
Defined functionality and increased luminescence of nanodiamonds for sensing and diagnostic applications by targeted high temperature reactions and electron beam irradiation†
Ch.
Laube
ab,
Y. M.
Riyad
a,
A.
Lotnyk
a,
F. P.
Lohmann
 a,
C.
Kranert
c,
R.
Hermann
a,
W.
Knolle
a,
Th.
Oeckinghaus
d,
R.
Reuter
d,
A.
Denisenko
d,
A.
Kahnt
e and
B.
Abel
*ab
a,
C.
Kranert
c,
R.
Hermann
a,
W.
Knolle
a,
Th.
Oeckinghaus
d,
R.
Reuter
d,
A.
Denisenko
d,
A.
Kahnt
e and
B.
Abel
*ab
aLeibniz-Institut für Oberflächenmodifizierung (IOM), Permoserstr. 15, D-04303 Leipzig, Germany. E-mail: bernd.abel@iom-leipzig.de
bWilhelm-Ostwald-Institute for Physical and Theoretical Chemistry, University of Leipzig, Permoserstr. 15, 04318 Leipzig, Germany
cUniversität Leipzig, Institut für Experimentelle Physik II, Halbleiterphysik, Linnéstr. 5, 04103 Leipzig, Germany
dInstitute of Physics, Research Center SCoPE and IQST, University of Stuttgart, 70569 Stuttgart, Germany
eDepartment of Chemistry and Pharmacy & Interdisciplinary Center for Molecular Materials, Friedrich-Alexander-Universitat Erlangen-Nürnberg, Egerlandstrasse 3, 91058 Erlangen, Germany
First published on 21st June 2017
Abstract
Nanodiamonds have excellent mechanical and optical properties, high surface areas and tunable functional surfaces. They are also non-toxic, which makes them well suited for biomedical applications. Here we highlight an integrated and scalable surface functionalization approach by a high temperature gas–solid phase reaction protocol monitored via thermogravimetry for very controlled and precise degraphitization, as well as hydrogen, oxygen and nitrogen (–NH2) functionalization in a high temperature reactor. In particular, we discuss the rational and precise control of chemical functionalization through introduction of functional groups and of an increased photoluminescence from additional nitrogen-vacancy defects (NV-centers) produced via controlled electron beam irradiation. We have shown that multiple surface analytical methods such as IR-, Raman, photoelectron spectroscopy, light scattering, and electron microscopies allow for quality control of the surface functionalization. The approach together with controlled electron beam irradiation is well suited to produce large amounts of functionalized bright fluorescent nanodiamond probes, which can easily be further chemically modified. Employing this integrated and scalable approach well defined (multi-) functionalized bright fluorescent nanodiamonds are available in large quantities and with high quality enabling a wide range of potential applications as novel sensors and for bioimaging beyond the lab scale.
Introduction
Nanoscale diamond particles have recently received considerable attention due to various emerging applications of nanodiamonds (NDs) in luminescence imaging, nuclear magnetic resonance imaging (MRI), drug delivery, quantum computing, surface coatings, and sensing.1–7 NDs show low cytotoxicity, unique biocompatibility in biological environments and they display extreme mechanical properties, which make them an ideal material in the area of implants, as well as coatings for micromechanical devices.8–11 Their luminescence properties are unique due to the presence of so called nitrogen-vacancy-centers (NV-centers). For many application fields, e.g. nano-sensors, defined surface termination and functionalization of the diamond nanomaterial, accompanied by superior and good luminescence properties, are prerequisites.12,13 In the present contribution we will highlight improvements in the surface functionalization and fluorescence properties that both limit current applications significantly.Most of the available diamond nanoparticles carry an oxygenated surface.14 However, sp2 carbon is also detectable on nanodiamond surfaces.2,14,15 This surface coverage is responsible for the grey color of nanodiamond powder. The surfaces exhibit different zeta potentials ranging from −50 mV to +50 mV which is also related to their agglomeration behavior.16 Before any defined modification the surface has to be cleaned, i.e., graphite layers (sp2-content) have to be removed efficiently and in a gentle way.15 In addition, the surface may be contaminated by metal impurities as a result of the high temperature and high pressure (HTHP) synthesis.14
In the last decade, different experimental strategies have been proposed to remove such impurities and to control ND surfaces. A common approach is the surface treatment of NDs using strong oxidizing acids making use of the higher oxidation rate of sp2 carbon compared to sp3-carbon.17–19 This harsh approach, although being efficient, has also limitations and drawbacks such as low purity of NDs. Other more or less efficient approaches include catalyst-assisted oxidation and ozone-enriched air oxidation,20,21 however, also with disadvantages such as low ND surface purity. A promising oxidative method is oxidation in an air (oxygen rich) atmosphere at high temperatures.15,22–24 The method as used so far has two major drawbacks. First, air oxidation in a temperature range of 400–500 °C still shows residual graphitic sp2-carbon.15 Second, metal and oxide impurities could not be removed.
Modification of the diamond surfaces starting from the oxidized surface can be done in two ways: the classical wet chemistry route or via dry chemistry by using high temperature gas or plasma techniques. Instead of hydrogen termination/modification strongly reducing agents like LiAlH4 often result in partly reduced hydroxylated diamond surfaces.25 Alternative and more efficient ways have been reported employing high temperature hydrogen gas or plasma. Ida et al. used a hydrogen atmosphere at 900 °C for the formation of C–H bonds on the surface of micrometer sized diamonds.26 Unfortunately, for diamond nanoparticles at this temperature partial graphitization is observed. Jiang et al. showed the formation of C–H bonds after treatment of detonation diamonds with hydrogen at 800 °C for 4 hours.27 Zhu et al. detected hydrogen-termination of NDs with a diameter of 125 nm at 750 °C.28 Girard et al. suggested the microwave plasma chemical vapor deposition (MPCVD) approach as a fast and efficient method for hydrogenation, but their results were not convincing in terms of the amount of non-diamond carbon.29 While a number of studies have been reported for the hydrogen termination of diamonds (with very different results and often with limited success), there have been only very few reports about the surface nitrogen modification – especially the functionalization and termination of the surface of NDs with amine groups. Here, amino groups on the surface of NDs are obtained either via the reaction of gaseous ammonia with chlorinated NDs or by a plasma treatment of the diamonds with ammonia.30,31 As a somewhat simpler alternative to create amine groups at nanodiamond surfaces, that even works for graphite layers near ND-surfaces, the reaction of ND with aminated silanes or different linker groups was suggested.32,33 Stanishevsky et al. employed nitrogen plasma on graphitized NDs, where the starting material already contains nitrogen.34 Even if a suitable protocol can be found this approach requires low pressure, such that upscaling could be technically difficult.
Functionalized diamond nanoparticles are mostly used as luminescent nanoparticle probes. In these applications high photoluminescence yields are desired – often much beyond their natural photoluminescence (PL). Additional nitrogen-vacancy centers are typically produced from single substitutional nitrogen centers by irradiation followed by annealing at temperatures above 700 °C.7 A wide range of high-energy particles are suitable for such irradiation, including electrons, protons, neutrons, ions, and gamma photons.35–37
In this paper we present a fast and reproducible strategy and optimized protocols for the modification of NDs starting from standard commercial HTHP-nanodiamonds (dark grey or black powder).
Results and discussion
General approach and strategy
According to Fig. 1, our strategy is to start with a nanomaterial containing a carbon layer and many unspecific oxygen containing functional groups (termed GND). We then remove the outermost graphite layer and transform the particles into oxygen terminated diamond nanoparticles (termed OND) containing COOH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and OH groups in addition to –O– groups. Then, in a second step we change the gas and temperature and re-functionalize the diamond nanoparticles containing mostly NH2-groups or through hydrogen termination (C–H). In particular, in the first step we remove graphite by a controlled oxidative high temperature air treatment at temperatures close to the degradation temperature (see the reactor design in the ESI† (S9)). This is a critical process, such that the degraphitization process had to be controlled and pre-investigated with thermogravimetric and differential scanning calorimetric methods, and we conducted an extensive parameter study. Our results show that a careful selection of the temperature set point for degraphitization has the biggest impact on the quality of the ND in terms of a low sp2-content. Employing the optimal temperature and gas settings for degraphitization we were able to produce high quality oxygen terminated NDs (white powder) characterized by full removal of non-diamond carbon and pronounced oxygen termination. These nanodiamonds served as a standard material for hydrogen and nitrogen termination by high temperature solid phase–gas reactions under different conditions. The special feature of the approach is that for all modifications, we used a wide range of different analytical methods like Raman spectroscopy, attenuated total reflection IR-spectroscopy, transmission electron microscopy, X-ray photoelectron spectroscopy and pH-value dependent zeta potential measurements to characterize their surface sp2 carbon content, (multi)functionality, density of functional groups, as well as other properties. Our results demonstrate that the approach can be used to successfully clean and specifically modify the diamond surface in a very controlled, reproducible and scalable way. In particular, this efficient method of obtaining a nitrogen (NH2) terminated surface offers a new route towards biofunctionalization of nanodiamonds.
O, and OH groups in addition to –O– groups. Then, in a second step we change the gas and temperature and re-functionalize the diamond nanoparticles containing mostly NH2-groups or through hydrogen termination (C–H). In particular, in the first step we remove graphite by a controlled oxidative high temperature air treatment at temperatures close to the degradation temperature (see the reactor design in the ESI† (S9)). This is a critical process, such that the degraphitization process had to be controlled and pre-investigated with thermogravimetric and differential scanning calorimetric methods, and we conducted an extensive parameter study. Our results show that a careful selection of the temperature set point for degraphitization has the biggest impact on the quality of the ND in terms of a low sp2-content. Employing the optimal temperature and gas settings for degraphitization we were able to produce high quality oxygen terminated NDs (white powder) characterized by full removal of non-diamond carbon and pronounced oxygen termination. These nanodiamonds served as a standard material for hydrogen and nitrogen termination by high temperature solid phase–gas reactions under different conditions. The special feature of the approach is that for all modifications, we used a wide range of different analytical methods like Raman spectroscopy, attenuated total reflection IR-spectroscopy, transmission electron microscopy, X-ray photoelectron spectroscopy and pH-value dependent zeta potential measurements to characterize their surface sp2 carbon content, (multi)functionality, density of functional groups, as well as other properties. Our results demonstrate that the approach can be used to successfully clean and specifically modify the diamond surface in a very controlled, reproducible and scalable way. In particular, this efficient method of obtaining a nitrogen (NH2) terminated surface offers a new route towards biofunctionalization of nanodiamonds.
 | ||
| Fig. 1 Scheme and general strategy for the synthesis of functionalized nanoparticles (GND: NDs with a graphite layer; OND: oxygen terminated nanoparticles, e.g., oxygen containing functional groups; HND: hydrogen termination (C–H); NND: nitrogen termination, i.e., presence of NH2 groups). | ||
Furthermore we establish and report a protocol to increase the photoluminescence (PL) of nanodiamonds containing about 100 ppm nitrogen (natural abundance) through the generation/activation of NV-centers by employing a high power 10 MeV linear electron accelerator (Mevex) in the Hertz-Application Laboratory in Leipzig and subsequent annealing of the obtained material at high temperatures.
Oxidative nanodiamond graphite removal
To determine the optimal temperature for the degraphitization process thermogravimetric measurements (TGA) and differential scanning calorimetric measurements (DSC) in air of the ‘as received’ GNDs were performed. The resulting plots for different particle sizes corresponding to the commercial samples described in the materials section are displayed in Fig. 2 a. It becomes apparent that different processes play a role during the heating of diamond nanoparticles in air. Gogotsi et al. reported a thermogravimetric analysis for particle sizes around 5–8 nm produced by detonation synthesis. In agreement with their results we identify three different temperature intervals (RI–RIII) for the degraphitization process.15 For separation of the regions we determined local minima of the first derivative of TGA curves. The resulting size dependent minima are represented by the dotted lines that separate the temperature regions. First (RI), at temperatures below 400 °C the surface adsorbs water layers and weakly bound surface impurities are removed.38 For more stable carbon species including bigger amorphous, graphitic, and diamond phases the temperature is still too low for significant carbon oxidation. Due to the different surface area to volume ratios heating causes a stronger mass loss as a function of temperature for smaller particles. In the second region (RII) at temperatures above 400 °C initial oxidation of the carbon species of the diamonds occurs. Gogotsi et al. described this region as an effective temperature region for an effective removal of non-diamond carbon. To prove this they used samples with different amounts of non-diamond carbon. In general, it can be seen that the oxidation temperature depends on the particle size. So with increasing particle size the initial oxidation temperature increases. We attribute this trend to the higher surface energy in the case of smaller particle size resulting in a high surface reactivity. The initial oxidation region is followed by the total oxidation region (RIII) of the diamond species much above 400 °C. Here, we observed rapid oxidation and combustion of all carbon species.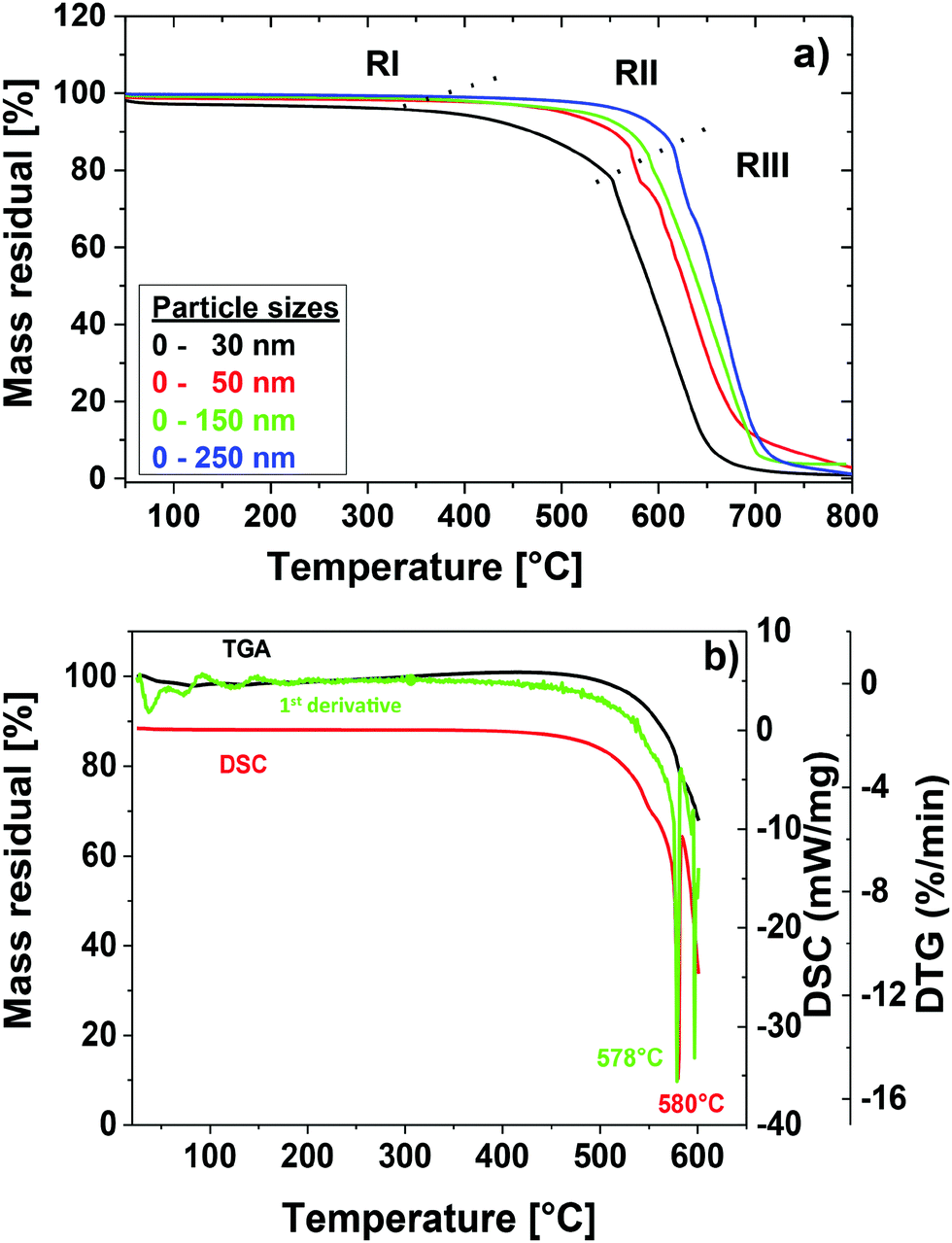 | ||
| Fig. 2 Cleaning process of NDs; (a) TGA of GNDs for different particle sizes; (b) TGA, DSC and first derivative of the TGA graph for GNDs (MSY 0–0.05) with averaged particle sizes around 52 nm. | ||
For the subsequent purification and modification experiments we used the protocol optimized for GND (MSY 0–0.05) particles with an average diameter around 52 nm determined by dynamical light scattering. Please note that for different particle sizes this protocol needs to be modified in terms of temperature and duration times. For the determination of the optimum of the initial oxidation for GND particles we used two complementary methods (DSC) and TGA as well as the first derivative of the thermogravimetric measurement as illustrated in Fig. 2b. Both complementary methods display a similar degradation temperature around 580 °C. The conclusion drawn from this is that 580 °C is the temperature, where degradation of the sp3 carbon starts, being also the point of the lowest possible sp2 content. In fact, this is the key to achieve high quality pre-cleaned NDs und subsequently well reproducible and well defined nanodiamonds that can be further modified and functionalized.
In order to obtain an overview and to conduct a parameter study of the kinetics of graphite removal from the diamond surface the influence of temperature (from 450 °C to 590 °C) and process time (1 hour to 24 hours) on the treatment of the GNDs in air was studied. Fig. 3 gives an overview of the optical appearance of the ND material powders after different treatment times and temperatures. It is obvious that temperature has a strong influence on the degraphitization. The material to start with displays a black color indicating the graphite content that is formed during the HTHP synthesis process. The air treatment at 450 °C shows no significant changes in the color of the nanodiamond powder independent of the process duration. Thus, the graphite around the diamond particles is probably only slightly reduced. At higher treatment temperatures of 550 °C and 575 °C a slight bleaching of the NDs after long process times occurs.
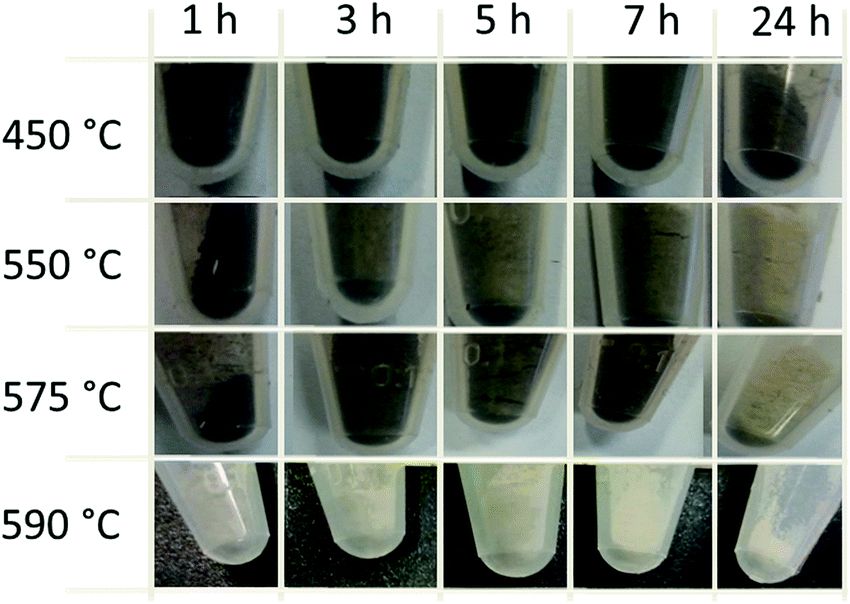 | ||
| Fig. 3 Images of heated GND (MSY 0–0.05) after different temperatures and treatment times under air. | ||
After 24 hours the color changes from black to grey that can be interpreted as the oxidative removal of weakly bound graphite ‘soot’. At 590 °C, which is a temperature close to the optimal degraphitization temperature, efficient degraphitization is observable. Even at short treatment times strong bleaching occurs. After 24 hours the white powder indicates successful removal of the carbon soot. To prove the degraphitisation Raman spectroscopy is a powerful tool. A detailed Raman study using excitation wavelengths of 532 nm for the 24 h treatment at different temperatures and duration dependent degraphitization at 590 °C is illustrated in the ESI† and described in the methods and materials section of this paper. Please note that under these conditions a pronounced decrease in the particle size is observed as well, as described by Stehlik et al.22 After the heating process at 590 °C for 24 hours an average size of d = 29.5 nm was determined using dynamic light scattering.
The decrease in the particle diameter correlates with a weight/mass loss during the oxidation process. The achieved weight yield can be easily well below 50% for very clean NDs. From this ‘standard’ of cleaned oxygen terminated diamond nanoparticles, we further proceed with subsequent functionalization employing the same reactor but changing the gas mixture and temperature. The following procedures for obtaining high quality oxygen, hydrogen and nitrogen termination have been developed.
Oxygen-termination by air oxidation at high temperatures (OND)
To achieve oxygen rich and sp2-carbon free nanodiamonds, GNDs were heated in a high temperature oven under air (1 atm) for oxidative degraphitization. The temperature range (450–590 °C) and the treatment time were optimized for the degraphitization process. The oxidation was performed without previous treatment of the as received GND. Due to the oxidative conditions oxygen termination/functionalization of the nanodiamond surface is unavoidable. The best results were obtained for ONDs heated at 590 °C for 24 hours under air. To remove silica impurities a treatment with a mixture of HF/NH4F (BOE 7-1(AF 87.5–12.5)) for one hour was successful. After cleaning steps (using water, CHCl3 and ether) an additional air heating step at 590 °C for 24 hours was performed. The oxygen terminated nanodiamonds produced under these conditions show a white color. As mentioned above, for all (following) modifications we used OND produced from commercial MSY 0.0–0.05 nanodiamond particle fractions (Microdiamant AG, Switzerland).Hydrogen termination (HND)
The hydrogen termination was performed by heating the ONDs at 700 °C under a permanent hydrogen flow of 40 ml min−1 for different times, 1 h, 3 h, and 5 h, in a tube furnace. The cooling step also takes place under a hydrogen atmosphere. The HND-powder shows a hydrophobic behavior compared to OND due to the removal of the hydrophilic oxygen containing functional groups.Nitrogen termination (NND)
For nitrogen termination ONDs were heated under a mixture of NH3 and argon. The flow rates were 40 ml min−1 (NH3) and 8 ml min−1 (Ar) for different times in a tube furnace at 550 °C, 600 °C, 650 °C and 700 °C. The duration was varied from 3 hours to 7 hours for 700 °C. The cooling step also occurred under a NH3/argon atmosphere. Interestingly, a color change to a red brownish powder occurs for the nitrogen terminated nanodiamond [NND].Characterization of surface functionalization and purity
Surface functionalization in the present approach must always be accompanied by a comprehensive quantitative analysis of (metallic) impurities, sp2-carbon content, as well as surface functionality. We employ a wide range of different analytical methods like Raman spectroscopy, attenuated total reflectance (ATR)-IR, transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and pH-value dependent zeta potential measurements as default and standard characterization techniques. The graphite content of the sample can be conveniently probed and observed by Raman spectroscopy and transmission electron microscopy (TEM). Raman spectroscopy allows for the estimation of the graphite content via the analysis of the G-band that is correlated with the carbon sp2-bond vibrations. Using the HRTEM a direct imaging of the graphite soot around the diamonds was possible.Using Raman spectroscopy the graphite content of the nanodiamonds can be estimated by the ratio of three different peaks: the diamond peak at around 1330 cm−1 and the D- and G bands at 1350 and 1580 cm−1, which correspond to disordered and ordered graphite layers around the diamond.40,41 The Raman spectra in the spectral range between 1300 cm−1 and 1750 cm−1 after excitation with 325 nm for all modifications (Fig. 4) are dominated by the diamond peak at 1335 cm−1.39 For the GND an additional broad band at 1580 cm−1 and a small band at 1360 cm−1 related to the D- and G-bands can be observed. The fact that the D- and G-bands are missing for the OND samples confirms removal of the graphite soot around the NDs. As shown in SI3 (ESI†) the loose graphite soot of the “as received” GND can be easily removed, as indicated by the complete removal of the D-band at low temperatures (450 °C, 24 h) or low duration times (590 °C, 15 min). Nevertheless, the GND sample appears to contain stable ordered graphite content indicated by the G-band. Therefore, complete removal of the G-band was only observed at high temperatures (T > 575 °C) and long duration times (t > 1 h). The Raman spectra are in good agreement with the observed color change presented in Fig. 3. This confirms the assumption that the white color of the diamond powder is related to a very high sp3 amount of the NDs. For HND we also did not observe D- and G- bands. Therefore, we assume that the modification process did not lead to significant regraphitisation. In the case of NND a small broad G-band indicating the formation of new graphitic structures on the diamond surface is observed. A complex surface reaction leading to a heteroaromatic surface could be the origin of the G-band. At temperatures over 700 °C Luo et al. reported the generation of N doped graphitic structures due to the pyrolysis of the carbon containing compounds.40 For Raman spectra of NND samples prepared at different temperatures (SI3, ESI†) (600–700 °C) employing 532 nm excitation a small G-band is observed that seems to be slightly increased with increasing temperature that agrees with the observations of Luo et al.40 The generation of nitrogen containing aromatic/graphitic structures can also be the reason for the color change during the nitrogen termination. Note that the difference between the signal intensities depending on the chosen excitation wavelength is due to different scattering efficiencies.41
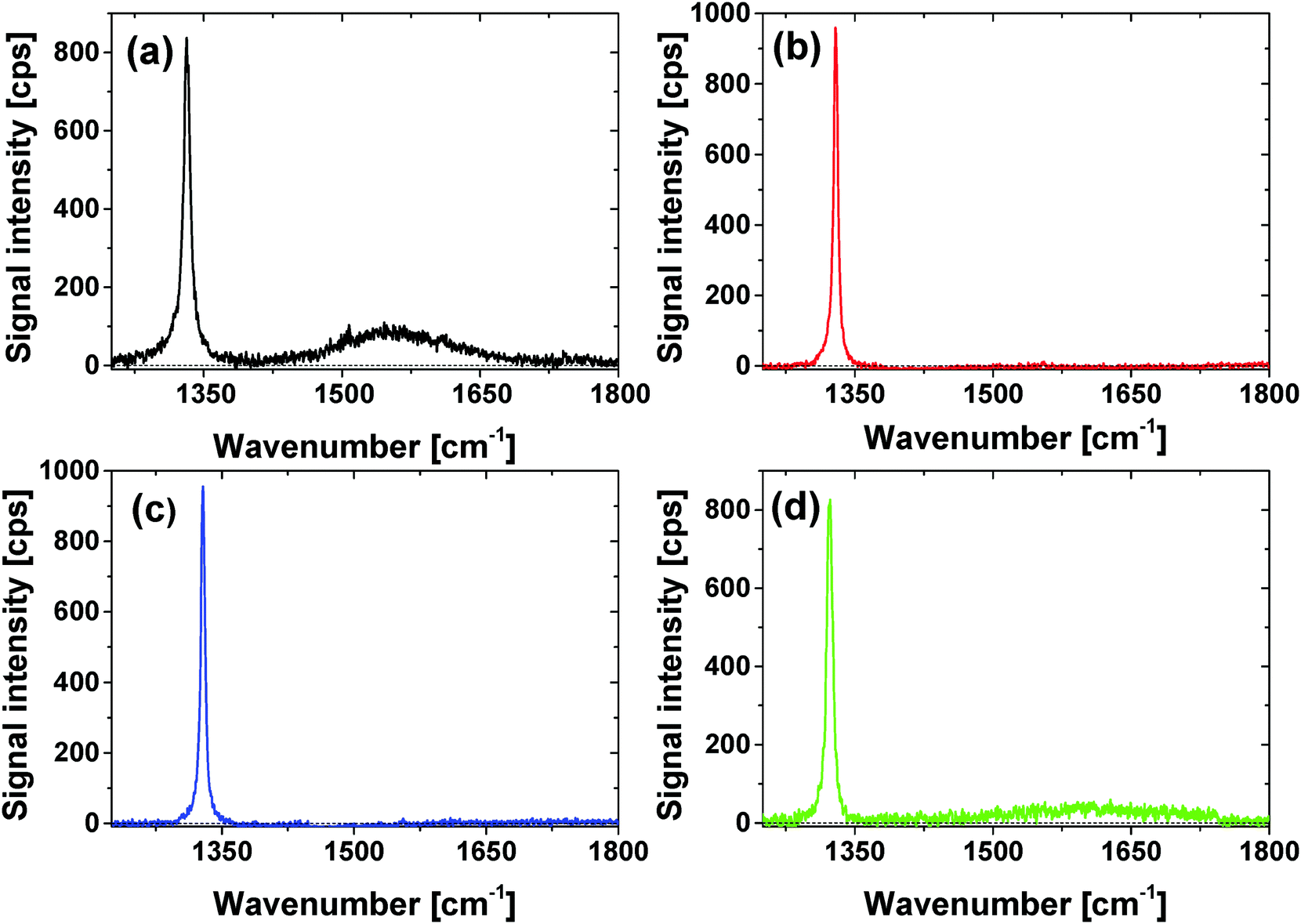 | ||
| Fig. 4 Raman spectra of (a) GND; (b) OND; (c) HND and (d) NND excited using λ = 325 nm, in the spectral range of 1300–1800 cm−1. | ||
HR-TEM images of GND, OND, HND and NND samples (see Fig. 5) display typical ND clusters. For the GND sample the graphite soot can be clearly discriminated due to the different contrast of sp3 and sp2 carbons in the image. The images suggest that a high content of amorphous carbon is existent in the GND samples. We observe loose graphite soot as well as graphite structures around the diamond in good agreement with the literature.42 For the OND and HND (Fig. 5b and c) nearly no graphite is visible in the diamond samples. The existence of small graphite structures can be explained by the graphitization caused by the electron irradiation.43 These results are in good agreement with the Raman spectra and confirm successful graphite layer removal. As a direct consequence, the functional groups for OND and HND are located directly at the sp3 diamond interface, whereas in the case of graphite containing ND it cannot be excluded that the functional groups are localized on top of the graphite layer. This makes the modification special in terms of further modifications and could lead to different surface reactivities compared to diamonds with non-diamond carbon layers. The images of NND prepared at 700 °C for 7 hours display some particles containing amorphous carbon as well as graphite surroundings of the diamond core in good agreement with the results from Raman spectroscopy. Note that we observed only graphite related structures for the minority of the NND particles. For further qualitative investigations of the observed graphitization of the NND-samples we performed high resolution transmission electron microscopy (HRTEM) as illustrated in the ESI† (SI4). The images correspond to NND samples prepared for 7 hours at 700 °C and for 5 hours at 600 °C. In both cases non-diamond carbon can be observed but with different morphologies. For the NND sample prepared at 700 °C particles totally covered with a high content of amorphous carbon structures can be found. The NND samples prepared at 600 °C display only a partial non-diamond carbon covering on the diamond surface of some particles. The qualitative images together with the Raman spectra (SI3, ESI†) indicate the trend of higher graphitization during nitrogen termination with increasing preparation temperature.
 | ||
Fig. 5 HR-TEM image with magnification of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000; (a) GND (zoomed in); (b) OND (zoomed in); (c) HND (prepared at 700 °C) and (d) NND (prepared at 700 °C for 7 hours). 000; (a) GND (zoomed in); (b) OND (zoomed in); (c) HND (prepared at 700 °C) and (d) NND (prepared at 700 °C for 7 hours). | ||
For further qualitative analysis of the surface functionalities attenuated total reflection infrared spectroscopy (ATR-IR) was employed. Please note that the spectra were normalized for a better qualitative overview. In Fig. 6a–c ATR-IR spectra of the modified ND are shown in characteristic wavelength regions. The resulting spectra of GND and OND have a characteristic carbonyl peak at 1782 cm−1, as well as a hydroxyl group peak at 1625 cm−1.44 The peak with a maximum at 3420 cm−1 can be attributed to the O–H vibration. The peak at 1440 cm−1 can be assigned to the corresponding OH in-plane bending vibration of the carboxylic groups.45 Moreover, the peak at 1305 cm−1 can be related to the C–O deformation vibration. The absence of C–H groups also shows the efficiency of the air treatment in terms of the production of clean ONDs.
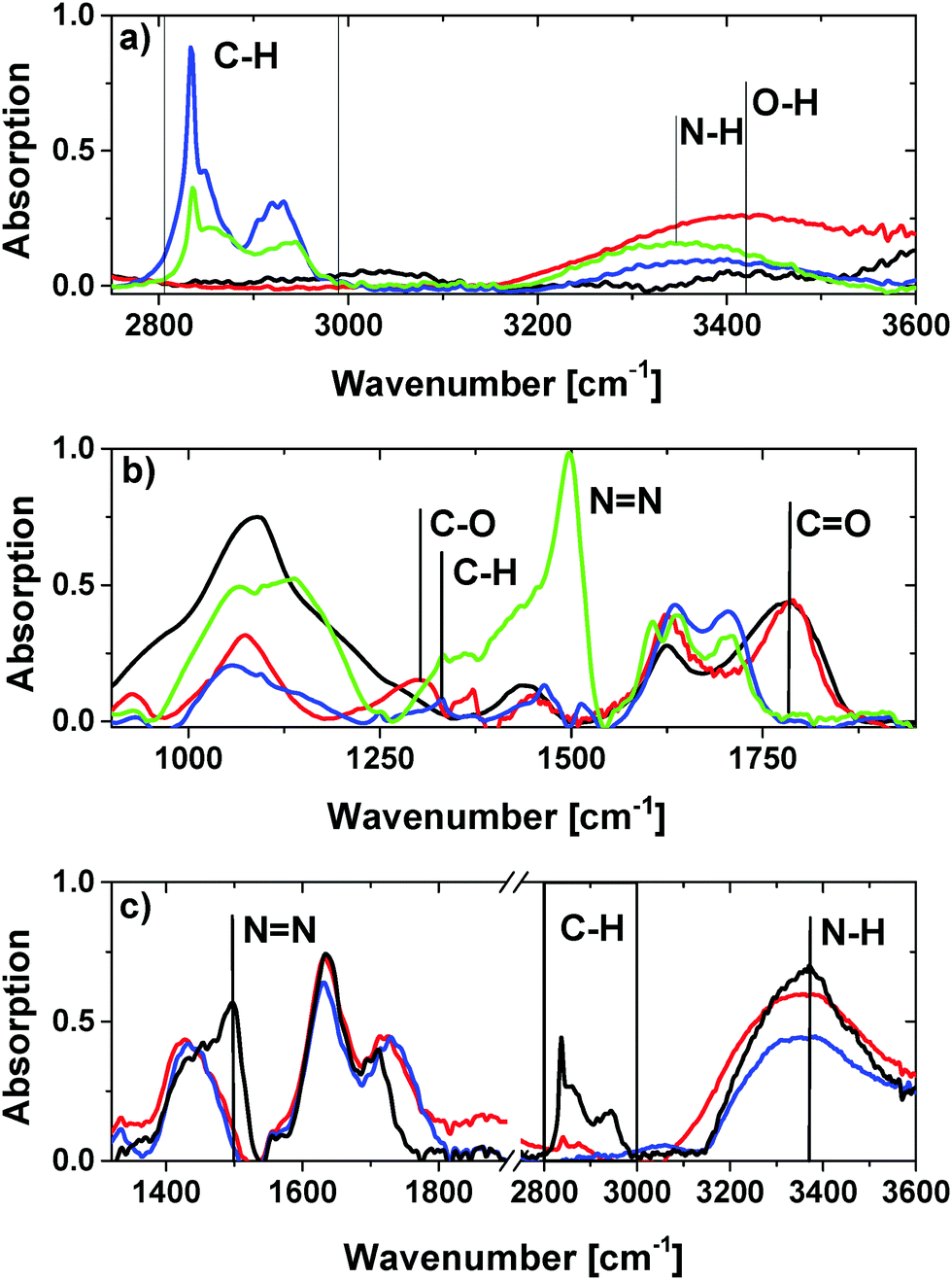 | ||
| Fig. 6 (a) Normalized ATR-FT-IR spectra at 2750–3600 cm−1 of NDs with different surfaces; GND (black), OND (red), HND (blue) and NND (7 h, 700 °C) (green); (b) normalized ATR-IR at 9001900 cm−1; GND (black), OND (red), HND (blue) and NND (green); (c) normalized ATR-IR spectra of NND produced at different temperatures for 5 hours at 700 °C (black), 600 °C (red), and 550 °C (blue). | ||
In contrast, for the hydrogen-terminated diamonds the absence of the carboxyl group peak and the existence of an intense C–H stretching vibration peak at 2800 cm−1–3000 cm−1 are observed. Also, the C–O deformation peak has vanished and a weak C–H deformation vibration band at 1325 cm−1 is visible. For HND the shift of the broad O–H vibration related peak to a maximum around 3400 cm−1 is assigned to the presence of water layers around the particle.
Nitrogen termination of the nanodiamond also results in the absence of carboxy-related vibrational peaks. The shift of the maximum of the broad band to 3350 cm−1 observed for NND samples relates to the H–N-stretching vibration and indicates the formation of amino groups.
Surprisingly, a sharp peak at 1490 cm−1 occurs for NND processed at 700 °C. We interprete this peak as a NN vibration band. In addition, the existence of an intense C–H peak at 2800–3000 cm−1 indicates that additional processes like decomposition of ammonia into nitrogen and hydrogen at high temperatures around 700 °C may take place.40,46 The generated hydrogen may cause competitive reactions with the NDs leading to partial hydrogen termination. Further experiments, where NNDs were produced at 600 °C and 550 °C, show no NN vibration related peak and a less intense C–H stretching band confirming the above decomposition assumption as shown in Fig. 6c. The H–N-stretching band is still existent for the 600 °C and 550 °C treatment. This illustrates that the amine group functionalization also works at lower temperatures. Note that without normalization of the spectra the hydroxyl group peak at 1630 cm−1 in the spectra of HND and NND is caused by the formation of a thin surface water layer and is much less intense compared to the OND and GND samples.47 The decrease in the intensity of this peak for NND and HND correlates with their hydrophobic behavior. This on the other hand fits well with the partial nitrogen and hydrogen termination of the NND surface. In addition, for NND the trend of an increased hydroxyl group peak and a decreased C–H stretch band with decreasing temperature indicates the existence of residual oxygen containing functional groups. Additionally, to confirm the existence of amino functionalities on the surface of NND as concluded from the ATR spectra we performed a ‘Kaiser’ test. The blue color of the NND suspension after treatment with the Ninhydrin solution (Sigma Aldrich 60017) and heating at 100 °C for 5 minutes proves the successful modification (see the ESI† (S5)). The positive results are in agreement with the ATR measurements.
For further evidence and proof for the surface functionalities, we applied pH dependent measurements of the zeta potential. The analysis is based on the fact that the removal of oxygen containing functional groups results in a decrease in the effective negative charge at the surface.48 The plots of ζ-potential against the pH-value for the different modified diamonds are illustrated in Fig. 7. For GND and OND a similar pH-value dependence of the ζ-potential is observed. The ζ-potential has a value of ζOND, GND = −30 mV for pH > 7. With decreasing pH-value only a small increase in the ζ-potential occurs and no isoelectric point is reached in the measurement range down to a pH value of 2.75. These results support the interpretation of the ATR-FT-IR measurements that GND and OND exhibit similar surface functionalities with a high amount of carboxylic groups. The curves of HND and NND exactly show the expected trend with the shift of the isoelectric point towards higher pH-values compared to OND and GND. The fact that the isoelectric point for HND (pH = 4) and NND (pH = 6) is reached at pH < 7 can be explained by the preferential adsorption of hydroxide ions compared to the adsorption of hydronium ions on the diamond surface for the neutral pH-region.49,50 The higher isoelectric point for NND can be explained by the alkaline behavior of nitrogen containing functional groups. The shift of the ζ-potential isoelectric point closer to neutral pH-values is also correlated with the lower stability of NND and HND suspensions. In the literature, several works describe HND suspensions with a high positive ζ-potential, and relatively stable suspensions were also described. Williams et al. measured the ζ-potential after diamond treatment with hydrogen at 500 °C.48 They described the resulting positive ζ-potential by an additional effect of residual graphite layers around the diamond. Measurements performed employing hydrogenated and graphitized NDs (GND treatment using hydrogen at 680 °C, duration time 5 hours) also show higher positive ζ-potentials around +30 mV at pH = 3.5 with a high isoelectric point around 8 that supports this assumption (S6, ESI†).
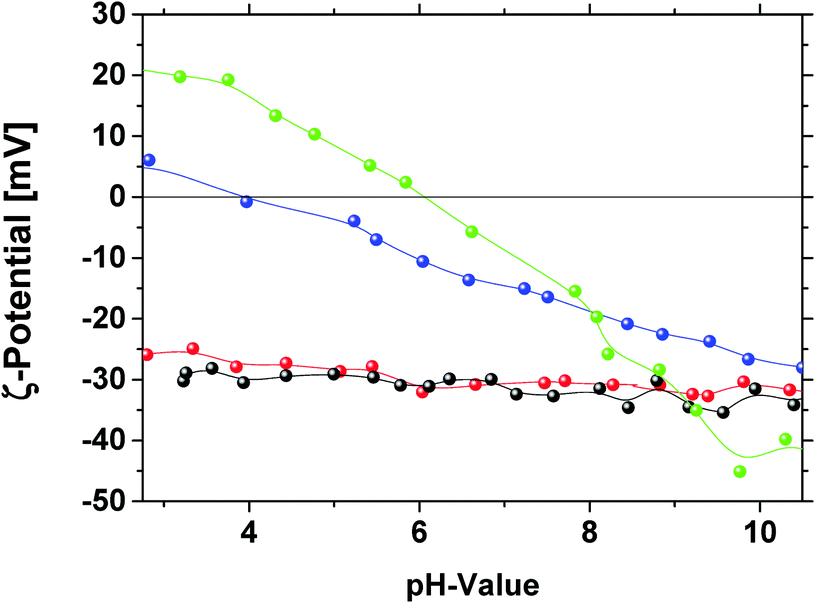 | ||
| Fig. 7 pH-Value dependent measurements of the ζ-potential of GND (black), OND (red), HND (blue) and NND (green). | ||
XPS is a powerful tool for quantitative surface element analysis. Therefore, the chemical composition of the NDs was determined using X-ray photoelectron spectroscopy. In addition to the illustrated results for the different modifications we also performed a parameter study for the preparation of HND and NND as shown in the ESI† (S7). The XPS spectra show no evidence of metallic impurities. Samples without a fluoric acid washing step do show contamination of silica, about 1% in GND and around 3% for OND. These impurities were identified to be residues from the HTHP synthesis of the NDs. The higher amount of silica in the OND – samples can be explained by an enrichment of silica due to the removal of the graphite. The resulting XPS and ATR-IR spectra (see the ESI† (S1–2)) show a significant decrease of the Si–O related peaks after HF washing as described in the experimental section. Another benefit of XPS is its potential in the quantitative analysis of surface functionalities.
This is due to the fact that electron-withdrawing functional groups lead to an increase in the binding energy of C1s core level electrons. High resolution XPS measurements were carried out for all modifications to analyze the surface functionality of the NDs. The obtained relative atomic contents for C1s, N1s, and O1s are collected in Table 1. Tests of the reproducibility for relevant samples including 4 preparations result in a standard deviation of atom percentage within C1s (σ < 1%), N1s (σ < 0.5%), O1s (σ < 2%). Please note that the relatively high standard deviation for the oxygen atom content is due to the necessary correction for oxygen related to residual SiO2. The removal of two oxygens per Si is unfortunately only a rough assumption leading to the higher error bars. For the C1s core level a deconvolution of the peak signal and fits (product of Gaussian and Lorentzian) was performed. The obtained atomic contributions of the fitted peak are listed separately in Table 1. The estimation of the deviations for the deconvoluted peaks is not given due to the auto-optimization function being used, which resulted in larger error bars. The corresponding high-resolution XPS spectra of the C1s core level region and the used deconvolution are illustrated in Fig. 8. As expected GND and OND have a high oxygen content, c(O1s)GND = 11.2% and c(O1s)OND = 10.8%, respectively. These results are in good agreement with our ATR-IR and pH-dependent ζ-potential measurements. In the literature, similar oxygen contents were published, for example Girard et al. reported an O/C ratio of about 18% for HTHP NDs with a similar diameter.29 Please note that the oxygen concentration in the sample is also increased by water and CO2 adsorbents on the surface. In the corresponding high-resolution XPS spectra an intense peak for all modifications is observed at 284.8 eV and corresponds to C–C sp3 diamond bonds. The peak in the GND spectra at a lower binding energy of 283.6 eV is related to the sp2 carbon.51
| Samples | GND | OND (24 h, 590 °C) | HND (5 h, 700 °C) | NND (7 h, 700 °C) |
|---|---|---|---|---|
| c(N1s) [%] | 0 | 0 | 0 | 3.8 ± 0.1 |
| c(O1s) [%] | 11.2 ± 0.5 | 10.8 ± 0.6 | 1.7 ± 1.4 | 2.3 ± 1 |
| c(C1s) C1 [%] | 61.1 | 65.3 | 73.1 | 61.2 |
| c(C1s) C2 [%] | 11.4 | 17.3 | 25.1 | 25.1 |
| c(C1s) C3 [%] | 12.4 | 5.6 | 0.0 | 7.4 |
| c(C1s) C4 [%] | 3.4 | 0.00 | 0.0 | 3.9 |
| c(C1s) total | 88.7 ± 0.5 | 88.2 ± 0.6 | 98.2 ± 1.4 | 93.9 ± 1.2 |
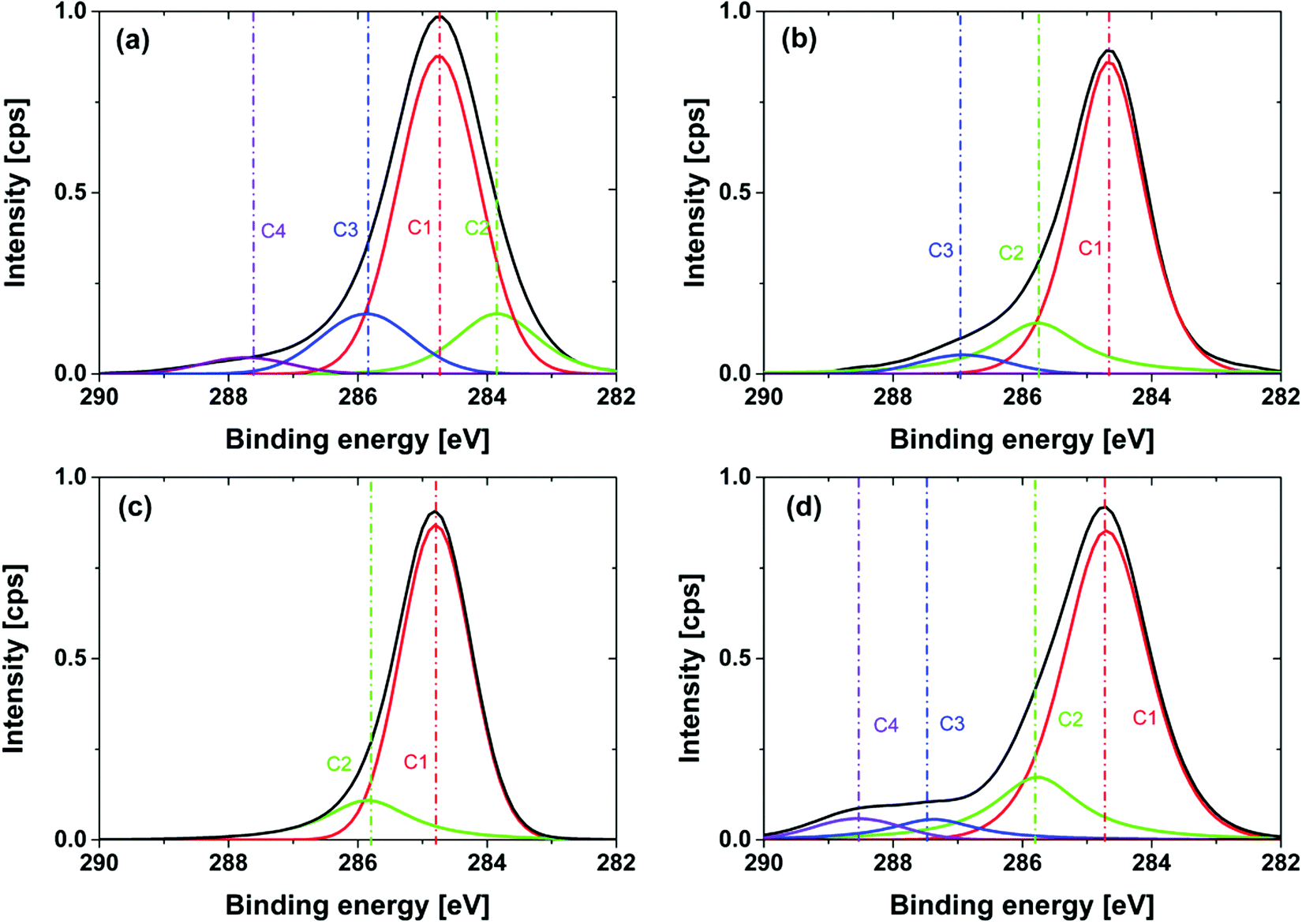 | ||
| Fig. 8 High resolution XPS of the C1s peak of (a) GND; (b) OND (590 °C, 24 hour); (c) HND (700 °C, 5 h) and (d) NND (700 °C, 7 h) immobilized on a silica wafer. | ||
Two additional peaks are present at higher binding energies of 286 eV and 287 eV and are associated with the oxygen–carbon component on the surface of the diamond. The peak at 286 eV can be related to C–O components due to the hydroxyl or ether groups, whereas the peak at 287 eV corresponds to highly oxidized carbon species like carboxy groups. For the hydrogenated diamonds, in agreement with the removal of the oxygen containing functionalities, a strong decrease in the oxygen content occurs, c(O1s)HND_5h = 1.7%, after 5 hours. For shorter reaction times of 1 and 3 hours a higher oxygen content was detected with 4.8% and 3.8%, respectively. From these results we assume that the oxygen removal kinetics has a fast and a slow component. The different functional groups have different oxidation potentials, so the reduction of carboxylic groups should be much more efficient than the reduction of hydroxyl groups. In the first step, the highly oxidized carboxylic and carbonyl groups are reduced resulting in a strong decrease in the oxygen content. In the second step carbons with lower oxidation number are reduced. This process appears to be much slower than the first reduction. The fact that hydroxyl groups are often still observable after the reduction of the diamond surface is in good agreement with this picture.14,16 A variation of the temperature (see the ESI† (S7)) demonstrates a larger influence on the oxygen content of the surface, so for reaction temperatures of 600 °C and 650 °C the atomic oxygen content is 6.3% and 5.5%, respectively. Samples prepared at 750 °C have a slight gray color, which is evidence for partial graphitization. Therefore, we consider 700 °C as the optimum treatment temperature in terms of oxygen removal and sp3/sp2 content. The C1s core level peak shows only two peaks at 284.8 eV and 285.7 eV. Several works assign those peaks to C–C and C–Hx but it is also possible that the second peak is related to C–O due to residual hydroxy groups of the reduction process.29,52 This ambiguity is difficult to resolve. The residual oxygen content may also be due to CO2 adsorption during the transfer of the modified diamonds from the tube reactor to the XPS chamber, which we find possible but unlikely. For NND produced at 700 °C in an ammonia atmosphere for 7 hours the concentration of nitrogen c(N1s)NND_7h = 3.8% suggests that only partial nitrogen termination took place even after a longer treatment time. The low oxygen content of c(O1s)NND 700 °C, 7h = 2.3% indicates the presence of surface carbons from C–H bonds, as identified using ATR-IR-spectroscopy. Shorter treatment showed that under the chosen conditions the maximum of nitrogen content was reached after 5 hours (4% ± 0.1%), correlating with a strong decrease in the oxygen content (see the ESI† (S7)).
The deconvolution of the C1s peak shows a main peak at 284.8 eV and three peaks at higher binding energies of 286 eV, 287.4 eV and 288.3 eV. The first peak at 286 eV can be related to C–N bonds.53 This agrees with the results of the ATR-IR measurement and the Kaiser test indicating that amino groups were formed on the surface of the diamond. A more detailed interpretation of the peaks at 287.4 eV and 288.3 eV is hardly possible at present. Due to the low oxygen content and the results of the ATR-IR-spectra the formation of amide groups can be excluded.
One possible explanation for the shift of the binding energy could be the formation of nitrogen heteroaromatic structures. The formation of nitrogen double bonds causes a strong C1s peak shift to higher energies as shown in the literature.54,55 For NNDs produced at a reaction temperature of 600 °C no peaks at higher binding energies occur, as illustrated in Fig. 9. This strengthens the conclusion that no ammonia decomposition and pyrolysis of surface functionalities happen at lower temperatures. The deconvolution of the C1s peak for NNDs produced at 550 °C shows again a small peak at a higher binding energy of 287.5 eV. The high oxygen content, c(O1s)NND 550 °C, 5h = 7.8%, indicates incomplete oxygen removal at this temperature. Therefore the peak probably results from C–O bonds. In general, due to the high nitrogen concentration measured by XPS in combination with the low graphite content assigned from the Raman probe, as well as the partial graphitization of the particles shown by HRTEM-images, led to the conclusion that we do not modify both the graphite content and degraphitized surface of the nanodiamond. With increasing preparation temperature the graphitization becomes more dominant. Nevertheless, the optimization of the nitrogen termination with regard to reduced surface graphitization will be a topic of further investigation.
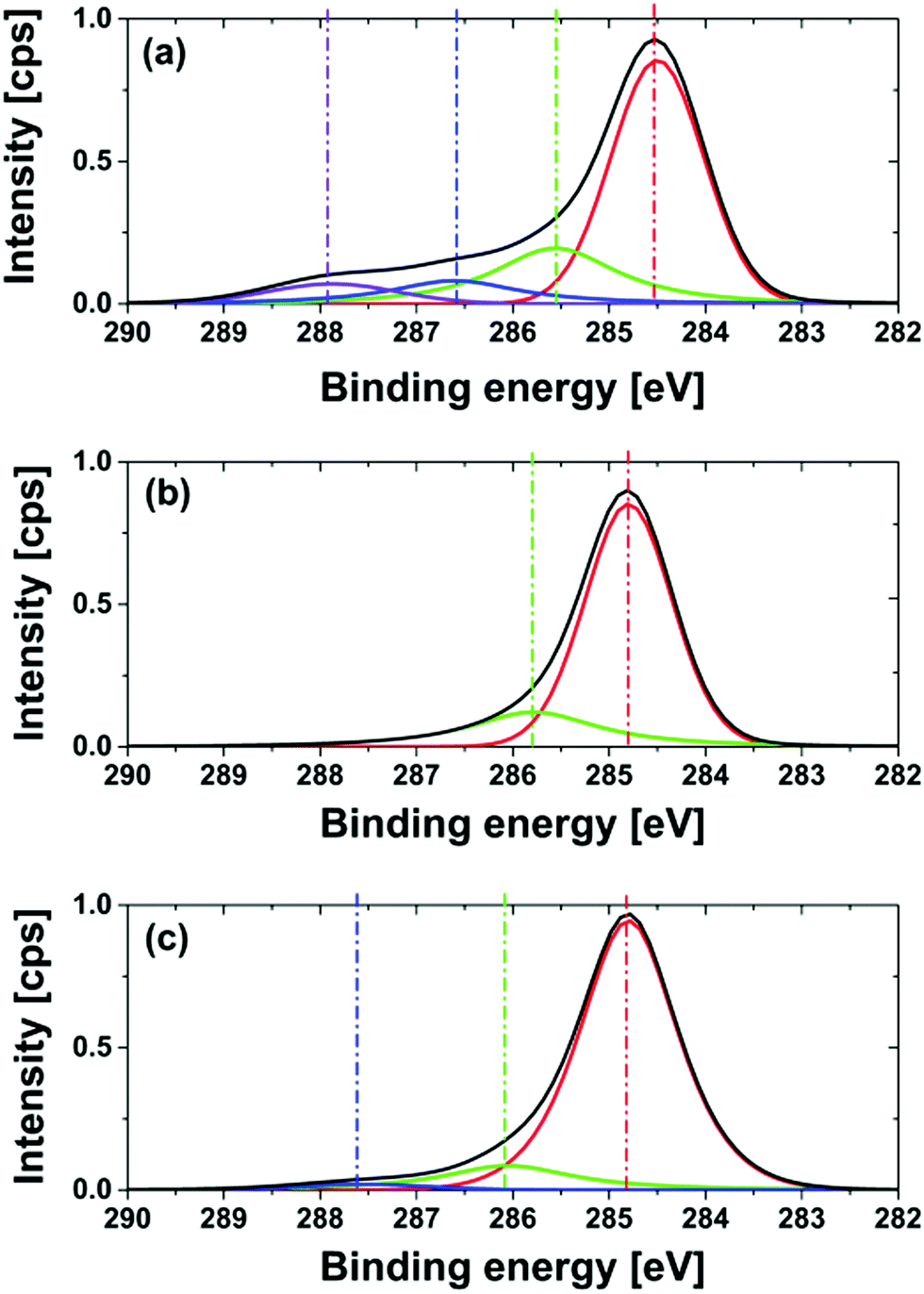 | ||
| Fig. 9 High resolution XPS and deconvolution of the C1s peak of NND produced for a treatment time of 5 hours, at different reaction temperatures: (a) 700 °C; (b) 600 °C; (c) 550 °C. | ||
Photoluminescent properties of nanodiamonds
The first photoluminescence measurements on ONDs illustrate already the formation/presence of NV-centers under the chosen conditions. The achieved emission spectra as well as fluorescence lifetime microscopy (FLIM) images are shown in the ESI† (S8).We want to emphasize here that the photoluminescence (PL) of graphite layer free and oxygen terminated nanodiamonds is in general significantly larger (up to a factor of 5–10, see S8, ESI†) than the PL of NDs with a graphite layer and untreated for the same average size. The fluorescence spectra of a cluster of ONDs as displayed in Fig. S8b (ESI†) consist of both NV0 and NV− as indicated by the zero phonon lines at 575 nm and 639 nm. The observed existence of both NV states is in agreement with the literature. Dantelle et al. estimated a ratio of NV−/NV0 = 60 to 40 for already electron irradiated samples treated with H2O2:H2SO4 (1/3 vol).35 The fluorescence spectra of GND (S8a, ESI†) show no signature of an NV-center and the fluorescence might come only from the graphite. Fig. (S8c, ESI†) shows a representative FLIM-image for a dispersed OND sample for a sampling time of 10 minutes to allow for significant photon accumulation. We used a 100× objective (UPLSAPO 100XO, Olympus); therefore, the illustrated structures are related to nanodiamond clusters (d < 10 μm) in contact with air. For the evaluation of fluorescence lifetime data we used a double exponential fit. We attribute the first fast decay (0.8 ns ± 0.5 ns) to surface defects such as interactions with different defect centers in agreement with reports in the literature.56–58 The fluorescence lifetimes of NV− centers were determined to be around 8.7 ns ± 0.6 ns being the average lifetime of the decay matrix for a set of 5 OND samples employing the SPCImage software (Becker&Hickl). A typical lifetime histogram of a decay matrix for OND is shown in (S8d, ESI†). For GND the determined lifetime was much shorter, i.e., 3.6 ns ± 1.2 ns. We assume that the emission may be attributed only to the graphite rather to NV-centers. Our determined lifetimes for ONDs are in good agreement with previous reports.59,60 However, in the literature a wide range of different fluorescence lifetimes of NV-centers up to 25 ns are described, but most of the papers deal with irradiation enhanced NV production or using different particle sizes, surfaces or suspensions of ND.61,62
Electron beam irradiation of diamond nanoparticles
Most diamond materials including nanodiamonds contain a certain concentration of nitrogen in the 100 ppm range and natural NV centers. Although the natural NV concentration is sufficient for some applications, most applications require much higher concentrations than those provided naturally. For example, in commercial nanodiamond powders, with a median particle size of ∼25 nm those particles that actually host an NV center are rare. Estimates range from 1 out of 10 to 1 out of 1000 NDs that in fact host an NV center.7 The concentration of NV centers can be enhanced considerably by activating the intrinsic nitrogen donors in the material using high-energy electron or ion irradiation and high-temperature (≥800 °C) annealing.35,37,63 High-energy irradiation creates a large number of vacancies, and annealing promotes the formation of NV centers through vacancy diffusion.64 Researchers have reported conversion efficiencies from nitrogen donor to NV in the 10% range and NV densities of tens of parts per million, corresponding to a few NV centers in a 10 nm particle.61 Alternatively, for bulk crystals, high concentrations of nitrogen can be introduced by ion implantation or by deliberate doping during CVD growth. Both ion implantation and doping provide excellent routes to create very shallow, near-surface NV centers. However, ion implantation is not trivial for nanoparticles. Since the concentration of abundant nitrogen in natural nanodiamonds is sufficient we find electron irradiation together with high temperature annealing to be a promising approach for the increase of photoluminescence in nanodiamonds. We therefore irradiated nanodiamonds with 2 radiation doses employing a 10 MeV linear electron accelerator (Mevex). Exposures were at 10 MeV with 2 doses: 1 × 1018 cm−2 (296 MGy) and 4 × 1018 cm−2 (1184 MGy).During the entire process the NDs were packed in graphite-based pellets for easy handling. After the irradiation the ND-containing pellets were annealed at 850 °C under high vacuum. Annealing was followed by graphite etching and chemical purification. The fluorescence intensity from individual nanoparticles was directly correlated with the particle size using a combined atomic force and scanning confocal fluorescence microscope. In Fig. 10 normalized photoluminescence is shown for the NDs of various sizes with the NDs with one NV-center being a reference. It has been found that nanodiamonds above 25 nm can be activated and new NV-centers can be generated with increasing irradiation dose. A PL increase by a factor of 5 can be achieved for NDs above 25 nm. This is a promising result; however, the procedure is certainly not optimized yet for the largest NV-center generation/activation in nanodiamonds. PL-enhancement for nanodiamonds well below 25 nm was difficult to observe/achieve under the present conditions.
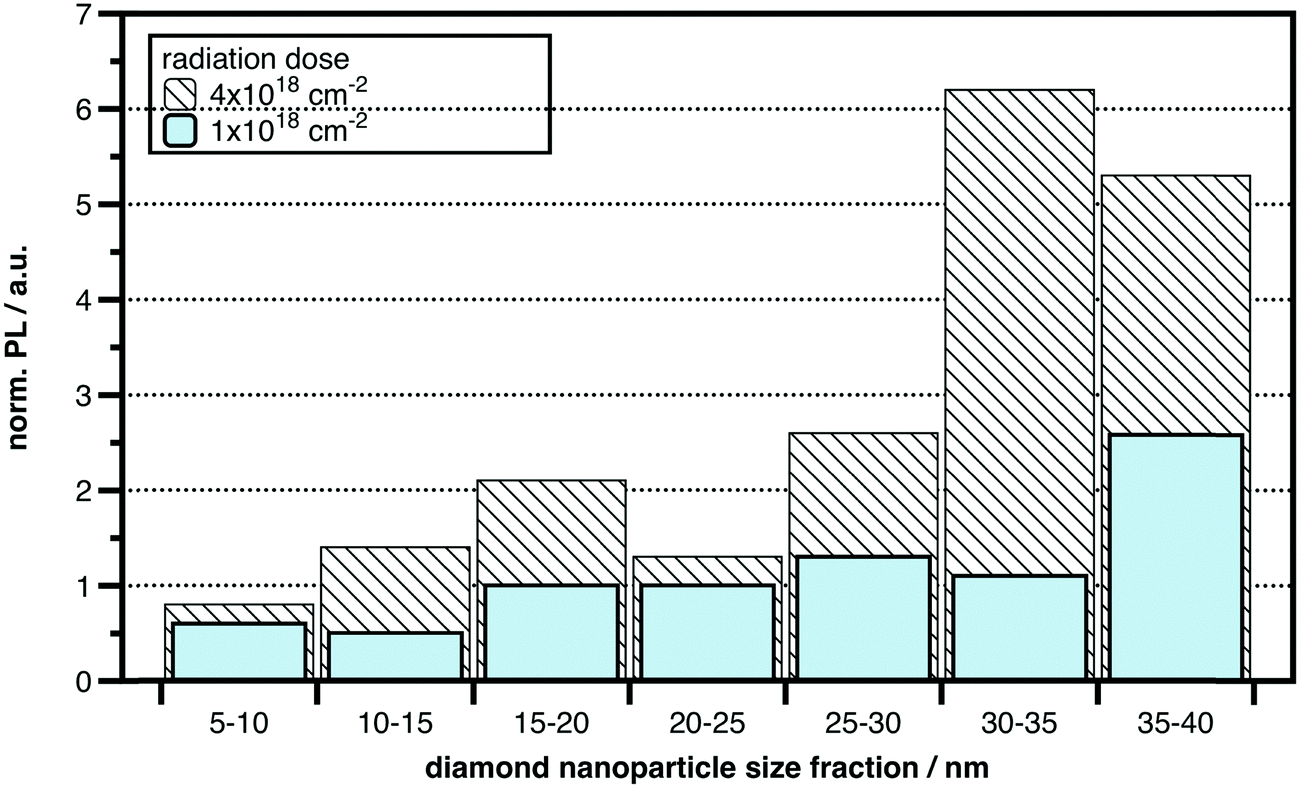 | ||
| Fig. 10 Distribution of photoluminescence intensity (e.g. the amount of NV centers) in diamond nanoparticles of different size ranges. Two bunches of nanoparticles were obtained by exposure to electron irradiation of two different fluences followed by thermal annealing and chemical purification (see text for details). | ||
Summary and conclusions
In this work we proposed and established a reliable procedure for tuning the surface functional groups of NDs. We performed well controlled and controllable solid phase–gas reactions for degraphitization and modification of commercially available NDs, as shown in Fig. 1. Each modification has been controlled and characterized comprehensively employing a number of surface analysis techniques. The purification and degraphitization of nanodiamonds make use of a controlled gas phase–solid state high temperature oxidation in air. The special feature of our approach is the higher process temperature being very close to the degradation threshold of the GNDs. This has to be sensitively monitored and controlled with sensitive thermogravimetric measurements. This results in a significant enhancement of the degraphitisation performance. For the treated OND samples nondiamond carbon layers were not detectable. An extension of this approach could also remove efficiently metallic impurities. The treatment of OND with hydrogen or ammonia leads to significant changes of the surface functionality. A parameter study pointed out the optimal parameter for the reaction in terms of temperature and duration of the processes. For the production of HND the optimum process temperature set point is around 700 °C in terms of oxygen removal and sp3/sp2-ratio. To the best of our knowledge this is the first demonstration of a successful nitrogen termination using a dry high temperature gas phase – solid state reaction approach. The detected atomic nitrogen content up to 3.8% demonstrates a high loading of nitrogen on the surface. The XPS and ATR measurements indicate amino group functionality at the nanodiamond surface. Simple Kaiser tests confirm the spectroscopic results and the presence of amino groups at the surface. They also demonstrate the reactivity of the amino groups and the amine functionality at the surface of the NNDs. In contrast to the ‘clean’ hydrogenation the reaction with ammonia at temperatures around 700 °C may lead to a somewhat more diverse situation, namely the presence of amino groups, partial hydrogen termination and the presence of heteroaromatic structures, possibly due to high temperature pyrolysis processes at the surface. By decreasing the temperature, these processes may be avoided resulting in an amine group-containing surface with an increase in oxygen content due to the decreased efficiency of the oxygen removal process. As a result of the changed surface functionality other properties of the particles are also changed, e.g., ζ-potentials that correlate with the solubility properties of the particles in solution. The solid phase–gas reaction approach can be seen as a fast user and environmentally friendly method to produce large quantities of functionalized high quality NDs on the macroscopic scale in the gram or even 100 g range.Beyond efficient and reproducible surface functionalization it has been found that nanodiamonds above 25 nm can be activated and new NV-centers can be generated with electron irradiation employing a 10 MeV linear electron accelerator. In an initial attempt with a non-optimized protocol the PL of NDs above 25 nm has been found to be a factor of ca. 5 larger than the PL of NDs for a dose of 1 × 1018 cm−2. This is a promising result; however, the procedure is not optimized for NV-center generation/activation in nanodiamonds. The fact that activation or the PL-enhancement for nanodiamonds significantly below 25 nm is difficult to detect may be a limitation because some applications in the field of medical imaging require nanodiamonds with enhanced PL in the range significantly below 20 nm. It is unclear if this is a fundamental limitation of the approach or if it is only due to a lack of optimization of the electron irradiation protocol. We assume the latter here. Nevertheless, oxygen atmosphere high temperature ‘shrinking’ or ‘etching’ of larger activated nanodiamonds employing the protocols described above may indeed circumvent this problem in an elegant way in the future. Activities in this direction are underway in the laboratory of the authors at present.
Experimental section
Materials
Commercial nanodiamond samples [GND] (MSY 0.0–0.05) were purchased from Microdiamant AG (Switzerland). These diamonds were produced by a high-temperature high-pressure process. The nitrogen content is listed to be around 100 ppm. The size distribution of the achieved nanodiamonds was determined using dynamic light scattering (Malvern Zetasizer) giving an averaged size distribution of D(n = 0.5) = 52.2 nm. For the determination of the size effect we used 0–30 nm: (MSY 0.0–0.03), 0–50 nm: (MSY 0.0–0.05), 0–150 nm: (MSY 0.0–0.15) and 0–250 nm: (MSY 0.0–0.25).Raman spectroscopy
Raman scattering was excited by λex = 325 nm emitted by a HeCd laser (Kimmon). The light was focused and collected in backscattering geometry using a 50× microscope objective with a numerical aperture of 0.4. The spectra were recorded using a JobinYvon U1000 Double spectrometer equipped with two gratings with 2400 grooves per millimeter and a liquid–nitrogen–cooled charge coupled device. The spectral resolution was chosen to be 1 cm−1.Raman scattering excited by λex = 532 nm emitted by a second order Nd:YAG laser was measured using an inverted confocal microscope (IX71, Olympus) fiber coupled to a spectrometer (iHR320, synapse CCD, HORIBA JobinYvon). The light was focused and collected in backscattering geometry using a 40× microscope objective (Olympus LUCPlan FLN 40× NA 0.6). The spectra were recorded using a grating with 1800 grooves per millimeter. The spectral resolution was chosen to be 1 cm−1. The pure nanodiamond powder was placed on an acetone and ethanol purified glass slide for recording the spectra.
Attenuated total reflectance spectroscopy (ATR)
The infrared spectra were measured using a VECTOR 22 (Bruker) combined with an attenuated total reflectance module (“Golden Gate”, Graseby, Specac). For all samples 128 scans were averaged. For measurements the nanodiamond powder was deposited onto the diamond window without any pretreatments.Zeta potential measurements (ζ-potential)
The zeta potential (ζ-potential) depending on the pH-value of the nanodiamond-particle water (Millipore, 18.2 MΩ) suspensions (0.05 mg ml−1) was characterized using a Malvern Zetasizer (Zetasizer Nano ZS with multipurpose titrator MPT-2, Malvern Instruments, Worcestershire, UK).Transmission electron microscopy (TEM)
TEM observations were performed on a probe Cs-corrected Titan3 G2 60-300 microscope equipped with a high-brightness electron gun (X-FEG) in TEM-mode. The microscope was operated at 300 kV. For imaging we used a CCD, Gatan UltraScan 1000XP (2k × 2k), the exposure time was 1 s. The nanodiamonds were therefore dip-coated on Cu grids covered with lacey carbon.X-ray photoelectron spectroscopy (XPS)
XPS analysis was carried out on a Kratos Axis Ultra (KratosAnalytical, Ltd., Manchester, United Kingdom) with a monochromatized Al excitation source at 150 W (15 kV, 10 mA, with a pass energy of 40 eV). Surface spectra were collected over a range of 0–1200 eV. The nominal resolutions were 1.0 eV for the survey (pass energy = 160 eV) and 0.1 eV for the high-resolution scans (pass energy = 40 eV), respectively. The binding energies were corrected for static charging of the samples with reference to the C1s main peak fixed at a binding energy of 284.8 eV. For measurements the sample was suspended in water (Millipore, 18.2 MΩ) and the suspension was dried on a silica or aluminium oxide wafer.Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)
For the TGA measurement a Pyris 1 (PerkinElmer) Thermo gravimeter was used; the heating rate was 10 °C min−1 under an air atmosphere for the temperature range from 50 °C to 800 °C. The DSC measurements were performed using a DSC 8000/8500 (PerkinElmer); the heating rate was 10 °C min−1 under an air atmosphere.Fluorescence analysis
The Fluorescence spectra were recorded from 540–775 nm under 532 nm excitation using an inverted confocal microscope (IX71, Olympus) fiber coupled to a spectrometer (iHR320, synapse CCD, HORIBA JobinYvon). For fluorescence lifetime microscopy we used a confocal setup as described elsewhere.65 As pulsed laser excitation sources a diode laser (513 nm wavelength, 60 ps pulse duration, 20 MHz repetition rate, Becker Hickl) was used. The spectral band pass filter combinations were selected for the NV− fluorescence (transparent between 690 and 855 nm). The sample was spin-coated out of an ultrasonicated 0.02 mg ml−1 ethanol suspension on an oxygen plasma cleaned glass slide.Irradiation and annealing of nanodiamonds
Commercial nanodiamonds of the size range 0 to 50 nm were purchased from Microdiamant AG (Switzerland). The initial ND particles were packaged into graphite-based pellets using a patented method: German Patent Application: “Production of local defects in nano-diamond and diamond crystals”, Priority Date 01.04.2014, Registr. Nr. 10 2014 104 551.3. The ND containing pellets were exposed to electron irradiation and subjected to thermal annealing at 850 °C for 2 hours under high vacuum. The processed NDs were dissolved by graphite etching followed by multi-step chemical purification.The irradiation was done using the 10 MeV linear electron accelerator, ELEKTRONIKA (Toriy Company, Moscow) at a pulse repetition rate of 10 Hz (duration of a single pulse, 4 μs). The dose per pulse (∼1.5 kGy) was determined by chemical dosimetry, and the corresponding electron fluence of ∼5 × 1012 cm−2 was calculated using an average stopping power of 2 MeV cm2 g−1, which is a good approximation for light materials in the energy range around 10 MeV.66 The temperature during the irradiation did not exceed 100 °C. Total exposures were 1 × 1018 cm−2 and 4 × 1018 cm−2.
Photoluminescence analysis of irradiated and annealed NDs
The histograms in Fig. 10 comprise the analysis of approx. 600 nanoparticles for each dose of electron irradiation. The NDs were diluted in water to a final concentration of 5 μg ml−1, and PVA was added to a concentration of 0.15% (w/w). The samples were sonicated to avoid agglomeration and spin-coated on plasma cleaned glass cover slides at 3000 rpm for 20 s. The NDs were characterised on a confocal microscope combined with an atomic force microscope (Asylum MFP-3D). The prepared NDs were diluted in water, sonicated to avoid agglomeration and spin-coated on glass cover slides. The size of individual NDs was determined by measuring the height (z-axis) avoiding any influence of the AFM tip shape. Optical excitation was performed using a 532 nm laser and the collected fluorescence was filtered using a 650 nm low-pass filter. To estimate the amount of NV centers in each ND particle, the recorded fluorescence was normalized to the averaged intensity from single NV. For identification of NDs with a single NV center we employed a Hanbury–Brown–Twiss scheme to derive the g(2) correlation function.Acknowledgements
A. K. and B. A. gratefully acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG) via grant KA 3491/2-1 and AB 63/14-1. BA acknowledges funding from the Bundesministerium für Bildung und Forschung via Project “Nanotrace” (BMBF-03X0130). We thank Prof. Dr B. Kersting for initial diamond samples and Prof. Dr J. Meijer for helpful discussion.References
- S. Pezzagna, et al., Creation efficiency of nitrogen-vacancy centres in diamond, New J. Phys., 2010, 12(6), 065017 CrossRef.
- V. N. Mochalin, et al., The properties and applications of nanodiamonds, Nat. Nanotechnol., 2012, 7(1), 11–23 CrossRef CAS PubMed.
- Y. Sun, et al., Disaggregation and Anionic Activation of Nanodiamonds Mediated by Sodium Hydride – A New Route to Functional Aliphatic Polyester-Based Nanodiamond Materials, Part. Part. Syst. Charact., 2015, 32(1), 35–42 CrossRef CAS.
- T. J. Merkel and J. M. DeSimone, Dodging Drug-Resistant Cancer with Diamonds, Sci. Transl. Med., 2011, 3(73), 73ps8–73ps8 Search PubMed.
- J. M. Taylor, et al., High-sensitivity diamond magnetometer with nanoscale resolution, Nat. Phys., 2008, 4(10), 810–816 CrossRef CAS.
- L. T. Hall, et al., High spatial and temporal resolution wide-field imaging of neuron activity using quantum NV-diamond, Sci. Rep., 2012, 2, 401 CrossRef CAS PubMed.
- R. Schirhagl, et al., Nitrogen-Vacancy Centers in Diamond: Nanoscale Sensors for Physics and Biology, Annu. Rev. Phys. Chem., 2014, 65(1), 83–105 CrossRef CAS PubMed.
- O. A. Shenderova and G. E. McGuire, Science and engineering of nanodiamond particle surfaces for biological applications (Review), Biointerphases, 2015, 10(3), 030802 CrossRef PubMed.
- X.-Q. Zhang, et al., Polymer-Functionalized Nanodiamond Platforms as Vehicles for Gene Delivery, ACS Nano, 2009, 3(9), 2609–2616 CrossRef CAS PubMed.
- M. Alishiri, A. Shojaei and M. J. Abdekhodaie, Biodegradable polyurethane acrylate/HEMA-grafted nanodiamond composites with bone regenerative potential applications: structure, mechanical properties and biocompatibility, RSC Adv., 2016, 6(11), 8743–8755 RSC.
- M. Ullah, et al., Reinforcing Effects of Modified Nanodiamonds on the Physical Properties of Polymer-Based Nanocomposites: A Review, Polym. – Plast. Technol. Eng., 2015, 54(8), 861–879 CrossRef CAS.
- I. Kratochvílová, et al., Magnetical and Optical Properties of Nanodiamonds Can Be Tuned by Particles Surface Chemistry: Theoretical and Experimental Study, J. Phys. Chem. C, 2014, 118(43), 25245–25252 Search PubMed.
- J. Gong, N. Steinsultz and M. Ouyang, Nanodiamond-based nanostructures for coupling nitrogen-vacancy centres to metal nanoparticles and semiconductor quantum dots, Nat. Commun., 2016, 7, 11820 CrossRef CAS PubMed.
- A. Krueger and D. Lang, Functionality is Key: Recent Progress in the Surface Modification of Nanodiamond, Adv. Funct. Mater., 2012, 22(5), 890–906 CrossRef CAS.
- S. Osswald, et al., Control of sp2/sp3 Carbon Ratio and Surface Chemistry of Nanodiamond Powders by Selective Oxidation in Air, J. Am. Chem. Soc., 2006, 128(35), 11635–11642 CrossRef CAS PubMed.
- A. Krueger and T. Boedeker, Deagglomeration and functionalisation of detonation nanodiamond with long alkyl chains, Diamond Relat. Mater., 2008, 17(7–10), 1367–1370 CrossRef CAS.
- X. Huaqing, Y. Wei and L. Yang, Thermal performance enhancement in nanofluids containing diamond nanoparticles, J. Phys. D: Appl. Phys., 2009, 42(9), 095413 CrossRef.
- V. Pichot, et al., An efficient purification method for detonation nanodiamonds, Diamond Relat. Mater., 2008, 17(1), 13–22 CrossRef CAS.
- S. Turner, et al., Determination of Size, Morphology, and Nitrogen Impurity Location in Treated Detonation Nanodiamond by Transmission Electron Microscopy, Adv. Funct. Mater., 2009, 19(13), 2116–2124 CrossRef CAS.
- O. Shenderova, et al., Surface Chemistry and Properties of Ozone-Purified Detonation Nanodiamonds, J. Phys. Chem. C, 2011, 115(20), 9827–9837 CAS.
- D. Mitev, et al., Surface peculiarities of detonation nanodiamonds in dependence of fabrication and purification methods, Diamond Relat. Mater., 2007, 16(4–7), 776–780 CrossRef CAS.
- S. Stehlik, et al., Size and Purity Control of HPHT Nanodiamonds down to 1 nm, J. Phys. Chem. C, 2015, 119(49), 27708–27720 CAS.
- R. Silbajoris, et al., Detonation nanodiamond toxicity in human airway epithelial cells is modulated by air oxidation, Diamond Relat. Mater., 2015, 58, 16–23 CrossRef CAS.
- X. Xu, et al., Influence of surface modification adopting thermal treatments on dispersion of detonation nanodiamond, J. Solid State Chem., 2005, 178(3), 688–693 CrossRef CAS.
- W.-W. Zheng, et al., Organic functionalization of ultradispersed nanodiamond: synthesis and applications, J. Mater. Chem., 2009, 19(44), 8432–8441 RSC.
- S. Ida, et al., Chemical reaction of hydrogenated diamond surface with peroxide radical initiators, Diamond Relat. Mater., 2003, 12(3–7), 601–605 CrossRef CAS.
- T. Jiang and K. Xu, FTIR study of ultradispersed diamond powder synthesized by explosive detonation, Carbon, 1995, 33(12), 1663–1671 CrossRef CAS.
- D. Zhu, et al., Photo-illuminated diamond as a solid-state source of solvated electrons in water for nitrogen reduction, Nat. Mater., 2013, 12(9), 836–841 CrossRef CAS PubMed.
- H. A. Girard, et al., Hydrogenation of nanodiamonds using MPCVD: A new route toward organic functionalization, Diamond Relat. Mater., 2010, 19(7–9), 1117–1123 CrossRef CAS.
- H. Koch, et al., Plasma amination of ultrananocrystalline diamond/amorphous carbon composite films for the attachment of biomolecules, Diamond Relat. Mater., 2011, 20(2), 254–258 CrossRef CAS.
- K.-I. Sotowa, et al., Effect of treatment temperature on the amination of chlorinated diamond, Diamond Relat. Mater., 2004, 13(1), 145–150 CrossRef CAS.
- A. Kruger, et al., Surface functionalisation of detonation diamond suitable for biological applications, J. Mater. Chem., 2006, 16(24), 2322–2328 RSC.
- Y. Liu, et al., Functionalization of Nanoscale Diamond Powder: Fluoro-, Alkyl-, Amino-, and Amino Acid-Nanodiamond Derivatives, Chem. Mater., 2004, 16(20), 3924–3930 CrossRef CAS.
- A. V. Stanishevsky, M. J. Walock and S. A. Catledge, Surface modification and stability of detonation nanodiamonds in microwave gas discharge plasma, Appl. Surf. Sci., 2015, 357(Pt B), 1403–1409 CrossRef CAS.
- G. Dantelle, et al., Efficient production of NV colour centres in nanodiamonds using high-energy electron irradiation, J. Lumin., 2010, 130(9), 1655–1658 CrossRef CAS.
- Y.-R. Chang, et al., Mass production and dynamic imaging of fluorescent nanodiamonds, Nat. Nanotechnol., 2008, 3(5), 284–288 CrossRef CAS PubMed.
- E. Kim, V. Acosta, E. Bauch, D. Budker and P. Hemmer, NV-Diamond Magnetometer Using Electron Irradiation, MRS Online Proc. Libr., 2013, 1511 DOI:10.1557/opl.2013.22.
- Q. Zou, et al., Analysis of structures and surface states of the nanodiamond particle synthesised by detonation, IET Micro & Nano Letters, 2009, 4(3), 133–141 CAS.
- V. Mochalin, S. Osswald and Y. Gogotsi, Contribution of Functional Groups to the Raman Spectrum of Nanodiamond Powders, Chem. Mater., 2009, 21(2), 273–279 CrossRef CAS.
- W. Luo, et al., Pyrolysis of Cellulose under Ammonia Leads to Nitrogen-Doped Nanoporous Carbon Generated through Methane Formation, Nano Lett., 2014, 14(4), 2225–2229 CrossRef CAS PubMed.
- P. Klar, et al., Raman scattering efficiency of graphene, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(20), 205435 CrossRef.
- A. Krueger, The structure and reactivity of nanoscale diamond, J. Mater. Chem., 2008, 18(13), 1485–1492 RSC.
- Z. Qiao, et al., Graphitization and microstructure transformation of nanodiamond to onion-like carbon, Scr. Mater., 2006, 54(2), 225–229 CrossRef CAS.
- S. Stehlik, et al., Water interaction with hydrogenated and oxidized detonation nanodiamonds – Microscopic and spectroscopic analyses, Diamond Relat. Mater., 2016, 63, 97–102 CrossRef CAS.
- J. Lambert, Introduction to Organic Spectrocscopy, Macmillan, 1987 Search PubMed.
- R. Lan and S. Tao, Ammonia as a suitable fuel for fuel cells, Frontiers in Energy Research, 2014, 2, 35 CrossRef.
- S. Ji, et al., FTIR study of the adsorption of water on ultradispersed diamond powder surface, Appl. Surf. Sci., 1998, 133(4), 231–238 CrossRef CAS.
- O. A. Williams, et al., Size-Dependent Reactivity of Diamond Nanoparticles, ACS Nano, 2010, 4(8), 4824–4830 CrossRef CAS PubMed.
- R. Zimmermann, S. Dukhin and C. Werner, Electrokinetic Measurements Reveal Interfacial Charge at Polymer Films Caused by Simple Electrolyte Ions, J. Phys. Chem. B, 2001, 105(36), 8544–8549 CrossRef CAS.
- A. Härtl, et al., The Ion Sensitivity of Surface Conductive Single Crystalline Diamond, J. Am. Chem. Soc., 2007, 129(5), 1287–1292 CrossRef PubMed.
- S. Ferro, M. Dal Colle and A. De Battisti, Chemical surface characterization of electrochemically and thermally oxidized boron-doped diamond film electrodes, Carbon, 2005, 43(6), 1191–1203 CrossRef CAS.
- L. Ley, et al., Electronic properties of single crystalline diamond surfaces, Carbon, 1999, 37(5), 793–799 CrossRef CAS.
- S. Bhattacharyya, J. Hong and G. Turban, Determination of the structure of amorphous nitrogenated carbon films by combined Raman and X-ray photoemission spectroscopy, J. Appl. Phys., 1998, 83(7), 3917–3919 CrossRef CAS.
- A. P. Dementjev, et al., X-Ray photoelectron spectroscopy reference data for identification of the C3N4 phase in carbon–nitrogen films, Diamond Relat. Mater., 2000, 9(11), 1904–1907 CrossRef CAS.
- M. Diani, et al., Search for carbon nitride CNx compounds with a high nitrogen content by electron cyclotron resonance plasma deposition, Diamond Relat. Mater., 1994, 3(3), 264–269 CrossRef CAS.
- D. Gatto Monticone, et al., Systematic study of defect-related quenching of NV luminescence in diamond with time-correlated single-photon counting spectroscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88(15), 155201 CrossRef.
- B. R. Smith, D. Gruber and T. Plakhotnik, The effects of surface oxidation on luminescence of nano diamonds, Diamond Relat. Mater., 2010, 19(4), 314–318 CrossRef CAS.
- G. Laporte and D. Psaltis, STED imaging of green fluorescent nanodiamonds containing nitrogen-vacancy-nitrogen centers, Biomed. Opt. Express, 2016, 7(1), 34–44 CrossRef CAS PubMed.
- R. Fudala, et al., FRET Enhanced Fluorescent Nanodiamonds, Curr. Pharm. Biotechnol., 2014, 14(13), 1127–1133 Search PubMed.
- R. Beams, et al., Nanoscale Fluorescence Lifetime Imaging of an Optical Antenna with a Single Diamond NV Center, Nano Lett., 2013, 13(8), 3807–3811 CrossRef CAS PubMed.
- R. Schirhagl, K. Chang, M. Loretz and C. L. Degen, Nitrogen-Vacancy Centers in Diamond: Nanoscale Sensors for Physics and Biology, Annu. Rev. Phys. Chem., 2014, 65(1), 83–105 CrossRef CAS PubMed.
- M. W. Doherty, et al., The nitrogen-vacancy colour centre in diamond, Phys. Rep., 2013, 528(1), 1–45 CrossRef CAS.
- B. Grotz, et al., Charge state manipulation of qubits in diamond, Nat. Commun., 2012, 3, 729 CrossRef PubMed.
- I. Konstantin and J. A. Guy, Trapping of vacancies by defects in diamond, J. Phys.: Condens. Matter, 2001, 13(26), 6015 Search PubMed.
- Y. M. Riyad, et al., Chemical Modification of a Tetrapyrrole-Type Photosensitizer: Tuning Application and Photochemical Action beyond the Singlet Oxygen Channel, J. Phys. Chem. B, 2014, 118(40), 11646–11658 CrossRef CAS PubMed.
- ISO/ASTM51401-13, Standard Practice for Use of a Dichromate Dosimetry System, ASTM International, West Conshohocken, PA, 2013, http://www.astm.org.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7qm00241f |
| This journal is © the Partner Organisations 2017 |