
Direct amination of the antiaromatic NiII norcorrole†
Takuya
Yoshida
and
Hiroshi
Shinokubo
 *
*
Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: hshino@chembio.nagoya-u.ac.jp
First published on 10th May 2017
Abstract
We have discovered a facile amination reaction of NiII dimesitylnorcorrole, which is an antiaromatic porphyrinoid with a 16π-electron conjugation system. The amination reaction proceeded at the β-positions of the norcorrole with high regioselectivity upon treatment with various amines used as solvents. The reaction requires neither catalysts nor pre-functionalization of the norcorrole. The optical and electrochemical properties of the amino norcorroles were substantially modulated by electron-donating nitrogen atoms. According to theoretical studies, the regioselectivity of amination can be accounted for by molecular orbitals of the norcorrole.
Introduction
The chemistry of antiaromatic compounds with 4nπ-electrons in their planar cyclic conjugation pathways has attracted increasing attention in recent years.1 They show distinctly different properties from conventional aromatic compounds, such as narrow HOMO–LUMO gaps2 and stable redox processes.3 In addition to these marked physical properties, antiaromatic compounds sometimes exhibit unique and enhanced reactivity because of their inherently unstable 4nπ-electron systems.4 Such high reactivity is potentially useful for selective functionalization of antiaromatic compounds toward further manipulation of their physical properties, which should be important for future applications.We have reported a gram scale synthesis of NiII dimesitylnorcorrole 1, which is a ring-contracted porphyrin with clear antiaromaticity stemmed from its planar 16π-electronic system.5 Although various types of antiaromatic porphyrinoids have been reported to date,6 their reactivity has rarely been investigated.7 From this point of view, we have explored the reactivity of norcorroles,5,8 as well as their intriguing physical properties and functions.9 We have developed nucleophilic C–H functionalization for norcorroles, in which cyano, phenylthio and phenoxy groups were introduced to the pyrrole β-positions of norcorroles without any catalysts. In particular, the nucleophilic thiolation reaction enabled the synthesis of a tethered norcorrole dimer, which experimentally demonstrated the emergence of three-dimensional aromaticity in π-stacked antiaromatic compounds.10 Li and Chmielewski et al. also investigated the reaction of norcorroles with several reagents to yield norcorrole derivatives with unique properties and structures.11 In the present work, we have attempted a reaction of norcorrole 1 with several amines to develop a facile and regioselective nucleophilic functionalization method for norcorroles.
Results and discussion
Initially, we found that butylamine underwent an addition reaction with NiII dimesitylnorcorrole 1 upon simple heating of a solution of 1 in butylamine (Scheme 1, entry 1). On the basis of 1H NMR and MS analyses, a bisamination product was obtained in 48% yield as a single regioisomer. X-ray structural analysis unambiguously identified the bisamination compound as 2b, in which two butylamino groups were introduced at the diagonal 3- and 12-positions of 1 (Fig. 1). Notably, the present direct amination reaction of 1 requires only butylamine: neither the addition of any catalysts nor pre-functionalization of the norcorrole skeleton is necessary. In entry 2, the use of degassed butylamine resulted in a decrease in the yield of 2b to 26% and monoamino product 2a was isolated in 23% yield. This result suggested that a trace amount of dioxygen in butylamine was crucial in the amination process. Consequently, we attempted the amination reaction of 1 under air at room temperature (entry 3). Under these conditions, 1 was totally consumed within 1 h to afford 2a and 2b in 45% and 52% yields, respectively. The amination under air at 80 °C provided 2b selectively in 82% yield in 20 min (entry 4). | ||
| Scheme 1 Synthesis of the mono- and bisaminonorcorroles. a The reaction was conducted under a N2 atmosphere. b The reaction was conducted under a N2 atmosphere with degassed butylamine. c A drop of tributylamine was added as a base. | ||
 | ||
| Fig. 1 (a) Top view and (b) side view of the X-ray crystal structure of bisaminonorcorrole 2b. | ||
Besides primary alkylamines, secondary alkylamines and primary arylamines could be employed as the reaction partner in amination: the reaction with dibutylamine at 80 °C afforded monoamino product 3a in 70% yield, while the yield of bisamino product 3b was low (entry 5). The reaction with piperidine at room temperature only furnished 4b as a major product (80%) with a trace amount of monoamino product 4a (entry 6). These results suggest that the nucleophilicity of amines is critical in this amination reaction. The amination of 1 with aniline also provided only monoamino product 5a in 55% yield (entry 7). In this case, the addition of tributylamine as a base enhanced the reactivity to provide bisamino product 5b along with 5a after stirring for 10 h (entry 8). Unfortunately, none of the amination products was obtained in the reaction of 1 with diphenylamine, probably due to its low nucleophilic character. In all entries, norcorroles substituted with three or four amino groups were not detected.
Although the reaction mechanism is still not completely understood at this moment, we propose the plausible reaction mechanism as depicted in Scheme 2. The nucleophilic addition of an amine to 1 provides an anionic intermediate A. Then, proton migration from the amine nitrogen to the anionic carbon atom yields a chlorin-type intermediate B, which is oxidized to aminonorcorrole C by atmospheric oxygen to restore the cyclic π-conjugation of norcorroles. Air oxidation of a chlorin-type dihydronorcorrole to norcorroles has been disclosed.8c
 | ||
| Scheme 2 Plausible reaction mechanism for the amination of 1. | ||
To elucidate the origin of the observed regioselectivity, we conducted theoretical calculations using the density functional theory (DFT) method at the B3LYP/6-31G(d)+SDD level. To reduce the calculation costs, the butylamino groups in 2a and 2b were replaced with methylamino groups in model compounds 2a′ and 2b′. The regioselectivity of the present amination reaction is clearly accounted for by the distribution of the LUMOs of norcorroles 1, 2a′ and 2b′ (Fig. 2). The LUMO of 1 suggests that the initial nucleophilic attack of the amine should occur at one of the four β-positions near to the meso-aryl substituents (3-, 7-, 12-, and 16-positions) to afford 2a, because the orbital is substantially distributed at the 3-, 7-, 12-, and 16-positions. In the LUMO of 2a′, a larger orbital coefficient lies at the 12-position than at other unsubstituted β-positions. This anisotropic distribution is the reason why the second amine attacks at the diagonal site to form 2b as the dominant bisamino product. Furthermore, 2b is no longer reactive to amines, because the remaining β-positions have fairly small orbital coefficients on the LUMO of 2b′. The strong electron-donating effects of two amino groups also reduce the reactivity of 2b.
 | ||
| Fig. 2 LUMOs of norcorroles 1, 2a′ and 2b′ calculated at the B3LYP/6-31G(d)+SDD level. The red arrows indicate the reactive positions. | ||
In order to evaluate the effects of introduced amino groups for norcorroles, we investigated the electrochemical properties of aminonorcorroles using cyclic voltammetry. The redox potentials of 2–5 in CH2Cl2 are summarized in Table 1 along with those of 1 (see also Fig. S13, ESI†). All NiII norcorrole derivatives show reversible reduction and oxidation waves. All potentials are cathodically shifted with the introduction of amino groups. Aminonorcorroles consequently exhibit significantly low first oxidation potentials (0.02 to −0.17 V) due to the inherent low oxidation potential of the antiaromatic norcorrole core, as well as the electron-donating effect of amines. Consequently, the present amination method is useful to create electron-rich motifs on the basis of norcorroles. The energy gap between the first oxidation and reduction potentials (ΔE) increased in the cases of bisaminonorcorroles 2b, 4b, and 5b (1.28, 1.16 and 1.20 V, respectively), because of the cathodic shift in the first reduction potentials. This tendency matched well with the results of computational studies, in which the LUMO of 2b′ is particularly destabilized by the electron-donating effect of the amino group to increase the HOMO–LUMO gap (Fig. S14, ESI†).
| Compounds | E ox2 | E ox1 | E red1 | E red2 | ΔEb |
|---|---|---|---|---|---|
| a Solvent: CH2Cl2, electrolyte: Bu4NPF6, working electrode: glassy carbon, counter electrode: Pt, reference electrode: Ag/AgClO4, scan rate: 0.1 V s−1. The potentials (V) versus the value for the ferrocene/ferrocenium cation couple are shown. b ΔE = Eox1 − Ered1. | |||||
| 1 | 0.79 | 0.16 | −0.92 | −1.70 | 1.08 |
| 2a | 0.51 | −0.07 | −1.18 | −1.87 | 1.11 |
| 3a | 0.49 | −0.06 | −1.14 | −1.81 | 1.08 |
| 5a | 0.58 | 0.02 | −1.07 | −1.75 | 1.09 |
| 2b | 0.40 | −0.17 | −1.45 | −2.03 | 1.28 |
| 4b | 0.39 | −0.14 | −1.30 | −1.90 | 1.16 |
| 5b | 0.52 | −0.04 | −1.24 | −1.82 | 1.20 |
The UV-vis-NIR absorption spectra of these norcorroles in CH2Cl2 are displayed in Fig. 3. The whole absorption envelopes distinctly depend on the number of amino groups attached to the norcorrole core: aminonorcorroles 2a, 3a and 5a exhibit broadened and relatively weak absorption bands, while bisaminonorcorroles 2b, 4b and 5b show sharp absorption bands similar to unsubstituted norcorrole 1.5 Furthermore, 2b, 4b and 5b have increased absorption bands in the NIR region compared with that of 1. We conducted TD-DFT calculations for 2a′ and 2b′ to obtain their theoretical absorption bands (Fig. 4 and Table S1, ESI†). The theoretical absorption spectra of 2a′ shows complex features with many transitions, which is in good agreement with the broadened experimental absorption spectra of 2a, 3a and 5a. The lower symmetric structure of monoaminonorcorrole 2a′ increases the number of its transition bands. The absorption of 2b′ is composed of fewer transitions because of its C2-symmetric structure, and thus 2b, 4b and 5b exhibit relatively simple absorption bands. The theoretical spectrum of 2b′ also suggests that the near IR absorption bands of bisaminonorcorroles can be assigned to the transition from HOMO−1 to LUMO, which are significantly intensified after the introduction of amino groups. On the other hand, the HOMO–LUMO transitions of both mono- and bisaminonorcorroles remain forbidden.
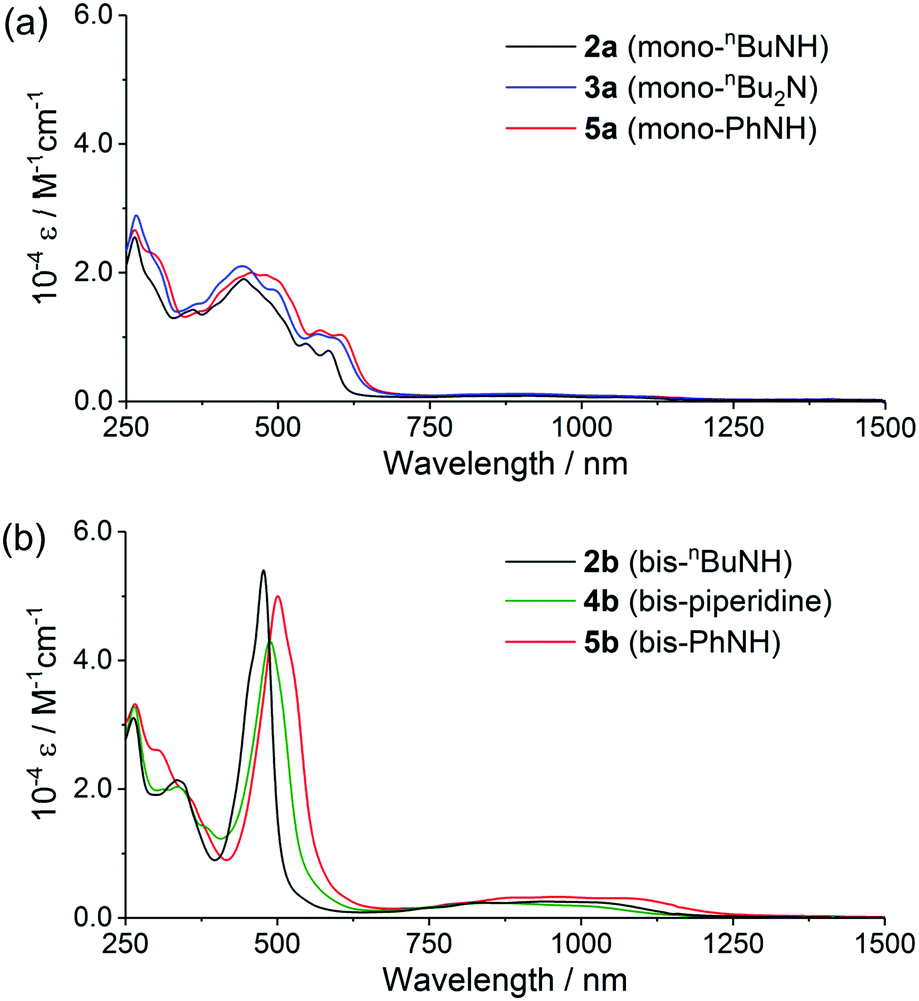 | ||
| Fig. 3 UV-vis-NIR absorption spectra of (a) mono- and (b) bisaminonorcorroles in CH2Cl2. | ||
 | ||
| Fig. 4 Theoretical absorption bands and oscillator strengths of (a) 2a′ and (b) 2b′ calculated at the B3LYP/6-31G(d)+SDD (sticks) with experimental spectra of (a) 2a and (b) 2b. | ||
Conclusions
In conclusion, we have successfully synthesized mono- and bis-aminonorcorroles with perfect regioselectivity through the catalyst-free amination reaction. The reaction requires atmospheric oxygen as an oxidant. This functionalization procedure represents the high reactivity of antiaromatic porphyrinoids induced by their unstable electronic structure. Aminonorcorroles exhibited highly electron-rich character and considerably altered optical properties because of strong electronic perturbation by the introduced amino groups. This facile functionalization methodology enables us to design various novel norcorroles, which would be useful for device applications.Experiments
Instrumentation and materials
1H NMR (500 MHz) and 13C NMR (126 MHz) spectra were recorded on a Bruker AVANCE III HD spectrometer. Chemical shifts were reported on the delta scale in ppm relative to CDCl3 (δ = 7.26 ppm) for 1H NMR and CDCl3 (δ = 77.16 ppm) for 13C NMR. UV/vis/NIR absorption spectra were recorded on a JASCO V670 spectrometer. Mass spectra were recorded on a Bruker microTOF using the ESI-TOF method for acetonitrile solutions. X-ray data were taken on a Bruker D8 QUEST X-ray diffractometer equipped with a PHOTON 100 CMOS active pixel sensor detector and an IμS microfocus source using Mo-Kα radiation (λ = 0.71073 Å). Redox potentials were measured using the cyclic voltammetry method on an ALS electrochemical analyser model 612C. Unless otherwise noted, materials obtained from commercial suppliers were used without further purification.General procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 444 (19000), 546 (9000), 908 (880) nm. HR-MS (ESI-MS): m/z = 647.2567, calcd for (C40H39N5Ni)+ = 647.2554 [(M)+]. 2b: 1H NMR (500 MHz, CDCl3): δ 6.67 (s, 4H, Mes), 4.05 (d, J = 3.7 Hz, 2H, β-H), 3.50 (d, J = 3.7 Hz, 2H, β-H), 3.00 (s, 2H, β-H), 2.57 (s, 12H, ortho-Me), 2.38 (t, J = 5.3 Hz, 2H, NH), 2.27 (td, J = 6.0, 5.3 Hz, 4H, alkyl), 2.09 (s, 6H, para-Me), 0.84–0.69 (m, 8H, alkyl), 0.64 (t, J = 7.0, 6H, alkyl) ppm, 13C NMR (126 MHz, CDCl3): δ 172.1, 160.3, 146.3, 143.2, 137.8, 136.0, 135.9, 135.8, 129.0, 128.4, 114.9, 112.9, 87.6, 44.0, 30.3, 21.0, 19.4, 18.8, 13.5 ppm, UV/vis/NIR (CH2Cl2): λmax (ε [M−1 cm−1]) = 264 (31000), 336 (21000), 477 (54000), 939 (2600) nm, HR-MS (ESI-MS): m/z = 718.3331, calcd for (C44H48N6Ni)+ = 718.3288 [(M)+], single crystals were obtained by vapor diffusion of hexane into a chloroform solution of 2b. C22H24N3Ni0.5, Mw = 359.80, triclinic, P
000), 444 (19000), 546 (9000), 908 (880) nm. HR-MS (ESI-MS): m/z = 647.2567, calcd for (C40H39N5Ni)+ = 647.2554 [(M)+]. 2b: 1H NMR (500 MHz, CDCl3): δ 6.67 (s, 4H, Mes), 4.05 (d, J = 3.7 Hz, 2H, β-H), 3.50 (d, J = 3.7 Hz, 2H, β-H), 3.00 (s, 2H, β-H), 2.57 (s, 12H, ortho-Me), 2.38 (t, J = 5.3 Hz, 2H, NH), 2.27 (td, J = 6.0, 5.3 Hz, 4H, alkyl), 2.09 (s, 6H, para-Me), 0.84–0.69 (m, 8H, alkyl), 0.64 (t, J = 7.0, 6H, alkyl) ppm, 13C NMR (126 MHz, CDCl3): δ 172.1, 160.3, 146.3, 143.2, 137.8, 136.0, 135.9, 135.8, 129.0, 128.4, 114.9, 112.9, 87.6, 44.0, 30.3, 21.0, 19.4, 18.8, 13.5 ppm, UV/vis/NIR (CH2Cl2): λmax (ε [M−1 cm−1]) = 264 (31000), 336 (21000), 477 (54000), 939 (2600) nm, HR-MS (ESI-MS): m/z = 718.3331, calcd for (C44H48N6Ni)+ = 718.3288 [(M)+], single crystals were obtained by vapor diffusion of hexane into a chloroform solution of 2b. C22H24N3Ni0.5, Mw = 359.80, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.3884(8) Å, b = 8.4668(5) Å, c = 14.2802(12) Å, α = 95.006(6)°, β = 94.479(7)°, γ = 113.925(7)°, V = 916.34(14) Å3, Z = 2, R = 0.0460 (I > 2.0σ(I)), Rw = 0.1197 (all data), GOF = 1.058.
000), 441 (29000), 566 (10000), 903 (1200) nm, HR-MS (ESI-MS): m/z = 703.3146, calcd for (C44H47N5Ni)+ = 703.3180 [(M)+].
000), 336 (20000), 488 (43000), 857 (2300) nm, HR-MS (ESI-MS): m/z = 742.3256, calcd for (C46H48N6Ni)+ = 742.3288 [(M)+].
000), 456 (20000), 569 (11000), 947 (1000) nm, HR-MS (ESI-MS): m/z = 667.2232, calcd for (C42H35N5Ni)+ = 667.2241 [(M)+]. 5b: 1H NMR (500 MHz, CDCl3): δ 6.93 (m, 4H, Ph), 6.73 (t, J = 7.5 Hz, 4H, Ph), 6.70 (s, 4H, Mes), 5.88 (dd, J = 8.8, 1.0 Hz, 4H, Ph), 4.02 (s, 2H, NH), 3.62 (d, J = 3.7 Hz, 2H, β-H), 3.29 (s, 2H, β-H), 3.14 (d, J = 3.7 Hz, 2H, β-H), 2.71 (s, 12H, ortho-Me), 2.11 (s, 6H, para-Me) ppm, 13C NMR (126 MHz, CDCl3): δ 173.1, 152.8, 148.9, 144.2, 139.8, 139.2, 138.4, 137.4, 136.1, 129.2, 129.1, 127.2, 122.7, 118.6, 116.8, 113.4, 91.8, 21.0, 18.6 ppm, UV/vis/NIR (CH2Cl2): λmax (ε [M−1 cm−1]) = 266 (33000), 501 (50000), 960 (3300) nm, HR-MS (ESI-MS): m/z = 758.2676, calcd for (C48H40N6Ni)+ = 758.2662 [(M)+].
, a = 8.3884(8) Å, b = 8.4668(5) Å, c = 14.2802(12) Å, α = 95.006(6)°, β = 94.479(7)°, γ = 113.925(7)°, V = 916.34(14) Å3, Z = 2, R = 0.0460 (I > 2.0σ(I)), Rw = 0.1197 (all data), GOF = 1.058.
000), 441 (29000), 566 (10000), 903 (1200) nm, HR-MS (ESI-MS): m/z = 703.3146, calcd for (C44H47N5Ni)+ = 703.3180 [(M)+].
000), 336 (20000), 488 (43000), 857 (2300) nm, HR-MS (ESI-MS): m/z = 742.3256, calcd for (C46H48N6Ni)+ = 742.3288 [(M)+].
000), 456 (20000), 569 (11000), 947 (1000) nm, HR-MS (ESI-MS): m/z = 667.2232, calcd for (C42H35N5Ni)+ = 667.2241 [(M)+]. 5b: 1H NMR (500 MHz, CDCl3): δ 6.93 (m, 4H, Ph), 6.73 (t, J = 7.5 Hz, 4H, Ph), 6.70 (s, 4H, Mes), 5.88 (dd, J = 8.8, 1.0 Hz, 4H, Ph), 4.02 (s, 2H, NH), 3.62 (d, J = 3.7 Hz, 2H, β-H), 3.29 (s, 2H, β-H), 3.14 (d, J = 3.7 Hz, 2H, β-H), 2.71 (s, 12H, ortho-Me), 2.11 (s, 6H, para-Me) ppm, 13C NMR (126 MHz, CDCl3): δ 173.1, 152.8, 148.9, 144.2, 139.8, 139.2, 138.4, 137.4, 136.1, 129.2, 129.1, 127.2, 122.7, 118.6, 116.8, 113.4, 91.8, 21.0, 18.6 ppm, UV/vis/NIR (CH2Cl2): λmax (ε [M−1 cm−1]) = 266 (33000), 501 (50000), 960 (3300) nm, HR-MS (ESI-MS): m/z = 758.2676, calcd for (C48H40N6Ni)+ = 758.2662 [(M)+].
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas “π-System Figuration (no. 2601)” (JSPS KAKENHI grant JP26102003) from MEXT (Japan) and by the Program for Leading Graduate Schools “Integrative Graduate Education and Research in Green Natural Sciences” from MEXT, Japan. T. Y. acknowledges the Japan Society for Promotion of Science for financial support.References
- For recent topics of antiaromatic compounds, see: (a) X. Shi, P. M. Burrezo, S. Lee, W. Zhang, B. Zheng, G. Dai, J. Chang, J. T. L. Navarrete, K.-W. Huang, D. Kim, J. Casado and C. Chi, Chem. Sci., 2014, 5, 4490 RSC; (b) M. Rosenberg, C. Dahlstrand, K. Kilsa and H. Ottosson, Chem. Rev., 2014, 114, 5379 CrossRef CAS PubMed; (c) Y. M. Sung, M.-C. Yoon, J. M. Lim, H. Rath, K. Naoda, A. Osuka and D. Kim, Nat. Chem., 2015, 7, 418 CrossRef CAS PubMed; (d) T. Furuyama, T. Sato and N. Kobayashi, J. Am. Chem. Soc., 2015, 137, 13788 CrossRef CAS PubMed; (e) J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schütt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jarolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley, Chem. Sci., 2016, 7, 5547 RSC; (f) C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827 CrossRef CAS PubMed; (g) H. Oshima, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2017, 56, 3270 CrossRef CAS PubMed.
- T. Nishinaga, T. Ohmae, K. Aita, M. Takase, M. Iyoda, T. Arai and Y. Kunugi, Chem. Commun., 2013, 49, 5354 RSC.
- (a) M. Ishida, S.-J. Kim, C. Preihs, K. Ohkubo, J. M. Lim, B. S. Lee, J. S. Park, V. M. Lynch, V. V. Roznyatovskiy, T. Sarma, P. K. Panda, C.-H. Lee, S. Fukuzumi, D. Kim and J. L. Sessler, Nat. Chem., 2013, 5, 15 CrossRef CAS PubMed; (b) T. Satoh, M. Minoura, H. Nakano, K. Furukawa and Y. Matano, Angew. Chem., Int. Ed., 2016, 55, 2235 CrossRef CAS PubMed.
- (a) L. Nyulászi, O. Hollóczki, C. Lescop, M. Hissler and R. Réau, Org. Biomol. Chem., 2006, 4, 996 RSC; (b) T. Nishinaga, T. Uto, R. Inoue, A. Matsuura, N. Treitel, M. Rabinovitz and K. Komatsu, Chem. – Eur. J., 2008, 14, 2067 CrossRef CAS PubMed; (c) C. Fan, L. G. Mercier, W. E. Piers, H. M. Tuononen and M. Parvez, J. Am. Chem. Soc., 2010, 132, 9604 CrossRef CAS PubMed.
- T. Ito, Y. Hayashi, S. Shimizu, J.-Y. Shin, N. Kobayashi and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 8542 CrossRef CAS PubMed.
- (a) S. Hiroto and H. Shinokubo, Handbook of Porphyrin Science, World Scientific Publishing, 2016, vol. 37, pp. 233–302 Search PubMed; (b) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342 CrossRef CAS PubMed; (c) M. Pawlicki and L. Latos-Grażyński, Chem. – Asian J., 2015, 10, 1438 CrossRef CAS PubMed; (d) B. K. Reddy, A. Basavarajappa, M. D. Ambhore and V. G. Anand, Chem. Rev., 2017, 117, 3420 CrossRef CAS PubMed.
- (a) C. Liu, D.-M. Shen and Q.-Y. Chen, J. Am. Chem. Soc., 2007, 129, 5814 CrossRef CAS PubMed; (b) V. L. Mishra, T. Furuyama, N. Kobayashi, K. Goto, T. Miyazaki, J.-S. Yang and T. Shinmyozu, Chem. – Eur. J., 2016, 22, 9190 CrossRef CAS PubMed.
- (a) T. Fukuoka, K. Uchida, Y. M. Sung, J.-Y. Shin, S. Ishida, J. M. Lim, S. Hiroto, K. Furukawa, D. Kim, T. Iwamoto and H. Shinokubo, Angew. Chem., Int. Ed., 2014, 53, 1506 CrossRef CAS; (b) R. Nozawa, K. Yamamoto, J.-Y. Shin, S. Hiroto and H. Shinokubo, Angew. Chem., Int. Ed., 2015, 54, 8454 CrossRef CAS PubMed; (c) R. Nozawa, K. Yamamoto, I. Hisaki, J.-Y. Shin and H. Shinokubo, Chem. Commun., 2016, 52, 7106 RSC.
- (a) J.-Y. Shin, T. Yamada, H. Yoshikawa, K. Awaga and H. Shinokubo, Angew. Chem., Int. Ed., 2014, 53, 3096 CrossRef CAS PubMed; (b) T. Yoshida, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Commun., 2017, 53, 1112 RSC.
- R. Nozawa, H. Tanaka, W.-Y. Cha, Y. Hong, I. Hisaki, S. Shimizu, J.-Y. Shin, T. Kowalczyk, S. Irle, D. Kim and H. Shinokubo, Nat. Commun., 2016, 7, 13620 CrossRef CAS PubMed.
- (a) B. Liu, X. Li, M. Stpień and P. J. Chmielewski, Chem. – Eur. J., 2015, 21, 7790 CrossRef CAS PubMed; (b) Z. Deng, X. Li, M. Stpień and P. J. Chmielewski, Chem. – Eur. J., 2016, 22, 4231 CrossRef CAS PubMed; (c) B. Liu, T. Yoshida, X. Li, M. Stpień, H. Shinokubo and P. J. Chmielewski, Angew. Chem., Int. Ed., 2016, 55, 13142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of compounds and details of calculations. CCDC 1545204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7qm00176b |
| This journal is © the Partner Organisations 2017 |