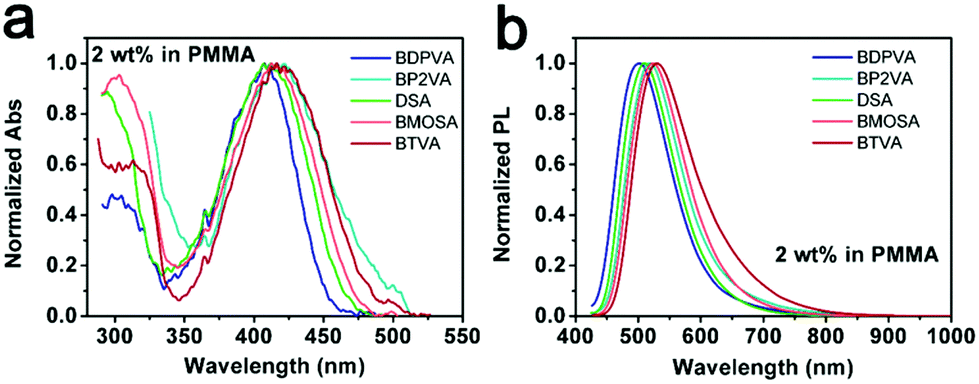 | ||
| Fig. 2 (a) Normalized absorption and (b) fluorescence spectra of the derivatives in PMMA films at 2 wt%. | ||
Ultrafast spectroscopy
Intramolecular vibration and rotation relaxation usually take place in picoseconds upon excitation, which is much faster than the radiative decay from S1 to S0 (ns).31,33 Hence, the fluorescence properties such as emission spectra and quantum yield are more related to the molecular configurations after initial relaxation. In order to show clearly what was happening in the sub-nanosecond scale, ultrafast transient absorption (TA) spectroscopy34 and the fluorescence up-conversion technique31,35 were carried out. BDPVA and DSA were taken as examples due to their unique structural characteristics. DSA has pristine phenyl moieties attached on the vinyl so as to eliminate the potential deviations from the electronic structure of substitutions on intramolecular rotation and vibration. Therefore, any change in the conjugation extent of DSA will be reflected by the differences in the spectra. On the other hand, the highly twisted configuration of BDPVA ensures that the biphenyl hardly contributes to the conjugation of anthracene through the vinyl even when α1 becomes pretty small. The vinyl itself has a small effect on the conjugation of anthracene in spite of α1. Thus it could be predicted that the conjugation extent of BDPVA is relatively unchanged. Furthermore, the spectra were recorded in toluene whose polarity is quite low to prevent the solutes from interacting with solvent molecules.TA spectra of DSA and BDPVA are shown in Fig. 3a and b. The pump pulse was at 400 nm to ensure that no transitions with higher energy (e.g. S0–S2) were involved. The characteristic stimulated emissions (SE) centering at ∼520 nm were observed for both DSA and BDPVA within a few ps after excitation, together with the excited state absorption (ESA) bands at 600–700 nm. The SE band of DSA deformed and vanished drastically after 5 ps, meanwhile the ESA band decreased and underwent a shift from 650 to 570 nm. In contrast, BDPVA has a nearly unchanged spectrum within 500 ps, which indicates a nearly unchanged conjugation in the excited state. By comparing the spectra with steady state fluorescence, the SE band could be attributed to the radiative S1–S0 decay of the DVA moiety. And the ESA band at 600–700 nm is from the transition of the DVA moiety to higher states. It is worth mentioning that the ESA band at 450 nm observed for both DSA and BDPVA is likely to be from the peripheral moieties since it has a higher energy than the S1–S0 transition of DVA, in consideration of the lowered energy gap between higher excited states. The spectral evidence of DSA indicates the possible ultrafast structural evolution that leads to a change in the conjugation extent. In order to prove this, DSA was dispersed in a rigid matrix (PMMA) with the corresponding TA spectra shown in Fig. 3c. DSA exhibited a constant decrease at the SE band which is in good agreement with its PL spectrum at 520 nm. In addition, the ESA band of DSA was merely shifted, in sharp contrast with the spectrum in toluene. Theoretical calculations based on time-dependent density functional theory (TD-DFT) predicts the structural change upon excitation. As indicated in Fig. S4 (ESI†), after excitation, the dihedral angle α1 of both BDPVA and DSA decreased rendering a more planer structure (from 54° to 36°) so as to extend the conjugation of the central anthracene. In a BDPVA molecule, the vinyl has small contribution since the biphenyl is nearly perpendicular to it. However, as for DSA the whole styryl would be more conjugated with anthracene owing to the small α2 and therefore a planer excited state configuration was formed in which the subsequent fluorescence took place. Furthermore, once the intramolecular rotation induced structural evolution was blocked in a rigid medium, DSA behaved more similarly to BDPVA indicating a similar conjugation extent of DVA. Consequently, the fluorescence of DSA at ∼600 nm should be originated from the quasi-planer configuration induced by the ultrafast structural evolution from the initial twisted structure.
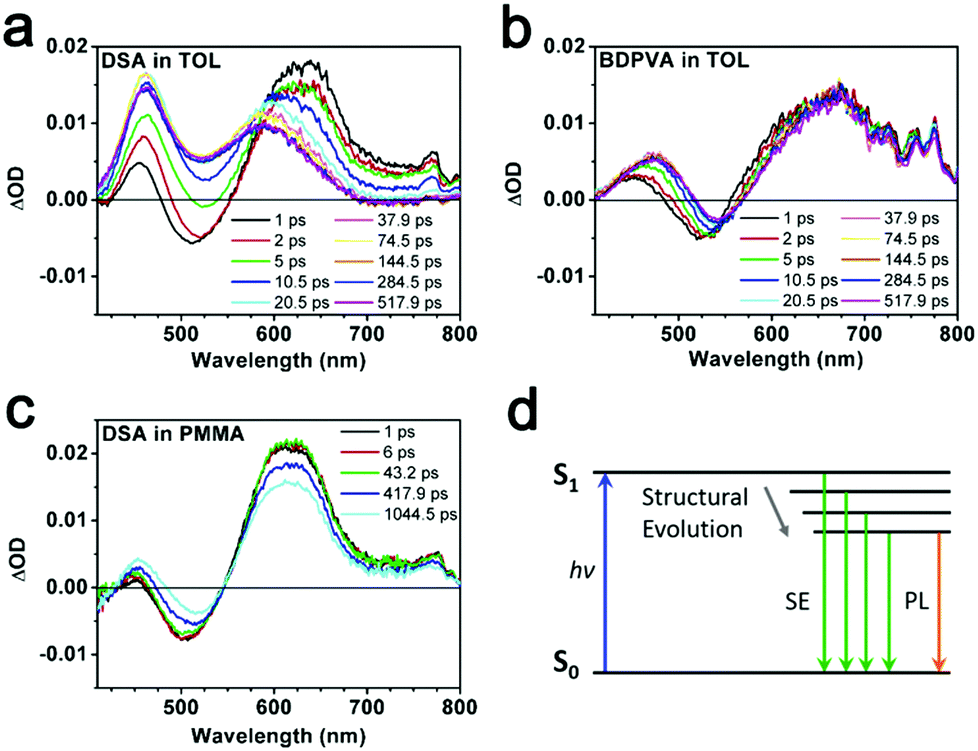 | ||
| Fig. 3 Transient absorption spectra of (a) DSA and (b) BDPVA in toluene. (c) Transient absorption spectrum of DSA in PMMA. (d) Illustration of the S1 energy of DSA accompanied by the structural evolution. The vibration states and evolution of S0 are omitted for clarity. | ||
A fluorescence up-conversion technique was employed to study the ultrafast process at specific wavelengths. Fig. 4 shows the time-resolved peak fluorescence of DSA and BDPVA in toluene. A variety of wavelengths covering the PL spectra were selected to study the structural evolution. As shown in Fig. 4a and b, the peak intensity of DSA at a shorter wavelength decreased much faster than that at a longer wavelength. For instance, a comparison between the intensity at 530 and 600 nm is shown in Fig. 4c. As indicated in the TA spectrum, 530 nm is the emission from an initially twisted structure right after excitation. Whereas, 600 nm is around the peak of the steady state PL spectrum of DSA. It was clearly shown that the emission at 530 nm was generated immediately after the excitation to reach the apex, and then underwent a fast decrease in 50 ps. In contrast, the emission at 600 nm was not directly pumped to the peak. It exhibited an increase in ∼20 ps after excitation and then remained nearly constant. This evidence shows that the species emitting at 600 nm is not preferably generated by the excitation pulse; instead it mainly originates from a fast evolution process, which is intramolecular rotation on this occasion.
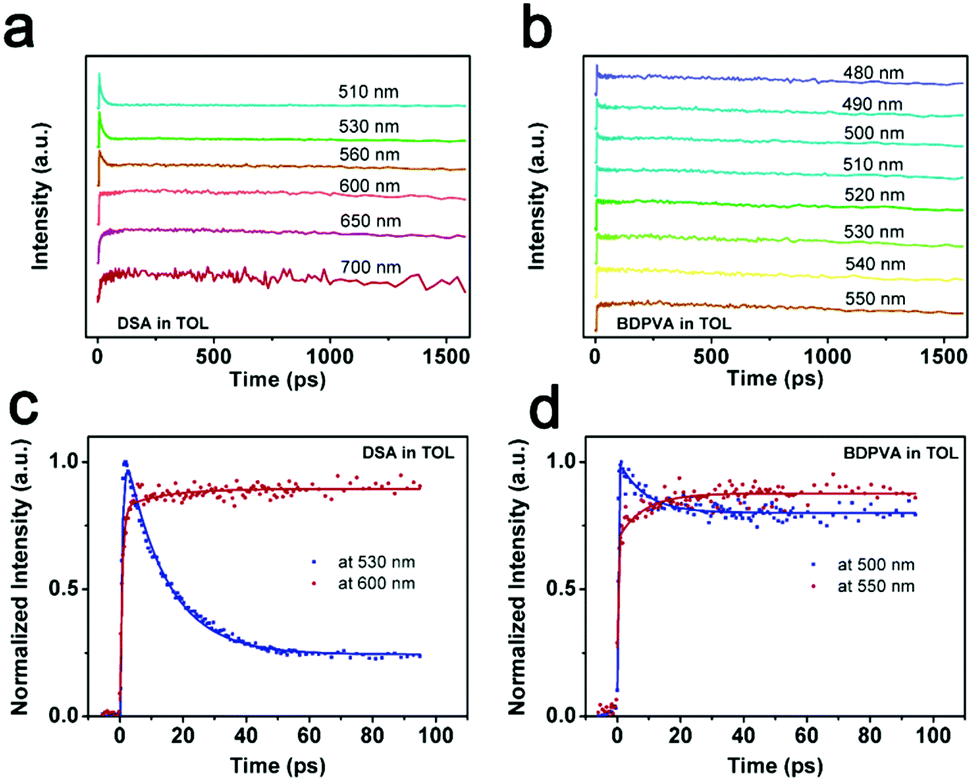 | ||
| Fig. 4 Time-resolved peak fluorescence of (a and c) DSA and (b and d) BDPVA at different wavelengths. | ||
Global fitting of the transients reveals the ultrafast decay kinetics of DSA and BDPVA as summarized in Table 2. DSA consists of three components with τ of 1.625, 15.29 and 3346 ps, respectively. The longest τ (3346 ps) can be attributed to the fluorescence process which is in agreement with the TCSPC data as shown below.21 The other two species with much shorter τ (1.625 and 15.29 ps) indicate the fast relaxation via intramolecular rotation after initial excitation. The amplitudes (Ai) demonstrate that the fast decay at 530 nm, where the stimulated emission occurs, is mainly induced by the species with short τ. In contrast, at 600 nm, which is around the peak wavelength of the PL spectra, the decay curve is dominated by the long τ species. And the slightly negative amplitudes of τ1 and τ2 indicate that the species emitting at 600 nm is generated not only by the excitation, but also by a transition from the short τ species.36,37 The intramolecular rotation around the vinyl facilitates the planarization and therefore the emitting wavelength is shifted from 530 nm to 600 nm due to the extended conjugation. A similar phenomenon is observed for BDPVA. Notably, the amplitudes of τ3 are much higher than those of τ1 and τ2 for both 500 and 550 nm. Taking the TA spectrum into account, the energy of the excited state is hardly affected by the rotation around the vinyl due to the huge steric hindrance around the biphenyl moiety. Although the intramolecular rotation does occur as indicated by the evolution of amplitudes, it has negligible effects on the conjugation so that the long τ species which is fluorescent becomes dominant.38
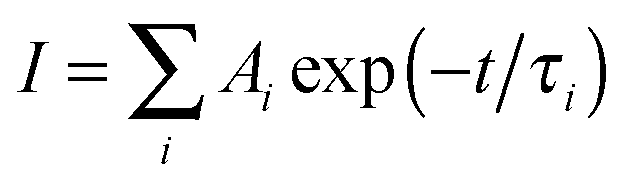 )
)
| Detection (nm) | A 1 | A 2 | A 3 | τ 1 (ps) | τ 2 (ps) | τ 3 (ps) | |
|---|---|---|---|---|---|---|---|
| DSA | 530 | 0.069 | 0.730 | 0.232 | 1.625 | 15.29 | 3346 |
| 600 | −0.117 | −0.039 | 0.899 | ||||
| BDPVA | 500 | 0.047 | 0.115 | 0.820 | 1.993 | 9.904 | 1550 |
| 550 | 0.021 | −0.178 | 0.902 | ||||
It is shown by ultrafast spectroscopy that the intramolecular rotation around the vinyl occurs for both DSA and BDPVA with different outcomes due to the geometrical difference in the molecular structures. It can be inferred that BP2VA, BMOSA and BTVA bearing a DVA structure will undergo a similar structural evolution and relax to a more planar structure than the ground state in dilute solution. The ones having a similar structure to BDPVA indeed will also relax to a planar structure, however, with much less conjugation from the peripheral moiety involved. The steric hindrance becomes significant for the biphenyl derived moieties such as BDPVA and therefore the excited state energy remains nearly unchanged in the presence of intramolecular rotation. Hopefully, the excited sate energy level and configuration can be precisely controlled by the proper design of substituents on vinyl.
Steady state fluorescence
Ultrafast spectroscopy reveals the intramolecular rotation within a few ps after photoexcitation, while the steady state fluorescence properties, such as PL spectra and quantum yield, are more related to the relaxed S1 state that takes place at an ns scale.31 The time-correlated single photon counting (TCSPC) technique was used to investigate the photophysical properties of DSA derivatives in their relaxed S1 state, from which the fluorescence was generated. The PL spectra become more comprehensible since the configurations of the derivatives have been figured out.In their excited states, BDPVA has a twisted structure, while the other 4 derivatives have quasi-planer structures, so the emission peak position of BDPVA is quite distinguishable because of the low conjugation extent. On the other hand, different substitutions have shown their effects on the emission of DSA as the electronic effect is conducted through the planer structure.39 DSA has a broad emission peaking at 615 nm in tetrahydrofuran. The electron-donating nature of methoxyl and thiophene lowers the bandgap of BMOSA and BTVA resulting in the red-shifted emission. The further shifted emission of BTVA originates from the stronger electron-donating properties of thiophene. In contrast, pyridinyl increases the bandgap by its electron-withdrawing ability to generate a blue-shifted emission of BP2VA. In addition, the fluorescence kinetics of the derivatives are revealed in addition to the quantum yield as indicated by the following equation:31

As shown in Fig. 5a and Fig. S5 (ESI†), the derivatives have different decay curves for the fluorescence. The lifetimes with the corresponding deactivation rates are summarized in Table 3. BDPVA, BP2VA and DSA possess a single exponential decay indicating that the S1–S0 transition is from a stable excited state. However, the best fitting parameters of BMOSA and BTVA exhibit a double-exponential decay, and the average lifetime (τavg) is much smaller than those of BDPVA, BP2VA and DSA. The short lifetime (τ1) species with few ages observed for both BMOSA and BTVA should be the residual of fast intramolecular rotation, meanwhile the smaller τ2 (as compared with the other derivatives) demonstrates the fast deactivation processes. kr and knr reveal the competing deactivation processes. kr is usually related to the intrinsic properties of the molecule, such as the conjugation, transition dipole moment (oscillator strength), spin etc.26,33knr is dominated by the nonradiative deactivation processes, for example, the most notorious intramolecular vibrational and rotational motions in AIE fluorophores.11,19 As shown in Table 3, kr is pretty similar for the derivatives varying in the same order of magnitude owing to the fact that S1–S0 transition primarily takes place on the DVA moiety. However, the knr values of different molecules differ a lot. BDPVA, BP2VA and DSA have knr values of ∼20–50 × 107 s−1, while BMOSA and BTVA have knr values that are higher than 100 × 107 s−1.
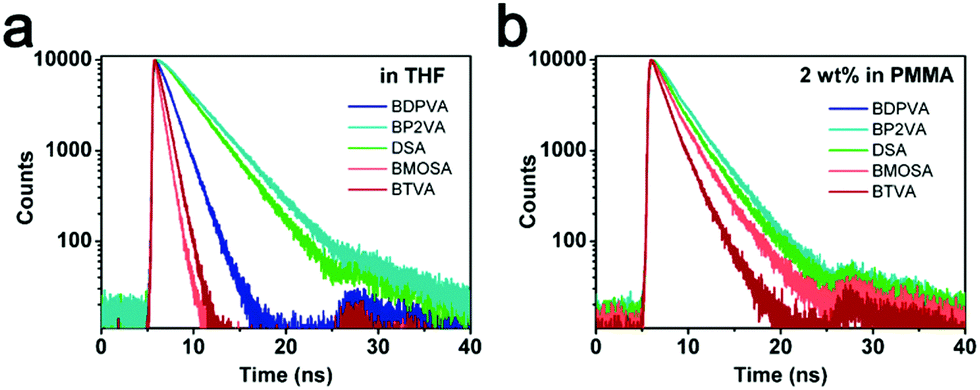 | ||
| Fig. 5 Time-resolved peak fluorescence of the derivatives in (a) tetrahydrofuran and (b) PMMA films (2% wt). | ||
| τ 1 (ns) | τ 2 (ns) | τ avg (ns) | ϕ | k r (107 s−1) | k nr (107 s−1) | |
|---|---|---|---|---|---|---|
| a The fluorescence is too weak to give an accurate quantum yield. | ||||||
| BDPVA | 1.41 | 1.41 | 0.17 | 12 | 59 | |
| BP2VA | 3.68 | 3.68 | 0.33 | 8.9 | 18 | |
| DSA | 3.22 | 3.22 | 0.21 | 6.5 | 25 | |
| BMOSA | 0.05 (18.5%) | 0.59 (81.5%) | 0.49 | <0.05a | <10 | >100 |
| BTVA | 0.02 (10.6%) | 0.76 (89.4%) | 0.68 | <0.05a | <7.3 | >100 |
We have shown that the derivatives tend to form a planer structure in the excited state to extend the conjugation in which the electron cloud is more delocalized and therefore the excited state is stabilized. By comparing the different values, it could be found that the derivatives with electron-donating groups (BMOSA and BTVA) have much higher knr, while the one with an electron-withdrawing group (BP2VA) has the smallest knr. Taking DSA as the standard, we would be able to draw a clear conclusion on the general tendency of the derivatives. A high knr indicates an unstable excited state that is vulnerable for a nonradiative process. As for BMOSA and BTVA, although the excited state energy is lowered due to the delocalization induced by the extended conjugation, the absolute energy level is still increased because of the electron-donating nature of methoxyl and thiophene. Hence BMOSA and BTVA molecules are more energetic than DSA molecules and undergo more rapid intramolecular vibrations/rotations to deplete the energy via a nonradiative process. It is noteworthy that the slightly higher knr of BDPVA as compared to DSA and BP2VA is a result of the increased steric hindrance. Fig. 5b shows the decay curves in PMMA films, where the derivatives have a similar τ of ∼2 ns. The average τ values are 2.89, 2.29, 2.64, 2.21 and 1.53 ns for BDPVA, BP2VA, DSA, BMOSA and BTVA, respectively (Fig. S6, ESI†). When the intramolecular rotation is restricted by the rigid matrix, the fluorescence is mainly from the DVA moiety so that the derivatives have similar fluorescence kinetics, which is in consistent with the previous discussion.
So far, the photophysical process of DSA derivatives upon excitation is somewhat clear. In order to minimize the potential energy in the ground state, the molecules are in a twisted structure which is maintained upon the photoexcitation according to the Franck–Condon principle.33 Afterwards the ultrafast intramolecular rotation takes place to induce molecular relaxation towards a planar structure to extend the conjugation and therefore lower the excited state energy. According to our results, the compilation of the DVA moiety with adjacent moieties is a general tendency for the DSA-structured molecules to stabilize the excited molecules.
From a single molecule to aggregates
Generally, DSA derivatives exhibit enhanced emission in molecular aggregates, which is in sharp contrast with the conventional organic dyes that suffer from concentration quenching.21,24,27 Various mechanisms explaining the enhanced emission have been proposed. Herein, we combine a photophysics study on single molecules with the crystal structures to establish a general understanding of the enhanced emission of DSA derivatives. The fluorescence properties of molecular clusters depend not only on the intrinsic properties of the molecule itself, but also on the supramolecular structure.The derivatives have nearly identical fluorescence emission from their single crystals. The peak position ranges slightly from 518 nm of BDPVA to 529 nm of BTVA. The dihedral angles (defined in Fig. S2, ESI†) shown in Fig. 6b demonstrate the twisted structures of the derivatives, which are thermodynamically reasonable to lower the potential energy for molecular packing. It should be noted that BDPVA molecules have two configurations in the crystals, corresponding to two α1 and four α2. The high α1 values of the derivatives indicate the small contribution from the peripheral groups to the conjugation of anthracene moieties, whereas the vinyl is nearly planar with the aryl groups. Subsequently, the molecular configurations have shown that the low-lying S1–S0 transitions mainly take place at the central anthracene moieties with the corresponding transition dipole moments oriented along the long axis of the molecules. The intramolecular effects have indicated that the PL emissions for all five derivatives are from a quite similar structure and therefore the spectra should be nearly identical to each other as observed, regardless of the intermolecular effects.
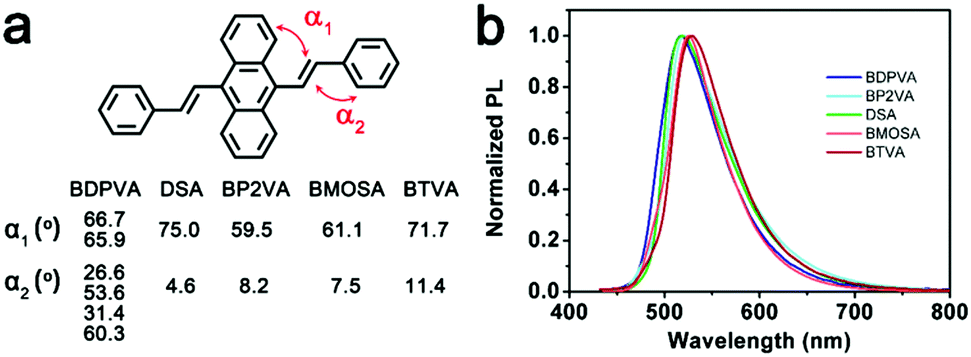 | ||
| Fig. 6 (a) Selected dihedral angles of the molecules and (b) PL spectra of the crystals. | ||
Intermolecular interactions have significant effects on the fluorescence of molecular aggregates as well.40 The molecules bearing vibronic transitions are usually regarded as molecular excitons and the intermolecular interactions are treated as excitonic coupling or more detailed exciton–polaron interactions for strongly coupled systems.41,42 As for a two-molecule system, the excitonic coupling will split the lowest excited state into two states that possess different transition dipole moments to the ground state according to the relative arrangement between the transition dipole moments of the adjacent molecules.43 And the one with higher transition dipole moment has a higher kr, in other words, it is more likely to be the state involved in the vibronic transition to the ground state. The excitonic coupling could be examined by the relative arrangement of the adjacent molecules since the transition dipole moment is dominated by the molecular structure. Molecular crystals have a definite supramolecular structure to serve as the ideal model for the study of the effect of intermolecular interactions as shown in Fig. 7 where the nearest two molecules are extracted from the corresponding structure.
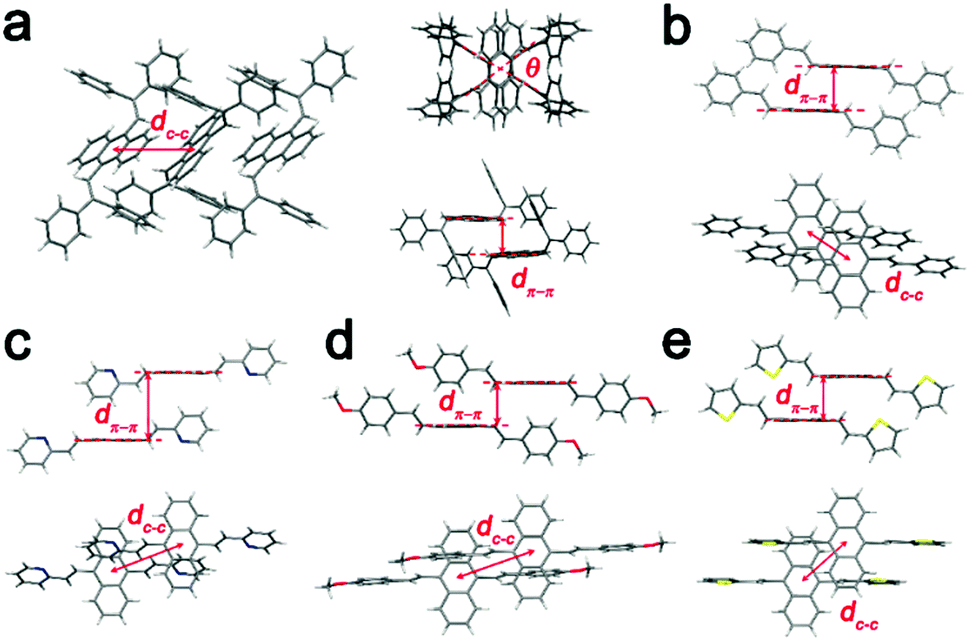 | ||
| Fig. 7 Molecular packing structures of (a) BDPVA, (b) DSA, (c) BP2VA, (d) BMOSA and (e) BTVA from the single crystal structures. | ||
The S1–S0 transition dipole moments of the derivatives are lying along the long axis and the arrangement of the transition dipole moments is evidently provided according to the crystal structure.18 BDPVA has a distinctive cross dipole stacking in which the rotational angle (Fig. 7a, θ = 67°) between neighboring transition dipoles reduces the energy splitting between the lowest two excited states of the dimer and a progressive transfer of intensity from the second excited state to the lowest excited state occurs.26,29 Therefore, it promotes a finite transition dipole moment between the ground state and the lowest excited state of the molecular aggregates indicating that the S1–S0 transition is prone to occur.26 The other four derivatives have similar stacking modes as shown in Fig. 7b–e. Each of the molecules is parallel to its neighbor with a slight offset along the long axis, favoring the J-aggregation nature (pitch angle θpitch higher than 54°) in which the transition dipole moment is concentrated in the lowest excited state and consequently results in a high kr value.44,45 In addition, no face-to-face facial π–π overlap is observed among the derivatives and the inter-centroid distance is quite large (>5 Å) eliminating the formation of an excimer as well as the possible intermolecular vibrational coupling.46 The intermolecular distance and fluorescence kinetics data summarized in Table 4 and Fig. S7 (ESI†) agree well with the prediction from crystal structures. Since the finite transition dipole moments are accumulated to the lowest ground state in the molecular aggregates, all the derivatives show increased kr as compared to single molecules (Table 3). Moreover, the nonradiative deactivation through vibrational and rotational motions are suppressed due to the molecular packing as indicated by the lowered knr values. As a result, the overall quantum yield is greatly increased in the crystals. It should be noted that the densest packing of BP2VA (the lowest dc–c and dπ–π) might lead to a strong excitonic coupling between the adjacent molecules that partially diminishes the transition dipole moment to the ground state. This is likely the main reason for the slightly low kr value of BP2VA.
| d c–c (Å) | d π–π (Å) | θ pitch (°) | τ avg (ns) | ϕ crystal | ϕ crystal/ϕsolution | k r (107 s−1) | k nr (107 s−1) | |
|---|---|---|---|---|---|---|---|---|
| BDPVA | 5.645 | 3.973 | n/a | 2.86 | 0.60 | 3.5 | 20 | 15 |
| BP2VA | 5.250 | 3.500 | 41.8 | 3.84 | 0.48 | 1.5 | 13 | 13 |
| DSA | 8.121 | 5.571 | 43.3 | 2.01 | 0.40 | 1.9 | 20 | 29 |
| BMOSA | 6.960 | 3.563 | 30.8 | 1.73 | 0.41 | >8 | 23 | 35 |
| BTVA | 5.641 | 3.683 | 40.8 | 1.20 | 0.25 | >5 | 21 | 62 |
The slender structure of the DVA moiety facilities J-aggregation along the long axis with the help of multiple supramolecular interactions (e.g. the C–H⋯π interaction between anthracene and the hydrogen atom on vinyl), allowing for various substitutions on the peripheral aryls without significant effects on the optically allowed S1–S0 transition. Generally speaking, the enhanced fluorescence of DSA derivatives in the molecular aggregates arises from two aspects. In addition to the common explanation for the AIE molecules, which is the blocked nonradiative deactivation from the restriction of intramolecular vibration and rotation, the key factor should be the promoted radiative deactivation process due to the concentrated transition dipole moments coupled to the ground state.
Conclusions
In summary, the photophysical properties of a series of DSA derivatives have been investigated in terms of steady state and ultrafast spectroscopy, molecular configurations and crystal structures. Intramolecular rotation around the vinyl moiety plays an important role in the whole photophysical process. In dilute solutions, the DSA derivatives possess a twisted structure in the ground state that eventually relaxes to a planar structure within several picoseconds. The excited state is therefore stabilized by the extended conjugation of the planar structure from which the large Stokes shift originates. The subsequent vibronic transition takes place in the relaxed excited state with a kr of ∼2 × 107 s−1. However, the competing nonradiative deactivation is dominated when the molecules are dispersed in solvents resulting in low quantum yields. In contrast, the nonradiative deactivation is greatly suppressed by molecular stacking in the aggregates together with the optically allowed S1–S0 transition, and therefore the fluorescence is significantly enhanced. Both of the intra- and intermolecular effects on the fluorescence of DSA derivatives are taken into account to clearly demonstrate the photophysical process in either the dispersed or aggregated state. These findings presented here could be applied to more DSA derivatives than the ones discussed, leading to further development of the DSA based materials as efficient fluorophores, and possibly initiating novel structures beyond DSA.Acknowledgements
This work was supported by 973 Program (2013CB834701), the Natural Science Foundation of China (No. 51573068, 51373063, and 21221063), Program for Chang Jiang Scholars and Innovative Research Team in University (No. IRT101713018), Program for Changbaishan Scholars of Jilin Province, and the Project of Jilin Province (20160101305JC).References
- K. D. Chaudhuri, Z. Angew. Phys., 1959, 154, 34 CAS.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318 CrossRef CAS PubMed.
- J. Dong, K. M. Solntsev and L. M. Tolbert, J. Am. Chem. Soc., 2009, 131, 662 CrossRef CAS PubMed.
- L. Chen, S. Xu, D. McBranch and D. Whitten, J. Am. Chem. Soc., 2000, 122, 9302 CrossRef CAS.
- S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS PubMed.
- H. Saigusa and E. C. Lim, J. Phys. Chem., 1995, 99, 15738 CrossRef CAS.
- P. N. Taylor, M. J. O'Connell, L. A. McNeill, M. J. Hall, R. T. Aplin and H. L. Anderson, Angew. Chem., Int. Ed., 2000, 39, 3456 CrossRef CAS PubMed.
- B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- S. Liu, F. Li, Q. Diao and Y. Ma, Org. Electron., 2010, 11, 613 CrossRef CAS.
- Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- Y. Hong, Methods Appl. Fluoresc., 2016, 4, 022003 CrossRef.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Pena-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845 CAS.
- J. He, B. Xu, F. Chen, H. Xia, K. Li, L. Ye and W. Tian, J. Phys. Chem. C, 2009, 113, 9892 CAS.
- J. Zhang, B. Xu, J. Chen, S. Ma, Y. Dong, L. Wang, B. Li, L. Ye and W. Tian, Adv. Mater., 2014, 26, 739 CrossRef PubMed.
- Z. Wang, B. Xu, L. Zhan, J. Zhang, T. Ma, J. Zhang, X. Fu and W. Tian, Nanoscale, 2013, 5, 2065 RSC.
- K. Ma, H. Wang, X. Li, B. Xu and W. Tian, Anal. Bioanal. Chem., 2015, 407, 2625 CrossRef CAS PubMed.
- X. Li, K. Ma, S. Zhu, S. Yao, Z. Liu, B. Xu, B. Yang and W. Tian, Anal. Chem., 2014, 86, 298 CrossRef CAS PubMed.
- J. Cornil, D. Beljonne, J. P. Calbert and J. L. Brédas, Adv. Mater., 2001, 13, 1053 CrossRef CAS.
- Y. Dong, B. Xu, J. Zhang, H. Lu, S. Wen, F. Chen, J. He, B. Li, L. Ye and W. Tian, CrystEngComm, 2012, 14, 6593 RSC.
- Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782 CrossRef CAS PubMed.
- J. Zhang, B. Xu, J. Chen, L. Wang and W. Tian, J. Phys. Chem. C, 2013, 117, 23117 CAS.
- J. B. Birks and L. G. Christophorou, Spectrochim. Acta, 1963, 19, 401 CrossRef.
- B. Valeur, Characteristics of Fluorescence Emission, Molecular Fluorescence, Wiley-VCH Verlag GmbH, 2001, pp. 34–71 Search PubMed.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef PubMed.
- B. Valeur, Absorption of Uv-Visible Light, Molecular Fluorescence, Wiley-VCH Verlag GmbH, 2001, pp. 20–33 Search PubMed.
- J. Cabanillas-Gonzalez, G. Grancini and G. Lanzani, Adv. Mater., 2011, 23, 5468 CrossRef CAS PubMed.
- H. Chosrowjan, S. Taniguchi and F. Tanaka, FEBS J., 2015, 282, 3003 CrossRef CAS PubMed.
- T. Yoshihara, S. I. Druzhinin and K. A. Zachariasse, J. Am. Chem. Soc., 2004, 126, 8535 CrossRef CAS PubMed.
- A. Douhal, M. Sanz, M. A. Carranza, J. A. Organero and L. Santos, Chem. Phys. Lett., 2004, 394, 54 CrossRef CAS.
- B.-R. Gao, H.-Y. Wang, Y.-W. Hao, L.-M. Fu, H.-H. Fang, Y. Jiang, L. Wang, Q.-D. Chen, H. Xia, L.-Y. Pan, Y.-G. Ma and H.-B. Sun, J. Phys. Chem. B, 2010, 114, 128 CrossRef CAS PubMed.
- X. Y. Shen, Y. J. Wang, E. Zhao, W. Z. Yuan, Y. Liu, P. Lu, A. Qin, Y. Ma, J. Z. Sun and B. Z. Tang, J. Phys. Chem. C, 2013, 117, 7334 CAS.
- K. Wang, H. Zhang, S. Chen, G. Yang, J. Zhang, W. Tian, Z. Su and Y. Wang, Adv. Mater., 2014, 26, 6168 CrossRef CAS PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- A. P. Demchenko, J. Heldt, J. Waluk, P.-T. Chou, P. K. Sengupta, L. Brizhik and J. C. del Valle, Angew. Chem., Int. Ed., 2014, 53, 14316 CrossRef CAS PubMed.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed.
- S. Choi, J. Bouffard and Y. Kim, Chem. Sci., 2014, 5, 751 RSC.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed.
- J. Gierschner, H.-G. Mack, D. Oelkrug, I. Waldner and H. Rau, J. Phys. Chem. A, 2004, 108, 257 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1526629. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00032d |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2017 |