DOI:
10.1039/C7QM00145B
(Research Article)
Mater. Chem. Front., 2017,
1, 1858-1865
Turning on the solid emission from non-emissive 2-aryl-3-cyanobenzofurans by tethering tetraphenylethene for green electroluminescence†
Received
29th March 2017
, Accepted 9th May 2017
First published on 9th May 2017
Abstract
Three green-emitting compounds 1–3 based on 2-aryl-3-cyanobenzofuran fluorophore tethered with aggregation-induced emission active (AIE-active) tetraphenylethene (TPE) were designed and synthesized. p-Methoxyphenyl (PMP) was installed on the 5-, 6-, and 7-positions of 2-aryl-3-cyanobenzofuran for systematically investigating the substituent effect on their photophysical properties in solutions, nanoparticles, films, crystals, as well as powders. The aggregation-caused quenching (ACQ) problem arising from the π–π stacking between fluorophores was surmounted by covalently bonding the TPE unit. Thus, these compounds emitted green light in solids, and the film's quantum yield approached 50.1%. Red-shifted mechanochromism was observed for the crystalline state with a small crystal density. The best OLED performance was achieved using the compound with PMP occupied at the 6-position of 2-aryl-3-cyanobenzofuran, which possessed the longest efficient conjugation length.
Introduction
Since 1987, organic light-emitting diodes (OLEDs) have attracted much attention because of their practical applications for flat panel displays and lighting.1,2 In this blooming research area, organic emissive materials with unique properties are in continuous demand and thus developed. From fluorescent compounds to phosphorescent heavy-metal complexes3 and to the thermally activated decayed fluorescent (TADF) emitters,4 a number of new emissive compounds have come into being. No matter how fast and how far the research is gone, the discovery of new fluorophores has always been the cornerstone for the development of OLEDs and is therefore of significant importance.
Aggregation-caused quenching (ACQ), or concentration-quenching effect (CQE), is a popular phenomenon for various aromatic compounds, typically for those compounds with planar structures, because these compounds enjoy π–π stacking when they get closer in aggregates or in solids. This phenomenon extremely limits their practical applications in the fields of optoelectronics and in biosensors because the applicable state would be solid. To surmount this obstacle, considerable efforts have been made, including adding bulky groups5 or tethering branched chains,6 to prevent π–π stacking between the conjugated fluorophores. In addition to these structural tunings, octasubstituted cyclooctatetraenes (COTs) with loss of the core planarity have been synthesized and fabricated for blue electroluminescence with excellent efficiencies.7 Although the ACQ problem is effectively inhibited, COTs emit light at a large energy gap because of the interrupted efficient conjugation length.
Aggregation-induced emission (AIE), or aggregation-induced emission enhancement (AIEE), has an opposite phenomenon to the aforementioned ACQ.8 These compounds are non-emissive or low-emissive in dilute solutions, but they emit light upon aggregate formation. In this category, important molecules are hexaphenylsilole (HPS),9 tetraphenylethene (TPE),10 cyanostilbene,11 and so on. The mechanism behind AIE or AIEE could be ascribed to the restriction of intramolecular rotation (RIR) of molecules in aggregates, which decreases the non-radiative decay process when a molecule is photo-excited and turns on the fluorescence accordingly. AIE characteristics light up the fluorophores in solids and make them extremely useful in biosensors12 and in material science.13 As the research proceeded in-depth, it was also discovered that bonding the AIE-active unit to the ACQ unit could effectively transform ACQ fluorogens into AIE fluorogens, and thus changed the fate of ACQ molecules from dark to bright without adverse effects.14 For instance, bonding AIE-active TPE to ACQ fluorogens, such as perylene, provides photostable probes for bioimaging with red-NIR emission.15
Cyanostilbene and its derivatives have attracted considerable attention since the first published example (CN-MBE) (Scheme 1).11,16CN-MBE shows no fluorescence in solution but emits bright blue light (480 nm) in nanoparticles. In our previous studies, we synthesized 2-aryl-3-cyanobenzofurans (Ar-CBFs) by the reaction of benzyl cyanides and ortho-hydroxybenzaldehydes.17 Because of the benzofuran ring formation, the free rotation of stilbene was forbidden. As a result, Ar-CBFs showed the bright bluish or greenish luminescence in dilute solutions under UV irradiation, up to 88.2% quantum yield. However, ACQ was observed in the meantime for these compounds in aggregates because of the strong π–π interactions between the 2-aryl-3-cyanobenzofuran cores (Fig. S1, ESI†). In order to solve this problem and extend the utilities of this new fluorophore in material science, we covalently bonded an AIE-active TPE in the target molecules (TPE-CBFs), as shown in Scheme 1. p-Methoxyphenyl (PMP) group was introduced on the 5-, 6-, and 7-positions of benzofuran in order to deduce the substituent effect on the photoluminescent property and electroluminescent performance. Thus, compounds 1, 2, and 3 have been synthesized (Scheme S1, ESI†). In comparison with previously reported aromatic cores fabricated for green electroluminescence, such as pyrene,18 coumarin,19 azafluoranthene,20 phenothiazine,21 azaacenes,22 indolodibenzothiophene,23 dibenzo[a,c]phenazine,24 heavy-metal complexes, and cationic iridium complexes,25TPE-CBFs exhibited unique advantages, including feasible preparation, solution-processed fabrication, commendable thermal stability, and high quantum yields in solids.
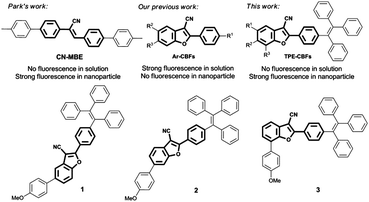 |
| | Scheme 1 Design of this study. | |
Results and discussion
Synthesis
The synthetic route to compounds 1, 2, and 3 is shown in Scheme S1 (ESI†). Reactions of 2-(4-(1,2,2-triphenylvinyl)phenyl)acetonitrile26 with corresponding 2-hydroxybenzaldehydes afforded target molecules 1, 2, and 3 in yields of 53.1%, 82.2%, and 75.1%, respectively. Compounds 1, 2, and 3 were fully characterized by 1H NMR, 13C NMR, HRMS, IR, and their single crystal analysis (Fig. S2–S15, ESI†).
Crystallographic analysis
Single crystals of 2 and 3 were obtained by slow vaporization of the mixed solvents of dichloromethane and n-hexane, and the single crystal of 1 was obtained from the mixed solvents of tetrahydrofuran and chloroform. Compounds 2 and 3 tended to crystallize; however, the crystallization of 1 was a difficult process. Crystal data are listed in Table S1 (ESI†). In all three cases, phenylene of TPE on the 2-position of 3-cyanobenzofuran is slightly twisted towards the benzofuran core, as shown in Fig. 1. These dihedral angles for compounds 1, 2, and 3 were found to be 10.84°, 15.94°, and 13.59°, respectively. The dihedral angle between PMP and the benzofuran core depended on the substitution type. The largest dihedral angle (45.17°) was observed for compound 2, which effectively prevented the π–π stacking between 2-aryl-3-cyanobenzofuran cores and also influenced the stacking model in the crystal.
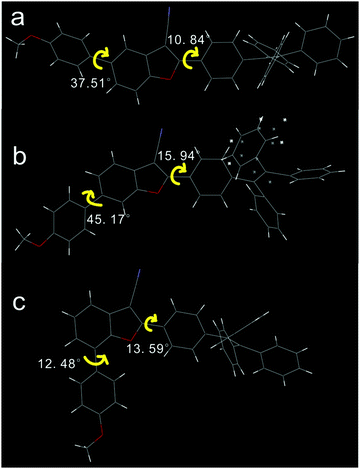 |
| | Fig. 1 Dihedral angles in compounds 1 (a), 2 (b), and 3 (c). | |
The stacking mode of three single crystals is shown in Fig. 2. To compound 1, two 2-aryl-3-cyanobenzofuran cores were anti-parallelly attracted by two C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N⋯H interactions and formed a planar structure. Afterwards, parallel but flip-over π–π stacking between two cores constructed these planes layer by layer. To compound 2, two cores were attracted by C–H⋯O and formed a cyclic structure without planarization. Subsequently, the two dimers assembled into a tetramer through C–H⋯π interactions. The large dihedral angle (45.17°) between PMP and 2-aryl-3-cyanobenzofuran prevented the cores’ π–π stacking. The strong C–H⋯π interactions made the single crystal of 2 possess the smallest volume and the largest density among these three single crystals. The density of the crystal was determined to be 1.223, 1.292, and 1.228 g cm−3 for compounds 1, 2, and 3, respectively. To compound 3, two cores were aligned anti-parallelly through a twisted π–π stacking, and the resulting dimer further assembled into a tetramer by C–H⋯π interactions. Powder XRD indicated that compounds 1 and 3 exhibit more ordered structure than compound 2 possessed (Fig. S16, ESI†). Clearly, both 1 and 3 adopted an ordered but loosely packed pattern, which facilitated structural re-assembly and might have induced different photophysical property under pressure.
N⋯H interactions and formed a planar structure. Afterwards, parallel but flip-over π–π stacking between two cores constructed these planes layer by layer. To compound 2, two cores were attracted by C–H⋯O and formed a cyclic structure without planarization. Subsequently, the two dimers assembled into a tetramer through C–H⋯π interactions. The large dihedral angle (45.17°) between PMP and 2-aryl-3-cyanobenzofuran prevented the cores’ π–π stacking. The strong C–H⋯π interactions made the single crystal of 2 possess the smallest volume and the largest density among these three single crystals. The density of the crystal was determined to be 1.223, 1.292, and 1.228 g cm−3 for compounds 1, 2, and 3, respectively. To compound 3, two cores were aligned anti-parallelly through a twisted π–π stacking, and the resulting dimer further assembled into a tetramer by C–H⋯π interactions. Powder XRD indicated that compounds 1 and 3 exhibit more ordered structure than compound 2 possessed (Fig. S16, ESI†). Clearly, both 1 and 3 adopted an ordered but loosely packed pattern, which facilitated structural re-assembly and might have induced different photophysical property under pressure.
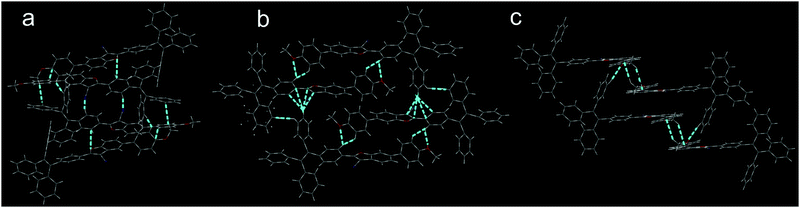 |
| | Fig. 2 Stacking models of compounds 1 (a), 2 (b), and 3 (c). | |
UV-vis absorption spectra
Absorption spectra were recorded in a concentration of 2 × 10−5 M in THF. Compounds 1, 2, and 3 absorbed light at about 370 nm (Fig. 3a). Maximum absorption wavelengths as well as molar absorptivities for each compound are listed in Table 1. Compound 2 presented the largest maximum absorption wavelength at 371 nm with the highest molar absorptivity, which was ascribed to the π–π* transition, implying that compound 2 possessed the longest efficient conjugation length from PMP to TPE even with the largest dihedral angle between PMP and benzofuran (Fig. 1). It also implied that the electron donating nature of MeO, resulting from the conjugative effect of MeO, became apparent when PMP occupied the 6-position of benzofuran.
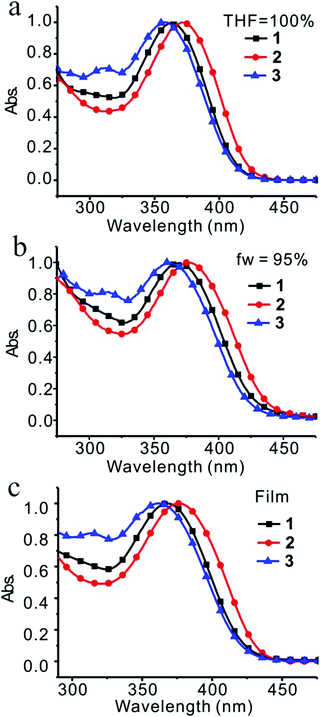 |
| | Fig. 3 Absorption of compounds 1, 2 and 3 in THF (a), in H2O/THF = 95/5 (v/v) (b), and in film (c). | |
Table 1 Optical properties of compounds 1, 2, and 3
| Compound |
λ
Abs.
(nm) |
λ
Em (nm) |
| THF (ε × 104) |
Nanob (ε × 104) |
Film |
Nano (Φ %) |
Filmc (Φ %) |
Powder |
Mole absorption coefficient (ε): M−1 cm−1.
Determined at mixed solution (c = 2 × 10−5 mol L−1, Vwater![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VTHF = 95:5).
Obtained by peak electroluminescence. :VTHF = 95:5).
Obtained by peak electroluminescence.
|
|
1
|
363 (3.80) |
370 (2.89) |
368 |
499 (18.6) |
500 (43.2) |
481 |
|
2
|
371 (4.33) |
377 (3.02) |
375 |
503 (20.3) |
504 (50.1) |
491 |
|
3
|
356 (2.78) |
360 (2.35) |
363 |
498 (18.3) |
497 (43.1) |
459 |
In order to form aggregates in narrow size distribution, the different combination of solvents and the different concentrations of solutes were screened.27 Among the systems of THF/H2O (fw = 95%), THF/hexane (fh = 95%), CH3CN/H2O (fw = 95%), CHCl3/hexane (fh = 95%), and CHCl3/MeOH (fM = 95%), the combination of THF and H2O was the best based on the emission spectra (Fig. S17a, ESI†) and TEM images (Fig. S17b and c, ESI†). Moreover, the concentration of 2 × 10−5 mol L−1 was determined to be optimal for the formation of nanoparticles in comparison with others, such as the concentrations of 1 × 10−4 mol L−1 and 1 × 10−3 mol L−1 because needles and bars would form in these conditions (Fig. S18, ESI†).
With the established method for the preparation of nanoparticles, discrimination between H-aggregates and J-aggregates was achieved by absorption spectral examinations of these compounds in different ratios of THF and water in a concentration of 2 × 10−5 mol L−1. The size of nanoparticles and their distribution depended on the ratio of water in THF. It was confirmed by dynamic light scattering (DLS) (Fig. S19, ESI†) and transmission electron microscopy (TEM) (Fig. S20–S22, ESI†). Generally speaking, as the water fraction increased to 70–75%, nanoparticles started to form. Further increase in the water fraction led to a smaller size of nanoparticles and a narrower size distribution. Compound 1 absorbed light at 363 nm in THF solution and red-shifted to 370 nm as the water fraction increased to 95% (Table 1 and Fig. S23a, ESI†), whereas the diameter of the nanoparticle was determined to be 68 nm by DLS and 70 nm by TEM. The red-shifted absorption implied that compound 1 possessed J-aggregation mode in nanoparticles.
Similar situations were observed for compounds 2 and 3 (Table 1 and Fig. S23, ESI†). Formation of aggregates induced slightly red-shifted absorption of 2 and 3 with the shifted values of 6 nm and 4 nm, respectively. In fact, anti-parallel alignment was indeed observed in all three cases by crystallographic analysis, which might have induced H-aggregation, reduced dipole moment and resulted in blue-shifted absorption. However, this fact was not primary, and all three compounds adopted J-aggregation in majority and produced red-shifted absorptions by aggregate formation. When the absorption was examined in films, slightly blue-shifted absorptions in comparison with those in the nanoparticles were observed for compounds 1 and 2. Compound 3 presented opposite direction. A further red-shifted absorption was seen in the films (Fig. S24, ESI†).
Fluorescent spectra
The dilute THF solutions of 1, 2, and 3 emitted very weak fluorescence that could be ignored. However, as water was gradually added into the THF solution, a turn-on fluorescence was observed for all three cases. For compound 1, fluorescence began to show on adding water to 75%. With a further increase in the water fraction to 95%, the solution emitted light at 499 nm with the quantum yield of 18.6% (Fig. 4a). For compound 2, as the water fraction was increased to 65%, the fluorescence was turned on. With further increase of water fraction to 75%, a red-shifted emission but in decreased intensity was observed. A steady increment of the fluorescent intensity was shown as water fraction changed from 75% to 95%. Finally, quantum yield was tuned to 20.9%, which was 261 times its initial value (Fig. 4b). For compound 3, fluorescent spectra did not change as water fraction approached 60%. By increasing the water fraction to 65%, the solution emitted light at 430 nm. As the water fraction increased to 75%, the emission spectrum red-shifted and emitted light at 497 nm. From 75% to 95%, a steady increment of the fluorescent intensity was observed (Fig. 4c).
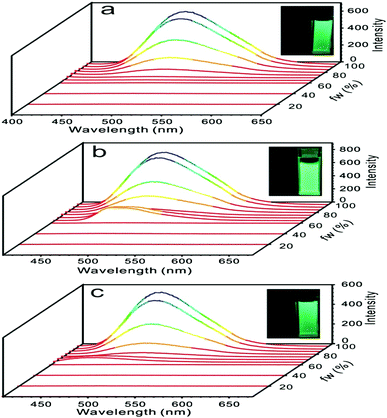 |
| | Fig. 4 Fluorescent spectra of compounds 1 (a), 2 (b), and 3 (c) in water/THF (from 0/100 to 95/5, v/v), inset: photograph of turn-on fluorescence by aggregate formation at 0/100 (left) and 95/5 (right). | |
Emission spectra of 1, 2, and 3 in water/THF (95/5, v/v) solutions were almost identical (Fig. S25, ESI†). Stokes’ shifts for 1, 2, and 3 were found to be 129 nm, 126 nm, and 138 nm, while quantum yields were determined to be 18.6%, 20.3%, and 18.3%, respectively. Film emissions of 1, 2, and 3 were similar to their emissions in nanoparticles. Stokes’ shifts were calculated to be 132 nm, 129 nm, and 134 nm, and quantum yields were determined to be 43.2%, 50.1%, and 43.1% for compounds 1, 2, and 3, respectively. Different emissions were observed for these compounds in powders. Powder emission of compound 1 presented a broader emission peak in comparison to that with its emission in nanoparticles (Fig. 5a), whereas 2 showed a narrower emission (Fig. 5b). 3 showed the most blue-shifted emission but with the same full-width at half-maximum (FWHM) (Fig. 5c). Thus, powders under UV irradiation showed different colors. Both 1 and 2 were green, whereas 3 was bright blue under UV irradiation. By finely grinding powder 1, a red-shifted emission was observed (Fig. 6a). Similar situation was observed for compound 3 (Fig. 6c). After grinding 3 into powder, the emissive color changed from blue to green under UV irradiation. For compound 2, the emissive color did not change before and after grinding (Fig. 6b). The loosely packed pattern of 1 and 3 rendered them as mechanochromic luminogens, which exhibited red-shifted emission under pressure because of the increased intermolecular interactions (Fig. S26, ESI†). It indicated that both 1 and 3 might be applied in mechanosensors, security paper, and optical storage for mechanical response.28,29
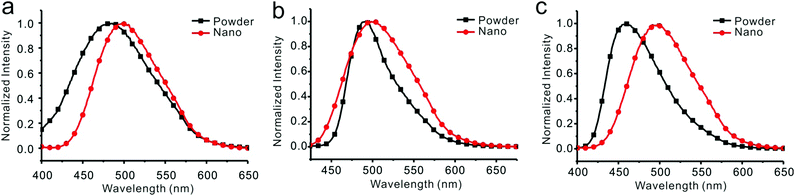 |
| | Fig. 5 Fluorescent spectra of compounds 1 (a), 2 (b), and 3 (c) in THF/water = 5/95 (v/v) and in powders. | |
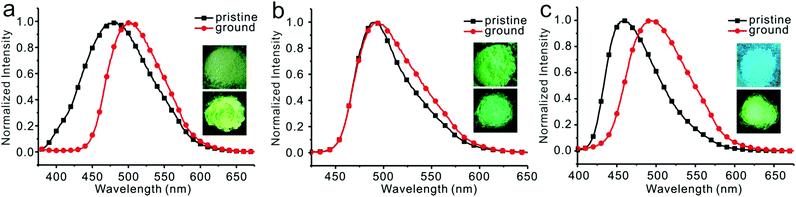 |
| | Fig. 6 Fluorescent spectra and photographs of compounds 1 (a), 2 (b), and 3 (c) before and after grinding, inset: photograph of color change before (top) and after (down) grinding. | |
Fluorescent lifetime measurement
Fluorescent decay was measured, and the results are listed in Table S2 (ESI†). Emission of three compounds in nanoparticles was composed of two components. As the water fraction changed from 75% to 95%, both components gave longer lifetimes (Fig. 7 and Table S2, ESI†). For instance, emission of compound 1 in the solution of THF/water (25/75) showed two components with 0.5582 ns and 2.3842 ns lifetimes, respectively. Both ratio and decay of these two components changed as water fraction increased. The component with longer lifetime changed from 15.04% to 36.54%, whereas the lifetime changed from 2.3842 ns to 3.3480 ns, accordingly.
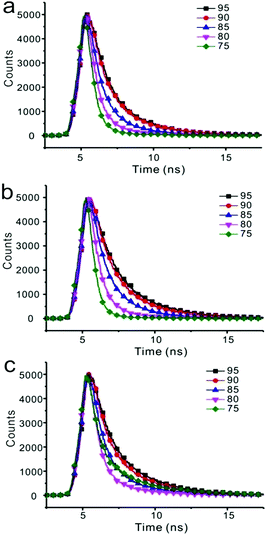 |
| | Fig. 7 The time-resolved fluorescence of compounds 1 (a), 2 (b), 3 (c) at varying fraction of water in THF solution. | |
Theoretical calculations
Structural optimization and the HOMO/LUMO orbitals were obtained from DFT using the B3LYP functional and the 6-31G* basis set. As shown in Fig. 8, the overall electron density of HOMOs of compounds 1 and 3 was localized on the benzofuran core and AIE-active TPE, whereas the overall electron density of HOMO of compound 2 extended from PMP to the TPE end. No big difference was observed for three LUMOs. Energy gaps were calculated to be 3.363 eV, 3.310 eV, and 3.373 eV for compounds 1, 2, and 3, respectively (Table 2).
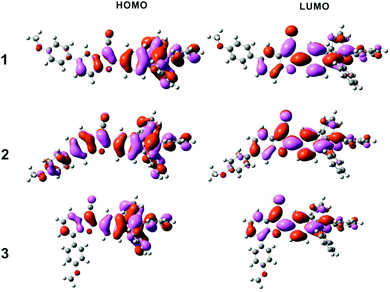 |
| | Fig. 8 The frontier orbital plots of the HOMO and LUMO of compounds 1, 2, and 3. | |
Table 2 The value of HOMO, LUMO, and energy gap of compounds 1, 2, and 3
| Compound |
HOMO |
LUMO |
E
g
|
| CVa (eV) |
Calcdb (eV) |
CVc (eV) |
Calcd (eV) |
CV (eV) |
Calcd (eV) |
UVd (eV) |
|
Potentials vs. reference electrode SCE, working electrode glassy carbon, auxiliary electrode Pt, c = 2 × 10−3 mol L−1, 0.1 M Bu4N+PF6−–MeCN, scan rate 100 mV s−1, HOMO = Eref − Eox(peak potential).
Obtained from DFT using the B3LYP functional and the 6-31G* basis set.
LUMO = Eref − Ered(peak potential).
E
g = 1241/λ(UV peak).
|
|
1
|
−5.907 |
−5.691 |
−2.711 |
−2.328 |
3.196 |
3.363 |
3.419 |
|
2
|
−5.887 |
−5.635 |
−2.705 |
−2.325 |
3.182 |
3.310 |
3.346 |
|
3
|
−5.921 |
−5.683 |
−2.677 |
−2.310 |
3.244 |
3.373 |
3.487 |
Cyclic voltammetry
Three compounds showed similar voltammograms (Fig. S27, ESI†), and their HOMO/LUMO energy potentials were calculated based on the equations of HOMO = Eref − Eox(peak potential) and LUMO = Eref − Ered(peak potential), respectively. Results were in good accordance with those obtained from the aforementioned theoretical calculations and the UV measurements (Table 2). HOMO/LUMO orbitals, calculated with their onset potential, are listed in Table S3 (ESI†) for comparison. HOMO of compound 2 possesses the highest energy potential among these three compounds. It also indicated that compound 2 possesses the longest efficient conjugation length and absorbs light at the lowest energy band as we observed.
Thermal stability
Thermal gravimetric analysis (TGA) was used to explore the thermal stabilities of compounds 1, 2, and 3 (Fig. S28, ESI†). All of these compounds were relatively stable, and the decomposition temperatures were determined to be 437 °C, 469 °C, and 436 °C for compounds 1, 2, and 3, respectively.
Device performance
Before fabricating the device, the film morphology was investigated by atomic force microscopy (AFM) as it was a key factor for the device performance. AFM images (Fig. S29, ESI†) indicated that these films, formed by spin-coating compounds 1–3 in chlorobenzene solution, were uniform and of excellent film-forming properties. A simple OLED structure, ITO/PEDOT:PSS (40 nm)/compound (1–3) (60 nm)/TBPI (25 nm)/Ca (10 nm)/Ag (80 nm), was screened and used to evaluate the electroluminescent properties of these new compounds as an emitting layer. Devices 1, 2, and 3 represented the devices fabricated with compounds 1, 2, and 3 as emitting materials, respectively. Three devices showed almost identical electroluminescent spectra (Fig. 9, Table 3 and Fig. S30 (ESI†)). Device 2 emitted light at 504 nm and achieved the best device performance with a turn-on voltage of 5.6 V, a brightness of 620 cd m−2, a current efficiency of 1.62 cd A−1, a power efficiency of 0.73 lm W−1, and the EQE of 0.63% at 7 V. Although these data are moderate in comparison with the literature results, the strategy, tethering an ACQ with AIE-active unit to overcome the inherent ACQ obstacle, is feasible and endows these fluorophores a bright future.
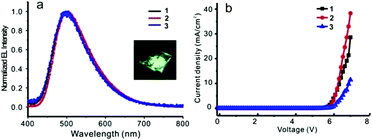 |
| | Fig. 9 (a) Normalized EL spectra of devices 1–3, inset: photograph of luminescent device (b) current density–voltage of devices 1–3. | |
Table 3 OLED performances of compounds 1, 2, and 3
| Device |
λ
EL
(nm) |
V
on
(V) |
L
(cd m−2) |
η
c
(cd A−1) |
η
p
(lm W−1) |
EQEf (%) |
|
Peak electroluminescence.
Turn-on voltage.
Brightness when the voltage is 7 V.
Current efficiency when the voltage is 7 V.
Power efficiency when the voltage is 7 V.
Obtained when the voltage is 7 V.
|
|
1
|
500 |
5.9 |
403 |
1.41 |
0.63 |
0.57 |
|
2
|
504 |
5.6 |
620 |
1.62 |
0.73 |
0.63 |
|
3
|
497 |
5.8 |
153 |
1.33 |
0.60 |
0.56 |
Conclusion
In conclusion, we changed an ACQ molecule, 2-phenyl-3-cyanobenzofuran, into an AIE-active molecule by covalently bonding tetraphenylethene at the appropriate position. This alternation lit up these molecules in solids under UV irradiation and made them useful for multiple purposes, such as mechanochromic materials and active emitting layers in organic light-emitting diodes. Although the substituent effect is not apparent in this series, several results could still be preliminarily concluded. Efficient conjugate length played a key role in absorption and emission. The one with PMP substituted on the 6-position of 2-aryl-3-cyanobenzofuran exhibited the smallest HOMO/LUMO energy gap because of the longest efficient conjugate length. As an effective complementation to the dipole moment used for predicting the mechanochromic property,28 crystal density might be applied to indicate the tendency of mechanochromic shift: the smaller the crystal density, the larger the mechanochromic shift.
Experimental section
General considerations
Unless otherwise mentioned, solvents and reagents were purchased from commercial sources and used as received. Tetrahydrofuran (THF) was distilled from sodium prior to use. Melting points were measured with a micro melting point apparatus. Infrared spectra were obtained with an FTIR spectrometer. NMR spectra were obtained on a Bruker AVANCE DMX 400 spectrometer operating at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Unless otherwise noted, all the NMR spectra were recorded at room temperature. High-resolution mass spectra (HRMS) data were obtained with an electron ionization time-of-flight (EI-TOF) mass spectrometer. Flash column chromatography was performed employing 300–400 mesh silica gel. Thin layer chromatography (TLC) was performed on silica gel HSGF254.
Synthesis and characterization of compound 1
4-Hydroxy-4′-methoxy-[1,1′-biphenyl]-3-carbaldehyde (115 mg, 0.5 mmol), 2-(4-(1,2,2-triphenylvinyl)phenyl)acetonitrile (186 mg, 0.5 mmol) and Na2CO3 (212 mg, 2 mmol) were dissolved in DMSO (4 mL). The resulting solution was stirred at 110 °C for 15 h. The solvent was poured into water and extracted twice with DCM. The combined organic layers were dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was subjected to silica gel column chromatography with DCM/petroleum ether (1/4, v/v) as eluent. In this way, compound 1 was obtained. Similar procedure produced compounds 2 and 3.
Compound 1.
Light yellow solid, mp 226–227 °C. 1H NMR (400 MHz, CDCl3): δ 7.97–7.94 (m, 2H), 7.80–7.80 (m, 1H), 7.58–7.53 (m, 4H), 7.22–7.19 (m, 2H), 7.17–7.03 (m, 15H), 7.02–6.99 (m, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 162.0, 159.3, 152.5, 147.2, 143.3, 143.2, 143.1, 142.7, 139.8, 138.1, 133.0, 132.2, 131.4, 131.3, 131.3, 128.5, 128.0, 127.9, 127.7, 126.8, 126.8, 125.8, 125.6, 117.6, 114.5, 114.4, 111.7, 87.8, 55.4. HRMS (EI-TOF): cacld for C42H29NO2 [M+], 579.2198; found: 579.2199. IR: 3054, 2224, 1609, 1577, 1519, 1492, 1465, 1443, 1248, 1204, 1180, 1139, 1112, 1037, 835, 699, 668 cm−1.
Compound 2.
Green solid, mp 223–224 °C. 1H NMR (400 MHz, CDCl3): δ 7.96–7.49 (m, 2H), 7.71–7.68 (m, 2H), 7.59–7.56 (m, 3H), 7.21–7.19 (m, 2H), 7.16–7.03 (m, 15H), 7.02–7.00 (m, 2H), 3.87 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 161.7, 159.5, 154.0, 147.1, 143.2, 143.2, 143.0, 142.6, 139.8, 139.8, 132.8, 132.1, 131.4, 131.3, 131.3, 128.4, 128.0, 127.9, 127.7, 127.0, 126.8, 126.8, 125.9, 125.7, 125.7, 123.9, 119.8, 114.4, 109.4, 87.7, 55.4. HRMS (EI-TOF): cacld for C42H29NO2 [M+], 579.2198; found: 579.2195. IR: 3054, 2225, 1607, 1520, 1482, 1443, 1297, 1250, 1173, 1145, 1111, 1075, 1036, 917, 835, 753, 699 cm−1.
Compound 3.
Light green solid, mp 254–254.9 °C. 1H NMR (400 MHz, CDCl3): δ 7.95–7.93 (m, 2H), 7.78–7.75 (m, 2H), 7.60 (dd, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.48 (dd, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.41 (t, J = 7.6 Hz, 1H), 7.20–7.18 (m, 2H), 7.15–7.03 (m, 17H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 161.4, 159.6, 150.3, 147.2, 143.2, 143.1, 143.1, 142.5, 139.8, 132.1, 131.3, 131.3, 131.3, 129.6, 128.1, 127.9, 127.9, 127.7, 127.6, 127.0, 126.8, 125.8, 125.7, 125.6, 125.3, 125.2, 118.1, 114.5, 114.3, 87.9, 55.3. HRMS (EI-TOF): cacld for C42H29NO2 [M+], 579.2198; found: 579.2195. IR: 3054, 2225, 1608, 1519, 1482, 1443, 1404, 1251, 1181, 1146, 1111, 1030, 833, 788, 737, 699 cm−1.
Spectroscopic characterization
The absorption spectra were measured using a UV-vis spectrometer (Shimadzu, UV-2450, Japan). FL spectra were recorded on a fluorescence spectro-photometer (Shimadzu, RF-5301PC, Japan) with a xenon lamp as excitation source. Solution concentration was 2 × 10−5 M. Nanoparticles were formed by adding a certain volume of water into its dilute THF solution with a vigorous stirring for 5 minutes, and all the solution concentration was the same. The absorption spectra, FL spectra and particle size distribution of the resulting solutions were then obtained immediately.
Particle size
DLS was determined at room temperature on a Brookhaven 90 Plus particle size analyzer. Images of particle size were obtained using a HT7700 transmission electron microscope with a copper net as a supporter.
Powder X-ray diffraction (XRD)
XRD was recorded at room temperature by a Xeuss with the SAXS/WAXS system. The data were recorded in the 2θ mode with a step size of 0.05021°.
Cyclic voltammetry
CV was performed on a CHI 600E electrochemical analyzer in acetonitrile (c = 2 × 10−3 mol L−1) at room temperature.
Thermogravimetric analysis
TGA results were obtained with a TGA 1100SF thermal analyzer at heating and cooling rates of 10 K min−1 under an N2 atmosphere.
Acknowledgements
The authors thank Prof. Yizhen Jin for the guidance in the fabrication of device and thank the National Natural Science Foundation of China (21472173).
Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- R. C. Kwong, S. Sibley, T. Dubovoy, M. Baldo, S. R. Forrest and M. E. Thompson, Chem. Mater., 1999, 11, 3709–3713 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed; J. J. Luo, G. H. Xie, S. L. Gong, T. H. Chen and C. L. Yang, Chem. Commun., 2016, 52, 2292–2295 RSC.
- J. L. Banal, J. M. White, K. P. Ghiggino and W. W. H. Wong, Sci. Rep., 2014, 4, 4635 CrossRef PubMed.
- H. Li, Y. Guo, G. X. Li, H. P. Xiao, Y. X. Lei, X. B. Huang, J. X. Chen, H. Y. Wu, J. C. Ding and Y. X. Cheng, J. Phys. Chem. C, 2015, 119, 6737–6748 CAS.
- P. Lu, H. P. Hong, G. P. Cai, P. Djurovich, W. P. Weber and M. E. Thompson, J. Am. Chem. Soc., 2000, 122, 7480–7486 CrossRef CAS.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- J. D. Luo, Z. L. Xie, J. W. Y. Lam, L. Cheng, H. Y. Chen, C. F. Qiu, H. S. Kwok, X. W. Zhan, Y. Q. Liu, D. B. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- M. Wang, G. X. Zhang, D. Q. Zhang, A. B. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed.
- D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed.
- J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- Q. L. Zhao, K. Li, S. J. Chen, A. J. Qin, D. Ding, S. Zhang, Y. Liu, B. Liu, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 15128–15135 RSC.
-
(a) S. Shin, S. H. Gihm, C. R. Park, S. Kim and S. Y. Park, Chem. Mater., 2013, 25, 3288–3295 CrossRef CAS;
(b) H.-J. Kim, D. R. Whang, J. Gierschner and S. Y. Park, Angew. Chem., Int. Ed., 2016, 55, 15915–15919 CrossRef CAS PubMed;
(c) X. J. Li, Y. X. Xu, F. Li and Y. G. Ma, Org. Electron., 2012, 13, 762–766 CrossRef CAS.
- L. P. Zhang, Z. X. Peng, Q. D. Wen, X. H. Li, P. Lu and Y. G. Wang, Org. Lett., 2016, 18, 728–731 CrossRef CAS PubMed.
-
(a) Z. J. Zhao, X. J. Xu, Z. T. Jiang, P. Lu, G. Yu and Y. Q. Liu, J. Org. Chem., 2007, 72, 8345–8353 CrossRef CAS PubMed;
(b) Y. Zhan, J. Peng, K. Q. Ye, P. C. Xue and R. Lu, Org. Biomol. Chem., 2013, 11, 6814–6823 RSC.
-
(a) N. Prachumrak, S. Potjanasopa, R. Rattanawan, S. Namuangruk, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Tetrahedron, 2014, 70, 6249–9257 CrossRef CAS;
(b) P. Kotchapradist, N. Prachumrak, T. Sunonnam, R. Tarsang, S. Namuangruk, T. Sudyoadsuk, T. Keawin, S. Jungsuttiwong and V. Promarak, Dyes Pigm., 2015, 112, 227–235 CrossRef CAS.
- P. Gasiorski, E. Gondek, K. S. Danel, M. Pokladko-Kowar, P. Armatys and A. V. Kityk, Opt. Mater., 2013, 35, 681–684 CrossRef CAS.
-
(a) R. K. Konidena, K. R. J. Thomas, S. Kumar, Y. C. Wang, C. J. Li and J. H. Jou, J. Org. Chem., 2015, 80, 5812–5823 CrossRef CAS PubMed;
(b) G. B. Bodedla, K. R. J. Thomas, S. Kumar, J. H. Jou and C. J. Li, RSC Adv., 2015, 5, 87416–87428 RSC;
(c) K. Shanmugasundaram, M. S. Subeesh, C. D. Sunesh, R. K. Chitumalla, J. Jang and Y. Choe, J. Phys. Chem. C, 2016, 120, 20247–20253 CrossRef CAS.
- K. Kotwica, P. Bujak, D. Wamil, A. Pieczonka, G. Wiosna-Salyga, P. A. Gunka, T. Jaroch, R. Nowakowski, B. Luszczynska, E. Witkowska, I. Glowacki, J. Ulanski, M. Zagorska and A. Pron, J. Phys. Chem. C, 2015, 119, 10700–10708 CAS.
- K. Kotwica, P. Bujak, D. Wamil, A. Pieczonka, G. Wiosna-Salyga, P. A. Gunka, T. Jaroch, R. Nowakowski, B. Luszczynska, E. Witkowska, I. Glowacki, J. Ulanski, M. Zagorska and A. Pron, J. Phys. Chem. C, 2015, 119, 10700–10708 CAS.
- Y. J. He, S. Yagi, T. Maeda and H. Nakazumi, Mol. Cryst. Liq. Cryst., 2015, 621, 64–69 CrossRef CAS.
-
(a) N. M. Shavaleev, G. H. Xie, S. Varghese, D. B. Cordes, A. M. Z. Slawin, C. Momblona, E. Ortí, H. J. Bolink, I. D. W. Samuel and E. Zysman-Colman, Inorg. Chem., 2015, 54, 5907–5914 CrossRef CAS PubMed;
(b) F. L. Zhang, L. Duan, J. Qiao, G. F. Dong, L. D. Wang and Y. Qiu, Org. Electron., 2012, 13, 1277–1288 CrossRef CAS;
(c) L. He, L. Duan, J. Qiao, G. F. Dong, L. D. Wang and Y. Qiu, Chem. Mater., 2010, 22, 3535–3542 CrossRef CAS;
(d) D. Tordera, M. Delgado, E. Ortí, H. J. Bolink, J. Frey, M. K. Nazeeruddin and E. Baranoff, Chem. Mater., 2012, 24, 1896–1903 CrossRef CAS.
- Q. Y. Lu, X. F. Li, J. Li, Z. Y. Yang, B. J. Xu, Z. G. Chi, J. R. Xu and Y. Zhang, J. Mater. Chem. C, 2015, 3, 1225–1234 RSC.
-
(a) A. Rananaware, R. S. Bhosale, K. Ohkubo, H. Patil, L. A. Jones, S. L. Jackson, S. Fukuzumi, S. V. Bhosale and S. V. Bhosale, J. Org. Chem., 2015, 80, 3832–3840 CrossRef CAS PubMed;
(b) Anuradha, D. D. La, M. A. Kobaisi, A. Gupta and S. V. Bhosale, Chem. – Eur. J., 2017, 23, 3950–3956 CrossRef CAS PubMed;
(c) A. Rananaware, D. D. La and S. V. Bhosale, RSC Adv., 2015, 5, 63130–63134 RSC.
- J. Wu, Y. Y. Cheng, J. B. Lan, D. Wu, S. Y. Qian, L. P. Yan, Z. He, X. Y. Li, K. Wang, B. Zou and J. S. You, J. Am. Chem. Soc., 2016, 138, 12803–12812 CrossRef CAS PubMed.
- Y. Q. Dong, J. W. Y. Lam and B. Z. Tang, J. Phys. Chem. Lett., 2015, 6, 3429–3436 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1521108–1521110. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00145b |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Yanguang
Wang
* and
Yanguang
Wang









