
Nickel-catalyzed direct formation of the C–S bonds of aryl sulfides from arylsulfonyl chlorides and aryl iodides using Mn as a reducing agent †
Yang
Wang
,
Xiaofeng
Zhang
,
Haixiong
Liu
,
Hui
Chen
and
Deguang
Huang
*
Sate Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: dhuang@fjirsm.ac.cn
First published on 8th October 2016
Abstract
Various unsymmetrical aryl sulfides were synthesized by nickel-catalyzed C–S bond formation in good to excellent yields. The reactions employed arylsulfonyl chlorides as an aryl thiol source and Mn dust as a reducing agent. The scope and versatility of the method have been successfully demonstrated with 42 examples. Mechanistic studies revealed the existence of an intermediate disulfide substance. A catalytic cycle was proposed including a three-step reduction by Mn for the achievement of the reaction, and Ni(0) and Ni(I) species were supposed to be involved in the reaction mechanism.
Introduction
Organosulfur compounds are indispensable chemical substances to life,1 and widely exist in many biological macromolecules,2 materials science,3 pharmaceutical, agricultural and chemical agents.4 Carbon–sulfur (C–S) bond-forming reactions play an important role in the construction of sulfur-containing compounds, and the formation of C–S bonds via cross coupling reactions is common in thioether chemistry and environmental science,5 especially direct coupling of organic halides with thiols.6 Diaryl sulfide is the main member of organosulfur compounds, and is known to provide excellent structural features for various chemical and biological agents such as LuAA2104 for the treatment of major depressive disorder, HIV-1 integrase inhibitor, fluorinated diaryl sulfides as serotonin transporter ligands, trypanothione reductase inhibitor.7 A number of results have been published in the synthesis of aryl sulfide by using thiols as starting material in reaction,6 but the development of synthetic sulfidization is challenged due to the volatile properties and stinking nature of thiols leading to strict environmental and health issues. The reduction of aryl sulfones was ever-applied to synthesize aryl sulfides on the premise that strong reducing agents such as diisobutyl aluminium hydride (DIBAH) or LiAlH4 were generally used.8 Recently, a couple of transition metal-catalyzed arylsulfonyl sulfidizations for the synthesis of aryl sulfides from arylsulfonyl hydrazides were reported,9 but the use of flammable and explosive sulfonyl hydrazide species, and therein strong base reaction conditions would limit their applications in large-scale processes. Therefore, the simplification of C–S bond-forming reactions for the synthesis of aryl sulfides becomes important and the development of concise, safe and efficient methods has been expected.Arylsulfonyl chlorides are inexpensive, safe and readily available compounds. They have been widely used as the protecting groups and sulfonylating agents in materials science and medicinal chemistry over a century.10 The utilization of arylsulfonyl chlorides concerning the C–S bond coupling reactions generally produced the congeneric species of aryl sulfones,11 or disulfides to the end.12 The transition-metal-catalyzed direct formation of aryl sulfides from arylsulfonyl chlorides was rarely reported except for the formation of di(hetero)aryl sulfide compounds derived from N-methylindoles and indolizines respectively, via C–H bond functionalization (Scheme 1a & b).13 So far, the synthesis of aryl sulfides through direct construction of arylsulfonyl chlorides with aryl halides has not been found. There is yet another attractive and topical project of the C–S cross-coupling reaction by sulfhydrylation of arylsulfonyl chlorides.
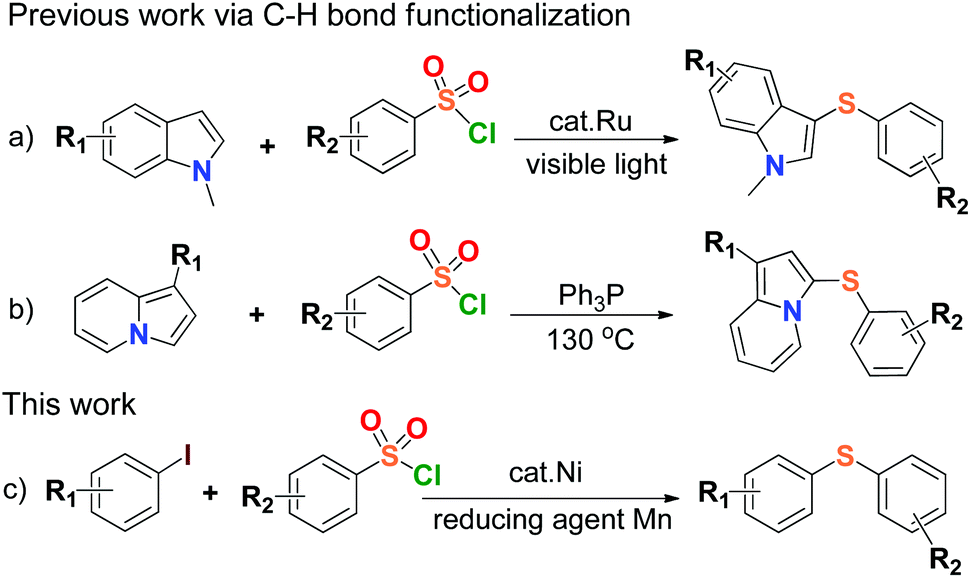 | ||
| Scheme 1 Direct synthesis of aryl sulfides from arylsulfonyl chlorides. | ||
On the other hand, research interest in low cost metal catalysis keeps on increasing with the great development of organometallic catalytic chemistry, and nickel salts have received much attention due to their efficiency in catalysis, environmental friendliness, and potential economic feasibility.14 In view of the above and as a part of our current research program, herein we develop a new, mild and efficient method for the formation of the C–S bonds by the coupling reaction between arylsulfonyl chlorides and aryl iodides in the presence of a nickel(II) complex as a catalyst and Mn dust as a reducing agent (Scheme 1c). This work might offer a clue to synthesize highly valuable unsymmetrical aryl sulfides by using simple and commercially available materials in practice. To our best knowledge, there is no report on sulfhydrylation using arylsulfonyl chlorides and aryl iodides, and it was also discovered that Mn dust could reduce arylsulfonyl chlorides to disulfides.
Results and discussion
We commenced the study by choosing p-toluenesulfonyl chloride and 4-iodoanisole as model substrates and investigated the reaction by varying different ingredients such as the catalyst, ligand, additive, reducing agent, substrate ratio and solvent to find out the most suitable conditions (Table 1). Upon a number of experimental data presented, it was found that the combination of NiCl2·glyme (10 mol%), L4 (15 mol%), Mn dust as the reducing agent, sodium iodide and potassium fluoride as the additive, and substrate ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in N,N-dimethylformamide (DMF) under a N2 atmosphere provided the best results, yielding the product, (4-methoxyphenyl) (4-methylphenyl) sulfane, in 92% based on 4-iodoanisole (entry 5, Table 1).
:1 in N,N-dimethylformamide (DMF) under a N2 atmosphere provided the best results, yielding the product, (4-methoxyphenyl) (4-methylphenyl) sulfane, in 92% based on 4-iodoanisole (entry 5, Table 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Additive | Reducing agent | m:nb |
Solvent | Yieldc (%) |
a Reaction conditions: 1a (0.6 mmol), 2a (0.2 mmol), catalysts (10 mol%), ligand (15 mol%), NaI (0.3 mmol), KF (0.3 mmol), reducing agent (1 mmol) and solvent (1 ml). Under a N2 atmosphere.
b m:n is the molar ratio of compound 1 to compound 2.
c Isolated yield based on aryl iodide after column chromatography.
d Sam = samarium dust.
|
|||||||
| 1 | None | L4 | NaI; KF | Mn | 3:1 |
DMF | 0 |
| 2 | NiCl 2 | L4 | NaI; KF | Mn | 3:1 |
DMF | 78 |
| 3 | NiBr 2 | L4 | NaI; KF | Mn | 3:1 |
DMF | 46 |
| 4 | Ni(OAc) 2 | L4 | NaI; KF | Mn | 3:1 |
DMF | Trace |
| 5 | NiCl 2 ·glyme | L 4 | NaI; KF | Mn |
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
DMF | 92 |
| 6 | NiCl2·glyme | L4 | NaI; KF | Zn | 3:1 |
DMF | 0 |
| 7 | NiCl2·glyme | L4 | NaI; KF | Sam | 3:1 |
DMF | 0 |
| 8 | NiCl2·glyme | L4 | NaI; KF | PPh 3 | 3:1 |
DMF | 0 |
| 9 | NiCl2·glyme | L4 | NaI; KF | None | 3:1 |
DMF | 0 |
| 10 | NiCl2·glyme | L4 | NaI; KF | Mn |
2:1
|
DMF | 74 |
| 11 | NiCl2·glyme | L4 | NaI; KF | Mn |
1:1
|
DMF | 35 |
| 12 | NiCl2·glyme | L4 | None | Mn | 3:1 |
DMF | 57 |
| 13 | NiCl2·glyme | L4 | NaI | Mn | 3:1 |
DMF | 66 |
| 14 | NiCl2·glyme | L4 | KF | Mn | 3:1 |
DMF | 54 |
| 15 | NiCl2·glyme | None | NaI; KF | Mn | 3:1 |
DMF | Trace |
| 16 | NiCl2·glyme | L 1 | NaI; KF | Mn | 3:1 |
DMF | 83 |
| 17 | NiCl2·glyme | L 2 | NaI; KF | Mn | 3:1 |
DMF | 85 |
| 18 | NiCl2·glyme | L 3 | NaI; KF | Mn | 3:1 |
DMF | 88 |
| 19 | NiCl2·glyme | L 5 | NaI; KF | Mn | 3:1 |
DMF | 87 |
| 20 | NiCl2·glyme | L4 | NaI; KF | Mn | 3:1 |
CH 3 CN | 0 |
| 21 | NiCl2·glyme | L4 | NaI; KF | Mn | 3:1 |
Toluene | 0 |
| 22 | NiCl2·glyme | L4 | NaI; KF | Mn | 3:1 |
CH 2 Cl 2 | 0 |

Modification of the experimental conditions by the absence of a catalyst, reducing agent, or ligand, respectively, confirmed that these species played a critical role in the C–S bond coupling reaction (entries 1, 9 and 15). Initial trails identified NiCl2·glyme as a highly efficient metal catalyst combined with Mn as a reducing agent. Other nickel sources such as NiBr2 (entry 3) and Ni(OAc)2 (entry 4) provided (4-methoxyphenyl)(4-methylphenyl) sulfane in much lower yields. Among all reducing agents analyzed, Mn was found to be essential for the reaction to occur (entry 5). Zn dust, Sam dust, PPh3 or the lack of reducing agents showed nonexistence of the product completely (entries 6–9). Chelate ligands with nitrogen donors have been successfully used for the cross-coupling reaction of alkyl halides in organometallic catalysis.15 Here higher yields of aryl sulfides could be achieved with the employment of bipyridines, di(2-pyridyl)ketone, o-phenanthroline, neocuproine or bathocuproin (L1–L5) as supported ligands, among which neocuproine (L4) worked the best (entry 5). Interestingly, the addition of sodium iodide and potassium fluoride as additives was found having a positive effect on the productivity (entries 5 and 12–14). Moreover, with regard to the solvent, this coupling reaction only proceeded efficiently in DMF (entries 5 and 20–22).
Encouraged by these results, we started to explore our reaction conditions by employing other substrates in this nickel-catalyzed cross-coupling reaction (Table 2). Aryl iodides were firstly applied to couple with p-toluenesulfonyl, and the experimental results showed an indication that the steric effect and electronic effect had clear impacts on the yield of the reaction. 4-Iodoanisole and 3-iodoanisole yielded the desired product in 92% (3f) and 81% (3g), with 2-iodoanisole giving only 22% yield (3h). The steric effect would reduce the yield apparently by comparison of the meta- (3i) and ortho-substituents (3j), and the para-positon substituents generally afforded a slightly better yield than the meta-positions’ in the reactions.
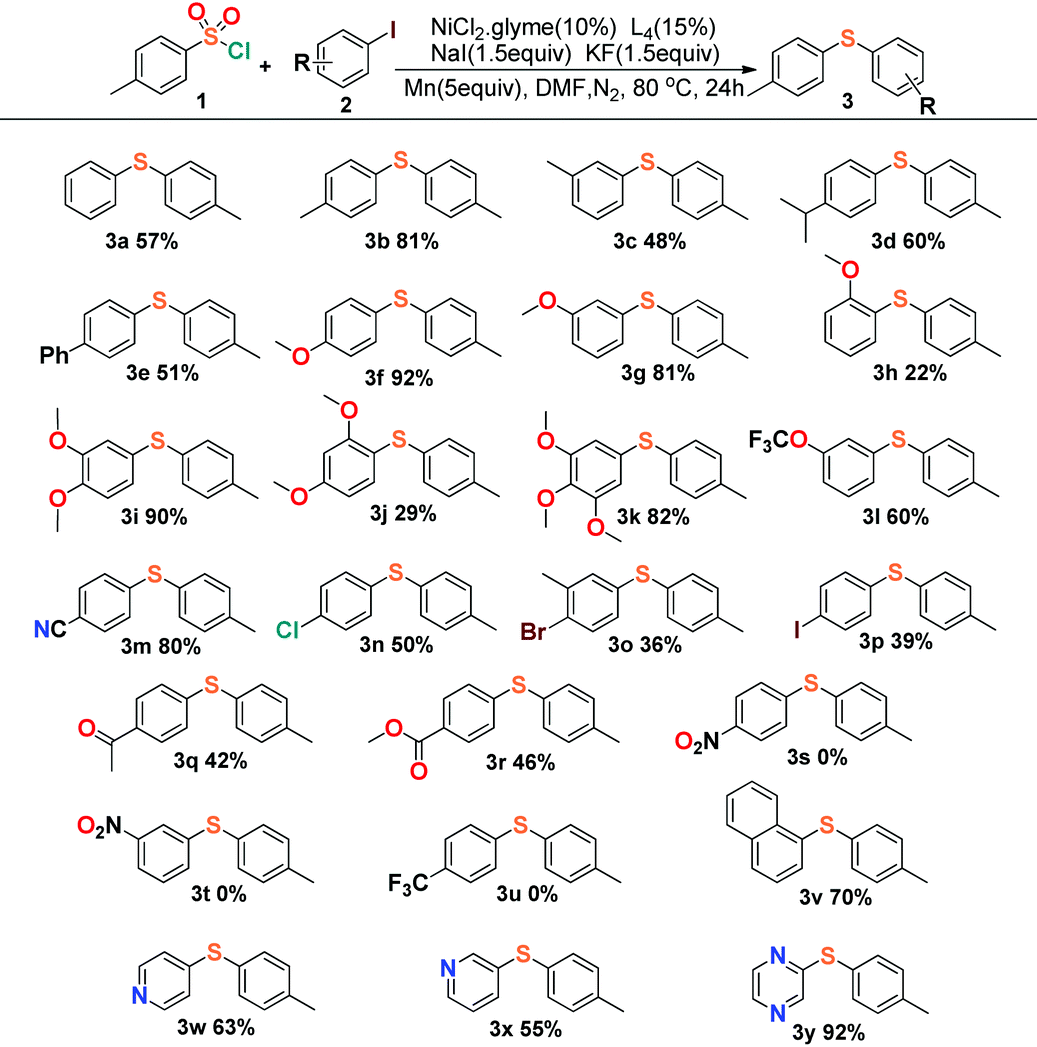
|
The investigation of the electronic effect demonstrated that electron-withdrawing groups showed an inhibiting effect on the reactions with yields all below 50% (3n–3u). The cross-coupling reaction could even vanish when the strong electron-withdrawing groups were applied such as nitryl (3s, 3t) or trifluoromethyl (3u) groups. Unexpectedly, the cyanide substituted 4-iodobenzonitrile (3m) displayed a different character from those strong electron-withdrawing groups with the desired product isolated in 80% yield, which could be compared to the yields of those reactions with electron-donating groups (>80%, 3f, 3i, 3k). This character was consistent with the result deduced from cyanide substituted 4-cyanobenzene-1-sulfonyl chloride (Table 3, 4n, 68%). The reason for the cyanide groups presenting a positive effect for C–S bond formation is not clear. Moreover, our reaction exhibited good tolerance towards the transformation of disubstituted aromatic C–X (X = Cl, Br, I) species to aryl sulfides. The reaction of 1,4-dihalide species with arylsulfonyl chlorides afforded the single substituted products (3n–3p) chemoselectively, leaving the other halide intact for further conjunctive manufacture. Finally, it was shown that not only a naphthalenyl group could be employed (3v) as a substrate for sulfidization, but also the pyridyl and pyrazinyl groups presented a positive effect in the preparation of aryl sulfide products (3w–3y).
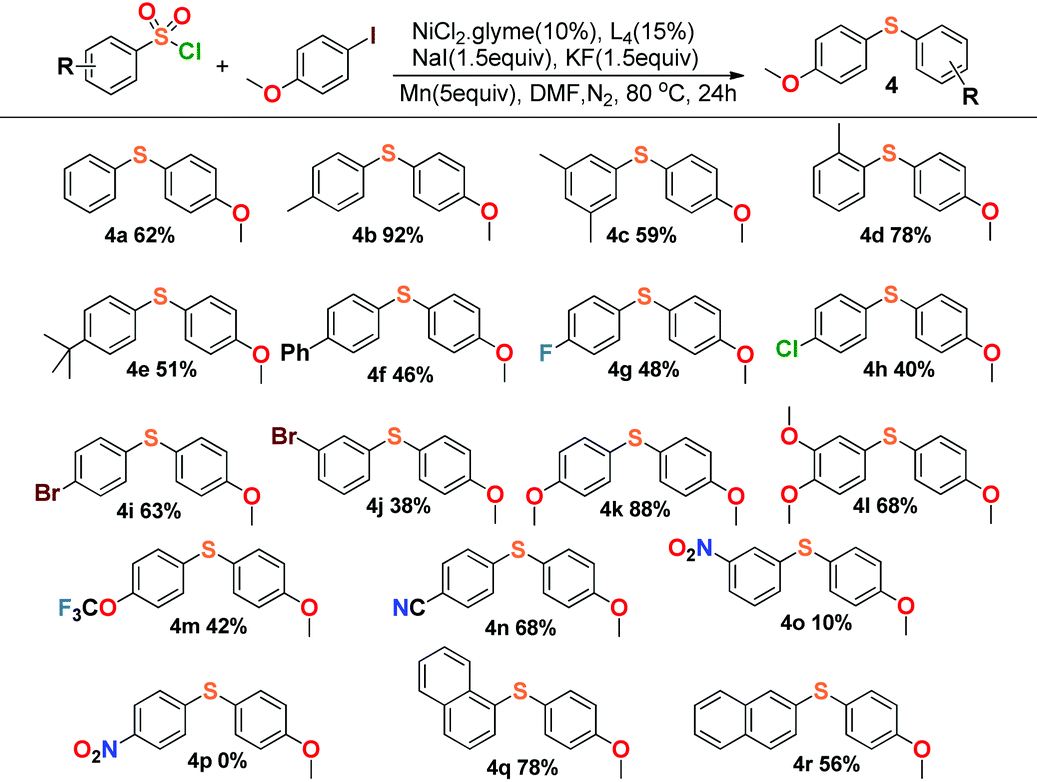
|
This reaction also exhibited good compatibility with a relatively broad range of arysulfonyl chlorides. As summarized in Table 3, arysulfonyl chlorides substituted with electron-donating or electron-withdrawing groups were successfully converted into the corresponding aryl sulfides except for the nitro group substituted substrates, 3-nitrobenzenesulfonyl chloride (4o) and 4-nitrobenzenesulfonyl chloride (4p). The former reaction produced only 10% yield and the latter gave no product at all. Perhaps the electron-withdrawing effect of the nitro group was so strong that the cross-coupling was restrained appropriately. In general, the substances with electron-donating groups (4k, 4l) afforded higher yields than those of the electron-withdrawing groups in our experiments (4g–4j), and α-naphthalenesulfonyl chloride (4q) offered higher yield than β-naphthalenesulfonyl chloride (4r).
For the purpose of exploration of the reaction mechanism, specific experiments were carried out intentionally (Scheme 2). The stirring of 4-cyanobenzene-1-sulfonyl chloride with Mn dust in DMF under a N2 atmosphere yielded the unique product bis(4-cyanophenyl) disulfide 5a in 63% yield (Scheme 2(a)). No reaction was observed when the disulfide precursor 5a was stirred with 4-iodoanisole in the absence of Mn dust (Scheme 2(b)) or NiCl2·glyme (Scheme 2(c)) respectively, which showed a clue that the Ni(II) would not be the original catalytic point heading for the occurrence of the reaction.16 Consequently, 83% of the desired product 4n was achieved when the combination of NiCl2·glyme and Mn dust was applied in DMF at 80 °C, in the presence of disulfide and 4-iodoanisole (Scheme 2(d)). The structures of compounds 5a and 4n were identified by X-ray crystallography (Fig. 1 and 2). Reduction of Ni(II) to Ni(0) by Mn dust was thereby supposed to be the starting point for the cross-coupling reaction, which was consistent with the reference that the reduced Ni(0) has the ability to insert into S–S bonds17 and the aryl sulfide species could be obtained via cleavage of the S–S bond of disulfide precursors.18
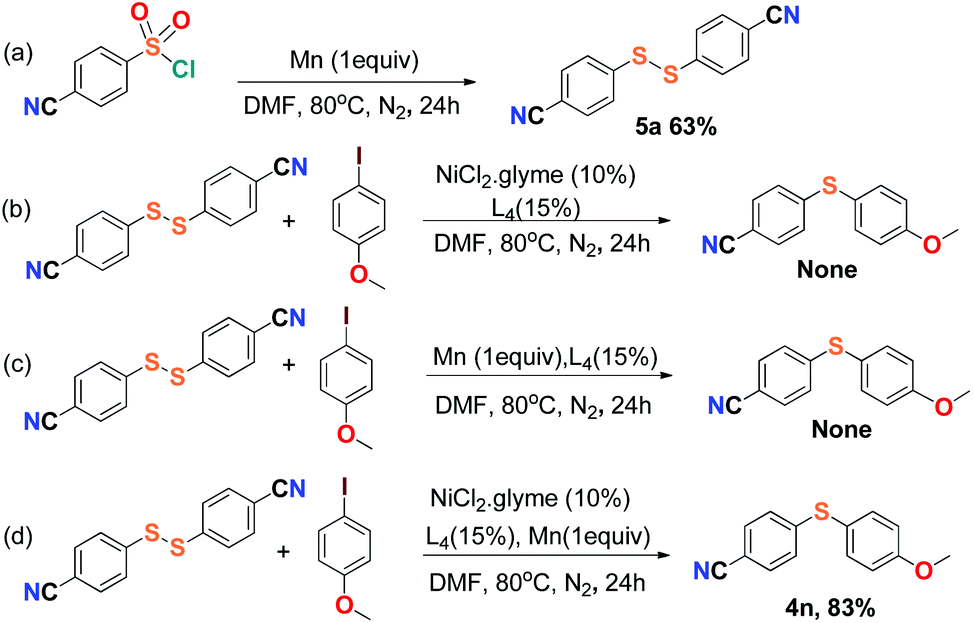 | ||
| Scheme 2 Mechanistic exploration of reductive sulfidization promoted by Mn dust. | ||
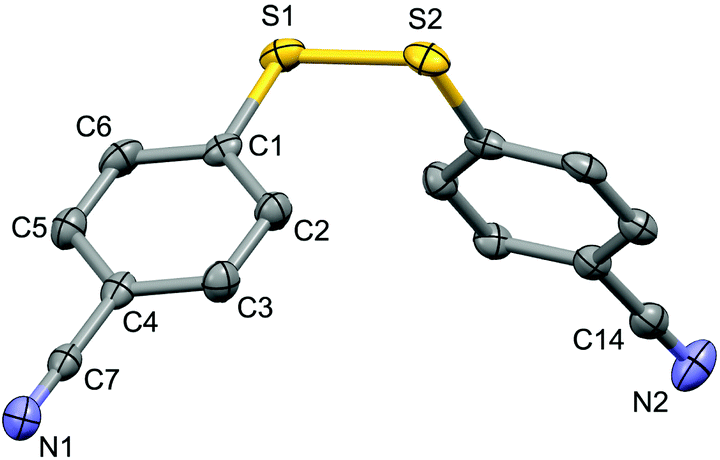 | ||
| Fig. 1 Structure of the complex 5a (CCDC 1494577) showing 50% probability ellipsoid. Selected bond lengths (Å): S1–S2 2.030(2), C7–N1 1.149(6), C14–N2 1.141(7). | ||
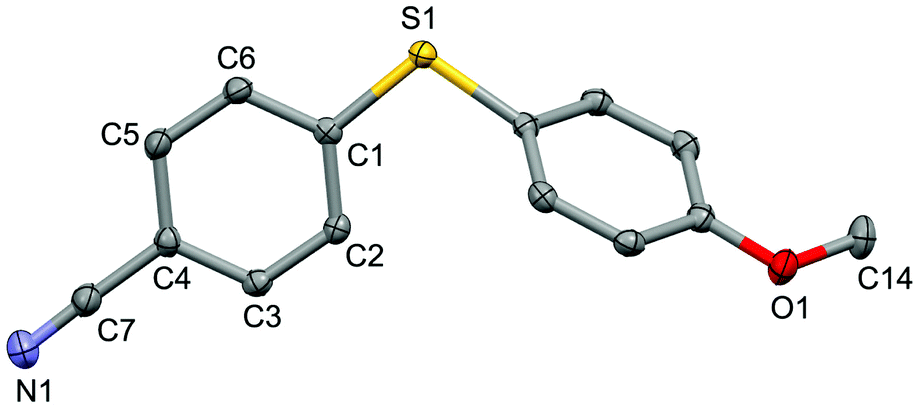 | ||
| Fig. 2 Structure of the complex 4n (CCDC 1494576) showing 50% probability ellipsoid. Selected bond lengths (Å): S1–C1 1.763(2), C7–N1 1.151(2), O1–C14 1.433(2). | ||
Based on the above results, a plausible mechanism is depicted in Scheme 3. Three steps of heterogeneous reduction by Mn dust were proposed to be involved in the catalytic cycle. Firstly, Mn reduces the initial precursors of L–Ni(II) (1) and arylsulfonyl chloride to form a highly reductive reagent L–Ni(0) (2) and disulfide precursor Ar–S–S–Ar simultaneously. They are followed to generate the intermediate L–Ni(II)(SAr)2 (3) via the insertion of L–Ni(0) into the S–S bond. Secondly, the intermediate (3) is one-electron reduced by Mn to produce a reactive species L–Ni(I)(SAr) (4). The species 4 undergoes a nucleophilic attack with aryl iodide (Ar′–I) to form a transition oxidant state L–Ni(III)(SAr)(SAr′)(I) (5), through which the desired product Ar–S–Ar′ is released subsequently, along with the formation of the reductant L–Ni(I)–I (6). Finally, the transporting species 6 undergoes a further reduction by Mn to give the starting reagent, L–Ni(0) (2). With respect to this catalytic cycle, a second equivalent of arylsulfonyl chloride would be consumed due to the generation of the by-product, Mn(SAr)2. This inference was supported by our reactions (Table 1, entries 5, 10 and 11) that the low ratios of arylsulfonyl chloride to aryl iodide led to a drastic decrease of yield consequently.
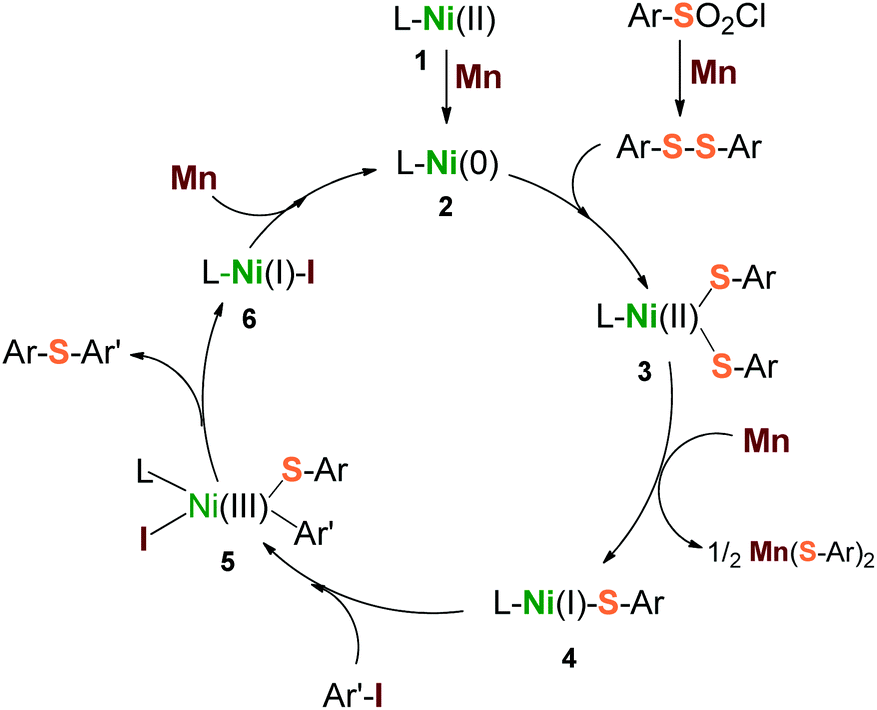 | ||
| Scheme 3 Proposed catalytic cycle for reductive sulfidization from arylsulfonyl chlorides and aryl iodides. | ||
Experimental section
NiCl2·glyme (4.4 mg, 0.02 mmol), neocuproine (L4) (6.3 mg, 0.03 mmol), NaI (45 mg, 0.3 mmol), KF (17.4 mg, 0.3 mmol), Mn dust (55 mg, 1 mmol), p-toluenesulfonyl chloride (114 mg, 0.6 mmol) and 4-iodoanisole (47.4 mg, 0.2 mmol) were mixed in dried DMF (1 ml) in a 35 ml Teflon screw-cap sealed tube. The tube was charged with N2 and the mixture was vigorously stirred at 80 °C for 24 h. After cooling to room temperature, the reaction mixture was diluted with dichloromethane (20 mL), filtered through a pad of silica gel and further concentrated under reduced pressure. The crude product was purified on a silica gel column eluted with petroleum ether/EtOAc (40:1 v/v) to afford products in yields up to 92% (3f). Detailed experimental processes are given in the ESI.†
Conclusions
In summary, we have described a novel, safe, and efficient nickel-catalyzed reaction to synthesize unsymmetrical aryl sulfides from arysulfonyl chlorides and aryl iodides directly in 42 samples by using Mn dust as a reducing agent. Experimental and mechanistic studies revealed a catalytic cycle in which a three-step reduction by Mn was supposed to be the key point for the achievement of the reaction. This work afforded a user-friendly and operationally simple cross-coupling reaction for the preparation of aryl sulfides under mild reaction conditions, and might provide a chance to access a multi-step reduction mechanism for further catalytic investigations.Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant No. 21371171).Notes and references
- E. Block, Angew. Chem., Int. Ed. Engl., 1992, 31, 1135 CrossRef.
- N.-S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257 CrossRef CAS PubMed.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef CAS PubMed.
- (a) A. Casini, J.-Y. Winum, J.-L. Montero, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2003, 13, 837 CrossRef CAS PubMed; (b) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133 CrossRef CAS PubMed.
- (a) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (b) H. Wang, L. Jiang, T. Chen and Y. Li, Eur. J. Org. Chem., 2010, 12, 2324 CrossRef; (c) J. Zhu, Y. Chen, F. Lin, B. Wang, Z. Chen and L. Liu, Org. Biomol. Chem., 2015, 13, 3711 RSC; (d) J. R. DeBergh, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10638 CrossRef CAS PubMed; (e) G. Yuan, J. Zheng, X. Gao, X. Li, L. Huang, H. Chen and H. Jiang, Chem. Commun., 2012, 48, 7513 RSC.
- (a) E. Schaumann, Top. Curr. Chem., 2007, 274, 1 CrossRef CAS; (b) H.-J. Xu, Y.-Q. Zhao, T. Feng and Y.-S. Feng, J. Org. Chem., 2012, 77, 2878 CrossRef CAS PubMed; (c) Y. Zhang, K. C. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495 CrossRef CAS PubMed; (d) M. A. Fernández-Rodríguez, Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180 CrossRef PubMed; (e) S. Kovácsa and Z. Novák, Org. Biomol. Chem., 2011, 9, 711 RSC; (f) C. C. Eichman and J. P. Stambuli, J. Org. Chem., 2009, 74, 4005 CrossRef CAS PubMed.
- (a) B. Bang-Andersen, T. Ruhland, M. Jørgensen, G. Smith, K. Frederiksen, K. G. Jensen, H. Zhong, S. M. Nielsen, S. Hogg, A. Mørk and T. B. Stensbøl, J. Med. Chem., 2011, 54, 3206 CrossRef CAS PubMed; (b) S. Pasquini, C. Mugnaini, C. Tintori, M. Botta, A. Trejos, R. K. Arvela, M. Larhed, M. Witvrouw, M. Michiels, F. Christ, Z. Debyser and F. Corelli, J. Med. Chem., 2008, 51, 5125 CrossRef CAS PubMed; (c) Y. Huang, S. Bae, Z. Zhu, N. Guo, B. L. Roth and M. Laruelle, J. Med. Chem., 2005, 48, 2559 CrossRef CAS PubMed; (d) B. Stump, C. Eberle, M. Kaiser, R. Brun, R. L. Krauth-Siegel and F. Diederich, Org. Biomol. Chem., 2008, 6, 3935 RSC.
- (a) E. Akgün, K. Mahmood and C. A. Mathis, J. Chem. Soc., Chem. Commun., 1994, 761 RSC; (b) N. Atsuko and K. Tadahiro, Chem. Pharm. Bull., 1987, 35, 1770 CrossRef; (c) M. A. Desando, S. Walker and W. H. Baarschers, J. Chem. Phys., 1980, 73, 3460 CrossRef CAS; (d) J. M. Khurana, V. Sharma and S. A. Chacko, Tetrahedron, 2007, 63, 966 CrossRef CAS.
- (a) N. Singh, R. Singh, D. S. Raghuvanshi and K. N. Singh, Org. Lett., 2013, 15, 5874 CrossRef CAS PubMed; (b) R. Singh, B. K. Allam, N. Singh, K. Kumari, S. K. Singh and K. N. Singh, Adv. Synth. Catal., 2015, 357, 1181 CrossRef CAS; (c) T. T. Wang, F. L. Yang and S. K. Tian, Adv. Synth. Catal., 2015, 357, 928 CrossRef CAS.
- (a) B. M. Graybill, J. Org. Chem., 1967, 32, 2931 CrossRef CAS; (b) S. J. Nara, J. R. Harjani and M. M. Salunkhe, J. Org. Chem., 2001, 66, 8616 CrossRef CAS PubMed; (c) J. K. Laha, K. P. Jethava and N. Dayal, J. Org. Chem., 2014, 79, 8010 CrossRef CAS PubMed; (d) S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474 CrossRef PubMed; (e) R. E. Kirk, M. Grayson and D. Eckroth, Kirk-Othmer Concise Encyclopedia of Chemical Technology, Wiley-VCH, New York, 1985 Search PubMed.
- (a) S. S. Labadie, J. Org. Chem., 1989, 54, 2496 CrossRef CAS; (b) B. P. Bandgar, S. V. Bettigeri and J. Phopase, Org. Lett., 2004, 6, 2105 CrossRef CAS PubMed; (c) X. Zhao, E. Dimitrijevicé and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466 CrossRef CAS PubMed; (d) Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270 CrossRef CAS PubMed; (e) O. Saidi, J. Marae, A. E. W. Ledger, P. Liu, M. F. Mahon, G. Kociok-KÖhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS PubMed.
- (a) Y. Liu and Y. Zhang, Tetrahedron Lett., 2003, 44, 4291 CrossRef CAS; (b) G. A. Olah, S. C. Narang, L. D. Field and R. Karpeles, J. Org. Chem., 1981, 46, 4792 CrossRef; (c) W. Steinkopf, I. Schubart and S. Schmidt, Chem. Ber., 1928, 61, 680 Search PubMed; (d) A. Feigenbaum, J. P. Pete and D. Scholler, J. Org. Chem., 1984, 49, 2355 CrossRef CAS.
- (a) M. Chen, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2012, 48, 11686 RSC; (b) Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188 RSC.
- (a) N. A. Weires, E. L. Baker and N. K. Garg, Nat. Chem., 2016, 8, 75 CrossRef CAS PubMed; (b) L. Hie, N. F. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 525, 79 CrossRef PubMed; (c) L. K. G. Ackerman, M. M. Lovell and D. J. Weix, Nature, 2015, 524, 454 CrossRef CAS PubMed; (d) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- (a) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146 CrossRef CAS PubMed; (b) J. Choi and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 9102 CrossRef CAS PubMed; (c) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cŕdenas, Angew. Chem., Int. Ed., 2007, 119, 8946 CrossRef; (d) J. T. Binder, C. J. Cordier and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 17003 CrossRef CAS PubMed; (e) Y. Liu, J. Cornella and R. Martin, J. Am. Chem. Soc., 2014, 136, 11212 CrossRef CAS PubMed.
- (a) N. Taniguchi, J. Org. Chem., 2004, 69, 6904 CrossRef CAS PubMed; (b) M. Uchino, K. Asagi, A. Yamamoto and S. Ikeda, J. Organomet. Chem., 1975, 84, 93 CrossRef CAS; (c) T. Yamamoto and Y. Sekine, Inorg. Chim. Acta, 1984, 83, 47 CrossRef CAS.
- (a) V. H. Schorr and G. Wilke, Angew. Chem., Int. Ed. Engl., 1969, 81, 896 Search PubMed; (b) R. G. Hayter and F. S. Humiec, J. Inorg. Nucl. Chem., 1964, 26, 807 CrossRef CAS.
- (a) N. Taniguchi and T. Onami, J. Org. Chem., 2004, 69, 915 CrossRef CAS PubMed; (b) A. Kumar, B. S. Bhakuni, Ch. D. Prasad, S. Kumar and S. Kumar, Tetrahedron, 2013, 69, 5383 CrossRef CAS; (c) M. Wen, C. Shen, L. Wang, P. Zhang and J. Jin, RSC Adv., 2015, 5, 1522 RSC; (d) S. Kumar and L. Engman, J. Org. Chem., 2006, 71, 5400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization of compounds presented in Tables 2 & 3 and Scheme 2; crystal data and structure refinement for 7 compounds. CCDC 1494571–1494577. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00451b |
| This journal is © the Partner Organisations 2017 |