
Calcium mediated C–F bond substitution in fluoroarenes towards C–chalcogen bond formation†
Pintu
Maity
a,
Sabir
Ahammed
b,
Rabindra Nath
Manna
c and
Brindaban C.
Ranu
*a
aDepartment of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India. E-mail: ocbcr@iacs.res.in
bDepartment of Chemistry, Aliah University, IIA/27, Action Area II, Newtown, Kolkata 700156, India
cDepartment of Physical Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India
First published on 20th October 2016
Abstract
A new calcium chloride mediated C–F bond cleavage in electron deficient fluoroarenes followed by functionalisation via C–Se/Te bond formation has been achieved in the absence of any additive/ligand/organometallic reagent. A series of functionalised organo-selenides and -tellurides were obtained in high yields by this procedure. The precise role of calcium and the possible mechanism have been proposed based on DFT analysis.
Introduction
The C–F bond dissociation energy is the largest among all other C–X bonds (X = halogen).1 Moreover, the reluctance of the C–F moiety to co-ordinate with the metal centres makes the C–F bond relatively inert2 and thus cleavage of a C–F bond towards further functionalisation is a challenging task. In practice, the activation of a C–F moiety is initiated either by reductive cleavage of the C–F bond by a low valent transition metal3 or intermolecular hydrogen–fluorine exchange mediated by a trivalent organolanthanide complex.4 Several reports of C–F bond activation in fluoroarenes towards C–C bond formation catalyzed by Pd- and Ni-complexes are also available.3 Besides the transition metals, the use of group 2 alkaline earth metals such as calcium, strontium and barium have also been reported for specific organic transformations. These elements show excellent Lewis acid property because of their large radii and electropositive character. In addition to these, their low toxicity and cost efficiency have made them more attractive to be explored for new reactions. Recently calcium complexes have been reported to be employed for different types of organic reactions such as alkene hydroamination,5 hydrophosphonations,6 Michael reaction,7 aldol condensation,8 epoxidation,9 Tishchenko reactions,10 Sonogashira reaction,11 Pictet Spengler reactions,12 Friedel Crafts alkylation,13 cyclopropanation,14 dynamic multicomponent reaction,15 intermolecular carbohydroxylation of alkynes etc.16 Shu Kobayashi and others have applied chiral complexes of calcium in an enantioselective process too.17 Recently Hill and co-workers demonstrated the use of a calcium β-diketiminate complex for C–F bond cleavage via an intermediate heteroleptic calcium fluoride complex.18 To add a simpler procedure we report here a calcium chloride mediated C–F bond cleavage in electron deficient fluoroarenes for C–Se and C–Te functionalisation in the absence of any additive and ligand leading to the synthesis of a series of organoselenides and tellurides (Scheme 1a). Nevertheless, zinc dust has been used to cleave the Se–Se/Te–Te bond to make the PhSe/PhTe moiety available for the reaction and in no way has any role in C–F bond cleavage. To the best of our knowledge we are not aware of any direct Ca-mediated defluorination of fluoroarenes towards functionalisation in the absence of any additive/ligand/base. However, one report for Sonogashira-type cross-coupling of alkynes with fluoroarenes in the presence of excess sodium, NaOMe, Ca(OH)2 and n-butylmagnesium chloride was found (Scheme 1b).11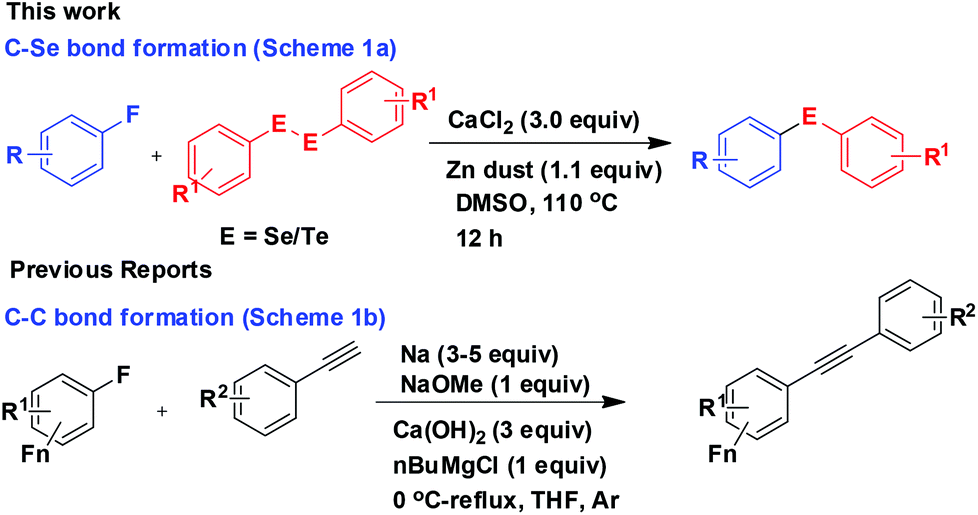 | ||
| Scheme 1 Calcium mediated C–F bond activation. | ||
Needless to say, the organoselenides are of much importance and of continued interest because of their presence as structural motifs in a variety of molecules of biological, pharmaceutical and material interest.19
They are used as catalysts for various reactions such as oxyselenation–elimination and valuable intermediates in organic synthesis.20 Thus a number of methods have been developed for their synthesis.21 These include transition metal catalyzed C–Se bond formation through cross coupling of aryl halides (mainly I, Br, Cl), aryl boronic acid, and aryl triflates with selenol/PhSeNa/diphenyl diselenides.22 Recently some metal free procedures23 (microwave irradiation, ball-milling, visible light photocatalysis etc.) have also been developed.
Results and discussion
To optimise the reaction conditions, a series of experiments were performed under varying reaction parameters such as different metal salts, solvents, temperature and time for a representative reaction of 4-fluorobenzonitrile and diphenyl diselenide. Among a variety of metal salts and solvents studied, calcium chloride in DMSO is the most effective combination and the best result was observed in a reaction at 110 °C for 12 h in the presence of Zn dust (Table 1, entry 4). A reaction at lower temperature and reduced time decreased the yield (Table 1, entries 1–3). The reaction did not initiate in the presence of other metal salts such as MnCl2, SnCl2, ZnCl2, SbCl5, NiCl2, AgF and AgOAc (Table 1, entries 5–11). Other calcium salts such as CaCO3, Ca(OAc)2 and Ca(NO3)2 have been investigated and none of these is considerably efficient although only Ca(OAc)2 provides 25% yield (Table 1, entries 12–14). DMF and NMP are also effective for the reaction (Table 1, entries 15 and 16). The reaction did not proceed at all in other solvents such as H2O, dimethyl carbonate (DMC) and xylene under similar conditions (Table 1, entries 17, 19 and 20). However a trace amount of the product was observed in CH3CN (Table 1, entry 18). Instead of Zn, the use of KOH and indium as cleaving agents failed to produce the product (Table 1, entries 21 and 22). The reaction did not proceed in the absence of calcium chloride (Table 1, entry 24). A reduction of the amount of CaCl2 decreased the yield (Table 1, entries 25 and 26). The reaction produced lower yields of the product on using group 1 metal salts such as LiCl, NaCl, KCl and CsF instead of CaCl2 (Table 1, entries 27–30).|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Time (h) | Temp. (°C) | Solvent | Yieldb (%) |
| a Reaction conditions: 4-fluorobenzonitrile (1.0 equiv.), diphenyl diselenide (0.6 equiv.), metal salt (3.0 equiv.), Zn dust (1.1 equiv.), argon atmosphere. b Isolated yield. c KOH (1.1 equiv.). d In (1.1 equiv.). e Without Zn dust. f Calcium salt (1.0 equiv.). g Calcium salt (2.0 equiv.). | |||||
| 1 | CaCl2 | 8 | 100 | DMSO | 45 |
| 2 | CaCl2 | 10 | 100 | DMSO | 57 |
| 3 | CaCl2 | 12 | 100 | DMSO | 65 |
| 4 | CaCl2 | 12 | 110 | DMSO | 92 |
| 5 | MnCl2 | 12 | 110 | DMSO | — |
| 6 | SnCl2 | 12 | 110 | DMSO | — |
| 7 | ZnCl2 | 12 | 110 | DMSO | — |
| 8 | SbCl5 | 12 | 110 | DMSO | — |
| 9 | NiCl2 | 12 | 110 | DMSO | — |
| 10 | AgF | 12 | 110 | DMSO | — |
| 11 | Ag(OAc) | 12 | 110 | DMSO | — |
| 12 | CaCO3 | 12 | 110 | DMSO | — |
| 13 | Ca(OAc)2 | 12 | 110 | DMSO | 25 |
| 14 | Ca(NO3)2 | 12 | 110 | DMSO | — |
| 15 | CaCl2 | 12 | 110 | DMF | 85 |
| 16 | CaCl2 | 12 | 110 | NMP | 81 |
| 17 | CaCl2 | 12 | 110 | H2O | — |
| 18 | CaCl2 | 12 | 110 | CH3CN | Trace |
| 19 | CaCl2 | 12 | 110 | DMC | — |
| 20 | CaCl2 | 12 | 110 | Xylene | — |
| 21c | CaCl2 | 12 | 110 | DMSO | 11 |
| 22d | CaCl2 | 12 | 110 | DMSO | — |
| 23e | CaCl2 | 12 | 110 | DMSO | Trace |
| 24 | — | 12 | 110 | DMSO | — |
| 25f | CaCl2 | 12 | 110 | DMSO | 65 |
| 26g | CaCl2 | 12 | 110 | DMSO | 84 |
| 27 | LiCl | 12 | 110 | DMSO | 31 |
| 28 | NaCl | 12 | 110 | DMSO | 12 |
| 29 | KCl | 12 | 110 | DMSO | 15 |
| 30 | CsF | 12 | 110 | DMSO | 38 |
Thus in a general experimental procedure a mixture of fluoroarene (1.0 mmol), diaryl diselenide (0.6 mmol), calcium chloride (3.0 mmol), and Zn dust (1.1 mmol) in DMSO (3 mL) was stirred under an argon atmosphere at 110 °C for 12 h. After evaporation of DMSO, the crude product was extracted with ethyl acetate. The pure product was isolated by column chromatography over silica gel. A wide range of diversely substituted diphenyl selenides have been obtained by this procedure. All the products are summarized in Table 2. Several (ten) of these compounds are reported for the first time.
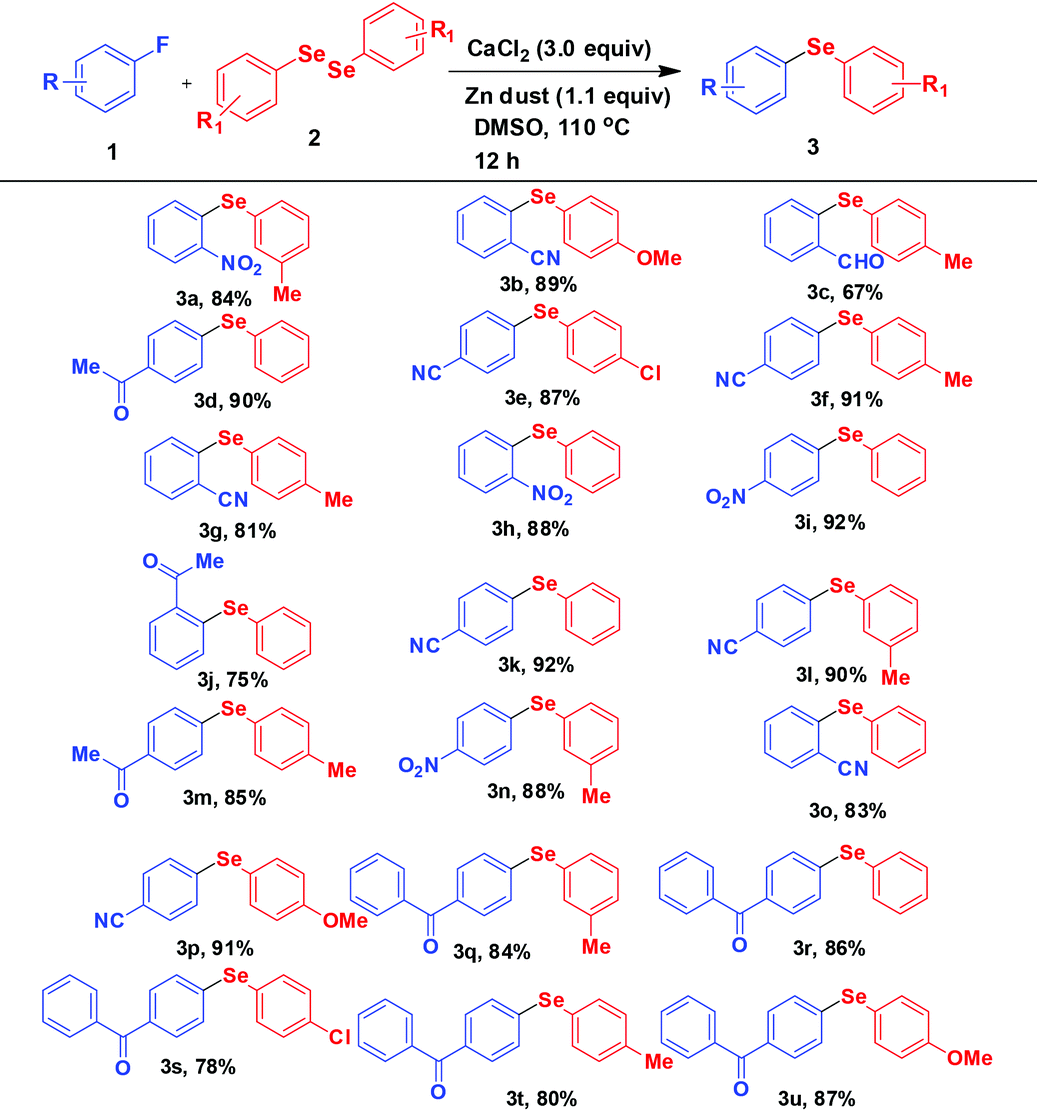
The fluoroarenes containing an electron withdrawing group (–NO2, –CN, –CHO, –COCH3, –COPh) underwent an efficient reaction with substituted diphenyl diselenides (–Me, –Cl, –OMe) to afford the corresponding selenides (3a–u). The unsubstituted and electron donating group substituted fluoroarenes are not effective for this reaction. Perfluoroarenes such as hexafluorobenzene and octafluorotoluene also did not produce the corresponding product under standardized conditions. It may be due to the less electrophilic nature of the arenes as well as less nucleophilic nature of PhSe under our experimental conditions. The other halogen (Cl, Br, I) substituted arenes also remained inert under these conditions. The Cl-substituted diphenyl diselenide reacted in the usual way keeping the Cl group unaffected during the reaction (3e and 3s) and it may be used for further functionalisation. In addition, the other functionalities (–NO2, –CN, –CHO, –COCH3, –COPh) present in the selenide products may be of much importance for further derivatisation. For example, 2-nitrophenyl-phenyl selenide obtained from 1-fluoro-2-nitrobenzene was successfully converted to 2-furylphenyl-phenyl selenide 4a by manipulation of nitro functionality (Scheme 2).24a,b
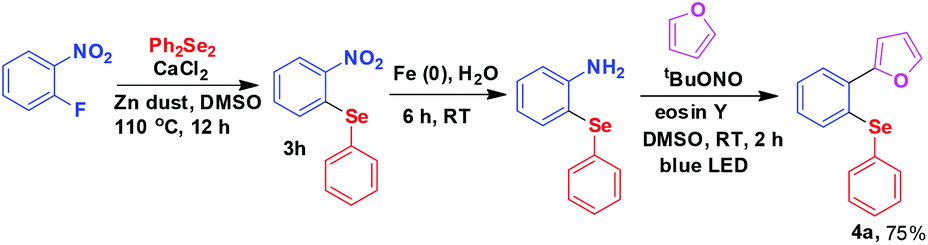 | ||
| Scheme 2 Synthesis of 2-furylphenyl-phenyl selenide from 2-fluoronitrobenzene. | ||
The unique feature of this protocol is the use of fluoroarenes for the C–Se bond formation involving diphenyl diselenide which was not addressed earlier except in one reaction with a specific substrate24c where the selenylation was performed using diphenyl diselenide and sodium borohydride. Notably, our method uses zinc dust and thus is compatible with reducible functionalities. Moreover, this has general applicability with all types of electron deficient fluoroarenes.
This strategy is also applied for the synthesis of unsymmetrical diaryl tellurides (Scheme 3). In general we obtained relatively low yield in tellurides. This may be due to the low reactivity of tellurium species probably dictated by the larger size of Te.
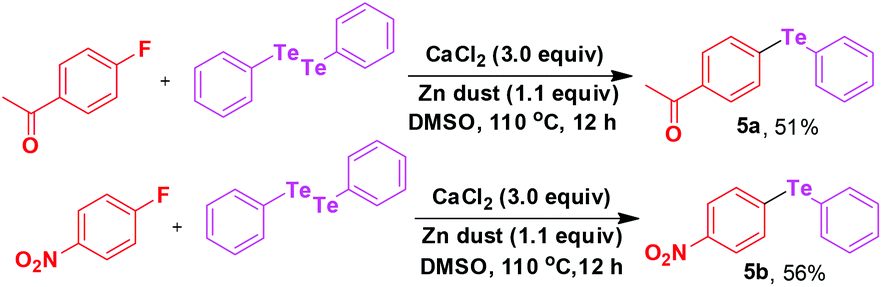 | ||
| Scheme 3 Calcium mediated C(sp2)–Te bond formation. | ||
We then turned our attention to find the mechanism for this Ca-mediated selenylation reaction. To check the involvement of a radical process, the reaction of 4-fluoro benzonitrile and diphenyl diselenide was performed in the presence of TEMPO (radical quencher), THF (electron receptor), and nitroarene (electron acceptor) separately. Notably, the reactions remained unaffected. So the possibility of a radical pathway is unlikely for this reaction. In this reaction, zinc dust has been used to cleave the Se–Se/Te–Te bond to make the PhSe/PhTe moiety available. It has been observed that no product was formed in the absence of calcium chloride (Table 1, entry 24). However, if a free nucleophile (PhSe/PhTe−) is available in the medium the reaction is expected to produce some amount of product. Thus the reaction is not likely to follow the classical Nucleophilic Aromatic Substitution (SNAr) pathway. To have a better understanding on the role of Ca in C–F bond cleavage the mechanism of the process has been investigated using quantum mechanical (QM) calculations. For this purpose, we have prepared six different models. Herein, models A, B, C, D and E denote 4-fluorobenzonitrile and Ph–Se–Zn–Se–Ph structures with Ca2+, without any metal cation, with Zn2+, with Ni2+ and with one explicit DMSO molecule ligating with Ca2+, respectively. On the other side, model F is represented by 4-fluorobenzonitrile and the Ph–Se–Ca–Se–Ph structure with a Ca2+ cation. The computational details of QM modeling approaches are described in the ESI.† Cyclic six membered transition state (TSA) structures for the formation of selenides are found more stable with lower activation free energy (ΔG‡ = 32.5 kcal mol−1) in the presence of Ca2+ than the four membered TSB (ΔG‡ = 41.1 kcal mol−1) structure obtained in the absence of metal salts. The C(sp2)–Se and C(sp2)–F bond distances for six membered TS structures for the models A, C, and D in the presence of Zn and metal salts (such as Ca2+, Zn2+, and Ni2+) are in the range of 2.77–2.94 Å and 1.84–2.18 Å, respectively. For the four membered TSB, C(sp2)–Se and C(sp2)–F bond distances are 2.37 and 1.70 Å, respectively, which are significantly shorter than the distances obtained from the six TS structures. On the other side, the predicted activation barriers for the formation of selenides in the presence of Zn2+ and Ni2+ are 37.4 and 35.1 kcal mol−1, respectively, at the SMDDMSO/B3LYP/6-311+G(d,p) level of theory,25 which are significantly higher than the activation energy barrier (32.5 kcal mol−1) obtained in the presence of calcium metal salts. Moreover, Ca2+ having a vacant d orbital is a hard Lewis acid and possesses a high charge to size ratio whereas Zn2+ and Ni2+ are border line Lewis acids. According to the hard soft acid base principle, the hard Lewis base F− prefers to bind to the hard Lewis acid Ca2+ than other border line Lewis acids. Therefore, Ca2+ makes F− a good leaving group compared to the other metal salts which have been used in our studies. Hence, calcium salt mediated C–F bond cleavage for the formation of diaryl selenides is favorable than other metal salts. Our experimental results also showed this trend. However, the possibility of transmetallation during the reaction is also investigated. In the model F, transmetallation leads to a Ph–Se–Ca–Se–Ph type intermediate from Ph–Se–Zn–Se–Ph. Moreover, in the TSF structure, Se–Ca distances are significantly elongated (2.87 and 2.97 Å) compared to those in other TS structures (Fig. 1). The free energy of activation for the model F via TSF for the formation of selenides is 26.6 kcal mol−1 at the SMDDMSO/B3LYP/6-311+G(d,p) level of theory which is lower than the value derived from the other models (Fig. 2). Moreover, the C(sp2)–Se and C(sp2)–F bond distances in the TSE are 2.67 and 1.57 Å, respectively, which are slightly shorter than the aforementioned distances in the other TS structures. The computed reaction free energy along the model E is −28.6 kcal mol−1 which indicates that the formation of selenides is energetically favorable. According to our study, transmetallation helps to decrease the free energy barrier from 32.5 (model A) to 26.6 kcal mol−1 (model F). Therefore, our results show that formation of diaryl selenides proceeds via the Ph–Se–Ca–Se–Ph intermediate than Ph–Se–Zn–Se–Ph. Furthermore, we have also explored the role of DMSO in the mechanism using model E, where one DMSO molecule possesses substantial ligating interaction with the Ca2+ metal centre. In the TSE, the DMSO molecule is interacting with the Ca2+ ion (Ca2+–O: 2.15 Å) although, the computed activation free energy for the model E is higher than the value obtained from the model F.
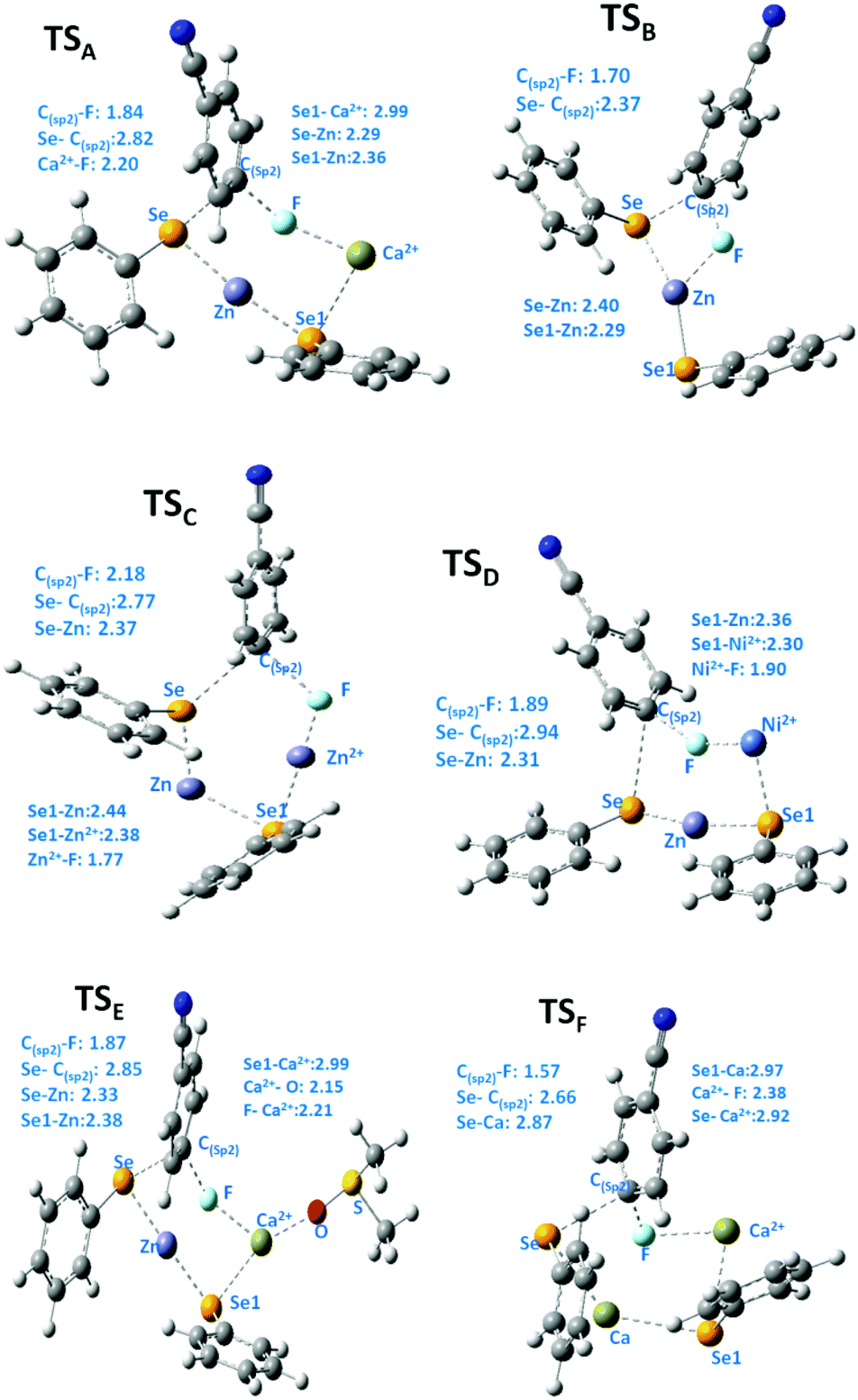 | ||
| Fig. 1 Key geometrical distances (Å) of the TS structures from different models for the formation of selenides are obtained at the SMD(DMSO)B3LYP/6-311+G(d,p) level of theory at 110 °C. | ||
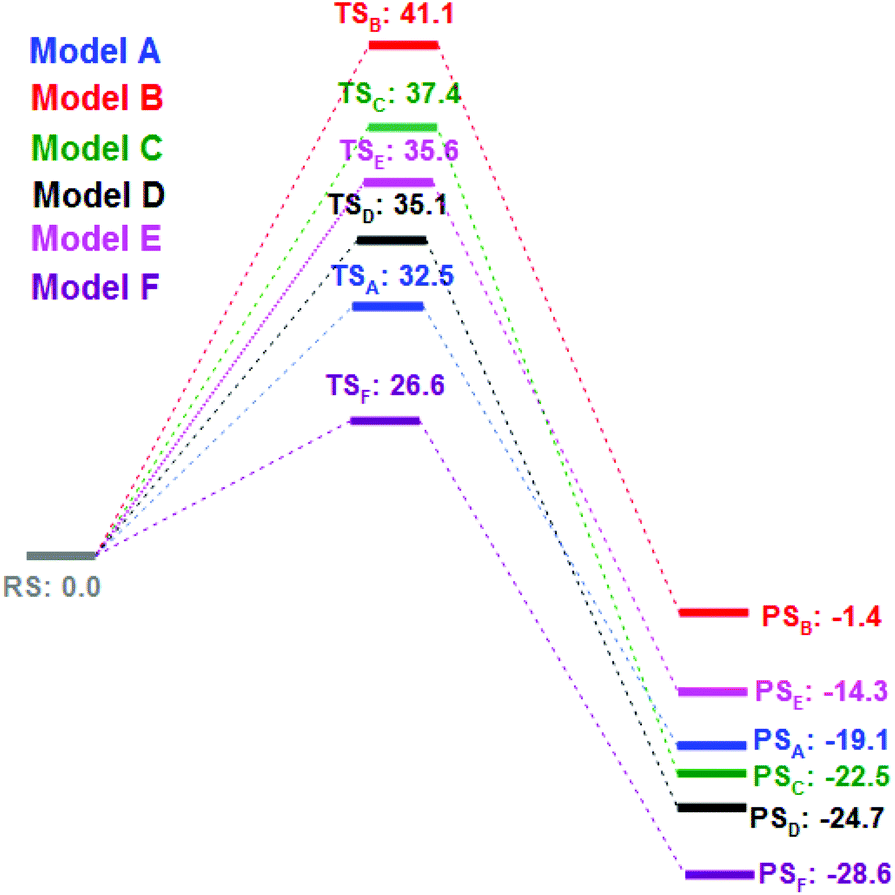 | ||
| Fig. 2 Computed Gibbs free energy (in kcal mol−1) profiles for the C(sp2)–F bond activation in fluoroarenes via C(sp2)–Se bond formation are obtained using different models at the SMDDMSO/B3LYP/6-311+G(d,p) level of theory at 110 °C. Moreover, RS, TS and PS represented the reactant state, transition state and product state, respectively. | ||
Based on the results of the computational study we have proposed a possible reaction pathway (Scheme 4). In the first step diphenyl diselenide reacts with Zn dust to produce (PhSe)2Zn (A). Then the (PhSe)2Zn (A) interacts with calcium to give an intermediate (B) through transmetallation. Subsequently, (PhSe)2Ca reacts with electron deficient fluoroarene in the presence of calcium chloride to form the stable six membered transition state C which ultimately leads to the corresponding diphenyl selenide.
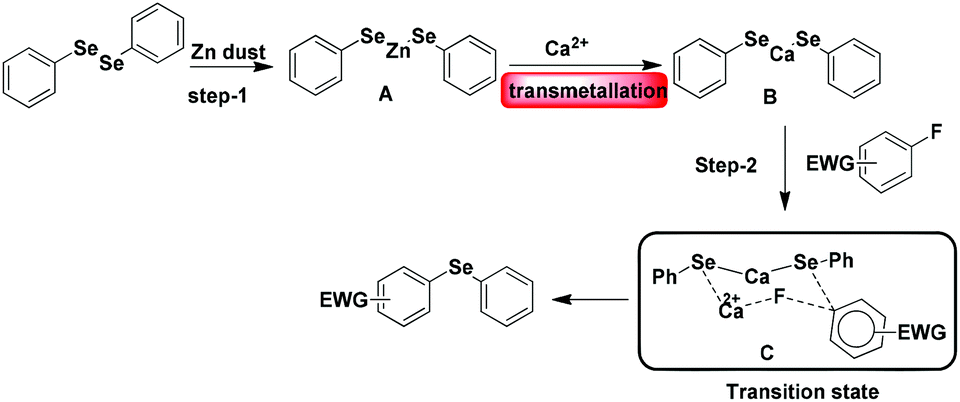 | ||
| Scheme 4 Possible reaction pathway. | ||
Conclusion
In conclusion, we have achieved direct calcium chloride mediated C–F bond activation in electron deficient fluoroarenes towards C–chalcogen bond formation in the absence of any additive/ligand leading to the synthesis of functionalised organo-selenides and tellurides. This protocol provides enormous scope for further manipulation to obtain new selenide derivatives. To the best of our knowledge this is the first report of C–F functionalisation of fluoroarenes using only Ca-salt in the absence of any strong base and organometallic reagent. The role of calcium in the cleavage of the C–F bond and the mechanism of the reaction has been established by using DFT analysis. This work thus opens a new vista for functionalisation of the apparently inert C–F bond by a simple operation using an inexpensive reagent, calcium chloride.Experimental section
General
IR spectra were taken as thin films for liquid compounds and as KBr pellets for solids. NMR spectra were recorded at 300, 400 and 500 MHz for 1H NMR spectra and at 75, 100 and 125 MHz for 13C NMR spectra in CDCl3 solutions. HRMS analysis was performed in a Qtof mass analyzer using an ESI ionization method. Elemental analyses are done at our institute using an autoanalyser. The all substituted diaryl diselenides were synthesized using previously reported procedures.26eRepresentative experimental procedure for the synthesis of (2-nitrophenyl)(m-tolyl)selane (Table 2, 3a)
A mixture of 2-fluoronitrobenzene (141 mg, 1.0 mmol), 3-methyl diphenyl diselenide (204 mg, 0.6 mmol), calcium chloride (333 mg, 3.0 mmol), and Zn dust (72 mg, 1.1 mmol) in DMSO (3 mL) was heated at 110 °C (bath temperature) for 12 h (TLC) in an oil bath under an argon atmosphere. The reaction mixture was then allowed to cool and DMSO was evaporated under vacuum. The residue was extracted with EtOAc (3 × 20 mL). The extract was washed with water (10 mL) and brine (10 mL). The organic phase was dried (Na2SO4) and evaporated to afford the crude product, which was purified by column chromatography over silica gel (hexane) to provide the pure (2-nitrophenyl)(m-tolyl)selane as a pale yellow solid (245 mg, 84%), mp 79–82 °C, 1H NMR (CDCl3, 300 MHz) δ 2.40 (s, 3H), 7.01–7.04 (m, 1H), 7.28–7.38 (m, 4H), 7.49–7.53 (m, 2H), 8.28–8.32 (m, 1H); 13C NMR (CDCl3, 75 MHz) δ 21.4, 125.8, 126.1, 127.9, 130.0, 130.4, 130.8, 133.7, 134.5, 136.2, 138.0, 140.1, 145.7; IR (KBr): 2984, 2883, 1587, 1512, 1330 cm−1; anal. calcd for C13H11NO2Se: C 53.44, H 3.79, N 4.79; found: C 53.42, H 3.82, N 4.75%.This procedure was followed for all the reactions listed in Table 2, Schemes 2 and 3. Many of these compounds have not been reported earlier and these unknown compounds were characterized suitably by their spectroscopic and spectrometric data (IR, 1H NMR, 13C NMR, HRMS and elemental analysis). The known compounds were identified by comparison of their spectroscopic and spectrometric data with those reported earlier. All these data are provided here.
Acknowledgements
We are pleased to acknowledge the financial support from DST, New Delhi through an award of JCB Fellowship to B. C. R. (Grant no. SR/S2/JCB-11/2008). BCR also thanks Indian National Science Academy, New Delhi for the offer of INSA Senior Scientist to him. P. M. thanks CSIR for his fellowship. We thank the supercomputing facility at IACS for computational support.Notes and references
- J. E. Huheey and R. L. Keiter, Inorganic Chemistry, Harper & Collins, 4th edn, 1995 Search PubMed.
- (a) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373 CrossRef CAS; (b) T. G. Richmond, Angew. Chem., Int. Ed., 2000, 39, 3241 CrossRef CAS; (c) U. Mazurek and H. Schwarz, Chem. Commun., 2003, 1321 RSC.
- (a) Y. Kiso, K. Tamao and M. Kumada, J. Organomet. Chem., 1973, 50, C12–C14 CrossRef CAS; (b) V. P. W. Bohm, C. W. K. Gstottmayr, T. Weskamp and W. A. Herrmann, Angew. Chem., Int. Ed., 2001, 40, 3387 CrossRef CAS; (c) F. Mongin, L. Mojovic, B. Guillamet, F. Trécourt and G. Quéguiner, J. Org. Chem., 2002, 67, 8991 CrossRef CAS PubMed; (d) K. Lamm, M. Stollenz, M. Meier, H. Gorls and D. Walther, J. Organomet. Chem., 2003, 681, 24 CrossRef CAS; (e) J. Dankwardt, J. Organomet. Chem., 2005, 690, 932 CrossRef CAS; (f) T. Saeki, Y. Takashima and K. Tamao, Synlett, 2005, 1771 CrossRef CAS; (g) L. Ackermann, R. Born, J. H. Spatz and D. Meyer, Angew. Chem., Int. Ed., 2005, 44, 7216 CrossRef CAS PubMed; (h) N. Yoshikai, H. Mashima and E. Nakamura, J. Am. Chem. Soc., 2005, 127, 17978 CrossRef CAS PubMed; (i) K. Inamoto, J. Kuroda, T. Sakamoto and K. Hiroya, Synthesis, 2007, 2853 CrossRef CAS; (j) N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 9590 CrossRef CAS PubMed; (k) L.-G. Xie and Z.-X. Wang, Chem. – Eur. J., 2010, 16, 10332 CrossRef CAS PubMed; (l) L. Ackermann, C. Wechsler, A. R. Kapdi and A. Althammer, Synlett, 2010, 294 CrossRef CAS; (m) W.-J. Guo and Z.-X. Wang, J. Org. Chem., 2013, 78, 1054 CrossRef CAS PubMed; (n) D. Wu and Z.-X. Wang, Org. Biomol. Chem., 2014, 12, 6414 RSC; (o) Y. Nakamura, N. Yoshikai, L. Ilies and E. Nakamura, Org. Lett., 2012, 14, 3316 CrossRef CAS PubMed; (p) F. Zhu and Z.-X. Wang, J. Org. Chem., 2014, 79, 4285 CrossRef CAS PubMed; (q) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505 CrossRef CAS PubMed; (r) T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964 CrossRef CAS PubMed; (s) X.-W. Liu, J. Echavarren, C. Zarate and R. Martin, J. Am. Chem. Soc., 2015, 137, 12470 CrossRef CAS PubMed; (t) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313 CrossRef CAS PubMed; (u) Y. M. Kim and S. Yu, J. Am. Chem. Soc., 2003, 125, 1696 CrossRef CAS PubMed; (v) D. W. Widdowson and R. Wilhelm, Chem. Commun., 2003, 578 CAS; (w) M. R. Cargill, G. Sandford, A. J. Tadeusiak, D. S. Yufit, J. A. K. Howard, P. Kilickiran and G. Nelles, J. Org. Chem., 2010, 75, 5860 CrossRef CAS PubMed.
- (a) L. Maron, L. Perrin and O. Eisenstein, Dalton Trans., 2003, 4313 RSC; (b) L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279 CrossRef CAS PubMed.
- (a) M. R. Crimmin, I. J. Casely and M. S. Hill, J. Am. Chem. Soc., 2005, 127, 2042 CrossRef CAS PubMed; (b) S. Datta, P. W. Roesky and S. Blechert, Organometallics, 2007, 46, 4392 CrossRef.
- (a) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2007, 26, 2953 CrossRef CAS; (b) M. R. Crimmin, A. G. M. Barrett, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Organometallics, 2008, 27, 497 CrossRef CAS.
- G. Kumaraswamy, M. N. V. Sastry and N. Jena, Tetrahedron Lett., 2001, 42, 8515 CrossRef CAS.
- T. Suzuki, N. Yamagiwa, Y. Matsuo, S. Sakamoto, K. Yamaguchi, M. Shibasaki and R. Nyori, Tetrahedron Lett., 2001, 42, 4669 CrossRef CAS.
- (a) G. Kumaraswamy, N. Jena, M. N. V. Sastry and G. Ramakrishna, ARKIVOC, 2005, 15, 53 Search PubMed; (b) G. Kumaraswamy, M. N. V. Sastry, N. Jena, K. Ravi Kumar and M. Vairamani, Tetrahedron: Asymmetry, 2003, 14, 3797 CrossRef CAS.
- M. R. Crimmin, A. G. M. Barrett, M. S. Hill and P. A. Procopiou, Org. Lett., 2007, 9, 331 CrossRef CAS PubMed.
- G. Jin, X. Zhang and S. Cao, Org. Lett., 2013, 15, 3114 CrossRef CAS PubMed.
- (a) M. J. V. Eynden and J. P. Stambuli, Org. Lett., 2008, 10, 5289 CrossRef PubMed; (b) M. J. V. Eynden, K. Kunchithapatham and J. P. Stambuli, J. Org. Chem., 2010, 75, 8542 CrossRef PubMed.
- M. Niggemann and M. J. Meel, Angew. Chem., Int. Ed., 2010, 49, 3684 CrossRef CAS PubMed.
- T. Haven, G. Kubik, S. Haubenreisser and M. Niggemann, Angew. Chem., Int. Ed., 2013, 52, 4016 CrossRef CAS PubMed.
- S. Gao, T. Stopka and M. Niggemann, Org. Lett., 2015, 17, 5080 CrossRef CAS PubMed.
- T. Stopka and M. Niggemann, Org. Lett., 2015, 17, 1437 CrossRef CAS PubMed.
- (a) Y. M. A. Yamada and S. Ikegami, Tetrahedron Lett., 2000, 41, 2165 CrossRef CAS; (b) G. Kumaraswamy, N. Jena, M. N. V. Sastry, M. Padmaja and B. Markondaiah, Adv. Synth. Catal., 2005, 347, 867 CrossRef CAS; (c) S. Saito, T. Tsubogo and S. Kobayashi, J. Am. Chem. Soc., 2007, 129, 5364 CrossRef CAS PubMed; (d) S. Kobayashi, T. Tsubogo, S. Saito and Y. Yamashita, Org. Lett., 2008, 10, 807 CrossRef CAS PubMed; (e) T. Poisson, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2005, 132, 7890 CrossRef PubMed; (f) S. Shimizu, T. Tsubogo, P. Xu and S. Kobayashi, Org. Lett., 2015, 17, 2006 CrossRef CAS PubMed; (g) W. Zheng, Z. Zhang, M. J. Kaplan and J. C. Antilla, J. Am. Chem. Soc., 2011, 133, 3339 CrossRef CAS PubMed; (h) A. Alix, C. Lalli, P. Retailleau and G. Masson, J. Am. Chem. Soc., 2012, 134, 10389 CrossRef CAS PubMed; (i) T. Tsubogo, S. Saito, K. Y. Seki, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2008, 130, 13321 CrossRef CAS PubMed; (j) T. Poisson, T. Tsubogo, Y. Yamashita and S. Kobayashi, J. Org. Chem., 2010, 75, 963 CrossRef CAS PubMed.
- A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Angew. Chem., Int. Ed., 2007, 46, 6339 CrossRef CAS PubMed.
- (a) G. Mugesh, W. W. duMont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS PubMed; (b) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed; (c) J. A. Woods, J. A. Hadfield, A. T. Mcgown and B. W. Fox, Bioorg. Med. Chem., 1993, 1, 333 CrossRef CAS PubMed; (d) L. Engman, D. Stern, H. Frisell, K. Vessman, M. Berglund, B. Ek and C. M. Anderson, Bioorg. Med. Chem., 1995, 3, 1255 CrossRef CAS PubMed; (e) J.-M. Bernardon and P. Diaz, PCT Int. Appl, WO1999065872A1, 1999 Search PubMed; (f) C. Millois and P. Diaz, Org. Lett., 2000, 2, 1705 CrossRef CAS PubMed.
- T. Wirth, Tetrahedron, 1999, 55, 1 CrossRef CAS.
- (a) N. Miyaura, in Metal-Catalyzed Cross Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, vol. 1, p. 41 Search PubMed; (b) G. Zeni, D. S. Ludtke, R. B. Panatieri and A. L. Braga, Chem. Rev., 2006, 106, 1032 CrossRef CAS PubMed; (c) G. Perin, E. J. Lenardao, R. G. Jacob and R. B. Panatieri, Chem. Rev., 2009, 109, 1277 CrossRef CAS PubMed.
- (a) Y. Nishiyama, K. Tokunaga and N. Sonoda, Org. Lett., 1999, 1, 1725 CrossRef CAS; (b) T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587 CrossRef CAS PubMed; (c) H. J. Cristau, B. Chaband, A. Chene and H. Christol, Synthesis, 1981, 892 CrossRef CAS; (d) K. Takagi, Chem. Lett., 1987, 2221 CrossRef CAS; (e) H. Suzuki, H. Abe and A. Osuka, Chem. Lett., 1981, 151 CrossRef CAS; (f) F. Y. Kwong and S. L. Buchwald, Org. Lett., 2002, 4, 3517 CrossRef CAS PubMed; (g) R. K. Gujadhur and D. Venkataraman, Tetrahedron Lett., 2003, 44, 81 CrossRef CAS; (h) M. Wang, K. Ren and L. Wang, Adv. Synth. Catal., 2009, 351, 1586 CrossRef CAS; (i) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 120, 2922 CrossRef; (j) I. P. Beletskaya, A. S. Sigeev, A. S. Peregudov and P. V. Petrovskii, Mendeleev Commun., 2000, 10, 213 CrossRef.
- (a) D. Kundu, S. Ahammed and B. C. Ranu, Green Chem., 2012, 14, 2024 RSC; (b) N. Mukherjee, T. Chatterjee and B. C. Ranu, J. Org. Chem., 2013, 8, 11110 CrossRef PubMed; (c) R. A. Balaguez, V. G. Ricordi, C. S. Freitas, G. Perin, R. F. Schumacher and D. Alves, Tetrahedron Lett., 2014, 55, 1057 CrossRef CAS; (d) D. Kundu, S. Ahammed and B. C. Ranu, Org. Lett., 2014, 16, 1814 CrossRef CAS PubMed.
- (a) R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982 RSC; (b) P. Maity, D. Kundu and B. C. Ranu, Eur. J. Org. Chem., 2015, 1727 CrossRef CAS; (c) J. Thomas, Z. Dong, W. Dehen and M. Smet, Macromol. Rapid Commun., 2012, 33, 2127 CrossRef CAS PubMed.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (d) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- (a) S. Zhang, A. Koe, C. Heintz, A. Senior and J. Jin, Tetrahedron Lett., 2016, 57, 260 CrossRef CAS; (b) J. V. Comasseto, A. T. Omori, A. L. M. Porto and L. H. Andrade, Tetrahedron Lett., 2004, 45, 473 CrossRef CAS; (c) Y. Mechehoud, F. Benayache, S. Benayache and P. Mosset, E-J. Chem., 2010, 7, S143 CrossRef CAS; (d) H. Zhao, Y. Jiang, Q. Chen and M. Cai, New J. Chem., 2015, 39, 2106 RSC; (e) D. Singh, A. M. Deobald, L. R. S. Camargo, G. Tabarelli, O. E. D. Rodrigues and A. L. Braga, Org. Lett., 2010, 12, 3288 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR spectra of all the synthesized compounds and DFT calculation data. See DOI: 10.1039/c6qo00515b |
| This journal is © the Partner Organisations 2017 |