
Diastereo- and enantioselective construction of biologically important pyrrolo[1,2-a]indole scaffolds via catalytic asymmetric [3 + 2] cyclodimerizations of 3-alkyl-2-vinylindoles†
Zi-Qi
Zhu‡
,
Lei
Yin‡
,
Yang
Wang‡
,
Yang
Shen
,
Can
Li
,
Guang-Jian
Mei
* and
Feng
Shi
*
Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, School of Chemistry and Chemical Engineering, Jiangsu Normal University, Xuzhou, 221116, China. Fax: +86 (516) 83500065; Tel: +86 (516) 83403183E-mail: fshi@jsnu.edu.cn; GuangjianM@jsnu.edu.cn
First published on 19th October 2016
Abstract
A catalytic asymmetric [3 + 2] cyclodimerization of 3-alkyl-2-vinylindoles has been established, which efficiently constructed a pyrrolo[1,2-a]indole scaffold with three contiguous stereogenic centers in a diastereo- and enantioselective fashion (up to 98% yield, >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 dr, 98:2 er). This reaction not only represents the first catalytic asymmetric version of this type of [3 + 2] cyclodimerization, but also supplies important examples for using 2-vinylindoles as nitrogen–carbon–carbon (NCC) building blocks in asymmetric catalysis and synthesis. More importantly, this reaction also provides an efficient method for enantioselectively constructing biologically significant pyrrolo[1,2-a]indole scaffolds.
:5 dr, 98:2 er). This reaction not only represents the first catalytic asymmetric version of this type of [3 + 2] cyclodimerization, but also supplies important examples for using 2-vinylindoles as nitrogen–carbon–carbon (NCC) building blocks in asymmetric catalysis and synthesis. More importantly, this reaction also provides an efficient method for enantioselectively constructing biologically significant pyrrolo[1,2-a]indole scaffolds.
Introduction
A chiral pyrrolo[1,2-a]indole scaffold exists in many biologically important molecules and natural products (Fig. 1).1 For instance, compound I acts as an S1P1 functional antagonist;1f flinderoles A–C (compounds II–IV) have been identified as antimalarial agents and have wide application in medicinal chemistry.1d,e Thus, the catalytic asymmetric construction of pyrrolo[1,2-a]indole scaffolds has attracted great attention from the organic chemists.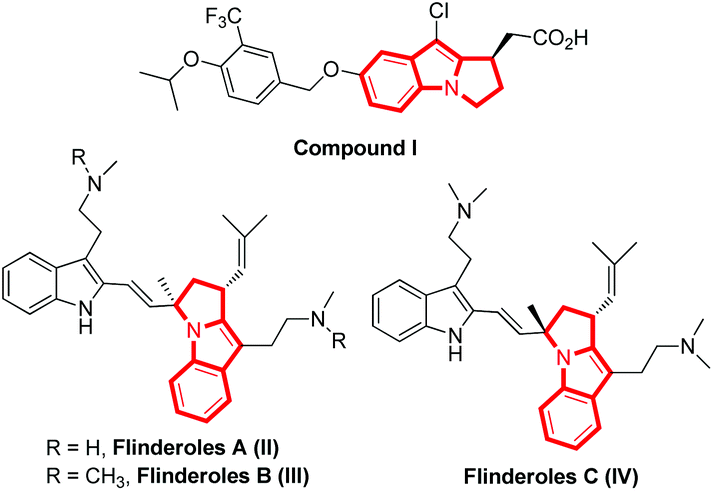 | ||
| Fig. 1 Selected biologically important compounds and natural products containing the pyrrolo[1,2-a]indole scaffold. | ||
To date, there are three typical methods for the catalytic asymmetric construction of pyrrolo[1,2-a]indole scaffolds (Scheme 1).2–4 One is enantioselective cascade reactions of indole derivatives in the presence of either chiral organocatalysts or chiral metal/ligands (Scheme 1a),2 another is chiral metal-catalyzed intramolecular hydroacylation or C–H alkylation of indole derivatives (Scheme 1b),3 and the other is organocatalytic [3 + 2] cycloaddition of indole derivatives (Scheme 1c), which was represented by the elegant work of Dieter Enders (eqn (1)).4 Although the first two methods have been well-developed, the last one is still underdeveloped, which is limited to chiral N-heterocyclic carbene (NHC) catalyzed asymmetric [3 + 2] cycloaddition of 2-nitrovinylindoles with α-functionalized aldehydes (eqn (1)).4 Nevertheless, the catalytic asymmetric [3 + 2] cycloaddition is one of the most powerful methods for the enantioselective construction of five-membered rings. So, it is highly valuable and desired to further develop catalytic asymmetric [3 + 2] cycloadditions for the construction of enantioenriched pyrrolo[1,2-a]indole scaffolds.
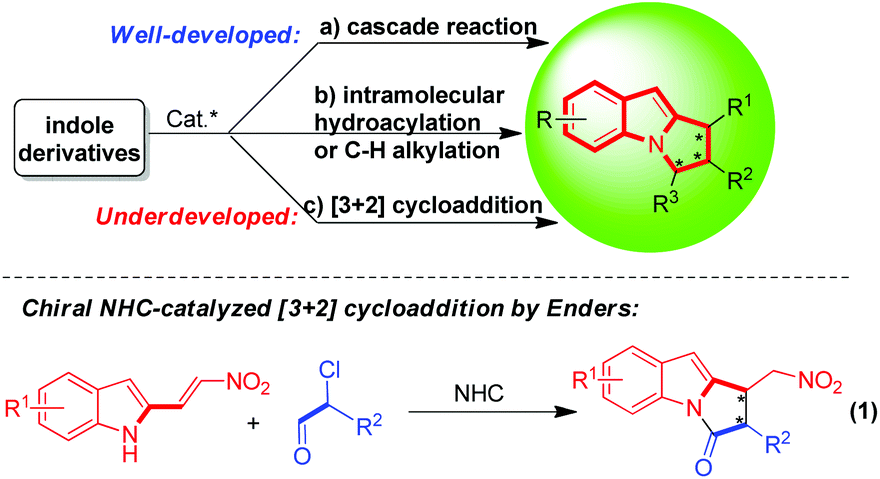 | ||
| Scheme 1 Profile of the catalytic asymmetric construction of pyrrolo[1,2-a]indole scaffolds. | ||
In the research field of asymmetric catalysis and synthesis, 2-vinylindoles have been widely utilized as a 4C or 2C building block for the enantioselective construction of six-membered cyclic rings fusing with an indole moiety.5 Recently, our group has designed and developed 3-alkyl-2-vinylindoles as a new class of 2-vinylindoles, which have accomplished some important transformations such as [3 + 2], [4 + 2], [5 + 2] cycloadditions and dearomatizations.6 However, 2-vinylindoles have seldom acted as nitrogen–carbon–carbon (NCC) building blocks for the construction of five-membered nitrogenous scaffolds.2a,b,4,7 Very recently, we have discovered an unexpected [3 + 2] cyclodimerization of 3-alkyl-2-vinylindoles in the presence of trifluoroacetic acid (TFA), wherein one of the 3-alkyl-2-vinylindole molecules acts as an NCC building block (eqn (2)).8
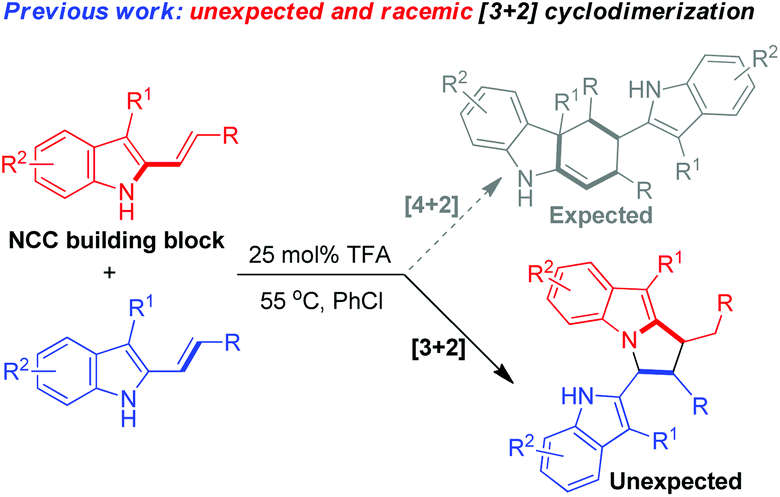 | (2) |
In order to develop catalytic asymmetric [3 + 2] cycloadditions for the construction of chiral pyrrolo[1,2-a]indole scaffolds, after we finished this work, we wondered whether this reaction could be performed in an enantioselective fashion in the presence of chiral phosphoric acids (CPAs), which have been proven to be a class of versatile organocatalysts for asymmetric catalysis and synthesis.9 In this design (Scheme 2), a CPA anion would form two hydrogen bonds with two molecules of 3-alkyl-2-vinylindoles, thus facilitating an enantioselective [3 + 2] cyclodimerization to generate a chiral pyrrolo[1,2-a]indole scaffold.
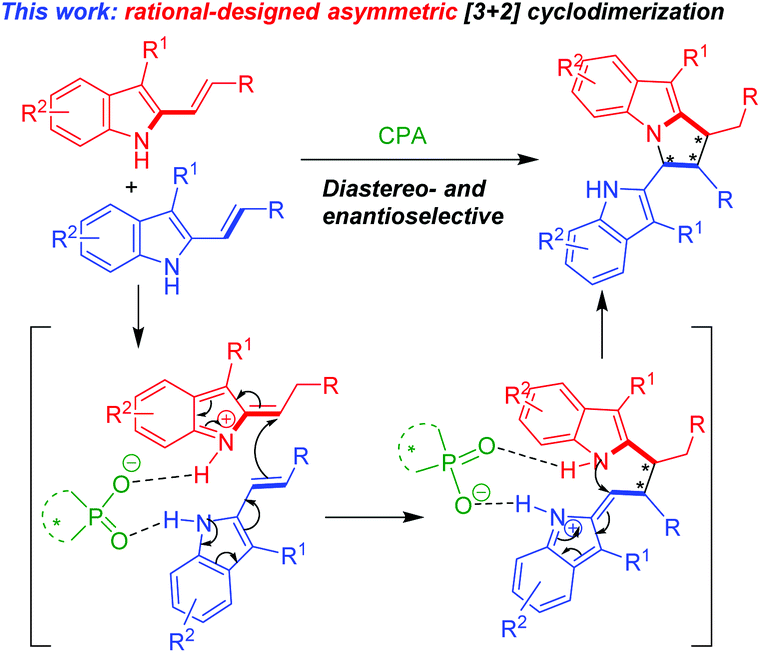 | ||
| Scheme 2 Design of the catalytic asymmetric [3 + 2] cyclodimerization. | ||
In this paper, we report the details of our investigation on this catalytic asymmetric [3 + 2] cyclodimerization of 3-alkyl-2-vinylindoles, which efficiently constructed biologically important pyrrolo[1,2-a]indole scaffolds in a diastereo- and enantioselective fashion (up to 98% yield, >95:5 dr, 98:2 er).
It should be mentioned that during our investigation, the group of Rodríguez developed a three-component [3 + 2] cyclization of indole-2-carboxaldehydes, amines and enol ethers under the catalysis of a chiral disulfonimide (CDS) (Scheme 3, eqn (3)).10 Schneider and co-worker established a CPA-catalyzed [3 + 2] cycloaddition of 2-indolyl carbinols with 3-alkyl-2-vinylindoles which were developed by our group (Scheme 3, eqn (4)).11 Both of the two creative approaches afforded pyrrolo[1,2-a]indoles in excellent diastereo- and enantioselectivities. Nevertheless, our strategy of catalytic asymmetric [3 + 2] cyclodimerization of 3-alkyl-2-vinylindoles still provides an alternative protocol for the enantioselective construction of pyrrolo[1,2-a]indole frameworks.
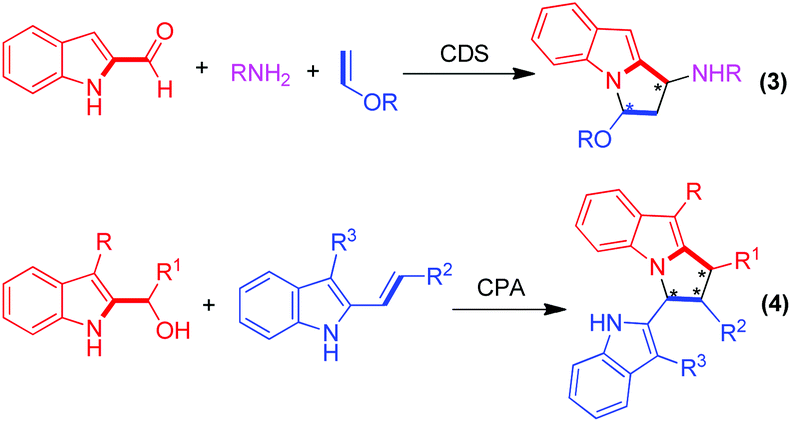 | ||
| Scheme 3 Recent studies on the enantioselective construction of pyrrolo[1,2-a]indole frameworks. | ||
Results and discussion
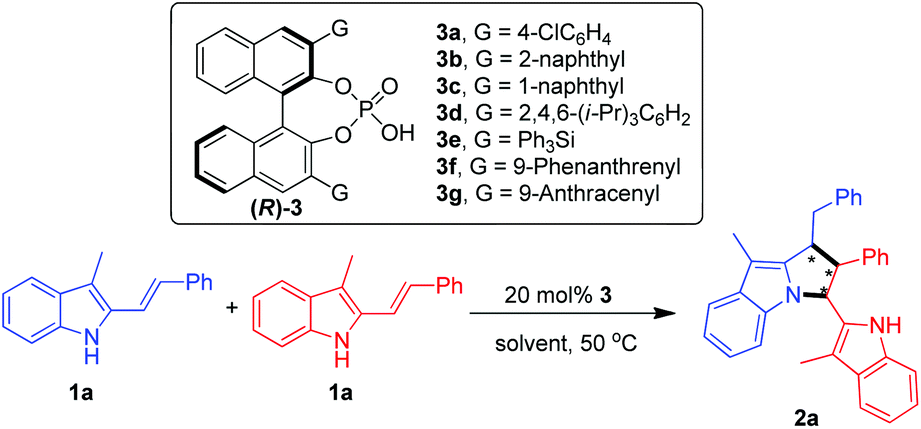
At the beginning, the [3 + 2] cyclodimerization of 3-methyl-2-vinylindole 1a was carried out under the catalysis of a series of chiral phosphoric acids 3 in chlorobenzene at 50 °C (Table 1, entries 1–7). The results revealed that the 3,3′-substituents of CPAs 3a–3g imposed an obvious effect on the catalytic activity of these chiral Brønsted acids. In detail, CPAs 3a–3c bearing smaller 3,3′-substituents could catalyze the [3 + 2] cyclodimerization to give the pyrrolo[1,2-a]indole product 2a in moderate yields but with low enantioselectivities (entries 1–3). In contrast, CPAs 3d–3f bearing larger 3,3′-substituents exhibited extremely low catalytic activity, which could hardly catalyze the [3 + 2] cyclodimerization (entries 4 and 5) although the enantioselectivities of the generated small amount of the products were considerable (entries 4 and 6). To our delight, CPA 3g with two bulky 3,3′-(9-anthracenyl) groups displayed the highest catalytic activity, which delivered the pyrrolo[1,2-a]indole product 2a at an acceptable yield of 56% and good enantioselectivity of 90:10 er (entry 7). So, CPA 3g was chosen as the optimal catalyst for subsequent screening of solvents (entries 8–17). At first, a wide range of halogenated arenes and arenes were evaluated (entries 8–13), which revealed that fluorobenzene was the most suitable reaction medium because the model reaction in fluorobenzene generated the desired product 2a at the highest yield of 64% along with the best enantioselectivity of 90:10 er (entry 8 vs. 9–13). Moreover, other representative solvents such as chloroalkane, ester, nitrile and ether were also examined (entries 14–17). However, most of these solvents failed to deliver the [3 + 2] cyclodimerization reaction (entries 15–17). Thus, fluorobenzene was selected as the optimal solvent for further optimization of conditions.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. | Solvent | Yieldb (%) | erc |
| a Unless otherwise indicated, the reaction was carried out at the 0.1 mmol scale and catalyzed by 20 mol% 3 in a solvent (1 mL) at 50 °C for 18 h. b Isolated yield and only one diastereomer were observed in all cases. c The enantiomeric ratio (er) value was determined by HPLC. N.R. = No reaction. | ||||
| 1 | 3a | PhCl | 50 | 54:46 |
| 2 | 3b | PhCl | 33 | 56:44 |
| 3 | 3c | PhCl | 30 | 72:28 |
| 4 | 3d | PhCl | 16 | 90:10 |
| 5 | 3e | PhCl | N.R. | — |
| 6 | 3f | PhCl | 7 | 90:10 |
| 7 | 3g | PhCl | 56 | 90:10 |
| 8 | 3g | PhF | 64 | 90:10 |
| 9 | 3g | PhBr | 46 | 90:10 |
| 10 | 3g | Toluene | 36 | 89:11 |
| 11 | 3g | o-Xylene | 34 | 90:10 |
| 12 | 3g | m-Xylene | 37 | 89:11 |
| 13 | 3g | p-Xylene | 39 | 89:11 |
| 14 | 3g | CH2ClCH2Cl | 35 | 89:11 |
| 15 | 3g | EtOAc | N.R. | — |
| 16 | 3g | CH3CN | N.R. | — |
| 17 | 3g | THF | N.R. | — |
In order to further improve the yield and the enantioselectivity, the reaction temperature was modulated (Table 2, entries 1–7). It was found that lowering the reaction temperature was detrimental to the yield although the enantioselectivity was maintained (entries 1–3), and no reaction occurred when the reaction temperature was lowered to 0 °C (entry 4). On the other hand, when the reaction temperature was elevated to 60 °C, a much higher yield of 91% was obtained with a slightly improved enantioselectivity of 91:9 er (entry 5). However, when the temperature was further increased, both the yield and the enantioselectivity were decreased to some extent (entries 6 and 7). So, the optimal reaction temperature was set at 60 °C. Besides, the catalyst loading was adjusted, which disclosed that either decreasing or increasing the catalyst loading could not benefit the yield and the enantioselectivity of this [3 + 2] cyclodimerization reaction (entries 8–10 vs. 5). Therefore, the final optimal condition was set in line with what entry 5 illustrated.
|
|
||||
|---|---|---|---|---|
| Entry | x | T (°C) | Yieldb (%) | erc |
| a Unless otherwise indicated, the reaction was carried out at the 0.1 mmol scale in a solvent (1 mL) for 18 h. b Isolated yield and only one diastereomer were observed in all cases. c The enantiomeric ratio (er) value was determined by HPLC. N.R. = No reaction. | ||||
| 1 | 20 | 50 | 64 | 90:10 |
| 2 | 20 | 40 | 40 | 90:10 |
| 3 | 20 | 30 | 28 | 90:10 |
| 4 | 20 | 0 | N.R. | — |
| 5 | 20 | 60 | 91 | 91:9 |
| 6 | 20 | 70 | 82 | 89:11 |
| 7 | 20 | 80 | 85 | 88:12 |
| 8 | 10 | 60 | 73 | 89:11 |
| 9 | 15 | 60 | 84 | 89:11 |
| 10 | 30 | 60 | 87 | 88:12 |
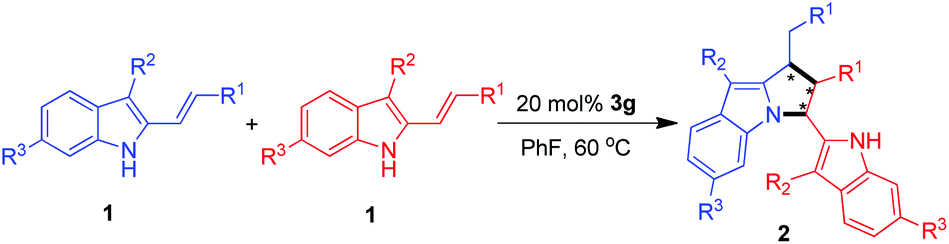
With the optimal conditions in hand, we then performed the investigation on the substrate scope of the catalytic asymmetric [3 + 2] cyclodimerizations. As shown in Table 3, this method was amenable to a wide range of 3-alkyl-2-vinylindoles 1 bearing different R3/R2/R1 substituents, which offered pyrrolo[1,2-a]indole derivatives 2 at generally high yields (up to 98%), excellent diastereoselectivities (all >95:5 dr) and good enantioselectivities (up to 98:2 er). Firstly, a series of 3-methyl-2-vinylindoles 1a–1h bearing a variety of terminal phenyl groups (R1) were employed to the reaction (entries 1–8), affording the [3 + 2] cyclodimerization products 2a–2h in overall excellent yields (84–98%) and high enantioselectivities (87:13 to 94:6 er). Apart from 3-methyl-2-vinylindoles, a number of 3-ethyl-2-vinylindoles 1i–1n bearing an ethyl group (R2) at the C3-position of the indole motif could also be utilized in the [3 + 2] cyclodimerization (entries 9–14), which gave the pyrrolo[1,2-a]indole products 2i–2n in excellent enantioselectivities (89:11 to 98:2 er). On comparison, it was found that 3-ethyl-substituted 2-vinylindoles exhibited higher capacity of controlling the enantioselectivity than their 3-methyl-substituted counterparts (entries 9–13 vs. 2–6). Notably, an alkyl group as exemplified by the methyl group could be employed as the terminal R1 substituent, and this substrate 1o smoothly underwent the [3 + 2] cyclodimerization at an acceptable yield of 60% albeit with a moderate enantioselectivity of 77:23 er (entry 15). We also employed substrates 1p–1r bearing an iso-propyl or an iso-butyl group as the terminal R1 substituent to the reaction (entries 16–18). However, substrate 1p bearing an iso-propyl group failed to undergo any reactions (entry 16), while substrates 1q–1r bearing an iso-butyl group smoothly underwent the [3 + 2] cyclodimerization to afford the corresponding products 2q–2r in good yields and excellent diastereoselectivities (entries 17 and 18). Unfortunately, the er values of the products 2q–2r were not determined (N.D.) because their HPLC methods could not be set up by using commonly used chiral columns. Moreover, the C6-chloro-substituted substrate 1s could successfully participate in the [3 + 2] cyclodimerization to afford the pyrrolo[1,2-a]indole product 2s in a good yield of 79% and a high enantioselectivity of 94:6 er (entry 19). Apart from methyl and ethyl groups, the benzyl group could serve as a suitable R2 substituent (entry 20), which delivered the corresponding product 2p in a high yield of 83% and a good stereoselectivity (>95:5 dr, 93:7 er).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 2 | R3/R2/R1 | Yieldb (%) | drc | erd |
| a Unless otherwise indicated, the reaction was carried out at the 0.1 mmol scale catalyzed by (R)-3g in fluorobenzene (1 mL) at 60 °C for 18 h. b Isolated yield. c The diastereomeric ratio (dr) was determined by 1H nuclear magnetic resonance (NMR) spectroscopy. d The enantiomeric ratio (er) value was determined by HPLC. e Catalyzed by (S)-3g. f The er value was not determined (N.D.) because the HPLC method could not be set up by using commonly used chiral columns. g The reaction time was 36 h. | |||||
| 1 | 2a | H/Me/C6H5 (1a) | 91 | >95:5 |
91:9 |
| 2 | 2b | H/Me/4-tBuC6H4 (1b) | 88 | >95:5 |
94:6 |
| 3 | 2c | H/Me/4-BrC6H4 (1c) | 87 | >95:5 |
91:9 |
| 4 | 2d | H/Me/4-ClC6H4 (1d) | 92 | >95:5 |
91:9 |
| 5 | 2e | H/Me/4-FC6H4 (1e) | 96 | >95:5 |
90:10 |
| 6 | 2f | H/Me/3-FC6H4 (1f) | 98 | >95:5 |
87:13 |
| 7e | ent-2g | H/Me/2-FC6H4 (1g) | 84 | >95:5 |
90:10 |
| 8e | ent-2h | H/Me/4-MeOC6H4 (1h) | 93 | >95:5 |
90:10 |
| 9e | ent-2i | H/Et/4-tBuC6H4 (1i) | 49 | >95:5 |
98:2 |
| 10 | 2j | H/Et/4-BrC6H4 (1j) | 78 | >95:5 |
96:4 |
| 11 | 2k | H/Et/4-ClC6H4 (1k) | 88 | >95:5 |
95:5 |
| 12e | ent-2l | H/Et/4-FC6H4 (1l) | 88 | >95:5 |
91:9 |
| 13 | 2m | H/Et/3-FC6H4 (1m) | 52 | >95:5 |
93:7 |
| 14 | 2n | H/Et/3-MeC6H4 (1n) | 79 | >95:5 |
89:11 |
| 15 | 2o | H/Me/Me (1o) | 60 | >95:5 |
77:23 |
| 16 | 2p | H/Me/iPr (1p) | N.R. | — | — |
| 17 | 2q | H/Me/iBu (1q) | 80 | >95:5 |
N.D.f |
| 18g | 2r | H/Et/iBu (1r) | 74 | >95:5 |
N.D.f |
| 19 | 2s | Cl/Me/C6H5 (1s) | 79 | >95:5 |
94:6 |
| 20e | ent-2t | H/Bn/C6H5 (1t) | 83 | >95:5 |
93:7 |
Moreover, under the standard conditions, we studied the possibility of catalytic asymmetric [3 + 2] heterodimerization by using two different 3-alkyl-2-vinylindoles 1b and 1e as substrates. As shown in Scheme 4, this reaction could afford the [3 + 2] heterodimerization products 2be and 2be′ in a total yield of 50% and good stereoselectivities. Meanwhile, we also found that a small amount of [3 + 2] homodimerization products 2b and 2e was simultaneously generated in good diastereo- and enantioselectivities. This result indicates that the catalytic asymmetric [3 + 2] heterodimerization is feasible albeit with unsatisfactory chemoselectivity.
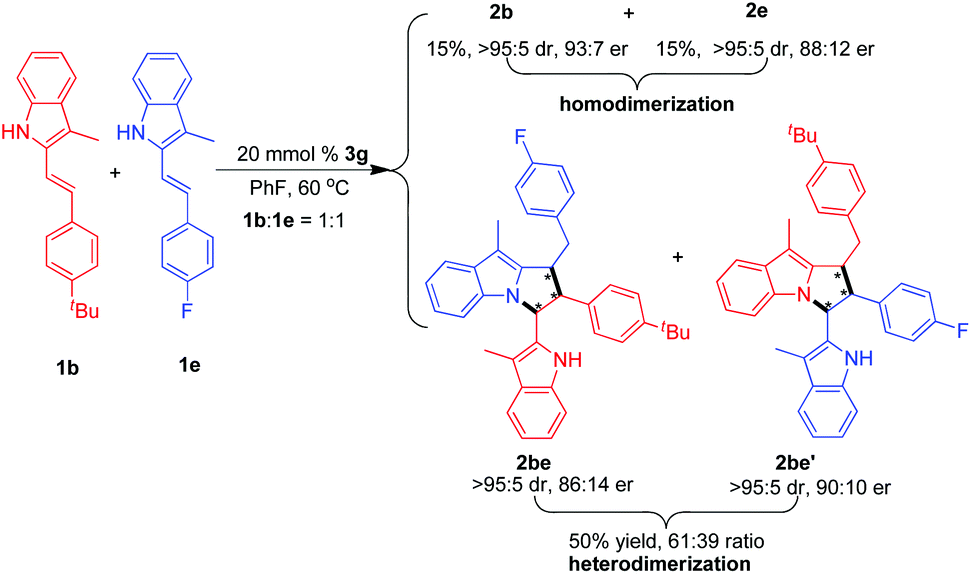 | ||
| Scheme 4 Investigation on catalytic asymmetric [3 + 2] heterodimerization. | ||
The relative configuration of product 2i was identified to be (trans, trans) by single-crystal X-ray diffraction analysis of its corresponding racemic product (Scheme 5).12 So, the relative configurations of other products 2 were assigned to be (trans, trans) by analogy. Besides, we made great efforts to cultivate single crystals from enantioselective products 2, but we failed. Fortunately, during the cultivation of single crystals from product 2l, which was generated in the presence of (R)-3g, we accidentally found that a small amount of single crystals was generated. Interestingly, the structure of the single crystal was finally discovered to be that of compound 4, whose absolute configuration was determined to be (R,S,R). We deduced that compound 4 was formed via oxidation of product 2l in air during the process of cultivating single crystals in a mixed solvent of petrol ether and dichloromethane (eqn (5)). Since the transformation of product 2l to compound 4 did not affect the three chiral centers, the absolute configuration of product 2l was deduced to be (R,S,R).
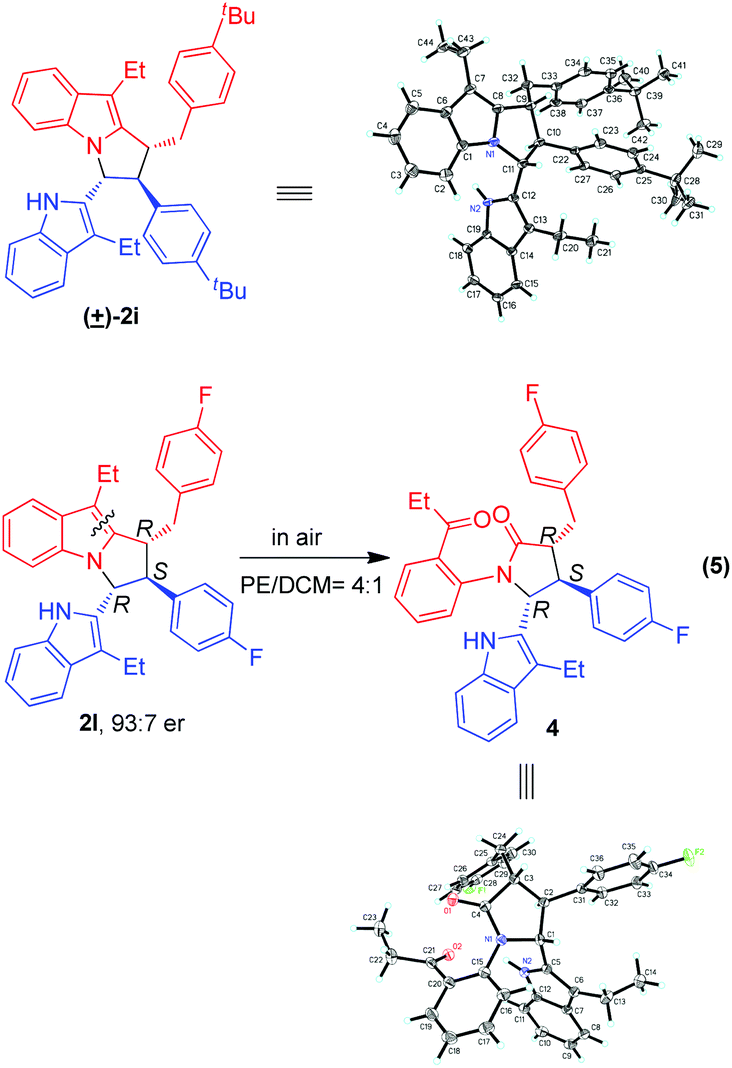 | ||
| Scheme 5 X-ray structures of racemic 2i and chiral compound 4 to determine the relative and absolute configurations of products 2. | ||
Before we obtained the single crystal of compound 4, in order to determine the absolute configuration of products 2, we also performed a conformational analysis for (R,S,R)-2k based on the relative configurations of products 2 by using the MMFF94S force field.13,14 All of the conformers were conducted for optimizations using density functional theory (DFT) at the B3LYP/6-311+G(d) level in the gas phase on the basis of the well-summarized reports. Totally 14 B3LYP/6-311+G(d)-optimized conformers with a relative energy of 0–2.5 kcal mol−1 were found and used for electronic circular dichroism (ECD) calculations using time-dependent DFT (TDDFT) at the B3LYP/6-311+G(d) level. The predicted ECD spectrum for (R,S,R)-2k fits the experimental ECD spectrum of (−)-2k well (Fig. 2) with the Boltzmann statistics. This exhibits that (−)-2k should have the (R,S,R) absolute configuration. Furthermore, optical rotation (OR) computations were then carried out at the same level in the gas phase.15 The simulated OR values were −38.6 for (R,S,R)-2k. The experimental OR value was −118.6 in acetone. Both have the same OR sign. Considering the evidence of ECD and OR, the absolute configuration of (−)-2k is assigned as (R,S,R), which is in accordance with the absolute configuration of product 2l. So, based on the experimental results and the theoretical calculations, the absolute configuration of products 2 was confirmed to be (R,S,R).
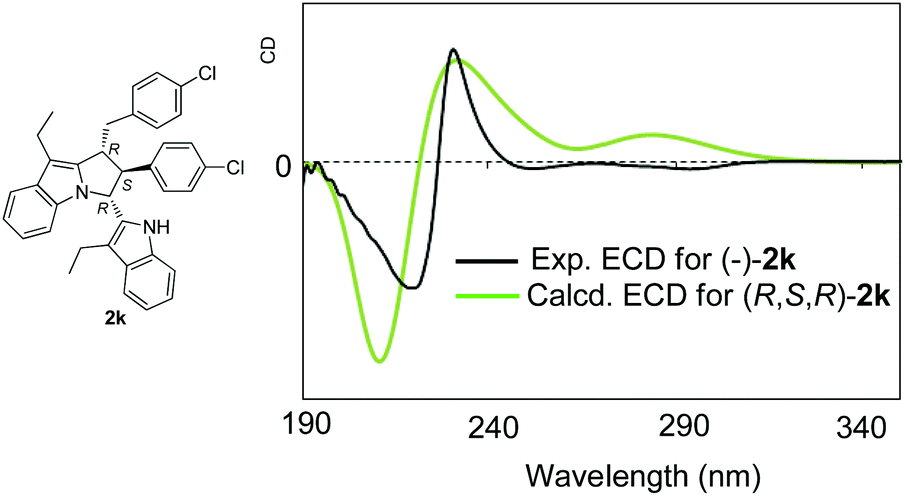 | ||
| Fig. 2 Comparison of the experimental ECD spectrum of (−)-2k with the predicted ECD of (R,S,R)-2k at the B3LYP/6-311+G(d) level. | ||
Based on the experimental results, a possible transition state and activation mode to explain the stereochemistry and the chirality induction of the reaction are illustrated in Scheme 6. In the presence of CPA (R)-3g, one molecule of the 3-alkyl-2-vinylindole was converted into a vinyliminium intermediate. Then, both the vinyliminium intermediate and another molecule of the 3-alkyl-2-vinylindole were simultaneously activated by the anion of (R)-3gvia the interaction of a hydrogen bond and an ion pair. Due to the rigid (R)-BINOL backbone and the bulky 3,3′-(9-anthracenyl) substituents of the catalyst (R)-3g, a diastereo- and enantioselective vinylogous Michael addition/intramolecular aza-Michael addition sequence occurred to accomplish the stereoselective [3 + 2] cyclodimerization and to construct the chiral pyrrolo[1,2-a]indole scaffold 2 with the observed (R,S,R)-configuration.
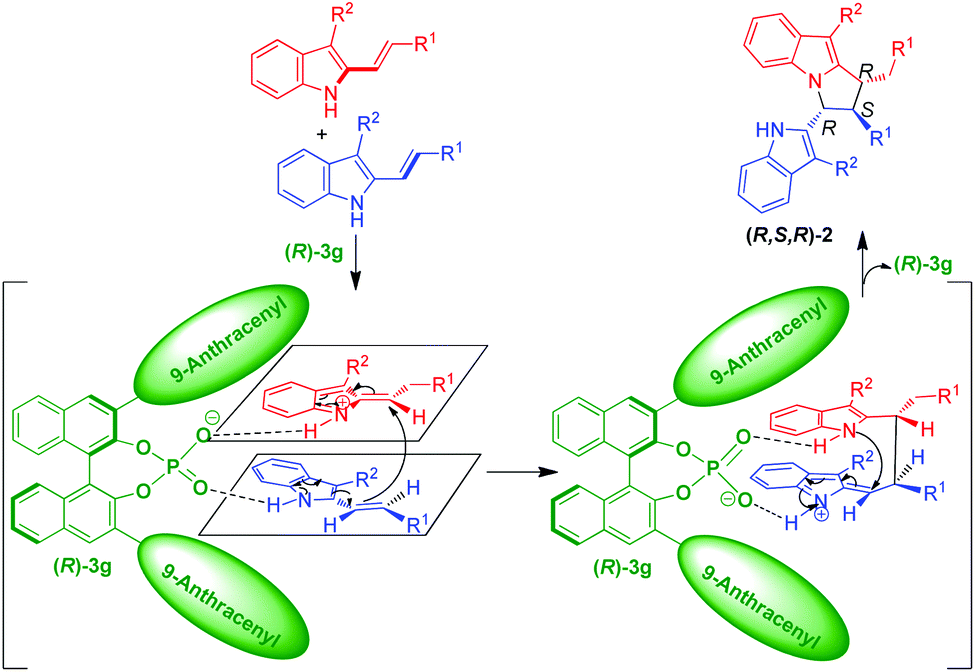 | ||
| Scheme 6 Suggested transition state to explain the stereochemistry. | ||
In order to gain additional insights into the reaction mechanism, some control experiments were carried out (Scheme 7). Firstly, to verify the possible reaction pathway proposed in Scheme 2, we performed a control experiment involving 2-indolyl carbinol as reported by Schneider.11 As illustrated in eqn (6), 2-indolyl carbinol 5 was utilized as a substrate to react with 3-alkyl-2-vinylindole 1a under the standard conditions. In the presence of CPA 3g, 2-indolyl carbinol 5 was easily transformed into carbocation and vinyliminium intermediates via dehydration, which could further react with 3-alkyl-2-vinylindole 1avia the cascade reaction of vinylogous Michael addition/intramolecular aza-Michael addition. As expected, this reaction smoothly occurred to afford the desired pyrrolo[1,2-a]indole product 6 in a high yield of 86%, which validated the proposed reaction pathway to some extent.
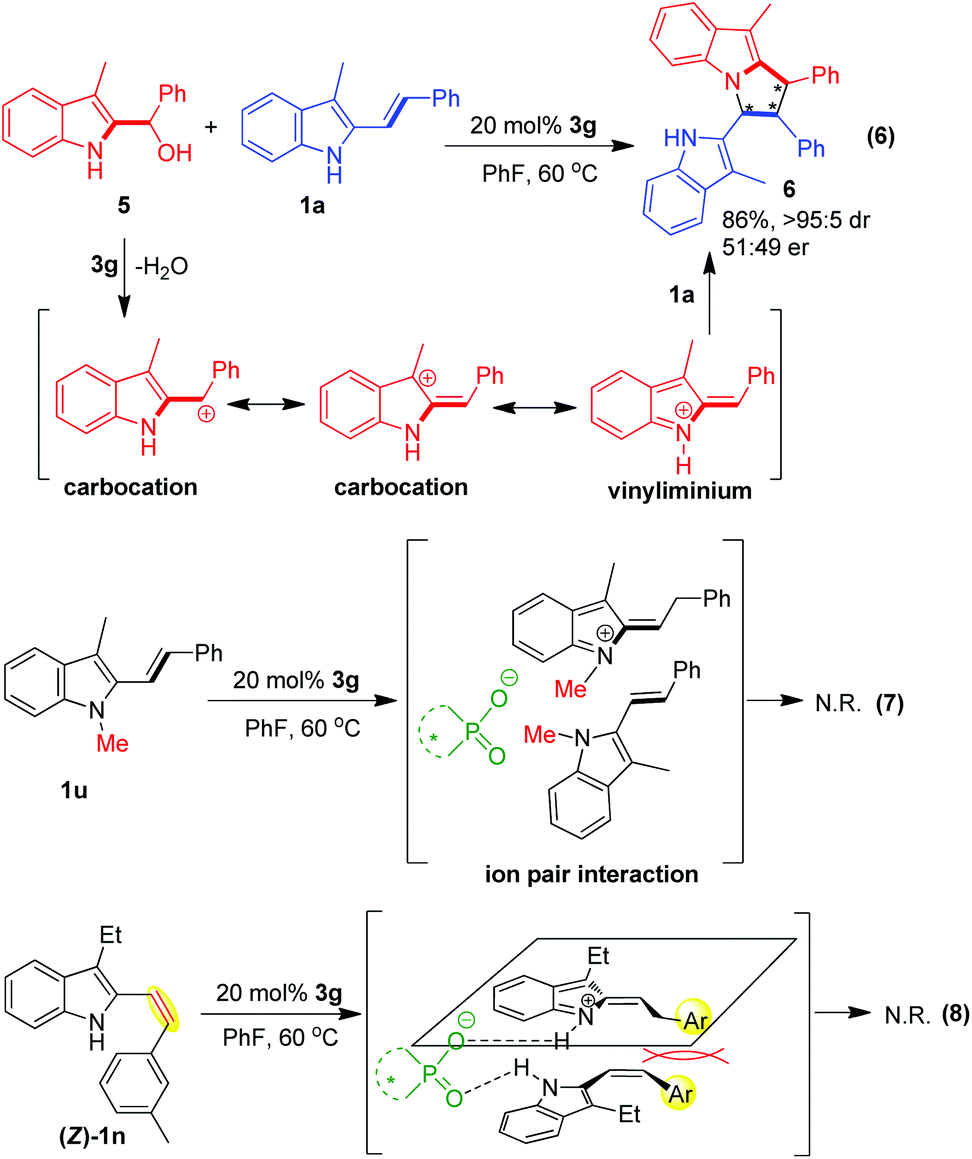 | ||
| Scheme 7 Control experiments. | ||
Secondly, to test the double hydrogen-bonding activation mode depicted in Scheme 2, another control experiment using N-methyl protected 3-alkyl-2-vinylindole 1u was performed under the standard conditions (eqn (7)). In this case, CPA 3g failed to form two hydrogen bonds with the two reaction partners, and only an ion pair interaction could be generated between the CPA anion and the vinyliminium intermediate. In addition, the existence of the N-methyl group would block the nitrogen atom of 1u to act as a NCC building block. So, it is not surprising that no reaction occurred in this case.
Thirdly, in order to investigate the effect of the (Z/E)-configuration of 3-alkyl-2-vinylindoles on the reaction, we carried out a control experiment by using (Z)-vinylindole 1n as a substrate under the standard conditions (eqn (8)). However, no reaction occurred, which implies that the (Z/E)-configuration of 3-alkyl-2-vinylindoles plays an important role in controlling the reactivity. In the suggested transition state, the two terminal aromatic groups of (Z)-vinylindoles were parallel to each other, which might generate a steric repulsion between them and therefore hampered the desired [3 + 2] cyclodimerization.
Finally, to demonstrate the applicability of this [3 + 2] cyclodimerization reaction, a relatively large-scale synthesis of product 2k was performed (Scheme 8), which offered the pyrrolo[1,2-a]indole product 2k in a good yield of 71%, an excellent diastereoselectivity of >95:5 dr and a high enantioselectivity of 95:5 er. Compared to the small-scale reaction (Table 3, entry 11), although the yield of the larger scale reaction was decreased but still on an acceptable level, the stereoselectivity was well maintained, which could be applicable to the preparative synthesis of chiral pyrrolo[1,2-a]indole products.
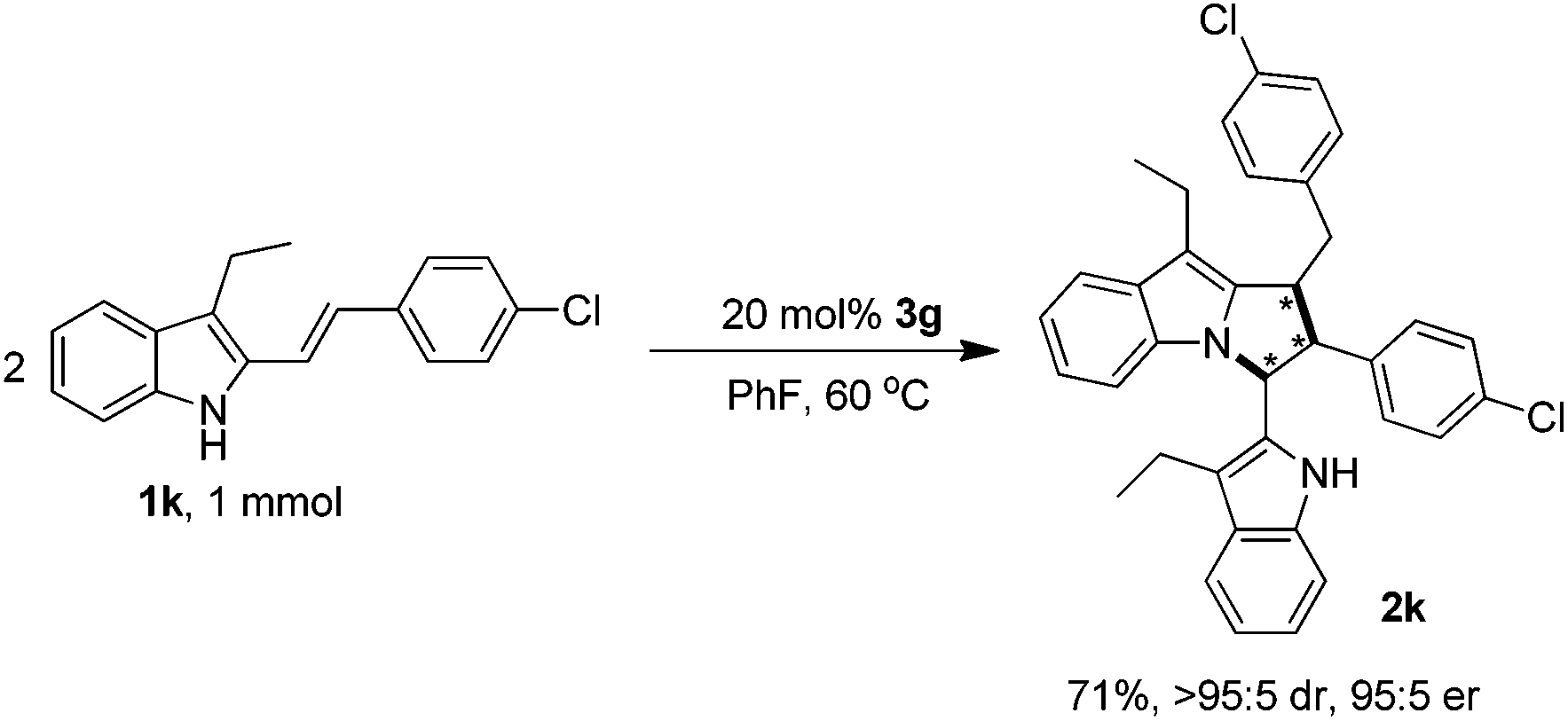 | ||
| Scheme 8 A large scale synthesis of product 2k. | ||
Conclusions
In summary, we have established a catalytic asymmetric [3 + 2] cyclodimerization of 3-alkyl-2-vinylindoles, which efficiently constructed the pyrrolo[1,2-a]indole scaffold with three contiguous stereogenic centers in a diastereo- and enantioselective fashion (up to 98% yield, >95:5 dr, 98:2 er). This reaction not only represents the first catalytic asymmetric version of this type of [3 + 2] cyclodimerization, but also supplies important examples for using 2-vinylindoles as NCC building blocks in asymmetric catalysis and synthesis. More importantly, this reaction also provides an efficient method for enantioselectively constructing biologically significant pyrrolo[1,2-a]indole scaffolds.
Experimental
General information
1H and 13C NMR spectra were measured at 400 and 100 MHz, respectively. The solvent used for NMR spectroscopy was CDCl3, using tetramethylsilane as the internal reference. HRMS (ESI) was determined by using a HRMS/MS instrument. Enantiomeric ratios (er) were determined by chiral high-performance liquid chromatography (chiral HPLC). The chiral columns used for the determination of enantiomeric excesses by chiral HPLC were Chiralpak OD-H, AD-H and AS-H columns. Optical rotation values were measured with instruments operating at λ = 589 nm, corresponding to the sodium D line at the temperatures indicated. Analytical grade solvents for the column chromatography were used after distillation. Substrates 1 were synthesized according to our previously reported method.6aGeneral procedure for the synthesis of pyrrolo[1,2-a]indoles 2 via catalytic asymmetric [3 + 2] cyclodimerizations
To the mixture of 3-alkyl-2-vinylindoles 1 (0.1 mmol) and CPA 3g (0.02 mmol) was added fluorobenzene (1 mL), which was stirred at 60 °C for 18 h. After the completion of the reaction, which was indicated by TLC, the reaction mixture was directly purified through preparative thin layer chromatography on silica gel to afford pure products 2.:5 dr; white sticky oil; [α]20D = −52.9 (c 0.24, acetone); 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.30–7.27 (m, 3H), 7.26–7.20 (m, 3H), 7.13–7.05 (m, 8H), 7.00 (s, 1H), 6.91 (t, J = 7.6 Hz, 1H), 6.64 (d, J = 8.1 Hz, 1H), 5.43 (d, J = 7.3 Hz, 1H), 4.02–3.97 (m, 1H), 3.71 (t, J = 7.4 Hz, 1H), 3.44 (dd, J = 13.9, 5.1 Hz, 1H), 3.08 (dd, J = 13.9, 5.5 Hz, 1H), 2.41 (s, 3H), 1.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 142.3, 141.1, 138.2, 135.7, 134.1, 132.7, 131.7, 130.1, 129.0, 128.8, 128.5, 127.9, 127.2, 126.7, 122.0, 120.8, 119.3, 119.1, 118.6, 118.5, 110.9, 110.0, 109.3, 102.4, 61.0, 60.6, 46.1, 37.6, 9.0, 8.0; IR (KBr): 3677, 3671, 3630, 2987, 1831, 1773, 1735, 1555, 1458, 1264, 896, 743 cm−1; ESI FTMS exact mass calcd for (C34H30N2 − H)− requires m/z 465.2330, found m/z 465.2339; enantiomeric ratio: 91:9, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 10.54 min (major), tR = 12.16 min (minor).
:5 dr; white sticky oil; [α]20D = −71.8 (c 0.31, acetone); 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 7.9 Hz, 1H), 7.53–7.48 (m, 1H), 7.39 (s, 1H), 7.30–7.27 (m, 2H), 7.25–7.21 (m, 2H), 7.14–7.01 (m, 8H), 6.94–6.88 (m, 1H), 6.64 (d, J = 8.1 Hz, 1H), 5.42 (d, J = 7.8 Hz, 1H), 3.97 (dd, J = 12.8, 5.6 Hz, 1H).3.68 (t, J = 7.9 Hz, 1H), 3.39 (dd, J = 14.0, 5.3 Hz, 1H), 3.07 (dd, J = 14.0, 5.7 Hz, 1H), 2.39 (s, 3H), 1.84 (s, 3H), 1.32 (d, J = 6.5 Hz, 18H); 13C NMR (100 MHz, CDCl3) δ 150.1, 149.4, 142.6, 137.3, 135.6, 135.2, 134.1, 132.8, 131.8, 129.6, 129.1, 127.6, 125.5, 125.3, 121.9, 120.7, 119.3, 119.1, 118.6, 118.5, 110.9, 110.1, 109.4, 102.6, 61.1, 60.3, 45.9, 36.8, 34.5, 34.4, 31.4, 31.4, 8.9, 7.9; IR (KBr): 3443, 2961, 2925, 1615, 1539, 1458, 1384, 1362, 1264, 826, 740, 699 cm−1; ESI FTMS exact mass calcd for (C42H46N2 − H)− requires m/z 577.3582, found m/z 577.3590; enantiomeric ratio: 94:6, determined by HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 99.5/0.5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 9.09 min (major), tR = 11.50 min (minor).
:5 dr; pale yellow sticky oil; [α]20D = −79.6 (c 0.48, acetone); 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.3 Hz, 2H), 7.21–7.07 (m, 5H), 6.95–6.90 (m, 5H), 6.64 (d, J = 8.1 Hz, 1H), 5.39 (d, J = 7.5 Hz, 1H), 4.04–3.82 (m, 1H), 3.60 (t, J = 7.6 Hz, 1H), 3.31 (dd, J = 14.0, 5.8 Hz, 1H), 3.10 (dd, J = 14.0, 5.5 Hz, 1H), 2.40 (s, 3H), 1.90 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.5, 139.5, 137.0, 135.8, 134.0, 132.8, 131.9, 131.6, 131.6, 131.0, 129.5, 129.0, 122.3, 121.3, 121.2, 120.7, 119.6, 119.4, 118.7, 118.7, 111.2, 110.1, 109.7, 102.9, 60.9, 60.6, 45.8, 37.3, 9.0, 8.2; IR (KBr): 3690, 3648, 3447, 2923, 1653, 1637, 1149, 1010, 799, 746, 670 cm−1; ESI FTMS exact mass calcd for (C34H28Br2N2 − H)− requires m/z 621.0540, found m/z 621.0550; enantiomeric ratio: 91:9, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 7.01 min (minor), tR = 10.09 min (major).
:5 dr; white sticky oil; [α]20D = −96.4 (c 0.42, acetone); 1H NMR (400 MHz, CDCl3) δ 7.60 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.28–7.26 (m, 1H), 7.25–7.24 (m, 1H), 7.20–7.09 (m, 7H), 7.01 (t, J = 7.9 Hz, 4H), 6.94 (t, J = 7.5 Hz, 1H), 6.64 (d, J = 8.1 Hz, 1H), 5.39 (d, J = 7.5 Hz, 1H), 3.97–3.90 (m, 1H), 3.62 (t, J = 7.6 Hz, 1H), 3.33 (dd, J = 14.0, 5.8 Hz, 1H), 3.12 (dd, J = 14.0, 5.5 Hz, 1H), 2.41 (s, 3H), 1.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.5, 139.0, 136.5, 135.8, 134.0, 133.2, 132.8, 132.6, 131.2, 131.1, 129.2, 129.0, 128.6, 122.3, 121.2, 119.6, 119.4, 118.7, 118.6, 111.1, 110.1, 109.7, 102.9, 61.0, 60.5, 45.9, 37.3, 9.0, 8.1; IR (KBr): 3646, 3626, 3443, 2915, 1696, 1682, 1651, 1634, 1457, 1150, 826, 743, 670 cm−1; ESI FTMS exact mass calcd for (C34H28Cl2N2 − H)− requires m/z 533.1551, found m/z 533.1562; enantiomeric ratio: 91:9, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 6.28 min (minor), tR = 8.82 min (major).
:5 dr; white sticky oil; [α]20D = −70.4 (c 0.41, acetone); 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 7.8 Hz, 1H), 7.25–7.21 (m, 1H), 7.20–7.10 (m, 4H), 7.07–7.00 (m, 4H), 6.98–6.88 (m, 5H), 6.64 (d, J = 8.1 Hz, 1H), 5.39 (d, J = 7.6 Hz, 1H), 3.95–3.87 (m, 1H), 3.64 (t, J = 7.7 Hz, 1H), 3.33 (dd, J = 14.1, 5.9 Hz, 1H), 3.15 (dd, J = 14.1, 5.5 Hz, 1H), 2.41 (s, 3H), 1.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.0 (J = 244.5 Hz), 161.7 (J = 243.8 Hz), 141.8, 136.3 (J = 3.2 Hz), 135.7, 134.0, 133.8 (J = 3.3 Hz), 132.8, 131.3 (J = 7.6 Hz), 131.2, 129.4 (J = 8.0 Hz), 129.0, 122.3, 121.1, 119.5, 119.3, 118.7, 118.6, 115.7 (J = 21.2 Hz), 115.3 (J = 20.9 Hz), 111.0, 110.1, 109.6, 102.8, 61.2, 60.5, 46.2, 37.1, 9.0, 8.0; 19F NMR (376 MHz, CDCl3) δ −115.07, −115.91; IR (KBr): 3646, 3626, 3444, 2963, 1645, 1634, 1508, 1456, 1223, 1158, 832, 744 cm−1; ESI FTMS exact mass calcd for (C34H28F2N2 − H)− requires m/z 501.2142, found m/z 501.2129; enantiomeric ratio: 90:10, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 5.95 min (major), tR = 8.58 min (minor).
:5 dr; white sticky oil; [α]20D = −60.9 (c 0.31, acetone); 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 8.0 Hz, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.42 (s, 1H), 7.24–7.09 (m, 6H), 6.98–6.74 (m, 7H), 6.64 (d, J = 8.1 Hz, 1H), 5.44 (d, J = 7.6 Hz, 1H), 3.95 (dd, J = 13.1, 6.3 Hz, 1H), 3.65 (t, J = 7.7 Hz, 1H), 3.33–3.18 (m, 2H), 2.37 (s, 3H), 1.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 162.9 (J = 245.3 Hz), 162.7 (J = 244.9 Hz), 143.0, 142.9 (J = 7.0 Hz), 141.5, 140.7 (J = 7.2 Hz), 135.7, 134.0, 132.7, 131.0, 130.3 (J = 8.3 Hz), 129.9 (J = 8.3 Hz), 129.1, 125.3 (J = 2.7 Hz), 123.7 (J = 2.8 Hz), 122.3, 121.2, 119.6, 119.4, 118.7, 116.7, 116.5, 114.7, 114.5, 114.4, 114.2, 113.7, 113.5, 111.0, 110.1, 109.8, 103.0, 61.4, 61.0, 45.9, 38.0, 8.9, 8.1; 19F NMR (376 MHz, CDCl3) δ −112.45, −112.9; IR (KBr): 3676, 3649, 3448, 2924, 1637, 1616, 1457, 1361, 1148, 1059, 890, 745, 689 cm−1; ESI FTMS exact mass calcd for (C34H28F2N2 − H)− requires m/z 501.2142, found m/z 501.2148; enantiomeric ratio: 87:13, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 98/2, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 7.90 min (minor), tR = 14.27 min (major).
:5 dr; white sticky oil; [α]20D = +78.1 (c 0.26, acetone); 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 7.9 Hz, 1H), 7.52–7.43 (m, 2H), 7.16–7.05 (m, 7H), 6.92–6.85(m, 6H), 6.66 (d, J = 8.1 Hz, 1H), 5.57 (d, J = 7.4 Hz, 1H), 4.16–4.05 (m, 1H), 3.86 (t, J = 7.3 Hz, 1H), 3.53 (dd, J = 13.9, 5.8 Hz, 1H), 3.09 (dd, J = 13.9, 7.6 Hz, 1H), 2.32 (s, 3H), 1.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 161.4 (J = 243.4 Hz), 160.7 (J = 245.0 Hz), 142.4, 135.7, 134.0, 132.7, 131.5, 131.4 (J = 7.4 Hz), 130.1 (J = 4.4 Hz), 129.2, 128.8 (J = 8.4 Hz), 128.4 (J = 8.2 Hz), 127.5, 125.5, 124.2 (J = 3.4 Hz), 123.9 (J = 3.5 Hz), 122.1, 121.0, 119.4, 119.2, 118.6 (J = 4.8 Hz), 115.7, 115.5, 115.3, 115.1, 111.0, 110.1, 109.6, 59.3, 57.3, 43.9, 32.4, 8.7, 7.9; 19F NMR (376 MHz, CDCl3) δ −115.68, −116.98; IR (KBr): 3648, 3628, 3587, 3566, 3446, 2963, 1716, 1647, 1490, 1458, 1231, 1105, 742, 669 cm−1; ESI FTMS exact mass calcd for (C34H28F2N2 − H)− requires m/z 501.2142, found m/z 501.2144; enantiomeric ratio: 90:10, determined by HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 98/2, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 5.78 min (minor), tR = 7.62 min (major).
:5 dr; yellow sticky oil; [α]20D = +27.5 (c 0.16, acetone); 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.9 Hz, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.11–6.97 (m, 7H), 6.95–6.86 (m, 3H), 6.83 (d, J = 8.5 Hz, 2H), 6.77 (d, J = 8.4 Hz, 2H), 6.63 (d, J = 8.1 Hz, 1H), 5.37 (d, J = 7.3 Hz, 1H), 3.91 (d, J = 5.9 Hz, 1H), 3.80 (d, J = 5.7 Hz, 6H), 3.66 (t, J = 7.3 Hz, 1H), 3.41 (dd, J = 14.1, 4.5 Hz, 1H), 2.97 (dd, J = 14.1, 5.2 Hz, 1H), 2.43 (s, 3H), 1.92 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.7, 158.3, 142.3, 135.7, 134.0, 133.2, 132.7, 131.9, 131.1, 130.1, 129.4, 128.9, 128.8, 121.9, 120.6, 119.2, 119.0, 118.5, 118.4, 114.1, 113.9, 113.8, 110.8, 109.9, 109.2, 102.1, 61.0, 59.5, 55.2, 55.2, 46.3, 36.3, 29.7, 8.9, 8.1; IR (KBr): 3680, 3341, 2865, 1802, 1735, 1594, 1420, 1132, 806, 754 cm−1; ESI FTMS exact mass calcd for (C36H34N2O2 − H)− requires m/z 527.2699, found m/z 577.2690; enantiomeric ratio: 90:10, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 22.990 min (minor), tR = 29.503 min (major).
:5 dr; white solid; [α]20D = +107.5 (c 0.28, acetone); 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.0 Hz, 1H), 7.56–7.49 (m, 1H), 7.36 (s, 1H), 7.25–7.20 (m, 4H), 7.15–7.03 (m, 6H), 7.00 (d, J = 8.3 Hz, 2H), 6.92–6.84 (m, 1H), 6.59 (d, J = 8.1 Hz, 1H), 5.42 (d, J = 8.2 Hz, 1H), 3.99–3.94 (m, 1H), 3.65 (t, J = 8.2 Hz, 1H), 3.38 (dd, J = 14.1, 5.4 Hz, 1H), 3.12 (dd, J = 14.2, 5.5 Hz, 1H), 2.93–2.86 (m, 2H), 2.59–2.25 (m, 2H), 1.38 (t, J = 7.5 Hz, 3H), 1.29 (d, J = 5.4 Hz, 18H), 0.68 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 150.1, 149.4, 142.3, 137.1, 135.9, 135.2, 133.2, 133.0, 131.1, 129.5, 128.1, 127.8, 125.5, 125.2, 121.9, 120.7, 119.3, 119.0, 118.9, 118.8, 116.7, 111.0, 110.2, 109.3, 60.9, 60.5, 46.1, 36.9, 34.5, 34.4, 31.4, 31.3, 17.7, 17.1, 15.5, 15.2; IR (KBr): 3651, 3630, 2962, 1458, 1390, 1360, 1275, 1261, 1050, 763, 749 cm−1; ESI FTMS exact mass calcd for (C44H50N2 − H)− requires m/z 605.3895, found m/z 605.3888; enantiomeric ratio: 98:2, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 4.16 min (major), tR = 6.04 min (minor).
:5 dr; white sticky oil; [α]20D = −83.4 (c 0.50, acetone); 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.3 Hz, 2H). 7.24–7.09 (m, 5H), 7.00 (d, J = 8.3 Hz, 2H), 6.97–6.88 (m, 3H), 6.66 (d, J = 8.1 Hz, 1H), 5.44 (d, J = 7.6 Hz, 1H), 3.95 (dd, J = 12.9, 6.0 Hz, 1H), 3.64 (t, J = 7.5 Hz, 1H), 3.31 (dd, J = 14.1, 6.2 Hz, 1H), 3.21 (dd, J = 14.1, 5.2 Hz, 1H), 3.04–2.85 (m, 2H), 2.55–2.35 (m, 2H), 1.44 (t, J = 7.5 Hz, 3H), 0.92 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.2, 139.7, 137.1, 136.1, 133.2, 131.0, 131.9, 131.6, 131.48, 130.5, 129.6, 128.0, 122.3, 121.2, 120.8, 119.6, 119.4, 119.0, 119.0, 116.9, 111.3, 110.2, 109.7, 60.8, 46.2, 37.7, 17.8, 17.2, 15.6; IR (KBr): 3690, 3649, 3450, 2961, 2925, 1637, 1488, 1458, 1149, 1072, 1010, 798, 743 cm−1; ESI FTMS exact mass calcd for (C36H32Br2N2 − H)− requires m/z 649.0853, found m/z 649.0855; enantiomeric ratio: 96:4, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 5.22 min (minor), tR = 7.22 min (major).
:5 dr; white sticky oil; [α]20D = −118.6 (c 0.35, acetone); 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.25–7.15 (m, 7H), 7.13–7.06 (m, 2H), 7.05–6.99 (m, 2H), 6.98–6.88 (m, 3H), 6.63 (d, J = 8.1 Hz, 1H), 5.40 (d, J = 7.5 Hz, 1H), 3.92 (dd, J = 12.9, 6.0 Hz, 1H), 3.62 (t, J = 7.5 Hz, 1H), 3.30 (dd, J = 14.1, 6.2 Hz, 1H), 3.19 (dd, J = 14.1, 5.2 Hz, 1H), 3.00–2.80 (m, 2H), 2.49–2.31 (m, 2H), 1.40 (t, J = 7.5 Hz, 3H), 0.88 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 141.2, 139.2, 136.5, 136.1, 133.1, 132.9, 132.7, 131.1, 130.5 129.2, 128.9, 128.6, 128.0, 122.3, 121.2, 119.6, 119.3, 119.0, 118.9, 116.9, 111.2, 110.2, 109.7, 60.8, 60.7, 46.3, 37.6, 17.8, 17.1, 15.5; IR (KBr): 3691, 3649, 3448, 2962, 1637, 1457, 1275, 1260, 1090, 798, 749, 669 cm−1; ESI FTMS exact mass calcd for (C36H32Cl2N2 − H)− requires m/z 561.1864, found m/z 561.1866; enantiomeric ratio: 95:5, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 16.72 min (minor), tR = 19.91 min (major).
:5 dr; yellow sticky oil; [α]20D = +91.2 (c 0.32, acetone); 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.0 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.24–7.03 (m, 7H), 7.02–6.85 (m, 7H), 6.62 (d, J = 8.1 Hz, 1H), 5.40 (d, J = 7.7 Hz, 1H), 3.93 (dd, J = 13.2, 5.9 Hz, 1H), 3.64 (t, J = 7.7 Hz, 1H), 3.38–3.15 (m, 2H), 2.96–2.87 (m, 2H), 2.46–2.34 (m, 2H), 1.41 (t, J = 7.5 Hz, 3H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 161.9 (J = 244.5 Hz), 161.7 (J = 244.0 Hz), 141.4, 136.4 (J = 3.2 Hz), 136.1, 133.8 (J = 3.3 Hz), 133.1, 132.9, 131.1 (J = 7.7 Hz), 130.6, 129.4 (J = 8.0 Hz), 128.0, 122.3, 121.1, 119.5, 119.3, 119.0, 118.9, 116.9, 115.6 (J = 21.3 Hz), 115.3 (J = 20.9 Hz), 111.1, 110.2, 109.6, 61.0, 60.7, 46.5, 37.5, 17.8, 17.1, 15.6, 15.5; 19F NMR (376 MHz, CDCl3) δ −115.13, −115.85; IR (KBr): 3690, 3649, 3629, 3448, 2935, 1637, 1508, 1458, 1264, 1223, 1157, 846, 747, 704 cm−1; ESI FTMS exact mass calcd for (C36H32F2N2 − H)− requires m/z 529.2455, found m/z 529.2459; enantiomeric ratio: 91:9, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 4.74 min (major), tR = 6.96 min (minor).
:5 dr; white sticky oil; [α]20D = −112.2 (c 0.25, acetone); 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.0 Hz, 1H), 7.56 (d, J = 7.7 Hz, 1H), 7.44 (s, 1H), 7.20–7.07 (m, 6H), 6.96–6.69 (m, 7H), 6.62 (d, J = 8.1 Hz, 1H), 5.45 (d, J = 7.8 Hz, 1H), 3.95 (dd, J = 13.0, 7.1 Hz, 1H), 3.66 (t, J = 7.8 Hz, 1H), 3.34–3.22 (m, 2H), 2.98–2.78 (m, 2H), 2.54–2.33 (m, 2H), 1.39 (t, J = 7.5 Hz, 3H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 162.8 (J = 245.2 Hz), 162.7 (J = 245.1 Hz), 142.8 (J = 7.1 Hz), 141.1, 140.6 (J = 7.2 Hz), 136.1, 133.1, 132.9, 130.4, 130.2 (J = 8.3 Hz), 129.9 (J = 8.2 Hz), 128.1, 125.2 (J = 2.8 Hz), 123.7 (J = 2.9 Hz), 122.3, 121.2, 119.6, 119.3, 119.1, 118.9, 117.1, 116.6, 116.4, 114.7 (J = 21.4 Hz), 114.2 (J = 21.0 Hz), 113.6 (J = 20.9 Hz), 111.2, 110.2, 109.8, 61.6, 60.8, 46.2 38.4, 17.7, 17.1, 15.6, 15.4; 19F NMR (376 MHz, CDCl3) δ −112.5, −112.8; IR (KBr): 3691, 3677, 3568, 3588, 3547, 3449, 1647, 1654, 1637, 1283, 890, 760, 698 cm−1; ESI FTMS exact mass calcd for (C36H32F2N2 − H)− requires m/z 529.2455, found m/z 529.2459; enantiomeric ratio: 93:7, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 9.79 min (minor), tR = 19.79 min (major).
:5 dr; white sticky oil; [α]20D = −81.7 (c 0.3, acetone); 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 7.7 Hz, 1H), 7.20–6.99 (m, 9H), 6.97–6.86 (m, 4H), 6.82 (s, 1H), 6.63 (d, J = 8.1 Hz, 1H), 5.45 (d, J = 7.4 Hz, 1H), 3.96 (dd, J = 12.5, 5.6 Hz, 1H), 3.68 (t, J = 7.3 Hz, 1H), 3.33 (dd, J = 13.9, 5.7 Hz, 1H), 3.15 (dd, J = 14.0, 5.4 Hz, 1H), 2.90 (q, J = 7.5 Hz, 2H), 2.50–2.35 (m, 2H), 2.26 (s, 3H), 2.22 (s, 3H), 1.41 (t, J = 7.5 Hz, 3H), 0.83 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 142.2, 141.3, 138.3, 138.2, 138.1, 136.0, 133.2, 132.9, 131.4, 131.1, 128.8, 128.6, 128.3, 128.0, 127.9, 127.4, 126.7, 124.8, 121.9, 120.7, 119.3, 119.0, 118.9, 118.8, 116.5, 111.0, 110.1, 109.1, 61.0, 60.9, 46.5, 38.3, 21.4, 21.4, 17.8, 17.1, 15.5; IR (KBr): 3650, 3630, 3504, 3483, 3396, 2962, 2850, 1458, 1389, 1260, 890, 741 cm−1; ESI FTMS exact mass calcd for (C38H38N2 − H)− requires m/z 521.2956, found m/z 521.2960; enantiomeric ratio: 89:11, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 5.81 min (minor), tR = 8.62 min (major).
:5 dr; yellow sticky oil; [α]20D = −58.4 (c 0.29, acetone); 1H NMR (400 MHz, CDCl3) δ 7.80 (s, 1H), 7.67–7.58 (m, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.21–7.11 (m, 3H), 7.04 (t, J = 7.5 Hz, 1H), 6.87 (t, J = 7.6 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 5.10 (d, J = 8.2 Hz, 1H), 3.06 (dd, J = 13.1, 5.4 Hz, 1H), 2.69 (dd, J = 14.8, 7.9 Hz, 1H), 2.45 (s, 3H), 2.35 (s, 3H), 2.04–1.99 (m, 2H), 1.32 (d, J = 6.7 Hz, 3H), 1.05 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 143.1, 135.7, 133.7, 132.5 132.1, 129.2, 122.2, 120.6, 119.4, 119.0, 118.7, 118.2, 111.0, 109.8, 109.7, 102.1, 59.9, 50.9, 45.7, 24.0, 17.4, 10.5, 8.7, 8.6; IR (KBr): 3732, 3443, 2960, 2922, 2854, 1637, 1548, 1384, 1331, 1298, 1261, 1080, 1028, 797, 745 cm−1; ESI FTMS exact mass calcd for (C24H26N2 − H)− requires m/z 341.2017, found m/z 341.2020; enantiomeric ratio: 77:23, determined by HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 98/2, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 4.94 min (minor), tR = 6.74 min (major).
:5 dr; yellow sticky oil; 1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.63–7.57 (m, 1H), 7.51 (d, J = 7.9 Hz, 1H), 7.23–7.11 (m, 3H), 7.04 (t, J = 7.4 Hz, 1H), 6.88 (t, J = 7.5 Hz, 1H), 6.62 (d, J = 8.0 Hz, 1H), 5.18 (d, J = 6.2 Hz, 1H), 3.12 (d, J = 5.2 Hz, 1H), 2.83–2.74 (m, 1H), 2.44 (s, 3H), 2.33 (s, 3H), 2.01–1.82 (m, 2H), 1.77–1.51 (m, 6H), 1.36–1.30 (m, 2H), 0.94–0.89 (m, 7H), 0.81 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 143.4, 135.7, 133.7, 133.2, 132.4, 129.3, 122.0, 120.5, 119.2, 119.0, 118.6, 118.1, 111.0, 109.6, 108.9, 101.7, 59.1, 53.6, 44.9, 43.5, 35.5, 30.6, 29.7, 28.2, 25.8, 23.1, 22.7, 22.6, 8.8, 8.8; IR (KBr): 3365, 3091, 2934, 1611, 1503, 1426, 1318, 1272, 794, 707 cm−1; ESI FTMS exact mass calcd for (C30H39N2 − H)− requires m/z 427.3113, found m/z 427.3108.
:5 dr; yellow sticky oil; 1H NMR (400 MHz, CDCl3) δ 7.76–7.62 (m, 2H), 7.56 (d, J = 7.9 Hz, 1H), 7.23–7.19 (m, 1H), 7.18–7.12 (m, 2H), 7.03 (t, J = 7.5 Hz, 1H), 6.87 (t, J = 7.6 Hz, 1H), 6.64 (d, J = 8.1 Hz, 1H), 5.17 (d, J = 6.2 Hz, 1H), 3.15 (dd, J = 10.7, 5.8 Hz, 1H), 2.99–2.87 (m, 2H), 2.85–2.74 (m, 2H), 2.00–1.84 (m, 2H), 1.80–1.71 (m, 1H), 1.71–1.50 (m, 5H), 1.41 (t, J = 7.5 Hz, 3H), 1.33 (d, J = 7.6 Hz, 3H), 0.91 (d, J = 6.6 Hz, 6H), 0.88 (d, J = 6.5 Hz, 3H), 0.84 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 143.0, 135.9, 132.8, 132.6, 128.3, 121.9, 120.5, 119.2, 119.0, 118.9, 118.4, 115.7, 111.2, 109.8, 108.8, 59.3, 53.4, 44.9, 43.5, 35.4, 30.8, 29.7, 28.3, 25.7, 23.1, 22.7, 22.6, 22.6, 17.7, 15.7, 15.4; IR (KBr): 3602, 3351, 3074, 1601, 1559, 1497, 1298, 827, 752 cm−1; ESI FTMS exact mass calcd for (C32H42N2 − H)− requires m/z 455.3426, found m/z 455.3418.
:5 dr; yellow sticky oil; [α]20D = −16.1 (c 0.24, acetone); 1H NMR (400 MHz, CDCl3) δ 7.48 (d, J = 8.5 Hz, 1H), 7.36 (d, J = 8.3 Hz, 1H), 7.32–7.27 (m, 4H), 7.26–7.22 (m, 2H), 7.11–7.02 (m, 7H), 6.73 (s, 1H), 6.57 (d, J = 1.8 Hz, 1H), 5.35 (d, J = 7.2 Hz, 1H), 3.99–3.94 (m, 1H), 3.68 (t, J = 7.1 Hz, 1H), 3.42 (dd, J = 13.9, 4.9 Hz, 1H), 3.05 (dd, J = 13.9, 5.4 Hz, 1H), 2.39 (s, 3H), 1.86 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 142.9, 140.9, 138.0, 136.1, 133.0, 132.7, 131.8, 130.2, 128.9, 128.6, 128.1, 127.7, 127.5, 127.4, 126.9, 126.8, 120.1, 120.0, 119.6, 119.4, 110.9, 109.7, 109.6, 102.8, 61.0, 60.4, 46.0, 37.5, 8.9, 8.0; IR (KBr): 3645, 3567, 3435, 3054, 2922, 2852, 1557, 1461, 1264, 1020, 896, 801, 742, 702, 699 cm−1; ESI FTMS exact mass calcd for (C34H28Cl2N2 − H)− requires m/z 533.1551, found m/z 533.1560; enantiomeric ratio: 94:6, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 98/2, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 20.06 min (major), tR = 34.18 min (minor).
:5 dr; light yellow oil; [α]20D = +80.5 (c 0.24, acetone); 1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 7.9 Hz, 1H), 7.42–7.33 (m, 3H), 7.30 (t, J = 7.5 Hz, 2H), 7.25–7.14 (m, 7H), 7.13–7.02 (m, 7H), 6.99 (d, J = 7.5 Hz, 4H), 6.90 (d, J = 4.2 Hz, 3H), 6.59 (d, J = 8.0 Hz, 1H), 5.48 (d, J = 6.8 Hz, 1H), 4.25 (dd, J = 34.3, 16.3 Hz, 2H), 3.82 (t, J = 10.3 Hz, 2H), 3.74 (t, J = 6.8 Hz, 1H), 3.62 (d, J = 16.1 Hz, 1H), 3.25 (dd, J = 13.8, 5.6 Hz, 1H), 3.03 (dd, J = 13.9, 5.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 143.2, 141.2, 141.1, 140.5, 138.1, 135.3, 133.7, 132.8, 132.6, 130.0, 128.8, 128.5, 128.4, 128.3, 128.2, 128.1, 127.7, 127.3, 126.6, 126.0, 125.7, 122.1, 120.9, 119.6, 119.4, 119.2, 119.1, 112.8, 111.1, 110.2, 106.0, 61.2, 60.8, 46.7, 38.1, 30.5, 29.7, 29.6; IR (KBr): 3568, 2843, 2760, 1655, 1532, 1479, 1361, 1206, 833, 781, 702 cm−1; ESI FTMS exact mass calcd for (C46H38N2 − H)− requires m/z 619.3133, found m/z 619.3139; enantiomeric ratio: 93:7, determined by HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 99/1, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 11.603 min (minor), tR = 20.940 min (major).
:39 molar ratio. 2be: yellow sticky oil; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 7.9 Hz, 1H), 7.52 (d, J = 7.6 Hz, 1H), 7.30 (d, J = 8.2 Hz, 2H), 7.23 (s, 1H), 7.18–7.05 (m, 6H), 7.01 (d, J = 8.2 Hz, 2H), 6.97–6.88 (m, 3H), 6.68 (d, J = 8.1 Hz, 1H), 5.46 (d, J = 7.3 Hz, 1H), 3.96 (dd, J = 12.5, 5.9 Hz, 1H), 3.67 (t, J = 7.3 Hz, 1H), 3.34 (dd, J = 14.0, 5.6 Hz, 1H), 3.13 (dd, J = 14.0, 5.6 Hz, 1H), 2.39 (s, 3H), 1.91 (s, 3H), 1.35 (s, 9H); 13C NMR (100 Hz, CDCl3) δ 162.7 (J = 243.5 Hz), 150.2, 142.2, 137.6, 135.6, 134.1 (J = 3.3 Hz), 134.0, 132.8, 131.7, 131.4 (J = 7.7 Hz), 129.1, 127.4, 125.6, 122.0, 120.9, 119.3 (J = 19.0 Hz), 118.6 (J = 6.4 Hz), 115.2 (J = 21.0 Hz), 110.9, 110.0, 109.3, 102.6, 61.0, 60.7, 46.1, 37.1, 34.5, 31.3, 8.9, 8.0; 19F NMR (376 MHz, CDCl3) δ −116.19; IR (KBr): 3629, 3445, 2964, 1637, 1544, 1456, 1388, 1320, 1239, 1141, 839, 745, 701 cm−1; ESI FTMS exact mass calcd for (C38H37FN2 − H)− requires m/z 539.2862, found m/z 539.2865; enantiomeric ratio: 86:14, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 98/2, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 17.06 min (minor), tR = 20.98 min (major). 2be′: yellow sticky oil; 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 7.9 Hz, 1H), 7.50 (d, J = 7.5 Hz, 1H), 7.46 (s, 1H), 7.20 (d, J = 8.1 Hz, 2H), 7.16–7.08 (m, 4H), 7.04–6.98 (m, 4H), 6.96–6.89 (m, 3H), 6.62 (d, J = 8.1 Hz, 1H), 5.39 (d, J = 8.2 Hz, 1H), 3.96 (dd, J = 13.5, 5.9 Hz, 1H), 3.66 (t, J = 8.3 Hz, 1H), 3.34 (dd, J = 14.0, 6.0 Hz, 1H), 3.18 (dd, J = 14.0, 5.4 Hz, 1H), 2.44 (s, 3H), 1.84 (s, 3H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 161.9 (J = 244.2 Hz), 149.5, 142.2, 135.9 (J = 3.2 Hz), 135.7, 134.8, 134.0, 132.7, 131.1, 129.6 (J = 7.9 Hz), 129.4, 129.0, 125.2, 122.1, 120.9, 119.3 (J = 17.2 Hz), 118.6 (J = 16.3 Hz), 115.4 (J = 21.2 Hz), 110.9, 110.0, 109.7, 102.7, 61.1, 60.4, 46.0, 37.0, 34.4, 31.3, 8.9, 7.9; 19F NMR (376 MHz, CDCl3) δ −115.49; IR (KBr): 3629, 3445, 2964, 1637, 1544, 1456, 1388, 1320, 1239, 1141, 839, 745, 701 cm−1; ESI FTMS exact mass calcd for (C38H37FN2 − H)− requires m/z 539.2862, found m/z 539.2865; enantiomeric ratio: 90:10, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 98/2, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 30.69 min (major), tR = 39.31 min (minor).
:5 dr; yellow sticky oil; [α]20D = −3.6 (c 0.69, acetone); 1H NMR (400 MHz, CDCl3) δ 7.98 (s, 1H), 7.61 (d, J = 7.9 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.35–7.27 (m, 6H), 7.24 (d, J = 7.5 Hz, 3H), 7.20–7.10 (m, 5H), 6.96 (t, J = 7.6 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 5.72 (d, J = 9.1 Hz, 1H), 4.80 (d, J = 9.5 Hz, 1H), 3.89 (t, J = 9.3 Hz, 1H), 2.06 (s, 3H), 1.91 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 141.4, 140.2, 138.0, 135.9, 133.7, 132.64 130.3, 129.2, 128.8, 128.7, 128.3, 127.8, 127.5, 127.1, 122.2, 121.2, 119.4, 119.3, 118.8, 118.7, 111.0, 110.6, 110.1, 103.5, 68.0, 60.9, 51.2, 8.5, 8.0; IR (KBr): 3478, 3031, 2974, 1605, 1489, 1418, 1362, 1284, 720, 685 cm−1; ESI FTMS exact mass calcd for (C33H28N2 − H)− requires m/z 452.2252, found m/z 452.2260; enantiomeric ratio: 51:49, determined by HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 95/5, flow rate 1.0 mL min−1, T = 30 °C, 254 nm): tR = 5.603 min (minor), tR = 7.743 min (major).
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21372002 and 21232007), PAPD, TAPP, and the Natural Science Foundation of Jiangsu Province (BK20160003).Notes and references
- (a) R. Faust, P. J. Garratt, R. Jones, L.-K. Yeh, A. Tsotinis, M. Panoussopoulou, T. Calogeropoulou, M.-T. Teh and D. Sugden, J. Med. Chem., 2000, 43, 1050 CrossRef CAS PubMed; (b) S. Rivara, M. Mor, C. Silva, V. Zuliani, F. Vacondio, G. Spadoni, A. Bedini, G. Tarzia, V. Lucini and M. Pannacci, J. Med. Chem., 2003, 46, 1429 CrossRef CAS PubMed; (c) C. Beaulieu, D. Guay, Z.-Y. Wang, Y. Leblanc, P. Roy, C. Dufresne, R. Zamboni, C. Berthelette, S. Day and N. Tsou, Bioorg. Med. Chem. Lett., 2008, 18, 2696 CrossRef CAS PubMed; (d) L. S. Fernandez, M. S. Buchanan, A. R. Carroll, Y.-J. Feng, R. J. Quinn and V. M. Avery, Org. Lett., 2009, 11, 329 CrossRef CAS PubMed; (e) R. M. Zeldin and F. D. Toste, Chem. Sci., 2011, 2, 1706 RSC; (f) D. J. Buzard, L. Lopez, J. Moody, A. Kawasaki, T. O. Schrader, M. Kasem, B. Johnson, X.-W. Zhu, L. Thoresen and S. H. Kim, ACS Med. Chem. Lett., 2014, 5, 1334 CrossRef CAS PubMed.
- (a) D. Enders, C. Wang, X. Yang and G. Raabe, Synlett, 2011, 469 CrossRef CAS; (b) D. Enders, A. Greb, K. Deckers, P. Selig and C. Merkens, Chem. – Eur. J., 2012, 18, 10226 CrossRef CAS PubMed; (c) M. Zeng, W. Zhang and S.-L. You, Chin. J. Chem., 2012, 30, 2615 CAS; (d) H.-G. Cheng, L.-Q. Lu, T. Wang, Q.-Q. Yang, X.-P. Liu, Y. Li, Q.-H. Deng, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 3250 CrossRef CAS PubMed; (e) Y.-L. Zhang, X.-H. Liu, X.-H. Zhao, J.-L. Zhang, L. Zhou, L.-L. Lin and X.-M. Feng, Chem. Commun., 2013, 49, 11311 RSC; (f) H. Lu, J.-B. Lin, J.-Y. Liu and P.-F. Xu, Chem. – Eur. J., 2014, 20, 11659 CrossRef CAS PubMed.
- (a) A. Ghosh and L. M. Stanley, Chem. Commun., 2014, 50, 2765 RSC; (b) T. Shibata, N. Ryu and H. Takano, Adv. Synth. Catal., 2015, 357, 1131 CrossRef CAS.
- Q.-J. Ni, H. Zhang, A. Grossmann, C. C. J. Loh, C. Merkens and D. Enders, Angew. Chem., Int. Ed., 2013, 52, 13562 CrossRef CAS PubMed.
- For some selected examples, see: (a) S. B. Jones, B. Simmons and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 13606 CrossRef CAS PubMed; (b) C. Zheng, Y. Lu, J. Zhang, X. Chen, Z. Chai, W. Ma and G. Zhao, Chem. – Eur. J., 2010, 16, 5853 CrossRef CAS PubMed; (c) X.-F. Wang, J.-R. Chen, Y.-J. Cao, H.-G. Cheng and W.-J. Xiao, Org. Lett., 2010, 12, 1140 CrossRef CAS PubMed; (d) G.-L. Bergonzini, L.-C. Gramigna, A. Mazzanti, M. Fochi, L. Bernardi and A. Ricci, Chem. Commun., 2010, 46, 327 RSC; (e) Y.-J. Cao, H.-G. Cheng, L.-Q. Lu, J.-J. Zhang, Y. Cheng, J.-R. Chen and W.-J. Xiao, Adv. Synth. Catal., 2011, 353, 617 CrossRef CAS; (f) T. Xu, H. Nora and M. Paolo, Angew. Chem., Int. Ed., 2014, 53, 2997 CrossRef PubMed; (g) Y. Wang, M.-S. Tu, L. Yin, M. Sun and F. Shi, J. Org. Chem., 2015, 80, 3223 CrossRef CAS PubMed.
- (a) W. Tan, X. Li, Y.-X. Gong, M.-D. Ge and F. Shi, Chem. Commun., 2014, 50, 15901 RSC; (b) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460 CrossRef CAS PubMed; (c) Y. Wang, M. Sun, L. Yin and F. Shi, Adv. Synth. Catal., 2015, 357, 4031 CrossRef CAS; (d) W. Dai, X.-L. Jiang, J.-Y. Tao and F. Shi, J. Org. Chem., 2016, 81, 185 CrossRef CAS PubMed; (e) J.-J. Zhao, M. Tang, H.-H. Zhang, M.-M. Xu and F. Shi, Chem. Commun., 2016, 52, 5953 RSC; (f) For a recent review: G.-J. Mei and F. Shi, Synlett, 2016 DOI:10.1055/s-0036-1588611.
- (a) C.-F. Gurtler, S. Blechert and E. Steckhan, Chem. – Eur. J., 1997, 3, 447 CrossRef CAS; (b) D.-H. Dethe, R.-D. Erande and A. Ranjan, J. Am. Chem. Soc., 2011, 133, 2864 CrossRef CAS PubMed; (c) D.-H. Dethe, R.-D. Erande and A. Ranjan, J. Org. Chem., 2013, 78, 10106 CrossRef CAS PubMed.
- L. Yin, Y. Wang, M. Sun and F. Shi, Adv. Synth. Catal., 2016, 358, 1093 CrossRef CAS.
- For some reviews, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS PubMed; (b) M. Terada, Chem. Commun., 2008, 4097 RSC; (c) M. Terada, Synthesis, 2010, 1929 CrossRef CAS; (d) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156 CrossRef CAS PubMed; (e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (f) H. Wu, Y.-P. He and F. Shi, Synthesis, 2015, 1990 CrossRef CAS. For some recent prominent examples, see: (g) Z. Zhang and J. C. Antilla, Angew. Chem., Int. Ed., 2012, 51, 11778 CrossRef CAS PubMed; (h) H. Qiu, M. Li, L.-Q. Jiang, F.-P. Lv, L. Zan, C.-W. Zhai, M. P. Doyle and W.-H. Hu, Nat. Chem., 2012, 4, 733 CrossRef CAS PubMed; (i) Z.-L. Tao, W.-Q. Zhang, D.-F. Chen, A. Adele and L.-Z. Gong, J. Am. Chem. Soc., 2013, 135, 9255 CrossRef CAS PubMed; (j) K. Saito, H. Miyashita and T. Akiyama, Org. Lett., 2014, 16, 5312 CrossRef CAS PubMed; (k) K. Saito, Y. Moriya and T. Akiyama, Org. Lett., 2015, 17, 3202 CrossRef CAS PubMed; (l) W. Zhao, Z. Wang, B. Chu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 1910 CrossRef CAS PubMed.
- A. Galván, A. B. González-Pérez, R. Álvarez, A. R. de Lera, F. J. Faňanás and F. Rodríguez, Angew. Chem., Int. Ed., 2016, 55, 3428 CrossRef PubMed.
- K. Bera and C. Schneider, Chem. – Eur. J., 2016, 22, 7074 CrossRef CAS PubMed.
- CCDC 1484268 for racemic 2i and CCDC 1508087 for compound 4, see ESI† for details.
- H. J. Zhu, J. X. Jiang, S. Saobo and C. U. Pittman, J. Org. Chem., 2005, 70, 261–267 CrossRef CAS PubMed.
- (a) H.-J. Zhu, Organic Stereochemistry - experimental and theoretical methods, Wiley-VCH, Verlag GmbH & Co. KGaA, 2015 Search PubMed; (b) H.-J. Zhu, Current Organic Stereochemistry, Science Presses of China, 2009 Search PubMed; (c) P. He, X. Wang, X. Guo, C. Zhou, S. Shen, D. Hu, X. Yang, D. Luo, R. Dukor and H. Zhu, Tetrahedron Lett., 2014, 55, 2965 CrossRef CAS; (d) S.-D. Zhao, L. Shen, D.-Q. Luo and H.-J. Zhu, Curr. Org. Chem., 2011, 15, 1843 CrossRef CAS; (e) H.-J. Zhu, W.-X. Li, D.-B. Hu and M.-L. Wen, Tetrahedron, 2014, 70, 8236 CrossRef CAS; (f) S.-S. Ding, C.-C. Zhang, W.-S. Shi, M.-M. Liang, Q. Yang and H.-J. Zhu, Tetrahedron Lett., 2016, 57, 75 CrossRef CAS; (g) D. Zhao, Z.-Q. Li, F. Cao, M.-M. Liang, C. U. Pittman Jr., H.-J. Zhu, L. Li and S.-S. Yu, Chirality, 2016, 28, 612 CrossRef CAS PubMed.
- (a) H.-J. Zhu, J. Ren and C. U. Pittman Jr., Tetrahedron, 2007, 63, 2292 CrossRef CAS; (b) Q.-M. Li, J. Ren, L. Shen, B. Bai, Q.-M. Li, X.-C. Liu, M.-L. Wen and H.-J. Zhu, Tetrahedron, 2013, 69, 3067 CrossRef CAS; (c) J. Ren, G.-L. Li, L. Shen, G.-L. Zhang, L. Nafie and H.-J. Zhu, Tetrahedron, 2013, 69, 10351 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Original NMR and HPLC spectra of products 2 and 6, theoretical calculations. CCDC 1484268 and 1508087. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00446f |
| ‡ These authors contributed equally to the work. |
| This journal is © the Partner Organisations 2017 |