
Autologous growth of nickel oxyhydroxides with in situ electrochemical iron doping for efficient oxygen evolution reactions†
Majid
Asnavandi
 and
Chuan
Zhao
*
and
Chuan
Zhao
*
School of Chemistry, The University of New South Wales, High St., Sydney, NSW 2052, Australia. E-mail: chuan.zhao@unsw.edu.au
First published on 6th September 2017
Abstract
Nanostructured Ni and Fe hydroxides and oxides have drawn significant attention for the oxygen evolution reaction (OER) because of their high catalytic efficiency and low cost. Herein, a facile cyclic voltammetry method is reported for the autologous growth of a highly integrated and efficient NiFe–OOH catalyst for the OER by electrochemical roughening of Ni foam substrates in 6 M KOH using an iron rod counter electrode. The electrochemical roughening created a nanostructured Ni surface with low interfacial electrical resistance and 25 times higher surface area, accompanied by in situ doping of Fe. Electrochemical characterization revealed that the formed NiFe–OOH catalysts exhibit an extremely low OER onset overpotential of 190 mV and a Tafel slope merely of 48.1 mV dec−1 in 1 M KOH. Furthermore, a NaBH4 reductive treatment was applied to create more surface defects on NiFe–OOH leading to further enhanced catalyst conductivity and a further decrease of the Tafel slope to 34.7 mV dec−1. The electrode demonstrated prolonged electrochemical and mechanical stability at high current densities in industrial 30 wt% KOH solutions.
Introduction
Hydrogen is a great energy carrier with no dependence on fossil fuels and zero carbon footprint1 and can be stored or used in chemical industrial processes such as petroleum refining, Haber–Bosch generation of ammonia and Fischer–Tropsch synthesis of hydrocarbons, or used as a fuel for fuel cells.1,2 Water electrolysis is a simple and effective industrial method for production of hydrogen from water. Nevertheless, development of more active, efficient, and low-cost electrode materials is essential for widespread application of these technologies. Significant recent research efforts have been devoted to developing low cost, non-precious metal (e.g. Ni, Co, Fe) based catalysts for catalysing the two half reactions of water splitting, such as NiCr, Co3O4, and NiSe for the oxygen evolution reaction (OER) and NiMo, CoNiFe, and CoW for the hydrogen evolution reaction (HER).3,4 As the kinetics of the OER are much slower than those of the HER, the design of efficient, low cost and robust OER electrodes is generally more challenging.NiFe hydroxides have been regarded as one of the most promising OER catalysts, which even outperform the benchmark Ir-based OER catalysts, due to strong synergistic effects between Ni and Fe. Studies have shown that Fe increases the catalyst conductivity5 and diminishes the charge recombination process of Ni4+ under OER conditions and enhances the faradic efficiency to more than 90%.6 Fe also is responsible for the high activity of Ni(OH)2 and NiOOH.7 Fe has been reported to be the catalytically active site in NiFe layered double hydroxide (LDH) materials, as well as active oxygen sites in NiFe–OOH.6 However, as is the case for many transition metal-based oxides and hydroxides, the NiFe hydroxide-based OER electrodes generally suffer from low electrical conductivity. In general, the low conductivity of OER electrodes stems from two sources: (i) the intrinsically low conductivity of the transition metal oxides and hydroxides (either catalysts or substrates) and (ii) the interfacial resistance when the catalysts are deposited or coated onto a substrate electrode.
Currently, most of the metal-based OER catalysts reported in the literature are electrodeposited or casted as foreign catalysts onto substrate surfaces for electrocatalysis. The electron transfer from the catalysts to the substrate current collector is inevitably limited. Furthermore, adhesion of foreign catalysts onto the substrate surface is usually not sufficiently strong and the use of a chemical binder, such as Nafion or hydrophobic polytetrafluoroethylene (PTFE) is often required. The use of binders can decrease the catalytic efficiency of the electrode by reducing the electrical conductivity and decreasing the active surface areas during operation,8 in addition to the exorbitant cost of Nafion. Furthermore, the catalyst/binder layer may peel off from the electrode surface at high current densities at which vigorous gas evolution occurs.9,10 Charge transfer between electrodeposited catalysts and electrode substrates is also limited. A decent strategy is the autologous growth of active species from the substrate to eliminate the catalyst–substrate interface resistance. Using an integrated electrode is a promising approach to address the interfacial resistance between catalysts and electrodes.11,12
In this work, we introduce a facile strategy for autologous growth of NiFe–OOH catalysts from a 3D nickel foam (NF) substrate by electrochemical roughening using cyclic voltammetry. Electrochemical roughening enables Ni–OOH nanostructures to grow directly from the NF substrate with an optimal catalyst–substrate contact. We further apply an iron rod counter electrode in the electrochemical cell, which provides a source of Fe for in situ doping into the roughened Ni foam working electrodes and inducing strong synergistic catalytic activity for the OER. The as-prepared electrode is highly integrated and robust with minimal interfacial electrical resistance between NiFe–OOH and the NF substrate. Furthermore, we introduce a reductive treatment process to create oxygen vacancies in the catalyst structure and enhance its conductivity (Fig. 1a). The synthesized electrodes exhibit extraordinary catalytic activity for the OER at high current densities and robustness in prolonged bulk water electrolysis in industrial 30 wt% KOH solutions.
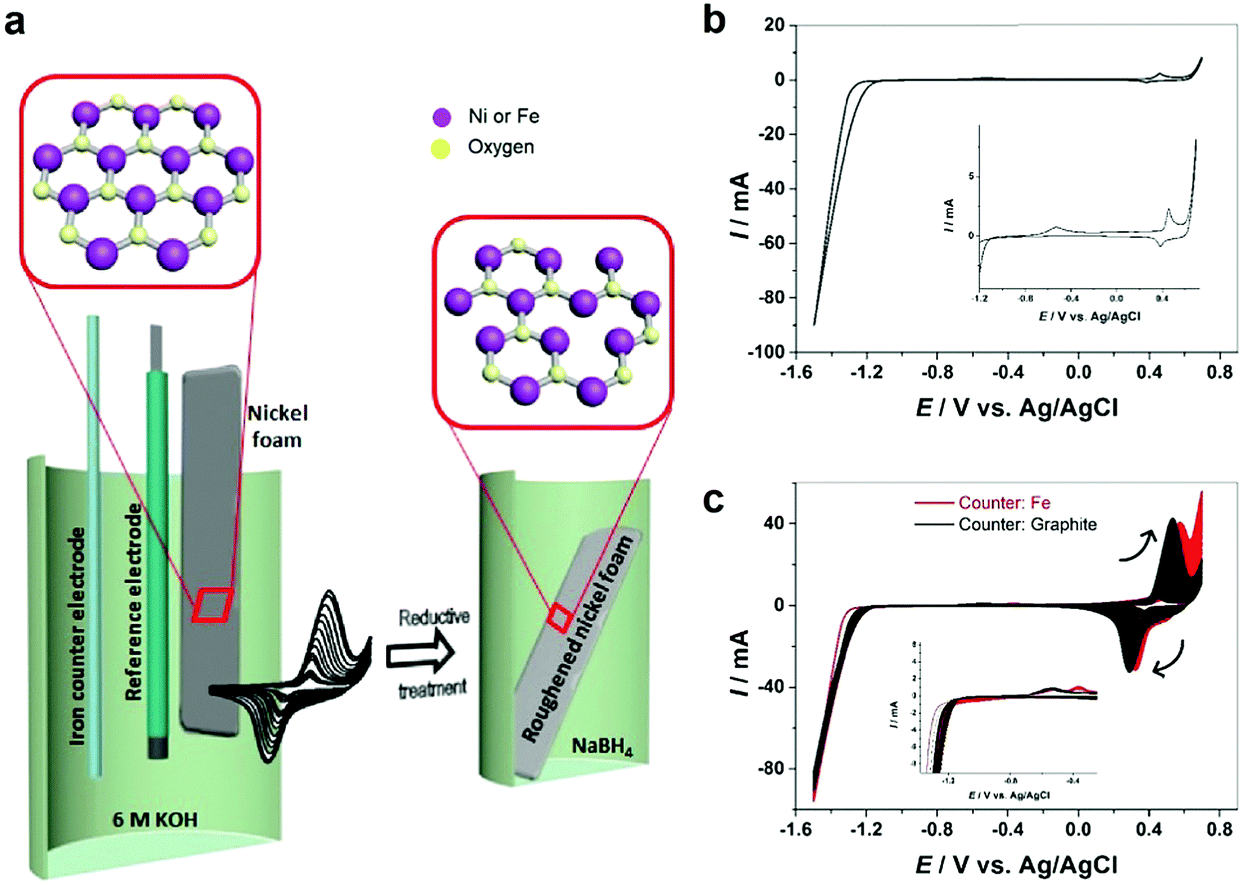 | ||
| Fig. 1 (a) Schematic of preparation steps of oxygen vacancy-rich NiFe–OOH on a nickel foam substrate, (b) voltammograms of Ni in 6 M KOH at 25 °C and scan rate of 200 mV s−1 with a graphite counter electrode, (c) next cycles with graphite and iron counter electrodes. The insets show more details about the zoomed-in areas of the CVs. | ||
Results and discussion
Electrochemical roughening of Ni foam
The electrochemical behavior of nickel depends on the structure and morphology as well as its production procedure.13 All electrochemical experiments in this work have been carried out on polycrystalline nickel. Commercially available nickel foam (NF) has been roughened by oxidation–reduction cycles. Fig. 1b demonstrates the cyclic voltammetry (CV) of nickel in 6 M KOH solution. Two anodic peaks observed at the potential of about −0.5 V and 0.4 V vs. Ag/AgCl are attributed to nickel oxidation and nickel hydroxide formation (eqn (1)) and subsequent formation of Ni(III) or Ni(IV) species such as β-Ni(OH)2 and NiOOH.14,15| Ni + 2OH− ↔ α-Ni(OH)2 + 2e−. | (1) |
On the cathodic scan, only the reduction peak for Ni(III) or Ni(IV) species is observed. The reduction of α-Ni(OH)2 to metallic Ni occurs concomitantly with hydrogen evolution,16 which is observed from ca. −1.2 V. Therefore, to ensure complete reduction of nickel hydroxide formed on anodic scans, it is important to scan to very negative potential regions where hydrogen evolution occurs. Otherwise, the Ni foam surface is covered with nickel hydroxide and gradually deactivated. Fig. S1 (ESI†) shows that when the cathodic scan is limited to −0.8 V vs. Ag/AgCl to avoid hydrogen evolution, the anodic peak at −0.5 V does not appear in the anodic scan and the peaks for nickel hydroxide formation and reduction decrease gradually with each cycle.
Electrochemical roughening of the NF is achieved in 6 M KOH solution by CV scanning between −1.5 and 0.7 V for 200 cycles using a graphite rod counter electrode. As it can be seen (Fig. 1c), by increasing the cycle numbers the peak current at ca. +0.5 V, as well as the corresponding cathodic peak increase gradually indicating that the NF surface area is increasing and the nickel foam surface has been roughened. Fig. S2 (ESI†) illustrates the color change of NF from pale grey to black after roughening.
SEM images of NF before and after the roughening process shown in Fig. 2a and b show that after the roughening process the previously smooth NF surface turns into a rough surface with high surface area. The electrochemical surface area (ECSA) measurement determined from the double layer capacitance (see the Experimental section and Fig. S3, ESI† for more information) suggests that the ECSA of the nickel foam is enhanced by more than 25 times after the roughening process (Table 1).
 | ||
| Fig. 2 SEM images of (a) pure NF, (b) NF after 200 cycles of CV with a graphite counter electrode, and (c) NF after 200 cycles of CV with an iron counter electrode; (d) image (c) at higher magnification; (e) EDS mapping of the NF electrochemically roughened by a Fe counter electrode. | ||
| OER electrode | Onset potentials (V vs. RHE) | Tafel slope (mV dec−1) | Roughness factor |
|---|---|---|---|
| NF | 1.55 | 88.0 | 21 |
| Roughened NF (graphite counter electrode) | 1.46 | 72.0 | 540 |
| Roughened NF (iron counter electrode) | 1.42 | 48.1 | 540 |
| Reduced roughened NF (iron counter electrode) | 1.42 | 34.7 | 540 |
In situ Fe doping of roughened Ni foam
It has been known that the presence of Fe in Ni-based catalysts can induce a very strong synergistic effect for the OER, by increasing the catalyst conductivity and modifying the electronic structure.5 To introduce Fe into the process for autologous growth of synergistic NiFe catalysts, we further employed an iron rod counter electrode for the electrochemical roughening of Ni foam and in situ doping of Fe into NF concurrently. During the cyclic voltammetry (shown in Fig. 1c), the iron dissolves from the counter electrode and diffuses to the NF working electrode and re-deposits onto the freshly formed nanostructured Ni foam electrode surface. Energy-dispersive X-ray spectroscopy (Fig. 2 and Fig. S4, ESI†) shows the presence of Fe in the nickel foam surface when the iron rod is used as the counter electrode in the roughening process. Fig. 2 also suggests that the introduction of the Fe component has no significant effect on the macroscopic appearance and microscopic morphology of the roughened NF. X-ray photoelectron spectroscopy (XPS) further revealed a small iron content of 1.96% atm. on the roughened NF surface. Fig. 3a displays the Fe 3p and Ni 3p spectra for roughened NFs, obtained with graphite and iron rod counter electrodes respectively. The Fe peak17 is clearly seen for the roughened NF surface using the iron rod counter electrode, but not the graphite counter electrode. There are also two major peaks for the Fe 2p photoelectron profile (Fig. S5a, ESI†) at 724.78 and 711.58 eV revealing that the iron atoms are present as Fe2+ and Fe3+ cations.18 Besides, the peaks at 68.2 and 71.3 eV in Fig. 3a are correlated to Ni2+ 3p1/2 and Ni3+ 3p3/2,19,20 respectively, while the Ni 2p1/2 peak at 855.8 eV (Fig. S5b, ESI†) is attributed to the Ni(OH)221 phase. This indicates the coexistence of Ni2+ and Ni3+ states on the roughened NF surface and suggests the formation of nickel and nickel–iron–oxyhydroxides.22 The X-ray diffraction (XRD) pattern of the roughened NF (Fig. 3b) shows that Ni and NiFe–oxyhydroxides formed on the NF surfaces are amorphous. This was further confirmed by high-resolution transmission electron microscopy (HRTEM) where no lattice fringes can be observed (Fig. 3c and Fig. S6, ESI†).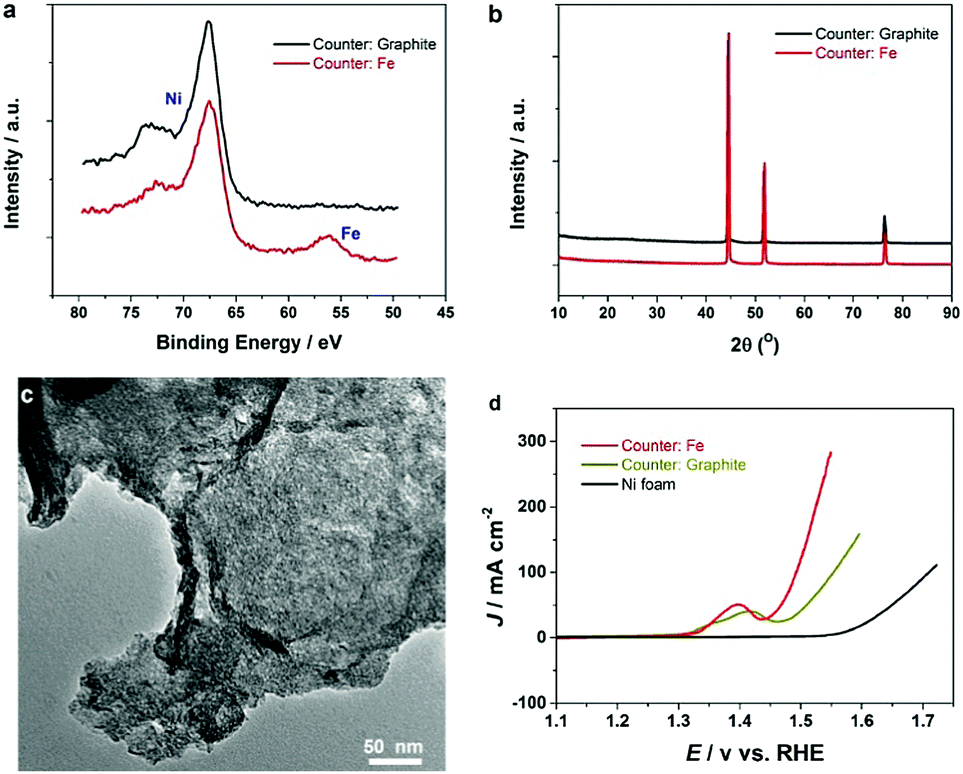 | ||
| Fig. 3 (a) High-resolution Ni 3p and Fe 3p XPS spectrum and (b) XRD pattern of NFs roughened with different counter electrodes, (c) TEM images of the nanostructured Ni–Fe oxyhydroxide formed on the NF roughened by an iron counter electrode, and (d) polarization curves of the different electrodes at a scan rate of 5 mV s−1 and 90% iR-compensation in 1 M KOH. | ||
Further studies of the roughening CV with a Fe counter electrode reveal an extra anodic peak at −0.38 V (vs. Ag/AgCl) and a correlating shoulder at the cathodic scan before the hydrogen evolution region (Fig. 1c). These peaks belong to Fe(OH)3− oxidation and Fe(OH)4− reduction which were also observed by Allanore et al. during Fe electrodeposition from 18 M NaOH solution.23 Furthermore, when Fe is used as the counter electrode, the Ni oxidation peak at 0.5 V slightly shifts to a more positive potential according to Fig. 1c, suggesting changes in the electronic structure and greater conductivity of the NF. This, in turn, reduces the average oxidation state and produces more active Ni sites.17 The Fe doping leads to significant enhancement of the OER performance. It is shown in Fig. 3d that the OER onset potential of the NF roughened by the Fe counter electrode is 1.46 V (vs. RHE) which is around 130 mV lower than for pure NF, and 40 mV lower than for NF roughened using a graphite counter electrode. It is noted that this onset overpotential (190 mV) for the OER is 40 mV lower than previously reported NiFe–LDH/NF prepared by the hydrothermal method24 and 20 mV lower than the electrodeposited NiFe/NF,25 attributed to high surface area and low interfacial catalyst–substrate resistance as a result of the autologous growth strategy.
Creating surface oxygen vacancies for further enhanced OER
To further improve the OER activity, we introduce a chemical reductive treatment process to create oxygen vacancies (OVs) on the NiFe–OOH catalysts and improve their conductivity. It has been reported that OVs can modify the electrical properties of metal oxides and improve the conductivity.26–28 Moreover, OVs generate low coordinated metals (LCMs) which act as active sites for the OER in metal oxides.29 In this study, the reductive treatment was performed by immersion of the roughened electrode in the NaBH4 solution for 15 minutes at room temperature. It is expected that the treatment changes neither the crystallinity nor morphology of the electrode (Fig. S7, ESI†). However, XPS spectra of Fe 2p, Ni 3p, Fe 3p and Ni 2p in Fig. 4a and b and Fig. S8 (ESI†) revealed small shifts to lower binding energies suggesting the reduction of Ni and Fe oxidation states30 by NaBH4 treatment. In addition, the oxygen status in NiFe–OOH before and after reduction was examined using O 1s spectra. There are three spin–orbit peaks including oxygen–metal bonds, oxygen loss and adsorbed oxygen on and within the surface in the lattice with lower binding energy, medium binding energy and higher binding energy, respectively.31,32 The ratio of peak area for medium to lower binding energy for the reduced electrode is larger than that of the electrode before the treatment (Fig. 4c–e), which indicates that the treatment led to the generation of oxygen vacancies.33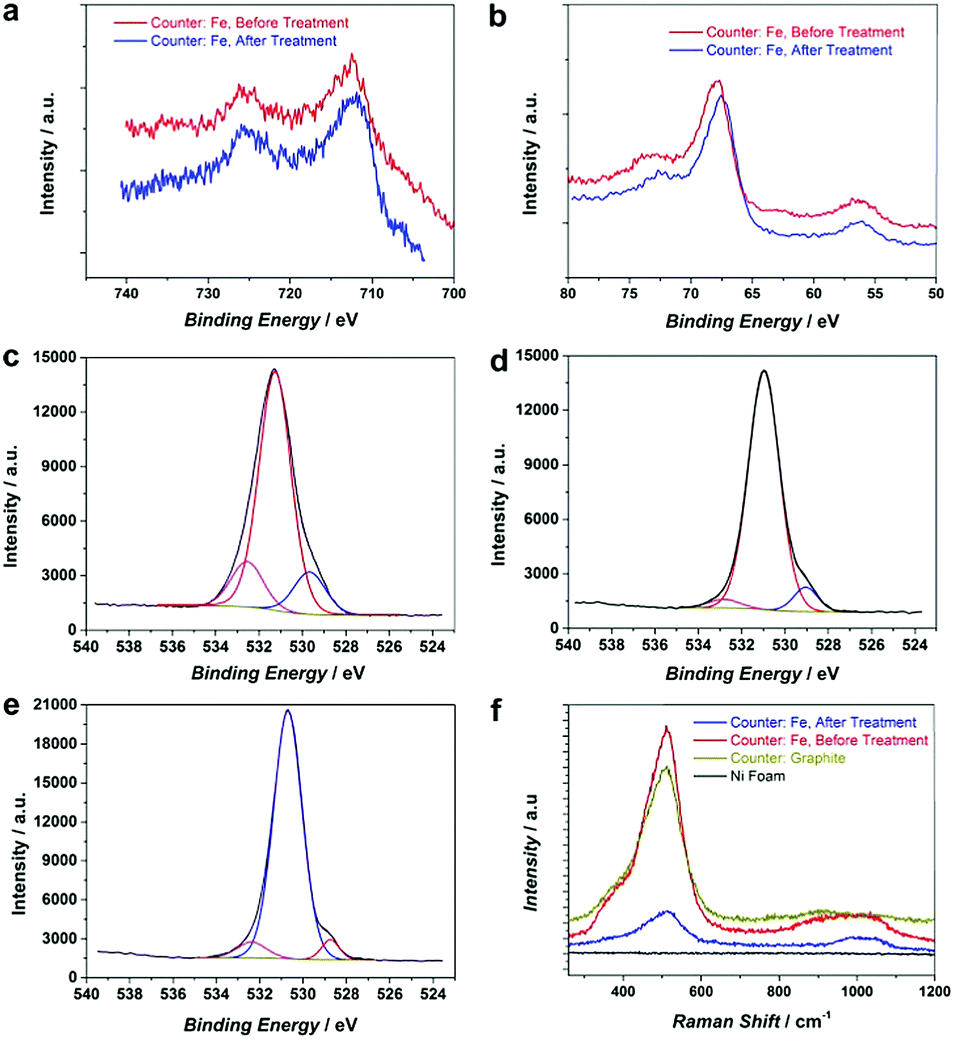 | ||
| Fig. 4 High resolution (a) Fe 2p and (b) Ni 3p and Fe 3p XPS spectrum of NiFe–OOH, before and after NaBH4 treatment, (c) high-resolution O 1s XPS spectrum of Ni–OOH, (d) high-resolution O 1s XPS spectrum of NiFe–OOH before the treatment, (e) high resolution O 1s XPS spectrum of NiFe–OOH after the treatment, and (f) Raman spectra of NF, Ni–OOH and NiFe–OOH, before and after the reductive treatment. | ||
This finding has been further confirmed by electron paramagnetic resonance (EPR) of electrodes. As Fig. S9 (ESI†) illustrates, the number of defects and oxygen vacancies in the reduced roughened NF is much higher than that in the as-prepared roughened one. The defective structure of the reduced NiFe–OOH is also examined by Raman spectroscopy in Fig. 4f. The observed bands centred at ca. 530 and 1000 cm−1 exhibit the stretching vibration of Ni–O and Ni–OH34,35 attributed to the layered double hydroxide (LDH) structures.34,36 The Raman scattering spectra showed broader and less intense peaks detected after the reductive treatment, suggesting the presence of defects in the material structure.37
Electrochemical impedance spectroscopy (EIS) results in Fig. S10 (ESI†) confirm that the fabricated NiFe–OOH electrode has higher conductivity than the electrodeposited NiFe–OOH one, mainly because of the low interfacial resistance of catalyst–substrate. EIS was also applied to gain insight into the effect of the NaBH4 treatment on the electrode conductivity of NiFe–OOH. Fig. 5a reveals that the presence of iron leads to a decrease in the interfacial impedance, which drops even further upon reductive treatment suggesting the increase of electrical conductivity upon the reductive treatment.
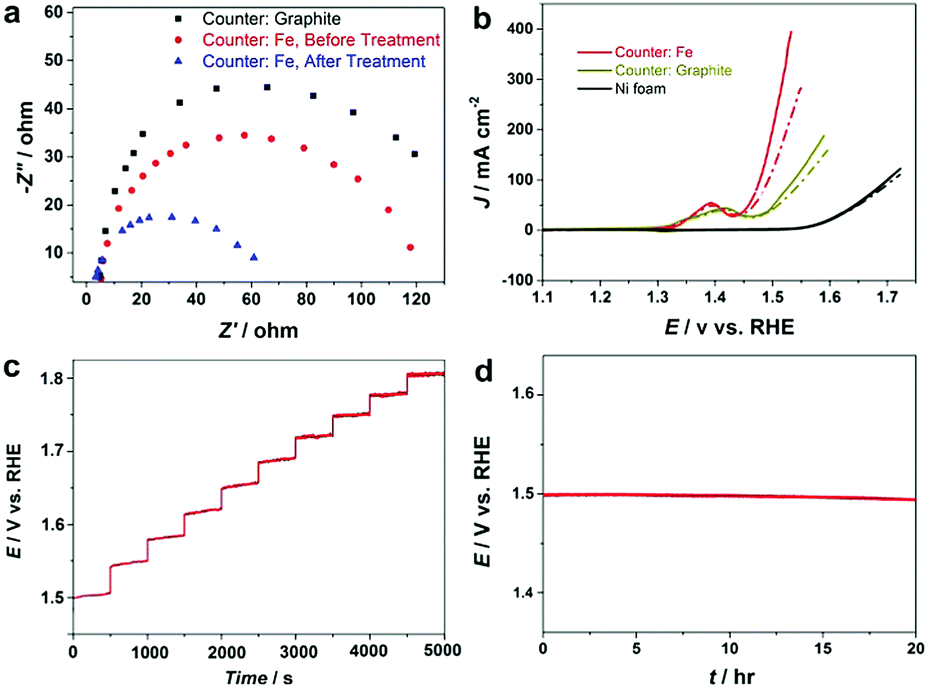 | ||
| Fig. 5 Electrochemical experiments in 1 M KOH at room temperature; (a) EIS response at potentials of 1.55 V vs. RHE of Ni–OOH/NF and NiFe–OOH/NF, before and after the reductive treatment, (b) OER activity of the electrodes before (dash lines) and after (solid lines) the reductive treatment at a scan rate of 5 mV s−1 and 90% iR-compensation, and (c) multi-step current process obtained for the NiFe–OOH/NF electrode after NaBH4 treatment. The current density started at 50 mA cm−2 and finished at 500 mA cm−2. (d) Chronopotentiometry of the NiFe–OOH/NF electrode after the reductive treatment at 50 mA cm−2. | ||
The increase of conductivity of the NiFe–OOH catalyst leads to significant improvement in the OER activity. Fig. 5b and Fig. S11 show the OER curves obtained with and without iR corrections. Significant enhancement in the OER polarisation curves of the NF after NaBH4 treatment is observed with similar onset potential. At a potential of +1.5 V (RHE), the OER current density obtained at the NiFe–OOH catalyst increases from 120 mA cm−2 to 200 mA cm−2 after the NaBH4 treatment. The Tafel slope also decreases from 48.1 to 34.7 mV dec−1, after the reductive treatment indicating enhanced conductivity and fast OER kinetics (Table 1 and Fig. S12, ESI†). Table S1 (ESI†) compares the fabricated electrode with other NiFe based OER electrodes. Furthermore, Fig. 5c displays a multi-step chronopotentiometric curve for the reduced NiFe–OOH electrode in 1 M KOH. In this experiment, the current is increased from 50 to 500 mA cm−2 with 500 s intervals and an increment rate of 50 mA cm−2. At the current density of 50 mA cm−2, the potential reaches 1.5 V vs. RHE and remains constant for the remaining 500 s. Similar behavior is observed for all current densities up to 500 mA cm−2 reflecting excellent mass transport and electrical conductivity, as well as the mechanical robustness of the electrode.
Finally, the long-term stability of the electrode is examined using the chronopotentiometric technique in 1 M KOH at a current density of 50 mA cm−2. According to Fig. 5d, it was found that the electrode is stable in the OER condition for more than 20 hours. We further examine the long-term electrochemical stability of the electrode in extremely corrosive 30 wt% KOH electrolyte that is used in industrial alkaline electrolysers. Fig. S13 (ESI†) exhibits that the electrode is mechanically and electrochemically stable at high current densities. No noticeable changes in the structure, surface morphology and chemical composition are detected (Fig. S14, ESI†) suggesting that the electrode is robust and can be used for prolonged alkaline water electrolysis under industrial conditions.
Experimental
First, nickel foam (Goodfellow, UK, 95% purity and 95% porosity) was sonicated in 5 M HCl for 30 minutes to remove the nickel oxide layer, and then rinsed with Milli-Q water and ethanol several times followed by drying in air. The roughening process has been carried out by cyclic voltammetry for 200 cycles in 6 M KOH solution at a potential window of +0.7 and −1.5 V vs. Ag/AgCl at a scan rate of 0.2 V s−1 using a three-electrode cell arrangement. Graphite or iron rods were utilized as counter electrodes to study the effect of the counter electrode material on the species formed onto the NF surface. After 200 cycles, the electrode was washed with Milli-Q water and immersed in 1 M NaBH4 solution for 15 minutes at room temperature followed by thorough rinsing with water. The NaBH4 reduction mechanism can be explained by providing free electrons in basic and neutral media according to eqn (2):38| BH4− + 8OH− → B(OH)4− + 4H2O + 8e−. | (2) |
For electrochemical experiments, a CH760 Electrochemical Workstation (CH Instrument, Texas, USA) was used under normal bench-top laboratory conditions with a Ag/AgCl (1 M KCl) reference electrode and a Pt wire counter electrode. CVs and LSVs were run at 5 mV s−1 scan rate while for Tafel slope determination this value was fixed at 0.1 mV s−1. The electrochemical impedance spectroscopy (EIS) test was performed by a BAS potentiostat in a frequency range of 100 kHz to 0.01 Hz. All current densities in this paper were calculated by using the geometric surface area of the working electrodes. The ECSA of each electrocatalyst is determined by double layer capacitance (CDL) according to eqn (3):39
| ECSA = CDL/CS, | (3) |
Material characterization has been carried out using an SEM, FEI Nova FESEM JEOL 7001F, to study the morphology. XRD was done on a PANalytical X’Pert instrument and TEM was performed using a Philips CM 200 microscope. Raman was performed by a Renishaw inVia Raman Microscope (514 nm). XPS results were achieved by a Thermo ESCALAB250i X-ray Photoelectron Spectrometer. A Bruker EMX X-Band EPR Spectrometer operating at a frequency of 9650 MHz and room temperature was utilized for EPR experiments. Since nickel has ferromagnetic properties and cannot be used directly in EPR, a copper plate was coated by a Ni layer electrodeposited from a Watts bath. Then the coated Ni was roughened via the aforementioned procedure and eventually reduced in NaBH4 solution.
Conclusions
In conclusion, a highly integrated, amorphous NiFe–OOH OER catalyst containing a high amount of oxygen vacancies with a high active surface area is prepared onto macroporous NF substrates via an autologous growth strategy. Concurrent nickel foam roughening and Fe doping was achieved in one step and in one pot by electrochemical roughening using a Fe counter electrode. The nanostructured electrode can catalyse the OER at an extremely low overpotential in industrial alkaline media with prominent durability under high current densities. It is anticipated that the facile, one-step roughening strategy can be easily adapted for industry-scale production. The Fe counter electrode also can be replaced by other metals such as Co, Cr, and Cu for facile preparation of a range of integrated multi-metallic catalysts for electrochemical energy storage and conversion applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to UNSW Mark Wainwright Analytical Center for providing access to their SEM, TEM, XRD, XPS, EPR and Raman facilities. We also thank Dr Bin Gong for his help regarding XPS experiments. The study was financed by the Australian Research Council (DP160103107).Notes and references
- M. Gong and H. Dai, Nano Res., 2014, 8, 23–39 CrossRef.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- C. Xiao, X. Lu and C. Zhao, Chem. Commun., 2014, 50, 10122–10125 RSC.
- M. Gorlin, P. Chernev, J. F. De Araujo, T. Reier, S. Dresp, B. Paul, R. Krahnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- G. Zhu, Z. He, J. Chen, J. Zhao, X. Feng, Y. Ma, Q. Fan, L. Wang and W. Huang, Nanoscale, 2014, 6, 1079–1085 RSC.
- F. Zhang, G. Chen, M. A. Hickner and B. E. Logan, J. Power Sources, 2012, 218, 100–105 CrossRef CAS.
- J. Ji, L. L. Zhang, H. Ji, Y. Li, X. Zhao, X. Bai and X. Fan, ACS Nano, 2013, 6237–6243 CrossRef CAS PubMed.
- D. Guo, J. Qi, W. Zhang and R. Cao, ChemSusChem, 2017, 10, 394–400 CrossRef CAS PubMed.
- W. Zhang, J. Qi, K. Liu and R. Cao, Adv. Energy Mater., 2016, 6, 1–6 CAS.
- J. Scherer, B. M. Ocko and O. M. Magnussen, Electrochim. Acta, 2003, 48, 1169–1191 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- J. Van Drunen, B. Kinkead, M. C. P. Wang, E. Sourty, B. D. Gates and G. Jerkiewicz, ACS Appl. Mater. Interfaces, 2013, 5, 6712–6722 CAS.
- A. Seghiouer, J. Chevalet, A. Barhoun and F. Lantelme, J. Electroanal. Chem., 1998, 442, 113–123 CrossRef CAS.
- K. L. Nardi, N. Yang, C. F. Dickens, A. L. Strickler and S. F. Bent, Adv. Energy Mater., 2015, 5, 1–10 Search PubMed.
- Ç. Üzüm, T. Shahwan, A. E. Eroǧlu, I. Lieberwirth, T. B. Scott and K. R. Hallam, Chem. Eng. J., 2008, 144, 213–220 CrossRef.
- X. Ning, Z. Wang and Z. Zhang, Sci. Rep., 2015, 5, 8460 CrossRef CAS PubMed.
- Y. Jeon, D.-H. Park, J.-I. Park, S.-H. Yoon, I. Mochida, J.-H. Choy and Y.-G. Shul, Sci. Rep., 2013, 3, 2902 CrossRef PubMed.
- L. M. Moroney, R. S. C. Smart and M. W. Roberts, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 1769 RSC.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- A. Allanore, H. Lavelaine, G. Valentin, J. P. Birat and F. Lapicque, J. Electrochem. Soc., 2007, 154, E187–E193 CrossRef CAS.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- T. Ling, D.-Y. Yan, Y. Jiao, H. Wang, Y. Zheng, X. Zheng, J. Mao, X.-W. Du, Z. Hu, M. Jaroniec and S.-Z. Qiao, Nat. Commun., 2016, 7, 12876–12883 CrossRef CAS PubMed.
- S. A. Ansari, M. M. Khan, S. Kalathil, A. Nisar, J. Lee and M. H. Cho, Nanoscale, 2013, 5, 9238–9246 RSC.
- Y. Wang, T. Zhou, K. Jiang, P. Da, Z. Peng, J. Tang, B. Kong, W.-B. Cai, Z. Yang and G. Zheng, Adv. Energy Mater., 2014, 4, 1400696 CrossRef.
- L. Han, S. Dong and E. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- Q.-H. Wu, A. Thissen, W. Jaegermann and M. Liu, Appl. Surf. Sci., 2004, 236, 473–478 CrossRef CAS.
- A. Tamilselvan, S. Balakumar, M. Sakar, C. Nayek, P. Murugavel and K. Saravana Kumar, Dalton Trans., 2014, 43, 5731–5738 RSC.
- X. Zhang, J. Qin, Y. Xue, P. Yu, B. Zhang, L. Wang and R. Liu, Sci. Rep., 2014, 4, 4596 CrossRef PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- J. Zhang, J. Tu, D. Zhou, H. Tang, L. Li, X. Wang and C. Gu, J. Mater. Chem. C, 2014, 2, 10409–10417 RSC.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- N. Han, F. Zhao and Y. Li, J. Mater. Chem. A, 2015, 3, 16348–16353 CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- P. Dechasit and W. Trakarnpruk, J. Met., Mater. Miner., 2011, 21, 19–27 CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- J.-P. D. Joel Fournier, L. Brosard, J.-Y. TiIquin, R. Côté and H. M. Daniel Guay, J. Electroanal. Chem., 1996, 143, 919–926 Search PubMed.
- C. C. L. Mccrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Asnavandi, B. H. R. Suryanto and C. Zhao, RSC Adv., 2015, 5, 74017–74023 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00367f |
| This journal is © the Partner Organisations 2017 |