
Ureasil organic–inorganic hybrids as photoactive waveguides for conjugated polyelectrolyte luminescent solar concentrators†
Ilaria
Meazzini
 a,
Camille
Blayo
a,
Jochen
Arlt
b,
Ana-Teresa
Marques
cd,
Ullrich
Scherf
d,
Hugh D.
Burrows
c and
Rachel C.
Evans
*ae
a,
Camille
Blayo
a,
Jochen
Arlt
b,
Ana-Teresa
Marques
cd,
Ullrich
Scherf
d,
Hugh D.
Burrows
c and
Rachel C.
Evans
*ae
aSchool of Chemistry, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
bCollaborative Optical Spectroscopy, Micromanipulation and Imaging Centre (COSMIC) and SUPA, School of Physics and Astronomy, King's Buildings, University of Edinburgh, EH9 3JZ, UK
cCentro de Química, Chemistry Department, University of Coimbra, 3004-535 Coimbra, Portugal
dMacromolecular Chemistry Group (buwmacro) and Institute for Polymer Technology, Bergische Universität Wuppertal, Wuppertal, Germany
eDepartment of Materials Science & Metallurgy, University of Cambridge, 27 Charles Babbage Road, Cambridge, CB3 0FS, UK. E-mail: rce26@cam.ac.uk
First published on 21st August 2017
Abstract
The development of an efficient luminescent solar concentrator (LSC), with minimised optical losses, requires careful consideration of its principal constituting materials, a waveguide and a luminophore, in tandem. Here, a series of LSCs are fabricated utilising a poly(fluorene-alt-phenylene) copolymer containing on-chain perylenediimide (PDI) chromophore units as the luminophore (PBS-PFP-PDI) immobilised within a poly(oxyalkylene)/siloxane organic–inorganic hybrid, known as a ureasil, as the waveguide. PBS-PFP-PDI and the ureasil both function as photoactive components, offering the possibility of energy transfer between the ureasil host and/or the PBS-PFP donor chains to the PDI acceptor, leading to reduced re-absorption losses and harvesting a broader wavelength range of the solar spectrum. A combination of studies using UV/vis absorption, Fourier transform infrared, steady-state and time-resolved photoluminescence spectroscopies revealed that the branching of the ureasil framework influences the packing of the polymer chains, with the tri-podal ureasil structure facilitating improved dispersion of the PBS-PFP-PDI chains, while the linear di-ureasil structure promotes more intimate mixing of the PBS-PFP-PDI and the ureasil. Picosecond time-correlated single photon counting measurements showed that strong spectral overlap, combined with efficient electronic coupling results in efficient excitation energy transfer from the ureasil to emissive trap sites localised on the PBS-PFP unit. This process inhibits subsequent energy transfer to the PDI chromophore, but leads to high solid-state photoluminescence quantum yields of >50%. The optical efficiency of the PBS-PFP-PDI–ureasil composites as LSCs was evaluated under AM1.5G solar simulated light delivering values of up to 5.6% using a scattering background, which could be boosted to 13.1% by increasing the percentage of PDI units per PBS-PFP chains using a model system. The results demonstrate that consideration of the combined photophysical properties of the luminophore and the waveguide are crucial to the design of next generation LSCs.
1. Introduction
The challenge facing many existing photovoltaic (PV) technologies is their inability to harvest all wavelengths of the solar spectrum efficiently,1 which, for example, restricts the power conversion efficiency of single junction crystalline silicon solar cells to ∼32%.2 To overcome this limitation, considerable interest has emerged in the development of spectral converter materials that transform the high- or low-energy regions of the solar spectrum into wavelengths than can be used more effectively by a given PV device.3,4 Spectral converters are attached to the surface of a finished solar cell, where they absorb solar photons and exploit a photoluminescence process (e.g. downshifting,1 singlet fission,5,6 upconversion7) to convert them to more useful energies. Moreover, as no modification to the standard cell architecture or the intrinsic device materials is required, the spectral converter can be tailored towards a specific type of solar cell based on the photophysical properties of the luminescent species used.Spectral converters can be deployed in a luminescent solar concentrator (LSC) architecture in which the luminophore is either coated on, or doped within, a transparent waveguide slab.8,9 Sunlight incident on the slab is absorbed by the luminophore, re-emitted at longer wavelengths, and transported via total internal reflection to the edges of the plate where it is collected by a PV cell. LSCs can capture both direct and diffuse light, and, as the incident surface is larger than the output aperture, they can concentrate light both spatially and spectrally.10 While historically LSCs were developed as a cheap replacement for large area solar cells, the rapid decline in the cost of PV modules has shifted the impetus for their development to other applications including building-integrated photovoltaics,8 solar windows11 and photobioreactors.12
Despite the relatively simple architecture, the efficiency of an LSC is often restricted due to loss mechanisms associated with both the luminophore and the waveguide. Luminophore losses include low absorption efficiencies, low emission quantum yields, and reabsorption of emitted photons by neighbouring molecules, particularly at high loading concentrations.13 Reabsorption losses can be minimised by using luminophores with a large Stokes' shift (e.g. quantum dots14–17) or by using distinct absorbing and emitting chromophores, localised either on the same molecule18–20 or different chemical species.21,22 In such multi-chromophoric systems, Förster resonance energy transfer (FRET) facilitates efficient non-radiative energy transfer between the absorbing and emissive species, providing they are electronically coupled through space and within the Förster radius.23 However, many π-conjugated organic molecules, including the archetypical perylene bisdiimide luminophores used in LSCs, exhibit a tendency to aggregate at the elevated concentrations required to achieve a suitable FRET distance.24 Aggregation may lead to either partial or complete emission quenching due to preferential relaxation via non-radiative channels. The combination of individual donor and acceptor chromophores within a single molecule may overcome this, but brings with it increased synthetic demands.25
A complementary approach is to use a photoactive waveguide, which, in addition to totally internally reflecting the emitted photons, can also participate in the FRET process. This strategy necessitates a move away from the conventional materials used in LSCs (e.g. poly(methacrylate) derivatives,26 poly(carbonate)27) towards alternative waveguide materials which afford additional functionality and synthetic flexibility. In this context, Camaioni et al. have investigated biodegradable polymers such as L-poly(lactic acid) (L-PLA)28 and silk fibroin from the Bombyx mori silkworm.29 We and others have reported the use of sol–gel processed organic–inorganic hybrid polymer waveguides from the ureasil family in LSCs.30–33 Ureasils are comprised of a siliceous skeleton that is chemically grafted to poly(alkylene oxide) chains through urea cross-linkages. In addition to satisfying the primary requirements of the waveguide (large optical window in UV/visible region, refractive index n ∼ 1.5), ureasils are intrinsically photoluminescent and can function as active hosts to tune the emission from luminescent dopants such as conjugated polymers,34,35 organic dyes33,36 and lanthanide complexes30 through energy transfer. Their facile sol–gel synthesis facilitates the controlled placement of luminophores within the ureasil framework via covalent grafting to the siliceous backbone, which can be used to both inhibit aggregation36 and/or promote specific packing.37
In this work, we investigate LSCs based on ureasil waveguides doped with a fluorene-phenylene conjugated polyelectrolyte (CPE), poly{1,4-phenylene-[9,9-bis(4-phenoxy-butylsulfonate)]fluorene-2,7-diyl}-based copolymer (PBS-PFP), functionalised with on-chain perylenediimide (PDI) moieties (PBS-PFP-PDI) (Fig. 1). Recently, we have shown that the incorporation of PBS-PFP into a di-ureasil host results in a dramatic increase in the photoluminescence quantum yield in the solid-state due to a synergistic electronic interaction between the photoactive host and the conjugated polyelectrolyte.34 We postulated that the related PBS-PFP-PDI luminophore may also exhibit this effect, whilst simultaneously offering the possibility for both on-chain intramolecular energy transfer between the PBS-PFP donor and the PDI acceptor38 and interspecies energy transfer between the ureasil donor and the PBS-PFP and/or PDI acceptor chromophores, all of which have the potential to reduce undesirable re-absorption effects and extend the light-harvesting window of the final LSC. Here, we examine both di- and tri-podal ureasil waveguides (Fig. 1) as active hosts and probe the influence of the local structural environment of the PBS-PFP-PDI luminophore on the steady-state photoluminescence and energy transfer kinetics. The optical conversion efficiencies of the resultant LSCs were evaluated with a view to understanding the key materials design parameters which determine the performance of this system.
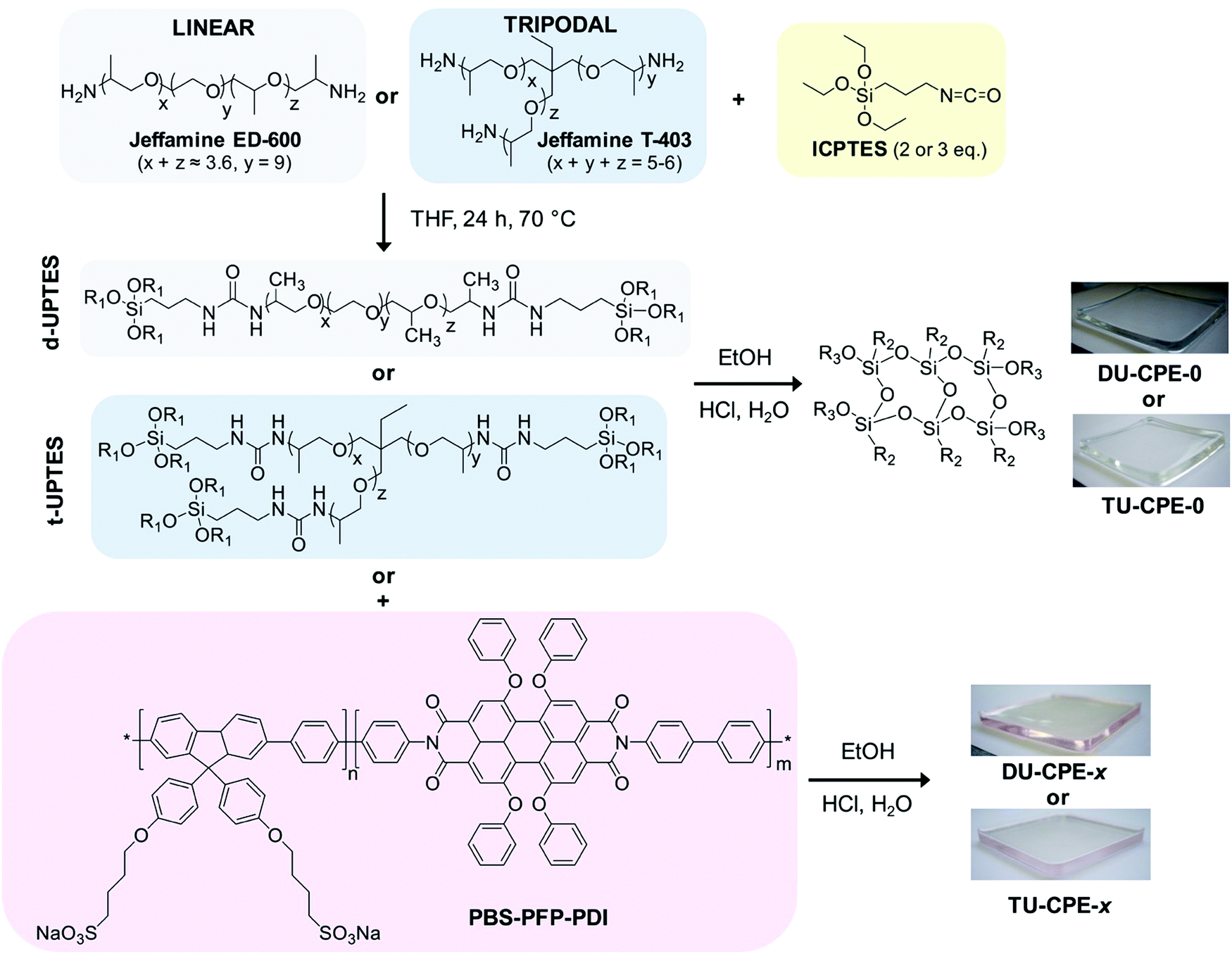 | ||
| Fig. 1 Synthetic route for the preparation of DU-CPE-x and TU-CPE-x (R1 = –CH2–CH3, R2 = –(CH2)3–NHCONH–Jeffamine and R3 = –Si–O–Si– or H). | ||
2. Experimental
Materials
The fluorene-phenylene poly{1,4-phenylene-[9,9-bis(4-phenoxy-butylsulfonate)]fluorene-2,7-diyl} copolymer (PBS-PFP) and PBS-PFP functionalised with on chain perylenediimine (PDI) units (PBS-PFP-PDI) were synthesised as previously described and the associated characterisation data for this batch of the polymer can be found in the literature.38,39 The Mw determined by gel permeation chromatography (GPC) (NMP/LiBr, UV detection 360 nm) was 2100 g mol−1 (Mw/Mn = 1.61). However, we note that GPC typically underestimates the Mw of conjugated polyelectrolytes due to polar interactions between the ionic side-chains with the column material (as can be seen by the cutoff of the dialysis membrane at 3500 g mol−1).39,40 Based on the monomer/PDI ratio in the starting reaction mixture, the copolymer is expected to contain on average 5% PDI units. However, single molecule wide-field imaging studies indicate that the average incorporation of the PDI chromophore is lower (1–2%).38 Lumogen Red F 305 was a kind gift from BASF, Germany. O,O′-Bis(2-aminopropyl) polypropylene glycol-block-polyethylene glycol-block-polypropylene glycol (Jeffamine ED-600), KBr (FT-IR grade, ≥99%), 3-(triethoxysilyl)propylisocyanate (ICPTES, 95%), tetrahydrofuran (THF, HPLC grade), EtOH (HPLC grade), 1,4-dioxane (HPLC grade) and HCl (37% puriss) were all purchased from Sigma. Jeffamine T-403 was a kind gift from Huntsman International (http://www.huntsman.com). All chemicals were used as received.Synthesis of di- and tri-ureasil organic–inorganic hybrids
Ureasil hybrids were prepared using a procedure previously reported in literature.36,41 In brief, Jeffamine (4.4 mmol) was dissolved in THF (12.5 mL) and ICPTES was added in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:3 molar ratio for Jeffamine ED-600 and Jeffamine T-403, respectively. Each mixture was then refluxed at 70 °C for 24 h, before cooling to room temperature (RT). To initiate the sol–gel process, EtOH, HCl (0.5 M) and water (molar ratio ICPTES:EtOH:HCl:H2O = 176:350:1:265) were added to either the di- or the triureapropyltriethoxysilane (d- and t-UPTES, respectively) intermediate (0.88 mmol). The sol was then stirred for 2 min, poured into a polypropylene mould and covered with Parafilm M®. Samples were aged for 2 days at RT, after which holes were pierced in the Parafilm M® and after a further 3 days the drying process was completed in an oven at 40 °C for 1 day. PBS-PFP-PDI-doped samples were prepared by addition of a stock solution of the conjugated polyelectrolyte (1 mg mL−1 in water/1,4-dioxane (1:1 v/v)) before the addition of the gelation reagents. For the preparation of larger monoliths, the gelation reagents and CPE stock solution were added to either d- or t-UPTES (7.04 mmol) in the same molar proportions reported above. The ageing process was carried for 5 days at RT, followed by 3 days in an oven at 30 °C. The di-ureasil sample containing Lumogen Red F 305 (DU-LR305) was prepared by addition of a volume (616.5 μL) of a solution of Lumogen Red F 305 (0.2 mg mL−1 in THF) to d-UPTES (7.04 mmol). The sample containing both Lumogen Red F 305 and PBS-PFP (DU-PBS-LR305) was prepared by addition of a volume (616.5 μL) of a solution of Lumogen Red F 305 (0.2 mg mL−1 in THF) and PBS-PFP (616.5 μL of a 1 mg mL−1 solution in water/1,4-dioxane (1:1 v/v)) to d-UPTES (7.04 mmol). For both samples, the gelation reaction and ageing process were carried out using the same quantities and procedures reported above.
:2 and 1:3 molar ratio for Jeffamine ED-600 and Jeffamine T-403, respectively. Each mixture was then refluxed at 70 °C for 24 h, before cooling to room temperature (RT). To initiate the sol–gel process, EtOH, HCl (0.5 M) and water (molar ratio ICPTES:EtOH:HCl:H2O = 176:350:1:265) were added to either the di- or the triureapropyltriethoxysilane (d- and t-UPTES, respectively) intermediate (0.88 mmol). The sol was then stirred for 2 min, poured into a polypropylene mould and covered with Parafilm M®. Samples were aged for 2 days at RT, after which holes were pierced in the Parafilm M® and after a further 3 days the drying process was completed in an oven at 40 °C for 1 day. PBS-PFP-PDI-doped samples were prepared by addition of a stock solution of the conjugated polyelectrolyte (1 mg mL−1 in water/1,4-dioxane (1:1 v/v)) before the addition of the gelation reagents. For the preparation of larger monoliths, the gelation reagents and CPE stock solution were added to either d- or t-UPTES (7.04 mmol) in the same molar proportions reported above. The ageing process was carried for 5 days at RT, followed by 3 days in an oven at 30 °C. The di-ureasil sample containing Lumogen Red F 305 (DU-LR305) was prepared by addition of a volume (616.5 μL) of a solution of Lumogen Red F 305 (0.2 mg mL−1 in THF) to d-UPTES (7.04 mmol). The sample containing both Lumogen Red F 305 and PBS-PFP (DU-PBS-LR305) was prepared by addition of a volume (616.5 μL) of a solution of Lumogen Red F 305 (0.2 mg mL−1 in THF) and PBS-PFP (616.5 μL of a 1 mg mL−1 solution in water/1,4-dioxane (1:1 v/v)) to d-UPTES (7.04 mmol). For both samples, the gelation reaction and ageing process were carried out using the same quantities and procedures reported above.
Optical efficiency measurements
The optical efficiency of the LSCs was determined under illumination with a Class ABB solar simulator (Abet Technologies, Model 10500), equipped with an AM 1.5 filter, using the previously reported procedure.32 The illumination spot was defined by a black mask with a circular aperture (area = 9.6 cm2). The emission from the four edges of the LSC was collected independently with an INS 125 integrating sphere connected to a spectroradiometer (International Light Technologies ILT950). The data were analysed over a range of 250–1050 nm using SpectrILight III software and the manufacturer provided calibration file ILT1007131U1INS125 for optical power measurements. The output of the solar simulator was calibrated to 1 Sun (995 ± 3 W m−2) using a reference Si solar cell from ReRa Technologies. For improved accuracy in calculating the optical efficiency, the input power i.e. the output of the solar simulator, was also measured using the spectroradiometer set-up, by illuminating the 2 cm diameter input port of the integrating sphere with the collimated light supplied by the solar simulator. This allowed for the power density from the entire collimated beam to be calculated accurately. Using this method, the integrated power of the solar simulator over the exposed area of 9.6 cm2 is 887.0 mW in the full spectrum range (250–1050 nm).3. Results and discussion
Fabrication of PBS-PFP-PDI-doped LSCs
Doped LSCs were prepared using a sol–gel chemistry route to incorporate PBS-PFP-PDI at different dopant concentrations (wt%) in either a di- or tri-branched organic–inorganic hybrid ureasil waveguide, using a previously reported procedure.32,36 In brief, the di-branched polyetheramine, Jeffamine ED-600 or the tri-branched Jeffamine T-403 is first reacted with ICPTES in THF to obtain the respective organic–inorganic hybrid precursor intermediates, di- or tri-ureapropyltriethoxysilane (d-UPTES and t-UPTES, respectively) (Fig. 1). A fixed volume of a stock solution of PBS-PFP-PDI in water:1,4-dioxane (1:1 v/v) was then added to the precursor solution to obtain the required dopant concentration (0.002–0.008 wt% with respect to dry monolith). Acid-catalysed hydrolysis and condensation of the siliceous network, followed by ageing, subsequently yields the doped LSC as a free-standing monolith (Fig. 1). The samples are designated as DU-CPE-x and TU-CPE-x, for di- and tri-ureasils, respectively, where x represents 1000 times the final concentration (in wt%) of PBS-PFP-PDI incorporated (i.e.DU-CPE-02 contains 0.002% w/w of CPE).
Steady-state spectroscopic characterisation
Fig. 2 shows the steady-state optical properties of PBS-PFP-PDI in solution and selected doped and updoped di- and tri-ureasil LSCs in the solid-state. In a good solvent (here, a mixture of water:1,4-dioxane (1:1 (v/v))), the optical properties of PBS-PFP-PDI are dominated by the contribution of the poly(fluorene-alt-phenylene) backbone, reflecting the low degree of on-chain incorporation of the PDI chromophore (∼1–2%).38 The absorption spectrum consists of a broad band centred at 358 nm, typical of poly(fluorene-alt-phenylene),42 and a weaker band between 450–610 nm, indicative of the presence of PDI. The contribution of the PDI chromophore is also observed in the corresponding excitation spectrum upon detection at 660 nm, which is the characteristic emission region for PDI.43,44 Upon selective excitation of the poly(fluorene) unit at 360 nm, a vibronically-structured emission band centred between 390–550 nm is detected, which is typical of the poly(fluorene) emission.45 However, no emission from the PDI chromophore is observed, either upon indirect or direct excitation, suggesting that fast non-radiative deactivation of the PDI excited state occurs in this solvent, possibly involving reversible electron transfer as previously reported.38
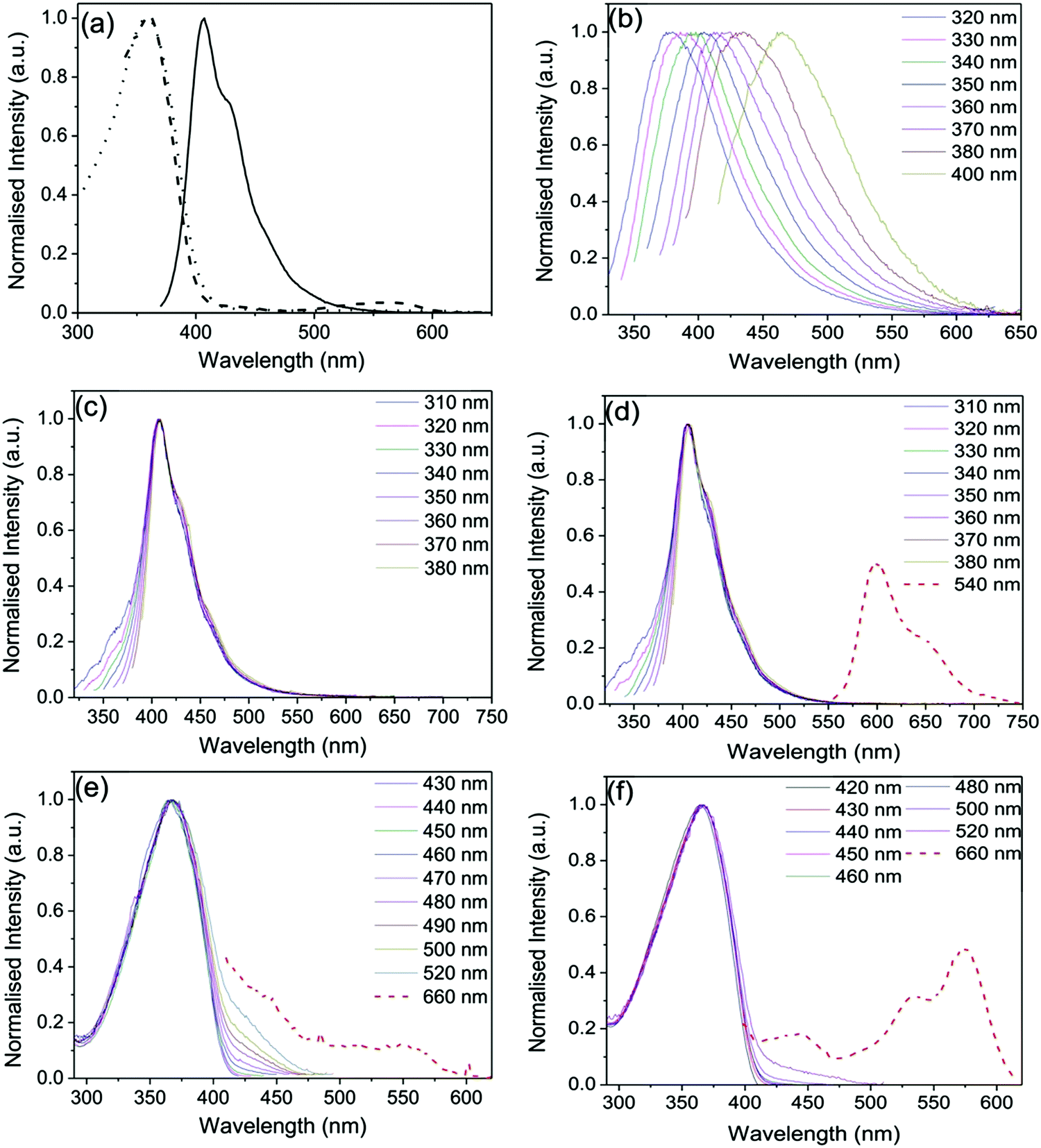 | ||
| Fig. 2 Steady-state optical properties of PBS-PFP-PDI in solution (water:1,4-dioxane (1:1 (v/v))) and selected doped and updoped di- and tri-ureasil LSCs in the solid-state. (a) Absorption (dotted line), excitation (dashed line) and emission (solid line) spectra of PBS-PFP-PDI (10−6 mol dm−3, λex = 360 nm, λem = 660 nm). Emission spectra of (b) DU-CPE-0, (c) DU-CPE-08 and (d) TU-CPE-08 at different excitation wavelengths. Excitation spectra of (e) DU-CPE-08 and (f) TU-CPE-08 at different emission wavelengths. | ||
The undoped DU-CPE-0 and TU-CPE-0 hybrids present excitation-wavelength dependent emission spectra characterised by a broad band centred between 380–470 nm (Fig. 2b and Fig. S1 (ESI†), respectively), which is characteristic of ureasils.36,46 For both samples, the emission band is red-shifted to longer wavelengths with increasing excitation wavelength and an analogous trend is observed for the corresponding excitation spectra (Fig. S1, ESI†). Each band can be deconvoluted into two separate components, which have previously been assigned to radiative recombination of donor–acceptor pairs mediated by one of two localised centres: (i) electron–hole recombination localised at oxygen defects in the siliceous nanoclusters and (ii) photoinduced proton transfer between the NH2+ and the N− moieties of the urea linkages.46,47
Upon doping with PBS-PFP-PDI, the emission spectra of the DU-CPE-x and TU-CPE-x series, are dominated by the characteristic poly(fluorene) emission band (Fig. 2c, d and Fig. S2, ESI†). However, the contribution of the ureasil can also be identified by the broadening of the blue-edge of the emission band upon varying the excitation wavelength between 310–340 nm (associated with siliceous-based defects) and the red-edge upon excitation between 350–380 nm (urea-centred emission). This excitation-wavelength dependent broadening of the emission band has previously been assigned to excitonic coupling between the ureasil and CPE components in a related system.34 We note that the emission maximum for the DU-CPE-x series is red-shifted (409 nm, Δλ = 2 nm) compared to that of PBS-PFP-PDI in solution, while for TU-CPE-x series the emission maximum is blue-shifted to 405 nm (Δλ = −2 nm). Previous studies have shown that due to the increased branching of the Jeffamine precursor, the tri-ureasil framework is less-condensed than the di-ureasil analogue, which leads to a reduction in the aggregation of conjugated polymer chains.37 The observed shifts in the emission maxima are in agreement with this trend, and suggest that the incorporated PBS-PFP-PDI chains are more aggregated in the case of the DU-CPE-x series.
Due to the low on-chain incorporation, the contribution of the PDI chromophore to the emission spectrum is extremely weak, and can only be clearly isolated for the TU-CPE-x series upon selective excitation at 540 nm and by increasing the emission and excitation slits (Fig. 2d and Fig. S2, ESI†). For the DU-CPE-x series measured under the same conditions, the PDI contribution is somewhat harder to isolate and exhibits a lower spectral resolution. The corresponding excitation spectra follow a similar trend for both series (Fig. 2e, f, and Fig. S3, ESI†) and exhibit the characteristic excitation profile of PBS-PFP-PDI upon detection between 420–520 nm, with the specific contribution of the PDI unit observed only upon detection at 660 nm. These results support the suggestion that PBS-PFP-PDI exists in a more aggregated state in the DU-CPE-x series, which leads to enhanced non-radiative relaxation of the PDI excited state. Moreover, the rigid TU-CPE-x matrix may partially switch-off the non-radiative channels that dominate relaxation of the excited PDI chromophore in solution. It is of note, however, that emission from the PDI chromophore can now be accessed upon incorporation of PBS-PFP-PDI into either the di- or tri-ureasil host, which is not possible in solution.
The photoluminescence quantum yields (ΦPL) were measured for each sample upon excitation at 360 nm. The ΦPL for the undoped DU-CPE-0 (6.3 ± 0.5%) is in excellent agreement with the literature,48 while for TU-CPE-0 the value is somewhat higher (15.7 ± 0.8%), but still within the expected range for this class of materials.49 In solution (water:1,4-dioxane, (1:1 (v/v))), PBS-PFP-PDI presents a ΦPL of 68.6 ± 1.2%, which is also slightly higher than previous reports (ΦPL = 55%).38 For the DU-CPE-x and TU-CPE-x series, the ΦPL values increase with the CPE concentration (from 42.3 to 60.5% and 39.8 to 51%, respectively), with the di-ureasil hybrids generally exhibiting a higher value for the equivalent concentration (Table 1). Due to the nature of these samples, re-absorption and waveguiding effects are commonly encountered while measuring the ΦPL using an integrating sphere.50 A correction for these effects has been performed using the method described by Ahn et al.,51 which leads to a 10–20% increase in the ΦPL values (Table 1). We note that these values are comparable to those obtained for PBS-PFP-PDI in a good solvent, which is unusual as CPEs typically exhibit a significantly decreased ΦPL upon transfer to the solid state.52–55 This is attributed to an electronic interaction between PBS-PFP chains and the ureasil and subsequent trapping at localised radiative sites as previously observed for the parent CPE (see also time-resolved photoluminescence studies below).34
| Sample name | Φ PL (measured) | Φ PL (corrected) |
|---|---|---|
| DU-CPE-0 | 6.3 (±0.5) | — |
| DU-CPE-02 | 42.3 (±1.4) | 47.7 (±1.5) |
| DU-CPE-04 | 56.4 (±1.5) | 59.9 (±1.5) |
| DU-CPE-08 | 60.5 (±1.3) | 65.1 (±1.7) |
| TU-CPE-0 | 15.7 (±0.8) | — |
| TU-CPE-02 | 39.8 (±2.0) | 51.1 (±2.5) |
| TU-CPE-04 | 48.8 (±1.4) | 56.4 (±2.1) |
| TU-CPE-08 | 51.0 (±0.9) | 55.5 (±0.9) |
The ΦPL of undoped ureasils has previously been shown to be related to the degree of hydrogen-bonding interactions between the urea groups on the organic backbone of the ureasil.49 A reduction in the urea-urea interaction leads to more efficient radiative relaxation associated with photoinduced proton transfer between donor–acceptor pairs located within the urea moieties.49 Fourier transform infrared (FTIR) spectroscopy is a convenient tool for analysing hydrogen bonding interactions associated with the stretching of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties in the urea groups, which are located within the amide I spectral region (1600–1800 cm−1).56 For the DU-CPE-x series, Gaussian deconvolution of the amide I band results in three components, one ascribed to self-organised urea-urea interactions centred at 1637 cm−1 (Peak 1), and two ascribed to urea-polyether disordered interactions centred at 1663 cm−1 (Peak 2) and 1716 cm−1 (Peak 3) (Fig. 3 and Fig. S4, ESI†).41,48 The relative contribution and peak centres do not show any remarkable changes upon incorporation of the CPE at any of the concentrations investigated, indicating that the local structure of the di-ureasil network is preserved upon doping (Table S1, ESI†). For the TU-CPE-x series, in addition to Peaks 1–3, Gaussian fits of the amide I region reveal the presence of a contribution centred at 1771 cm−1 (Peak 4), which has previously been assigned to urea moieties not participating in hydrogen bonding (Fig. 3b).41 This supports the belief that the tri-ureasils contain a more open framework36 and may indicate that the significantly higher ΦPL obtained for the undoped TU-CPE-0 sample is due to more efficient radiative recombination localised at the urea groups. However, as the ΦPL values are generally higher for the DU-CPE-x series, this suggests that the urea-centred radiative pathway makes only a minor contribution to the total emission in the doped samples (for both di- and tri-ureasils), which is in agreement with the steady-state PL spectra where the PBS-PFP-PDI emission dominates.
O moieties in the urea groups, which are located within the amide I spectral region (1600–1800 cm−1).56 For the DU-CPE-x series, Gaussian deconvolution of the amide I band results in three components, one ascribed to self-organised urea-urea interactions centred at 1637 cm−1 (Peak 1), and two ascribed to urea-polyether disordered interactions centred at 1663 cm−1 (Peak 2) and 1716 cm−1 (Peak 3) (Fig. 3 and Fig. S4, ESI†).41,48 The relative contribution and peak centres do not show any remarkable changes upon incorporation of the CPE at any of the concentrations investigated, indicating that the local structure of the di-ureasil network is preserved upon doping (Table S1, ESI†). For the TU-CPE-x series, in addition to Peaks 1–3, Gaussian fits of the amide I region reveal the presence of a contribution centred at 1771 cm−1 (Peak 4), which has previously been assigned to urea moieties not participating in hydrogen bonding (Fig. 3b).41 This supports the belief that the tri-ureasils contain a more open framework36 and may indicate that the significantly higher ΦPL obtained for the undoped TU-CPE-0 sample is due to more efficient radiative recombination localised at the urea groups. However, as the ΦPL values are generally higher for the DU-CPE-x series, this suggests that the urea-centred radiative pathway makes only a minor contribution to the total emission in the doped samples (for both di- and tri-ureasils), which is in agreement with the steady-state PL spectra where the PBS-PFP-PDI emission dominates.
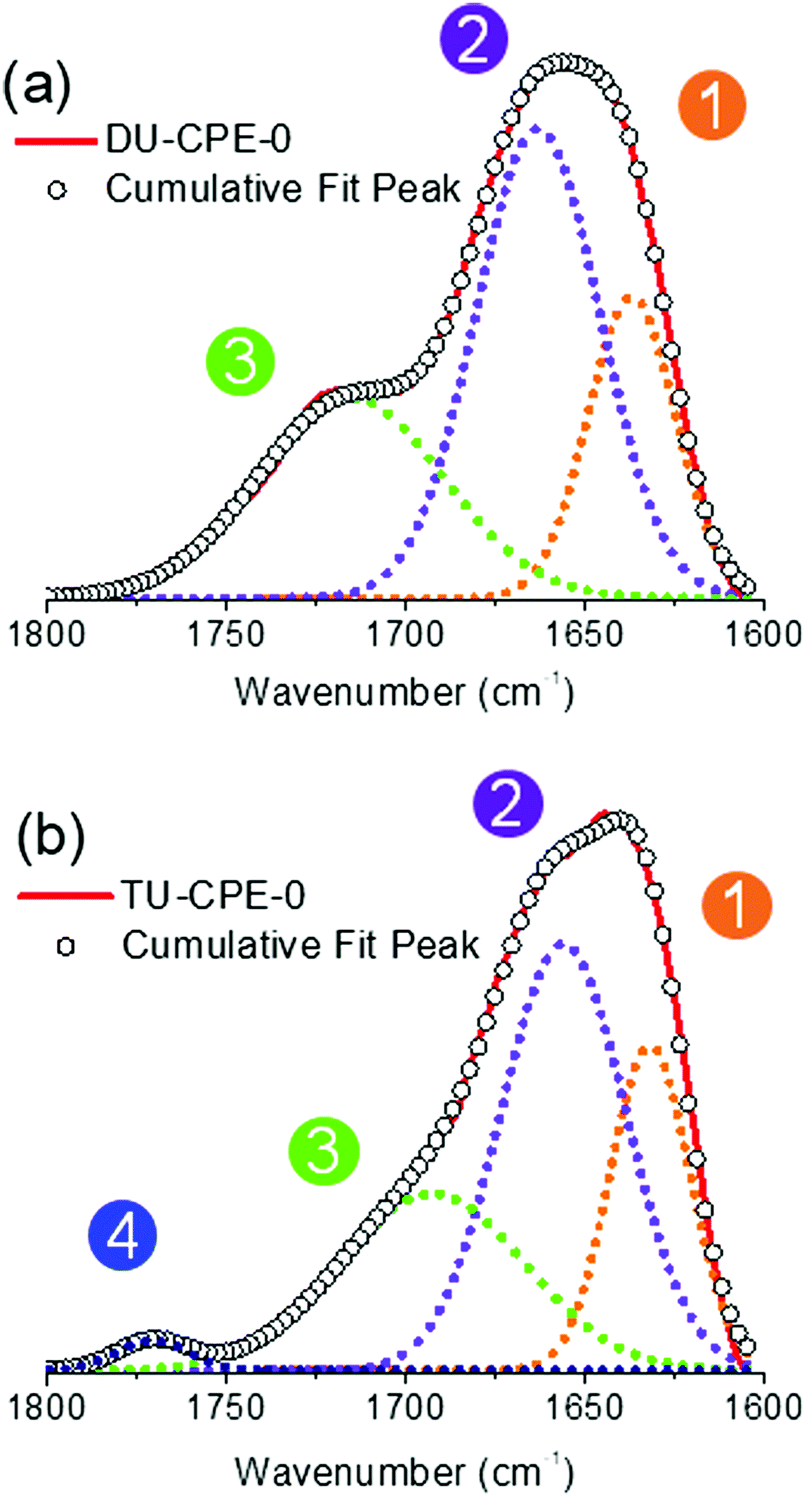 | ||
| Fig. 3 FTIR spectra and corresponding Gaussian curve-fits of the amide I region of (a) DU-CPE-0 and (b) TU-CPE-0. The circled numbers represent the peaks described in the text. | ||
Time-resolved photoluminescence studies
Despite good spectral overlap between the emission spectrum of the PBS-PFP homopolymer and the absorption spectrum of PDI, and density functional theory (DFT) calculations confirming that the PBS-PFP and PDI units are electronically coupled in PBS-PFP-PDI,38 the absence of emission from the PDI chromophore upon selective excitation of PBS-PFP suggests that CPE-centred FRET does not occur either in solution or upon incorporation within the ureasil waveguide. However, previous studies in our group have shown that ureasils themselves can behave as efficient excitation energy donors for both the PBS-PFP homopolymer34 and perylenediimides.32,36 Picosecond time-correlated single photon counting (TCSPC) emission lifetime measurements were thus performed to further investigate the excited-state behaviour of the PBS-PFP-PDI-doped ureasils and to determine if electronic coupling between the host waveguide and luminophore is significant in this system. The measurements were performed by selectively exciting the ureasil/poly(fluorene-alt-phenylene) components (λex = 370 nm) or the PDI chromophore (λex = 466 nm), while monitoring the emission at 420 nm (PBS-PFP/purplish-blue emission from ureasil) and 500 nm (blue emission from ureasil). Unfortunately, due to the weak emission, attempts to measure the decay curves at 600 nm (PDI emission) resulted in unsatisfactory signal-to-noise ratios and correspondingly poor fits (Fig. S5, ESI†). The fluorescence decays were in general best fitted with a multi-exponential decay law of the form: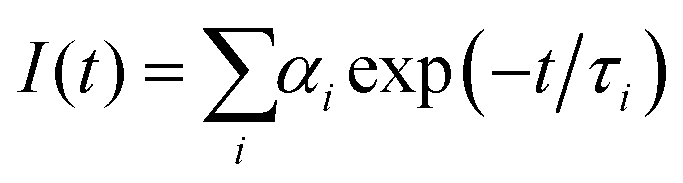 | (1) |
| Sample | τ 1 (ns) | τ 2 (ns) | τ 3 (ns) | τ 4 (ns) | α 1 | α 2 | α 3 | α 4 | χ 2 |
|---|---|---|---|---|---|---|---|---|---|
| CPE | 0.026 | 0.553 | 1.072 | −0.899 | 0.947 | 0.053 | 1.14 | ||
| DU-CPE-0 | 0.47 | 2.56 | 10.08 | 0.75 | 0.20 | 0.05 | 1.55 | ||
| DU-CPE-02 | 0.029 | 0.538 | 1.640 | −0.281 | 0.982 | 0.018 | 1.03 | ||
| DU-CPE-04 | 0.537 | 1.545 | 0.990 | 0.010 | 1.32 | ||||
| DU-CPE-08 | 0.548 | 1.224 | 0.983 | 0.017 | 1.20 | ||||
| TU-CPE-0 | 0.522 | 4.204 | 12.740 | 0.474 | 0.378 | 0.148 | 1.43 | ||
| TU-CPE-02 | 0.513 | 1.050 | 8.103 | 0.943 | 0.052 | 0.005 | 1.12 | ||
| TU-CPE-04 | 0.544 | 5.396 | 0.995 | 0.005 | 1.25 | ||||
| TU-CPE-08 | 0.550 | 3.223 | 0.996 | 0.004 | 1.31 |
For the PBS-PFP-PDI copolymer in water:1,4-dioxane (1:1 (v/v)), the fluorescence decay observed at 420 nm can be resolved in three independent components: τCPE1 ∼ 0.03 ns, which is believed to include contributions from fast intra-/inter-chain energy migration and conformational relaxation on the PFP backbone,57,58τCPE2 ∼ 0.55 ns, which is assigned to radiative relaxation associated with polymer clusters59 and τCPE3 ∼ 1.1 ns, which is attributed to radiative decay of isolated polymer chains.60 The longer lifetimes, τCPE2 and τCPE3, are somewhat longer than those previously reported for PBS-PFP-PDI in this solvent (∼0.10 ns and 0.70 ns, respectively38), but this is fully consistent with the higher ΦPL obtained here (∼69% vs. ∼55%38).
The emission decay curves for the DU-CPE-0 and TU-CPE-0 samples under the same measurement conditions (Fig. 4a) also exhibit three discrete exponential functions: τU1 <1 ns, τU2 ≈ 2.5–4.9 ns and τU3 ≈ 10–14.5 ns.34 The average lifetime, 〈τ〉, is longer for TU-CPE-0 (∼8.3 ns) than for DU-CPE-0 (∼4.8 ns), which is in agreement with the higher ΦPL. Moreover, as the detection wavelength is changed from 420 nm to 500 nm, 〈τ〉 increases to 8.8 ns for TU-CPE-0 and 5.9 ns for DU-CPE-0. This behaviour (and the relative amplitudes of the three lifetime contributions at each detection wavelength) reflects the spectral dependence of the two components responsible for the ureasil emission: at 420 nm the largest contribution to the emission originates predominantly from the siliceous domains (τU1), which switches to the urea-centred emission at 500 nm (τU3).34 The emission decay curves and corresponding fits for DU-CPE-x and TU-CPE-x obtained upon excitation at 370 nm and detection at 420 nm are presented in Fig. 4b and c, respectively. Each decay curve can be modelled with two lifetime components, with the exception of DU-CPE-02 and TU-CPE-02, which both require three. All samples exhibit an intermediate lifetime (τ2) of ∼0.50–0.55 ns which provides the largest contribution to the emission decay (α2). Due to the large spectral overlap between the emission of PBS-PFP-PDI and the emission of the ureasil at 420 nm, it is hard to unambiguously assign this lifetime to a single component, since it is of the same order of magnitude as both the shortest lifetime component of ureasil (τU1) and the contribution ascribed to radiative relaxation associated with clusters of polymer chains (τCPE2). Detailed examination of the relaxation kinetics in a related PBS-PFP–di-ureasil system has previously suggested that strong electronic coupling between the CPE and siliceous-based ureasil centres results in efficient excitation energy shuttling between the two species.34 The predominance of the intermediate τ2 component in the present PBS-PFP-PDI–ureasil systems suggests that a similar effect operates here.
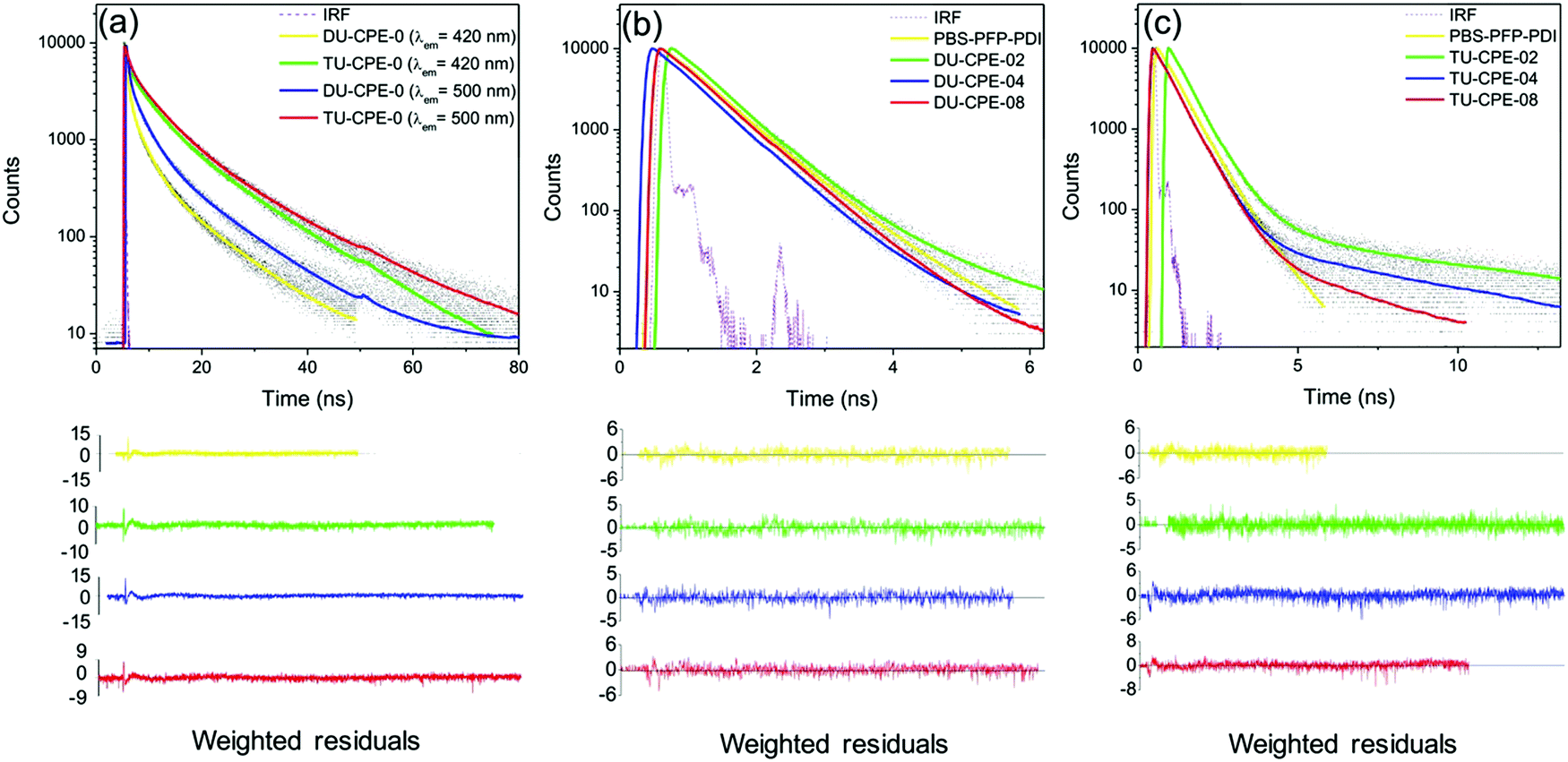 | ||
| Fig. 4 Emission decay curves (black dots), fits (coloured lines) and instrument response function (IRF) (dashed line) for PBS-PFP-PDI in solution (water/1,4-dioxane (1:1 (v/v))), DU-CPE-x and TU-CPE-x at selected excitation and emission wavelengths. (a) DU-CPE-0 and TU-CPE-0 (λex = 370 nm, λem = 420 nm and 500 nm), (b) PBS-PFP-PDI and DU-CPE-x (λex = 370 nm, λem = 420 nm) and (c) PBS-PFP-PDI and TU-CPE-x (λex = 370 nm, λem = 420 nm). The weighted residuals for each fit are also shown. | ||
For DU-CPE-02, an additional short-lived component (τ1 = 0.029 ns) was isolated, which is reminiscent of τCPE1 (on-chain/intrachain excitation migration and/or conformational relaxation). Since this contribution can only be resolved for DU-CPE-02, it may suggest that the PBS-PFP-PDI is present in isolated pockets within the ureasil. A similar characteristic is observed for TU-CPE-02, where τ3 (1.050 ns) is in good agreement with τCPE3, and a third, longer-lived component, τ4 = 8.10 ns is observed, which is reminiscent of the longest ureasil lifetime (τU3), suggesting again that the PBS-PFP-PDI and ureasil species emit discretely in this sample. However, a medium-long lifetime component, τ3 ∼ 1–6 ns, is also present in all samples, which varies with the PBS-PFP-PDI concentration and corresponds to the same timescale as both τCPE3 and τU2. For the DU-CPE-x series, τ3 decreases with increasing PBS-PFP-PDI concentration, from 1.6–1.2 ns. For the TU-CPE-x series, τ3 first increases, then decreases with concentration, but is significantly longer than for the di-ureasil series at the same concentration (e.g. τ3 = 5.4 ns and 1.5 ns for TU-CPE-04 and DU-CPE-04, respectively). As described above, we have previously showed that a di-ureasil matrix can behave as an active host for the PBS-PFP homopolymer, where efficient energy transfer from both the siliceous and the urea domains of the di-ureasil donor to the PBS-PFP acceptor leads to a general increase in the ΦPL of the system.34 It is tempting to suggest that a similar mechanism is responsible for the general decrease of τ3 with increasing CPE concentration observed here for both DU-/TU-CPE-04 and DU-/TU-CPE-08. However, this hypothesis is not consistent with the corresponding α values, which remain essentially unchanged in both series at these concentrations.
The decay curves collected at λem = 500 nm (λex = 370 nm) can be fitted with three components, with the exception of DU-CPE-02 which requires a fourth term (τ4 ∼ 7 ns), reinforcing our hypothesis of poor coupling between the ureasil and the CPE at this concentration (Table S2 and Fig. S6, S7, ESI†). For each di-ureasil sample three contributions can be isolated; τ1 ∼ 20–40 ps, which is reminiscent of the short-lived CPE component (τCPE1), τ2 ∼ 0.6 ns which can be ascribed to the combination of the ureasil (τU1) and the CPE (τCPE2), and τ3 ∼ 1.4–4.1 ns, which arises from a combination of the longer-lived ureasil contributions (τU2 and τU3) and emission from isolated CPE chains (τCPE3). Similarly, τ2 ∼ 0.6 ns and τ3 ∼ 1.5–2.1 ns can be identified for the TU-CPE-x samples, while a third decay term (τ4 ∼ 8.8–10.4 ns) is reminiscent of τU3 and is observed at all PBS-PFP-PDI concentrations. The general increase in the lifetimes observed at this emission wavelength predominantly originates from the ureasil emissive centres localised within the urea domains, which dominate at 500 nm. Although at this emission wavelength there is still some overlap between the PBS-PFP-PDI and ureasil emission, the semi-selectivity of this region for this emissive component of the ureasil can provide further insight. For DU-CPE-x, a short lifetime contribution (τ1) that is characteristic of the CPE (τCPE1) can now be isolated at all investigated concentrations. As the PBS-PFP-PDI concentration is increased, a concomitant increase in both τ2 and α2 is observed, which is accompanied by decrease in the longest lifetime and associated pre-exponential factor (τ3/τ4 decreases from ∼10.9 ns to 3.6 ns, Table S2, ESI†). For TU-CPE-x, the short τ1 is absent, but the same trend in the decrease of the longest lived lifetime component is observed with increasing PBS-PFP-PDI concentration (τ4 decreases from ∼14.0 ns to 8.8 ns, Table S2, ESI†). A few hypotheses can be proposed from these trends. Firstly, the observation of τ1 for DU-CPE-x but not TU-CPE-x supports the supposition that PBS-PFP-PDI shows a stronger tendency to form aggregated clusters in the di-ureasil host, as indicated by the red-shift in the emission maximum. As such, the probability of on-chain excitation energy migration is enhanced. 29Si magic-angle spinning nuclear magnetic resonance (MAS-NMR) studies have previously revealed that the tri-ureasil framework is generally less condensed and more open than that of di-ureasils.36 Moreover, the higher weight ratio of inorganic silica component to organic poly(ether) chains in the tri-ureasils compared to the di-ureasils imparts a greater rigidity to the network of TU-CPE-x samples, which can also be observed macroscopically (e.g.TU-CPE-0 is rigid and hard to bend, while DU-CPE-0 is flexible). These features, coupled with the absence of τ1 and the blue-shift in the emission maximum suggest that PBS-PFP-PDI is well-dispersed within the tri-ureasil host, facilitating excitation energy migration between the two components. Secondly, the longest lifetime component (and associated pre-exponential factor) with increasing CPE concentration for both DU-CPE-x and TU-CPE-x, supports our previous assignment of thermally-assisted excitation energy from NH-centred trap states to isolated CPE chains in these systems.34
Performance of PBS-PFP-PDI–ureasils as luminescent solar concentrators?
Our goal for this study was to elucidate if the combination of the photoactive ureasil waveguide with the PBS-PFP-PDI luminophore, which contains intramolecular donor–acceptor chromophores, could improve the performance of the resultant LSC, either by extending the light-harvesting window, facilitating energy transfer cascades between the various species or a combination of the two effects. While time-resolved emission studies confirmed efficient energy transfer between the ureasil host and the PBS-PFP chains, sensitisation of the PDI chromophore, either through on-chain energy transfer or from the ureasil, was not detected. The energy transfer cascade thus appears to stop at the blue-emitting components. However, as direct sensitisation of the PDI emission was observed upon incorporation of PBS-PFP-PDI in the ureasil, DU-CPE-08 and TU-CPE-08 were fabricated as large slabs (4 cm × 4 cm) to measure their performance as LSCs (Fig. 5). In addition to containing the highest PBS-PFP-PDI wt%, these samples also exhibit the highest ΦPL for each series.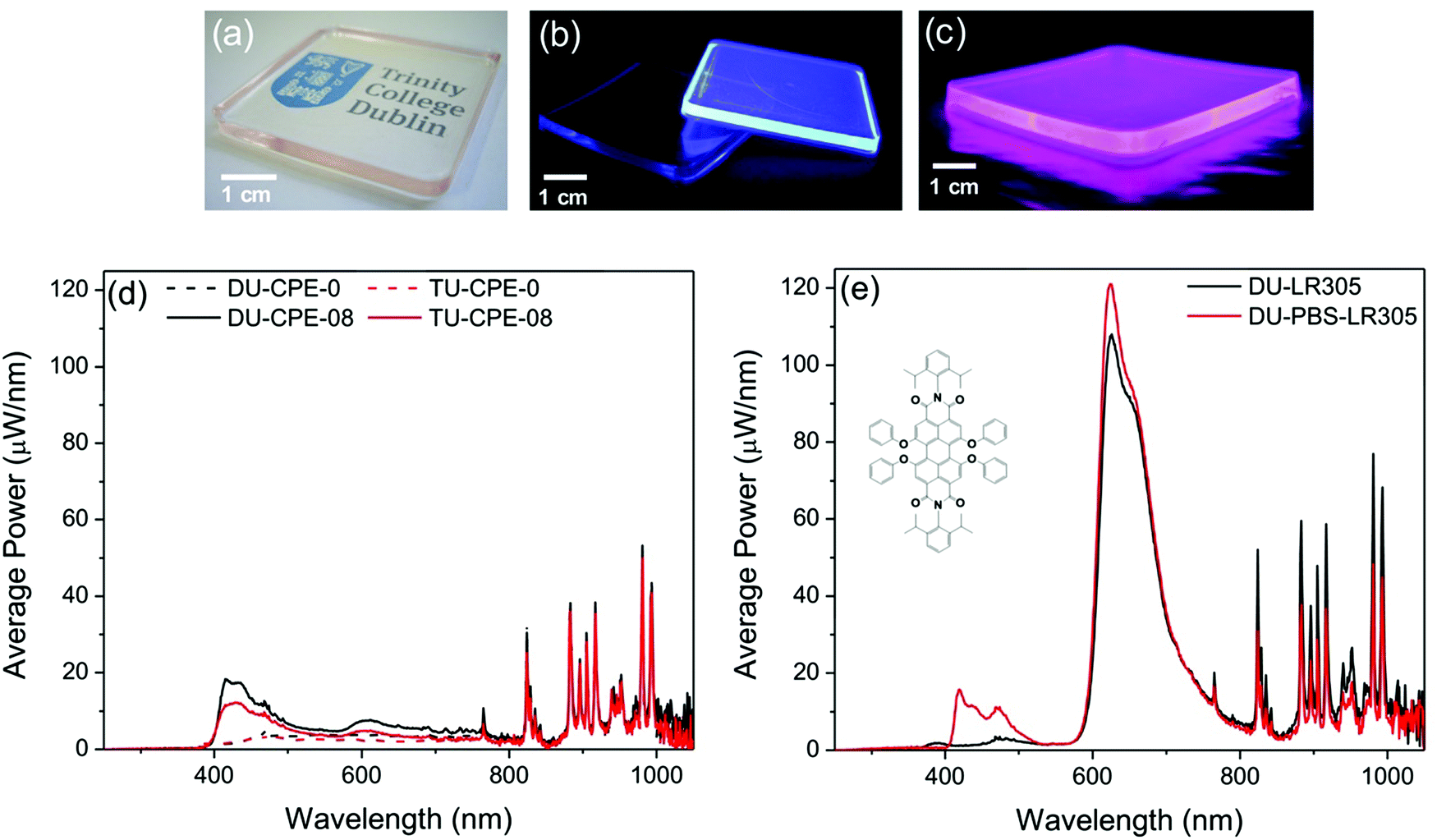 | ||
| Fig. 5 Evaluation of the performance of PBS-PFP-PDI–ureasils as luminescent solar concentrators. Photographs of (a) TU-CPE-08 in daylight, (b) DU-CPE-0 and DU-CPE-08 under UV irradiation (λex = 365 nm), and (c) DU-PBS-LR305 under UV irradiation (λex = 365 nm). (d) Optical power spectra of DU-CPE-0, DU-CPE-08, TU-CPE-0 and TU-CPE-08 with a dark absorbing background. (e) Optical power spectra of DU-LR305 and DU-PBS-LR305 with a dark absorbing background. The structure of the red luminophore LR305 is shown in the inset. | ||
The performance of an LSC is generally quantified by the optical conversion efficiency (ηopt), which is determined experimentally by measuring the optical power output summed over the four edges of the LSC (OPout) with respect to the total incident solar power on the surface of the LSC (OPin) according to:61
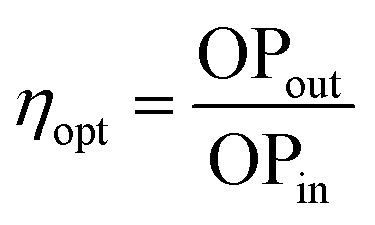 | (2) |
One of the major limitations of this system is the degree of on-chain substitution with the PDI chromophore, which is extremely low (∼1–2%). Despite the good spectral overlap, this inhibits efficient energy transfer from the PBS-PFP unit and/or the ureasil. Furthermore, significant emission in the red spectral region cannot be obtained even through direct excitation of the PDI unit due to its small contribution. Clearly, an increase in the percentage of on-chain PDI units in the CPE itself is thus required to counteract these constraints. To mimic this effect (without the need for significant synthetic efforts), a model system was created by mixing the PBS-PFP polymer with the perylene bisdiimide dye Lumogen F Red 305 (LR305) in solution. The model system was designed to contain an amount of LR305 equivalent to 10 times as much as that contained in PBS-PFP-PDI and was incorporated into a di-ureasil matrix (DU-PBS-LR305) at the same concentration used for DU-CPE-08. We note that this sample is not intended to represent a direct comparison between DU-CPE-08 and DU-PBS-LR305, but rather to provide a simulation of the potential of this system if the number of on-chain PDI units in the CPE were increased. A reference sample containing only the same amount of LR305 was also prepared (DU-LR305). As expected, incorporation of a higher ratio of the PDI component leads to a significant increase in the optical efficiency of the LSC (from 2.4 ± 0.4% to 6.2 ± 0.3% with an absorbing background and to 13.1 ± 0.5% with a scattering background) (Table 3).
| Sample name | Absorbing background | Scattering background | ||
|---|---|---|---|---|
| Single edge output (mW) | Total optical efficiency (%) | Single edge output (mW) | Total optical efficiency (%) | |
| DU-CPE-0 | 3.5 | 1.6 ± 0.1 | 14.6 | 6.6 ± 0.9 |
| TU-CPE-0 | 2.9 | 1.3 ± 0.3 | 12.3 | 5.6 ± 0.6 |
| DU-CPE-08 | 5.3 | 2.4 ± 0.4 | 12.5 | 5.6 ± 0.5 |
| TU-CPE-08 | 3.9 | 1.8 ± 0.1 | 12.2 | 5.5 ± 0.7 |
| DU-PBS-LR305 | 13.7 | 6.2 ± 0.3 | 29.1 | 13.1 ± 0.5 |
| DU-LR305 | 13.5 | 6.1 ± 0.6 | 36.9 | 16.7 ± 0.6 |
4. Conclusions
PBS-PFP-PDI–ureasil composites have been prepared and evaluated as luminescent solar concentrators. Steady-state photoluminescence studies revealed that incorporation of PBS-PFP-PDI within the ureasil host enables emission from the PDI chromophore to be isolated, which is not observed in solution, and is attributed to the elimination of a non-radiative decay channel within the solid-state environment. This effect is more evident in the TU-CPE-x series, which exhibit a more open and distorted internal framework which facilitates more effective dispersion of the polymer chains, and a more rigid macroscopic structure which hinders vibrational relaxation. Although, the emission primarily originates from the PBS-PFP species (∼400–500 nm) in both the DU-CPE-x and TU-CPE-x series, the excitation energy dependence and red- and blue-edge band broadening are indicative of the ureasil contribution. This is supported by the emission decay dynamics, which demonstrate that above a minimum concentration, there is intimate mixing of the two components, leading to efficient electronic coupling and energy transfer from both the siliceous and urea domains of the ureasil host to the PBS-PFP chromophore. For both series, the emission primarily originates from a lifetime component of ∼500–600 ps, which has previously been attributed to radiative trap sites associated with the CPE.34 This lifetime component is, on average, slightly longer for the DU-CPE-x series, which is consistent with its higher photoluminescence quantum yield. However, both DU-CPE-x and TU-CPE-x exhibit excellent solid-state ΦPL, which approach values (50–60%) close to those obtained for PBS-PFP-PDI in a good solvent. We note that while these values are high for CPEs in the solid-state, the ideal luminophore for a LSC should exhibit a ΦPL closer to unity,8 and our future work will be focussed at addressing this challenge.Our initial goal with this study was to determine if the photoactive ureasil could be utilised as both a waveguide and light-harvesting/energy donor component to fabricate efficient LSCs. Unfortunately, as PBS-PFP essentially functions as a radiative trap site and further energy transfer to the PDI chromophore is inhibited, this prevents the anticipated benefits of the energy transfer cascade and the obtained optical efficiencies of the resultant LSCs are modest. However, the degree of on-chain incorporation of the PDI chromophore in the present system is low (∼1–2%) and one could envisage that by increasing the effective content of PDI within the polymer, the energy transfer efficiency would improve. This scenario was simulated by preparing analogous LSCs, which contained the same effective PBS-PFP dopant wt%, blended with LR305 at ten times the on-chain concentration of the PDI unit. This led to a three-fold increase in the optical efficiency of the LSC, suggesting that this may be a viable approach to facilitate continuation of the energy transfer cascade.
The results suggest that ureasil-based organic–inorganic hybrids still have much to offer as waveguides for LSCs in diverse constructs. Their ease of processability using solution-phase chemistry offers huge potential for the fabrication of more unusual device architectures. Moreover, the ureasil framework effectively isolates π-conjugated luminophores, inhibiting aggregation induced quenching, thus ensuring high photoluminescence quantum yields in the solid-state. Ureasils are therefore valuable alternatives to the traditional base of poly(methyl methacrylate)-type waveguides in which these challenges are frequently encountered. However, it is clear that a step-change improvement in LSC performance will require consideration of the luminophore and waveguide components in tandem, and not as discrete entities. Advances in the design of new optical hybrid materials will clearly play a significant role in bringing LSC technology to the commercial market.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science Foundation Ireland under Grant No. 12/IP/1608 and 14/TIDA/2343. The authors acknowledge financial support from the European Commission under the Seventh Framework Programme by means of the grant agreement for the Integrated Infrastructure Initiative N. 262348 European Soft Matter Infrastructure (ESMI). HDB and ATM are grateful for funding from “The Coimbra Chemistry Centre” which is supported by the Fundação para a Ciência e a Tecnologia (FCT), Portuguese Agency for Scientific Research, through the programmes UID/QUI/UI0313/2013 and COMPETE.References
- B. S. Richards, Sol. Energy Mater. Sol. Cells, 2006, 90, 2329–2337 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173–201 RSC.
- B. McKenna and R. C. Evans, Adv. Mater., 2017, 29, 1606491 CrossRef PubMed.
- P. J. Jadhav, P. R. Brown, N. Thompson, B. Wunsch, A. Mohanty, S. R. Yost, E. Hontz, T. Van Voorhis, M. G. Bawendi, V. Bulović and M. A. Baldo, Adv. Mater., 2012, 24, 6169–6174 CrossRef CAS PubMed.
- B. Ehrler, B. J. Walker, M. L. Böhm, M. W. B. Wilson, Y. Vaynzof, R. H. Friend and N. C. Greenham, Nat. Commun., 2012, 3, 1019 CrossRef PubMed.
- J. C. Goldschmidt and S. Fischer, Adv. Opt. Mater., 2015, 3, 510–535 CrossRef CAS.
- M. G. Debije and P. P. C. Verbunt, Adv. Energy Mater., 2012, 2, 12–35 CrossRef CAS.
- J. L. Banal, B. Zhang, D. J. Jones, K. P. Ghiggino and W. W. H. Wong, Acc. Chem. Res., 2017, 50, 49–57 CrossRef CAS PubMed.
- W. G. J. H. M. van Sark, Renewable Energy, 2013, 49, 207–210 CrossRef CAS.
- F. M. Vossen, M. P. J. Aarts and M. G. Debije, Energy Build., 2016, 113, 123–132 CrossRef.
- H. Delavari Amrei, R. Ranjbar, S. Rastegar, B. Nasernejad and A. Nejadebrahim, J. Appl. Phycol., 2015, 27, 67–74 CrossRef CAS.
- B. C. Rowan, L. R. Wilson and B. S. Richards, IEEE J. Sel. Top. Quantum Electron., 2008, 14, 1312–1322 CrossRef CAS.
- F. Meinardi, A. Colombo, K. A. Velizhanin, R. Simonutti, M. Lorenzon, L. Beverina, R. Viswanatha, V. I. Klimov and S. Brovelli, Nat. Photonics, 2014, 8, 392–399 CrossRef CAS.
- L. R. Bradshaw, K. E. Knowles, S. McDowall and D. R. Gamelin, Nano Lett., 2015, 15, 1315–1323 CrossRef CAS PubMed.
- Y. Zhou, D. Benetti, Z. Fan, H. Zhao, D. Ma, A. O. Govorov, A. Vomiero and F. Rosei, Adv. Energy Mater., 2016, 6, 1501913 CrossRef.
- V. Sholin, J. D. Olson and S. A. Carter, J. Appl. Phys., 2007, 101, 123114 CrossRef.
- N. J. L. K. Davis, R. W. MacQueen, S. T. E. Jones, C. Orofino-Pena, D. Cortizo-Lacalle, R. G. D. Taylor, D. Credgington, P. J. Skabara and N. C. Greenham, J. Mater. Chem. C, 2017, 5, 1952–1962 RSC.
- O. Altan Bozdemir, S. Erbas-Cakmak, O. O. Ekiz, A. Dana and E. U. Akkaya, Angew. Chem., Int. Ed., 2011, 50, 10907–10912 CrossRef CAS PubMed.
- J. t. Schiphorst, A. M. Kendhale, M. G. Debije, C. Menelaou, L. M. Herz and A. P. H. J. Schenning, Chem. Mater., 2014, 26, 3876–3878 CrossRef.
- G. D. Gutierrez, I. Coropceanu, M. G. Bawendi and T. M. Swager, Adv. Mater., 2016, 28, 497–501 CrossRef CAS PubMed.
- J. L. Banal, K. P. Ghiggino and W. W. H. Wong, Phys. Chem. Chem. Phys., 2014, 16, 25358–25363 RSC.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
- L. R. Wilson, B. C. Rowan, N. Robertson, O. Moudam, A. C. Jones and B. S. Richards, Appl. Opt., 2010, 49, 1651–1661 CrossRef PubMed.
- C. L. Mulder, L. Theogarajan, M. Currie, J. K. Mapel, M. A. Baldo, M. Vaughn, P. Willard, B. D. Bruce, M. W. Moss, C. E. McLain and J. P. Morseman, Adv. Mater., 2009, 21, 3181–3185 CrossRef CAS.
- M. Zettl, O. Mayer, E. Klampaftis and B. S. Richards, Energy Technol., 2017, 5, 1037–1044 CrossRef.
- M. J. Kastelijn, C. W. M. Bastiaansen and M. G. Debije, Opt. Mater., 2009, 31, 1720–1722 CrossRef CAS.
- V. Fattori, M. Melucci, L. Ferrante, M. Zambianchi, I. Manet, W. Oberhauser, G. Giambastiani, M. Frediani, G. Giachi and N. Camaioni, Energy Environ. Sci., 2011, 4, 2849–2853 CAS.
- M. Melucci, M. Durso, L. Favaretto, M. L. Capobianco, V. Benfenati, A. Sagnella, G. Ruani, M. Muccini, R. Zamboni, V. Fattori and N. Camaioni, RSC Adv., 2012, 2, 8610–8613 RSC.
- M. M. Nolasco, P. M. Vaz, V. T. Freitas, P. P. Lima, P. S. André, R. A. S. Ferreira, P. D. Vaz, P. Ribeiro-Claro and L. D. Carlos, J. Mater. Chem. A, 2013, 1, 7339–7350 CAS.
- S. F. H. Correia, P. P. Lima, E. Pecoraro, S. J. L. Ribeiro, P. S. André, R. A. S. Ferreira and L. D. Carlos, Prog. Photovoltaics Res. Appl., 2016, 24, 1178–1193 CrossRef CAS.
- A. Kaniyoor, B. McKenna, S. Comby and R. C. Evans, Adv. Opt. Mater., 2016, 4, 444–456 CrossRef CAS.
- R. Rondão, A. R. Frias, S. F. H. Correia, L. Fu, V. de Zea Bermudez, P. S. André, R. A. S. Ferreira and L. D. Carlos, ACS Appl. Mater. Interfaces, 2017, 9, 12540–12546 Search PubMed.
- N. Willis-Fox, A.-T. Marques, J. Arlt, U. Scherf, L. D. Carlos, H. D. Burrows and R. C. Evans, Chem. Sci., 2015, 6, 7227–7237 RSC.
- N. Willis-Fox, M. Kraft, J. Arlt, U. Scherf and R. C. Evans, Adv. Funct. Mater., 2016, 26, 532–542 CrossRef CAS.
- I. Meazzini, N. Willis-Fox, C. Blayo, J. Arlt, S. Clement and R. C. Evans, J. Mater. Chem. C, 2016, 4, 4049–4059 RSC.
- I. Meazzini, J. M. Behrendt, M. L. Turner and R. C. Evans, Macromolecules, 2017, 50, 4235–4243 CrossRef CAS.
- A. T. Marques, H. D. Burrows, J. S. Seixas de Melo, A. J. M. Valente, L. L. G. Justino, U. Scherf, E. Fron, S. Rocha, J. Hofkens, E. W. Snedden and A. P. Monkman, J. Phys. Chem. B, 2012, 116, 7548–7559 CrossRef CAS PubMed.
- H. D. Burrows, V. M. M. Lobo, J. Pina, M. L. Ramos, J. Seixas de Melo, A. J. M. Valente, M. J. Tapia, S. Pradhan and U. Scherf, Macromolecules, 2004, 37, 7425–7427 CrossRef CAS.
- P. P. Nefedov, M. A. Lazareva, B. G. Belenkii, S. Y. Frenkel and M. M. Koton, J. Chromatogr. A, 1979, 170, 11–24 CrossRef CAS.
- V. de Zea Bermudez, L. D. Carlos and L. Alcácer, Chem. Mater., 1999, 11, 569–580 CrossRef.
- H. D. Burrows, M. J. Tapia, S. M. Fonseca, S. Pradhan, U. Scherf, C. L. Silva, A. A. C. C. Pais, A. J. M. Valente, K. Schillén, V. Alfredsson, A. M. Carnerup, M. Tomšič and A. Jamnik, Langmuir, 2009, 25, 5545–5556 CrossRef CAS PubMed.
- L. B. Å. Johansson and H. Langhals, Spectrochim. Acta, Part A, 1991, 47, 857–861 CrossRef.
- H. Quante, Y. Geerts and K. Müllen, Chem. Mater., 1997, 9, 495–500 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- L. D. Carlos, V. de Zea Bermudez, R. A. Sá Ferreira, L. Marques and M. Assunção, Chem. Mater., 1999, 11, 581–588 CrossRef CAS.
- L. D. Carlos, R. A. Sá Ferreira, R. N. Pereira, M. Assunção and V. de Zea Bermudez, J. Phys. Chem. B, 2004, 108, 14924–14932 CrossRef CAS.
- M. C. Gonçalves, V. de Zea Bermudez, R. A. Sá Ferreira, L. D. Carlos, D. Ostrovskii and J. Rocha, Chem. Mater., 2004, 16, 2530–2543 CrossRef.
- V. T. Freitas, P. P. Lima, V. de Zea Bermudez, R. A. S. Ferreira and L. D. Carlos, Eur. J. Inorg. Chem., 2012, 5390–5395 CrossRef CAS.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- T.-S. Ahn, R. O. Al-Kaysi, A. M. Müller, K. M. Wentz and C. J. Bardeen, Rev. Sci. Instrum., 2007, 78, 086105 CrossRef PubMed.
- R. Yang, A. Garcia, D. Korystov, A. Mikhailovsky, G. C. Bazan and T.-Q. Nguyen, J. Am. Chem. Soc., 2006, 128, 16532–16539 CrossRef CAS PubMed.
- T.-Q. Nguyen and B. J. Schwartz, J. Phys. Chem. B, 2002, 116, 8198–8208 CrossRef CAS.
- J. Stampfl, S. Tasch, G. Leising and U. Scherf, Synth. Met., 1995, 71, 2125–2128 CrossRef CAS.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CAS.
- L. Fu, R. A. Sá Ferreira, M. Fernandes, S. C. Nunes, V. de Zea Bermudez, G. Hungerford, J. Rocha and L. D. Carlos, Opt. Mater., 2008, 30, 1058–1064 CrossRef CAS.
- F. B. Dias, A. L. Maçanita, J. S. D. Melo, H. D. Burrows, R. Güntner, U. Scherf and A. P. Monkman, J. Phys. Chem. B, 2003, 118, 7119–7126 CrossRef CAS.
- E. Fron, A. Deres, S. Rocha, G. Zhou, K. Müllen, F. C. De Schryver, M. Sliwa, H. Uji-i, J. Hofkens and T. Vosch, J. Phys. Chem. B, 2010, 114, 1277–1286 CrossRef CAS PubMed.
- H. A. A. Attar and A. P. Monkman, Adv. Funct. Mater., 2008, 18, 2498–2509 CrossRef.
- M. Monteserín, H. D. Burrows, A. J. M. Valente, V. M. M. Lobo, R. Mallavia, M. J. Tapia, I. X. García-Zubiri, R. E. Di Paolo and A. L. Maçanita, J. Phys. Chem. B, 2007, 111, 13560–13569 CrossRef PubMed.
- S. Tsoi, D. J. Broer, C. W. M. Bastiaansen and M. G. Debije, Adv. Energy Mater., 2013, 3, 337–341 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, supporting photoluminescence and excitation spectra, FTIR fitting data, ps-TCSPC decay curves and fitting data and optical power spectrum of the solar simulator. See DOI: 10.1039/c7qm00264e |
| This journal is © the Partner Organisations 2017 |