
Controlling chemoselectivity in copper-catalyzed decarboxylative A3/A3 cross-couplings: direct formation of unsymmetrical 1,4-diamino-2-butynes†
Panfeng
Zhao
a,
Huangdi
Feng
*a,
Haoran
Pan
a,
Zhihua
Sun
a and
Minchao
Tong
*b
aCollege of Chemistry and Chemical Engineering, Shanghai University of Engineering Science, 333 Longteng Road, Shanghai, 201620, China. E-mail: hdfeng@sues.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, China. E-mail: minchaotong@sioc.ac.cn
First published on 10th October 2016
Abstract
A direct cross-coupling of propiolic acid with two kinds of in situ formed iminiums has been achieved via the CuI/CuCl2-catalyzed decarboxylative A3/A3 domino process. This novel protocol to synthesise unsymmetrical 1,4-diamino-2-butynes is operationally simple in an atom- and step-economical manner, and provides products in moderate to excellent yields with high chemoselectivity.
Unsymmetrical 1,4-diamino-2-butynes are of high synthetic interest due to their prevalence as the core intermediates for the construction of many bioactive molecules, organic materials and macromolecular materials.1,2 A number of methods have been developed for 1,4-diamino-2-butynes’ synthesis.3 Current synthetic strategies for accessing such frameworks focus on the construction of symmetrical 1,4-diamino-2-butynes by nucleophilic substitution reaction or transition metal-catalyzed homo-coupling reactions.4 For example, Nakamura and co-workers reported copper-catalyzed deacetylenative coupling of propargylamines to access symmetrical 1,4-disubstituted 1,4-diamino-2-butynes.5 However, the demand for commercially unavailable starting materials leads to elaborate synthetic procedures and increase in overall cost. Recently, it has been reported that unsubstituted symmetrical 1,4-diamino-2-butynes could be synthesised by a copper-catalyzed domino approach using calcium carbide.6 Despite these significant advances, the method is still limited to homo-coupling reactions. Therefore, the development of efficient, practical, atom and step economical methods for the synthesis of unsymmetrical 1,4-diamino-2-butynes has garnered significant attention and has always been a fantastic challenge.
One of the remarkable methods for the preparation of unsymmetrical motifs involves the transition-metal-catalyzed cross-coupling reaction.7 In recent years, the decarboxylative cross-coupling has enabled the development of modern synthetic chemistry that furnishes valuable chemicals.8,9 By and large, substituted propiolic acids have found widespread use in decarboxylative coupling reactions.10 However, the utility of unsubstituted propiolic acids has been relatively limited. Very recently, Cu/Pd bimetallic catalytic systems have shown to facilitate the cross-coupling between aryl halide electrophiles and unsubstituted propiolic acid nucleophiles, for example, decarboxylative and Sonogashira couplings (Scheme 1a),11 carbonylative and Sonogashira couplings (Scheme 1b) etc.12 Alternatively, the decarboxylative A3-coupling and A3-coupling enable the direct formation of 1,4-diamino-2-butynes from the C–COOH and C–H functionalizations of propiolic acid (Scheme 1c).13 Despite this potential, the application of this approach has been limited to homo-coupling reactions because the process of addition of activated acetylene–Cu species to iminiums is carried out very quickly with the lack of chemoselectivity. Herein we report cross-coupling under Cu(I)/Cu(II) catalysis that proceeds with simple experimental procedures in good yields and excellent chemoselectivities to obtain unsymmetrical 1,4-diamino-2-butynes (Scheme 1d).
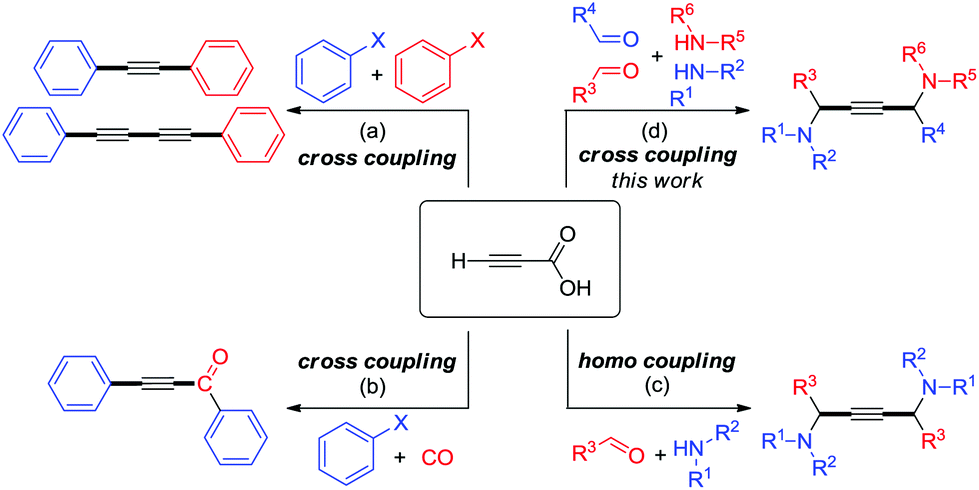 | ||
| Scheme 1 Overview of the coupling of unsubstituted propiolic acid. | ||
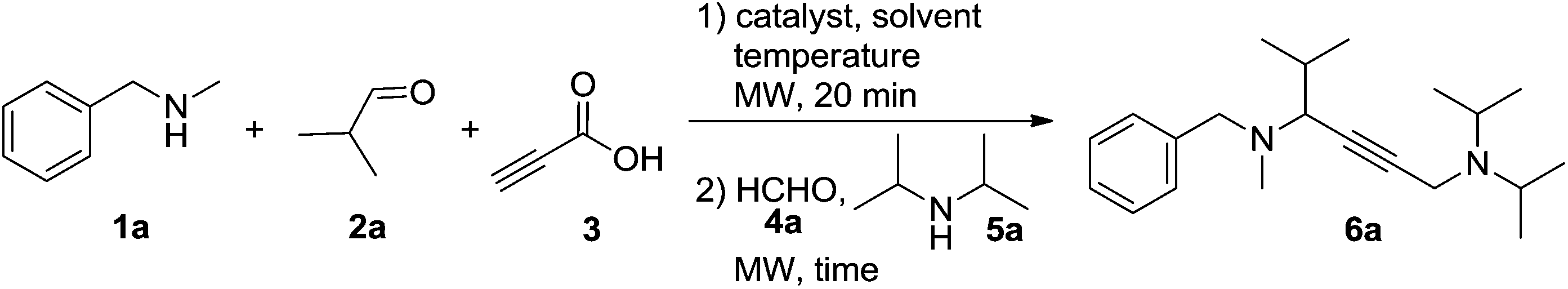
To develop this cascade approach, the conditions must be carefully selected to avoid the direct homo-coupling as an unwanted side reaction. Based on earlier work on the A3-coupling reactions,14 the one-pot procedure was initially studied by the treatment of N-methylbenzylamine 1a, isobutyraldehyde 2a and propiolic acid 3 in the presence of the CuI catalyst at 90 °C for 20 min in toluene, followed by adding formaldehyde solution 4a and diisopropylamine 5a to the reaction system, affording a moderate yield of 6a (Table 1, entry 1, 54% yield). Changing the catalyst to CuCl2 or CuBr2 was beneficial to this reaction (entries 2–6). Lowering the amount of the CuCl2 catalyst to 20 mol% still allowed the domino reaction to occur smoothly, affording the desired product 6a in 65% yield (entry 7). However, a further decrease of the catalyst to 10 mol% resulted in a striking descent in yield to 28% (entry 8). Subsequently, the systematic evaluation of solvents revealed that acetonitrile was the most effective, and the reaction also worked well in dioxane and DCE (entries 9–13). Notably, when 10 mol% CuI was used in combination with 10 mol% CuCl2, the highest yield was obtained (entry 14, 87%), the addition of CuBr or CuCl as the co-catalyst was found to be less effective for this reaction (entries 15 and 16). No improvement in yield was observed upon changing the reaction temperature (entries 17 and 18). We were pleased to find that a shorter activation time (15 min for step 1) gave a slightly higher yield of 6a (90%), meanwhile, a shorter reaction time (5 min for step 2) proved to be beneficial (entries 19–21). The product yield was decreased (entries 22 and 23) when the starting material concentration of propiolic acid was reduced to 0.7 mmol, or amine 5a was increased to 0.6 mmol.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catal. (mol%) | Solvent | Time (min) | Temp. (°C) | Yieldb (%) |
| a Reaction conditions: 1a (0.60 mmol), 2a (0.65 mmol), 3 (0.80 mmol), 10–30 mol% copper catalyst (5a as the basis) and solvent (0.6 mL) were activated for 20 min under microwave irradiation; then 4a (0.6 mmol), 5a (0.5 mmol) and solvent (0.4 mL) were added; the mixture was prepared under microwave irradiation. b Isolated yield (5a as the basis). c Reducing activation time to 15 min for step 1. d Reducing activation time to 10 min for step 1. e 0.7 mmol 3 was used. f 0.6 mmol 5a was used. | |||||
| 1 | CuI (30) | Toluene | 10 | 90 | 54 |
| 2 | CuBr (30) | Toluene | 10 | 90 | 49 |
| 3 | CuCl (30) | Toluene | 10 | 90 | 45 |
| 4 | Cu(OAc)2 (30) | Toluene | 10 | 90 | 20 |
| 5 | CuCl2 (30) | Toluene | 10 | 90 | 66 |
| 6 | CuBr2 (30) | Toluene | 10 | 90 | 60 |
| 7 | CuCl2 (20) | Toluene | 10 | 90 | 65 |
| 8 | CuCl2 (10) | Toluene | 10 | 90 | 28 |
| 9 | CuCl2 (20) | DCE | 10 | 90 | 57 |
| 10 | CuCl2 (20) | CH3CN | 10 | 90 | 73 |
| 11 | CuCl2 (20) | Dioxane | 10 | 90 | 62 |
| 12 | CuCl2 (20) | THF | 10 | 90 | 35 |
| 13 | CuCl2 (20) | H2O | 10 | 90 | 10 |
| 14 | CuCl2 (10) + CuI (10) | CH3CN | 10 | 90 | 87 |
| 15 | CuCl2 (10) + CuBr (10) | CH3CN | 10 | 90 | 70 |
| 16 | CuCl2 (10) + CuCl (10) | CH3CN | 10 | 90 | 71 |
| 17 | CuCl2 (10) + CuI (10) | CH3CN | 10 | 100 | 82 |
| 18 | CuCl2 (10) + CuI (10) | CH3CN | 10 | 70 | 74 |
| 19c | CuCl2 (10) + CuI (10) | CH3CN | 10 | 90 | 90 |
| 20d | CuCl2 (10) + CuI (10) | CH3CN | 10 | 90 | 86 |
| 21c | CuCl2 (10) + CuI (10) | CH3CN | 5 | 90 | 91 |
| 22c,e | CuCl2 (10) + CuI (10) | CH3CN | 5 | 90 | 85 |
| 23c,f | CuCl2 (10) + CuI (10) | CH3CN | 5 | 90 | 78 |
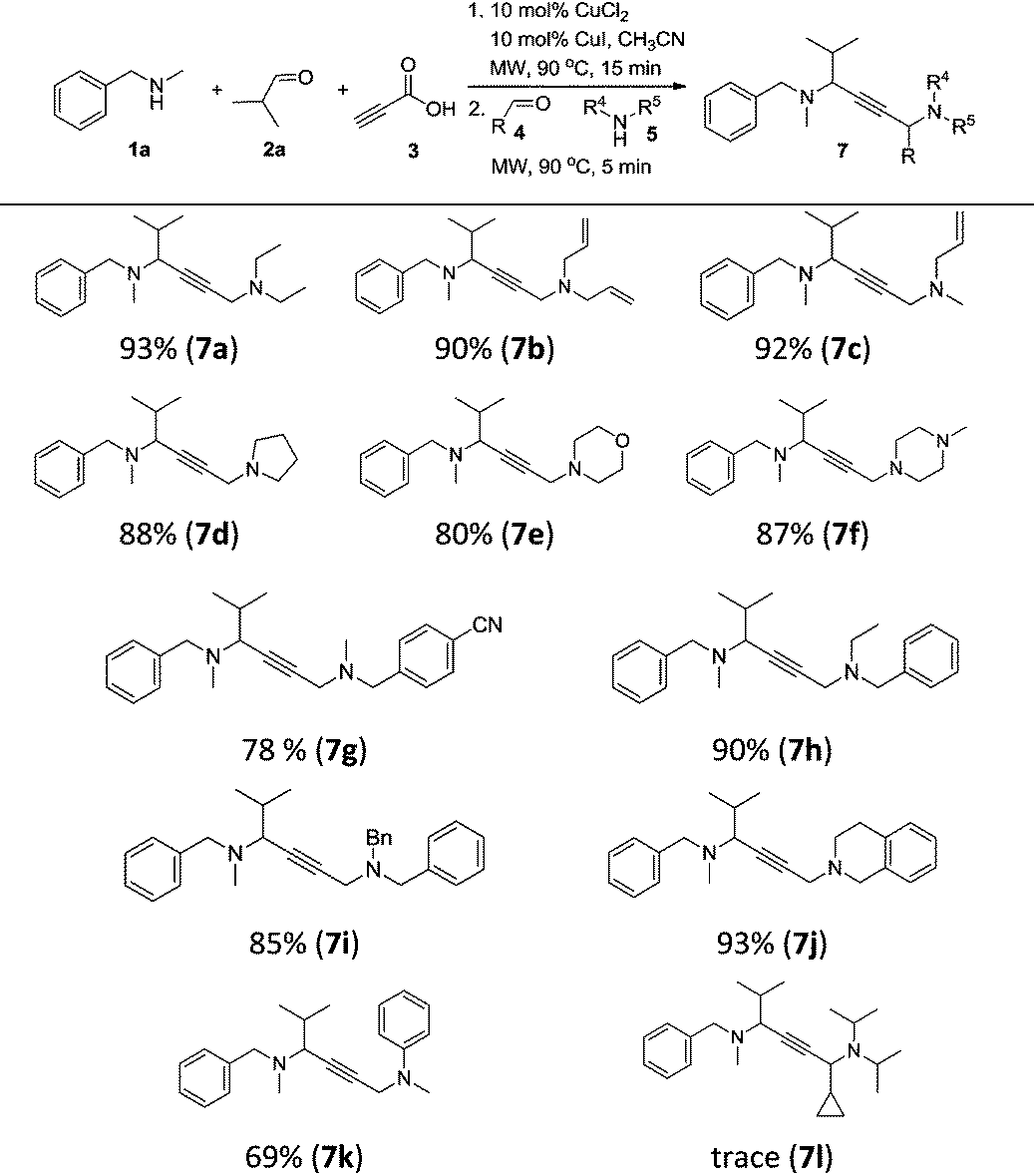
With the optimal conditions in hand, we turned our attention toward investigating the reaction substrate scope by exploring various amines 1 and aldehydes 2 (Table 2). First, a range of amines 1 was tested, affording the desired product in moderate to good yield. The results showed that increasing the steric hindrance of N-substituted benzylamines was detrimental to the reactivity (6a–6e). Meanwhile, relatively lower yields were obtained when alkylamines and heterocyclic amines were employed as the reaction substrates (6f–6h). We next looked to expand the scope of aldehydes, and were delighted to find that aliphatic aldehydes could perform very well, except 2-ethylbutanal (6i–6l). In contrast, using aromatic aldehydes led to a drop in the reaction yield (6m–6q). To our delight, formyl group substrates were also compatible, providing the desired products 6p and 6q in 62% and 68% yield, respectively.
| a Reaction conditions: CuCl2 (10 mol%), CuI (10 mol%), 1 (0.60 mmol), 2 (0.65 mmol), and 3 (0.80 mmol), in CH3CN (0.6 mL) at 90 °C for 15 min under microwave irradiation; then 4a (0.6 mmol), 5a (0.5 mmol) and CH3CN (0.4 mL) were added and kept at 90 °C for 5 min under microwave irradiation; isolated yield. |
|---|
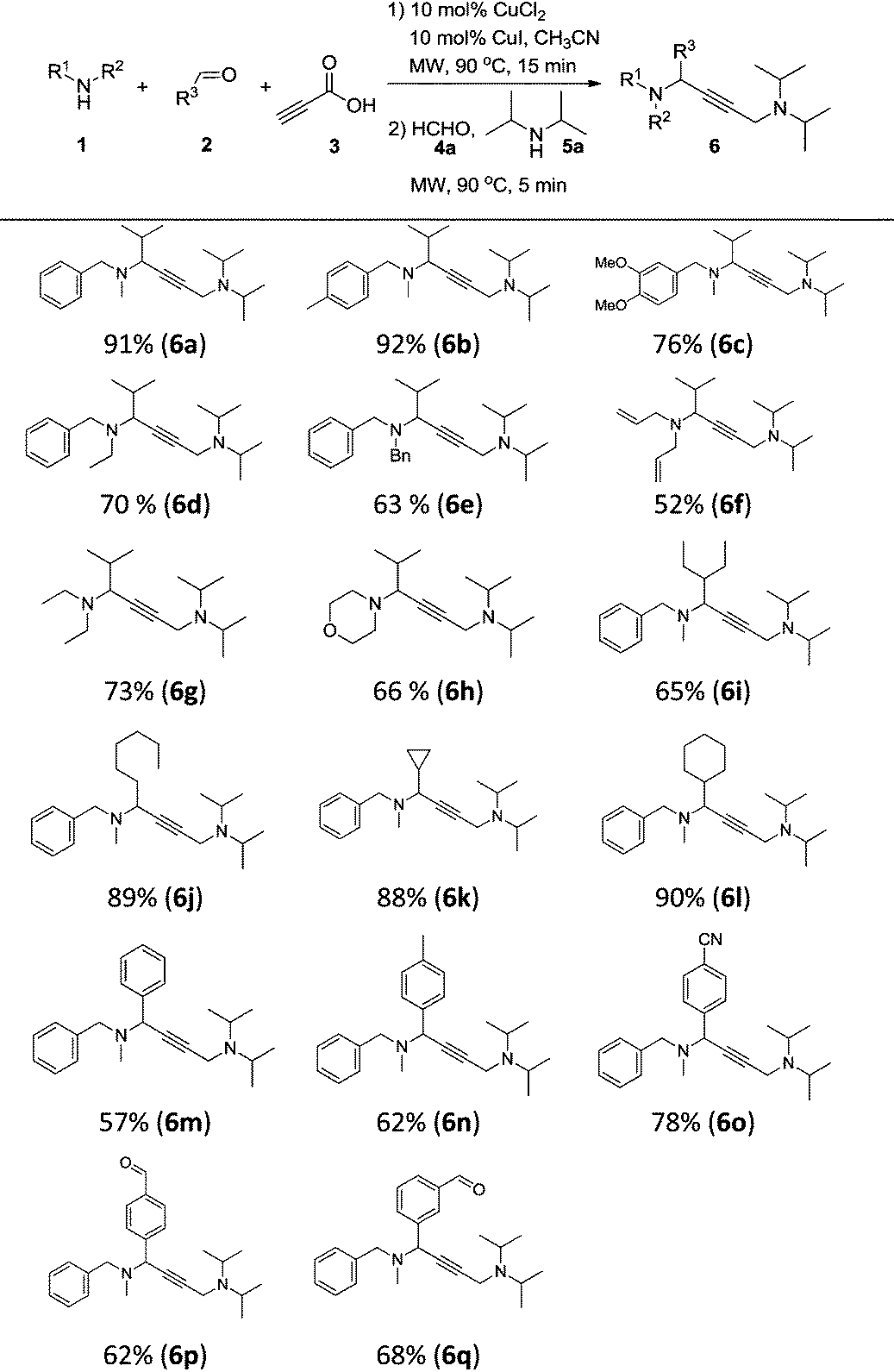
|
To achieve a more universal approach, other amines 5 were also evaluated (Table 3). An impressive scope was demonstrated with a broad range of amines. Alkyl, allyl, cyclic, heterocyclic, and benzyl secondary amines afforded good to excellent yields of the expected products under the standard reaction conditions (7a–7j). In addition, the introduction of an N-methylaniline was also suitable for this reaction, giving the desired product 7k in good yields. However, it should be noted that steric hindrance highly affects the reaction efficiency and only a trace amount of the corresponding product 7l was detected by 1H NMR when cyclopropanecarbaldehyde was employed. Instead, the corresponding symmetrical 1,4-diamino-2-butyne 8a and propargylamine 9a were obtained (Scheme 2, eqn (1)).
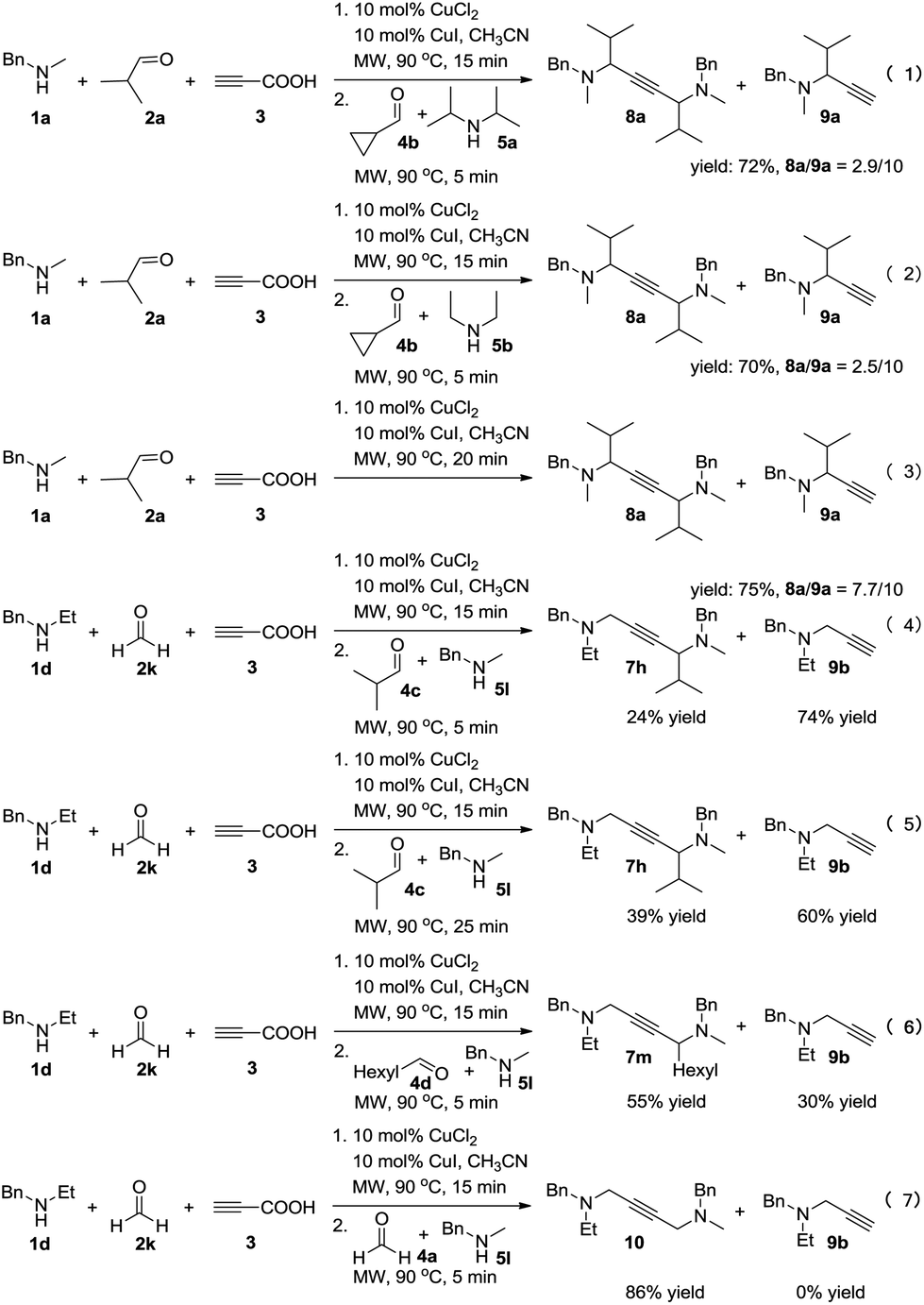 | ||
| Scheme 2 Control experiments. | ||
| a Reaction conditions: CuCl2 (10 mol%), CuI (10 mol%), 1a (0.60 mmol), 2a (0.65 mmol), and 3 (0.80 mmol), in CH3CN (0.6 mL) at 90 °C for 15 min under microwave irradiation; then 4 (0.6 mmol), 5 (0.5 mmol) and CH3CN (0.4 mL) were added and kept at 90 °C for 5 min under microwave irradiation; isolated yield. |
|---|
|
Control experiments were conducted in order to gain insight into the mechanism of this reaction (Scheme 2). The cross-coupling of 4b was strongly inhibited to constitute the desired product, and 72% of the byproducts 8a and 9a was obtained (eqn (1)). Furthermore, changing 5a into less sterically hindered 5b almost had no significant influence on the reaction (eqn (2)). This may due to the lower activity of the iminium intermediates for cyclopropanecarbaldehyde was not trapped by the key propargylamine–copper species. Remarkably, the yield of product 8a was increased without the addition of starting materials 4 and 5 (eqn (3)). It seems that during the process, a mixture of 8a and 9a was generated by deacetylenation or protonation of propargylamine–copper species. In order to alter the reactivity of the propargylamine–copper species to allow the reaction with the lower activity of iminium intermediates, we used formaldehyde solution 2k to replace 2a under the standard conditions, and the expected product 7h was obtained in 24% yield with 74% of the byproduct propargylamine 9b (eqn (4)). Prolonging the reaction time affects the efficiency of the reaction, increasing the yield of the desired product to 39% (eqn (5)). Much to our satisfaction, on reducing the steric hindrance of the in situ formed iminium from aldehydes 4 and amines 5, higher yields of the desired products 7m and 10 were obtained (eqn (6) and (7)).
Based on our experimental results and the previous publications, a plausible mechanism is illustrated in Scheme 3. The catalytic cycle of this copper-catalyzed reaction is possibly initiated by the cation exchange between propiolic acid 3 and the copper salt to form activation intermediate A, followed by decarboxylative addition to the in situ formed iminium salt (B1, B2) producing the active propargylamine–copper intermediates (C1, C2). It is worth mentioning that the stability of propargylamine–copper intermediate C is a key parameter. Meanwhile, the steric hindrance of the iminium salt is the driving force for the activation; less sterically hindered iminium salts (B2, B4) would provide the desired products (6, 7 and 10) and regenerate the copper catalyst. However, the lower activity and higher steric hindrance of iminium salts (B1, B3) lead to unsatisfied outcomes. The deacetylenation and protonation of C1 would occur to form the corresponding symmetrical 1,4-diamino-2-butynes 8 and propargylamines 9.5,15 Otherwise, we find significant steric hindrance effects of iminium salt B1 in this process, higher steric hindrance iminium salt B1 affords a lower yield of the expected products 6 and 7 in conjunction with the higher byproduct 9.
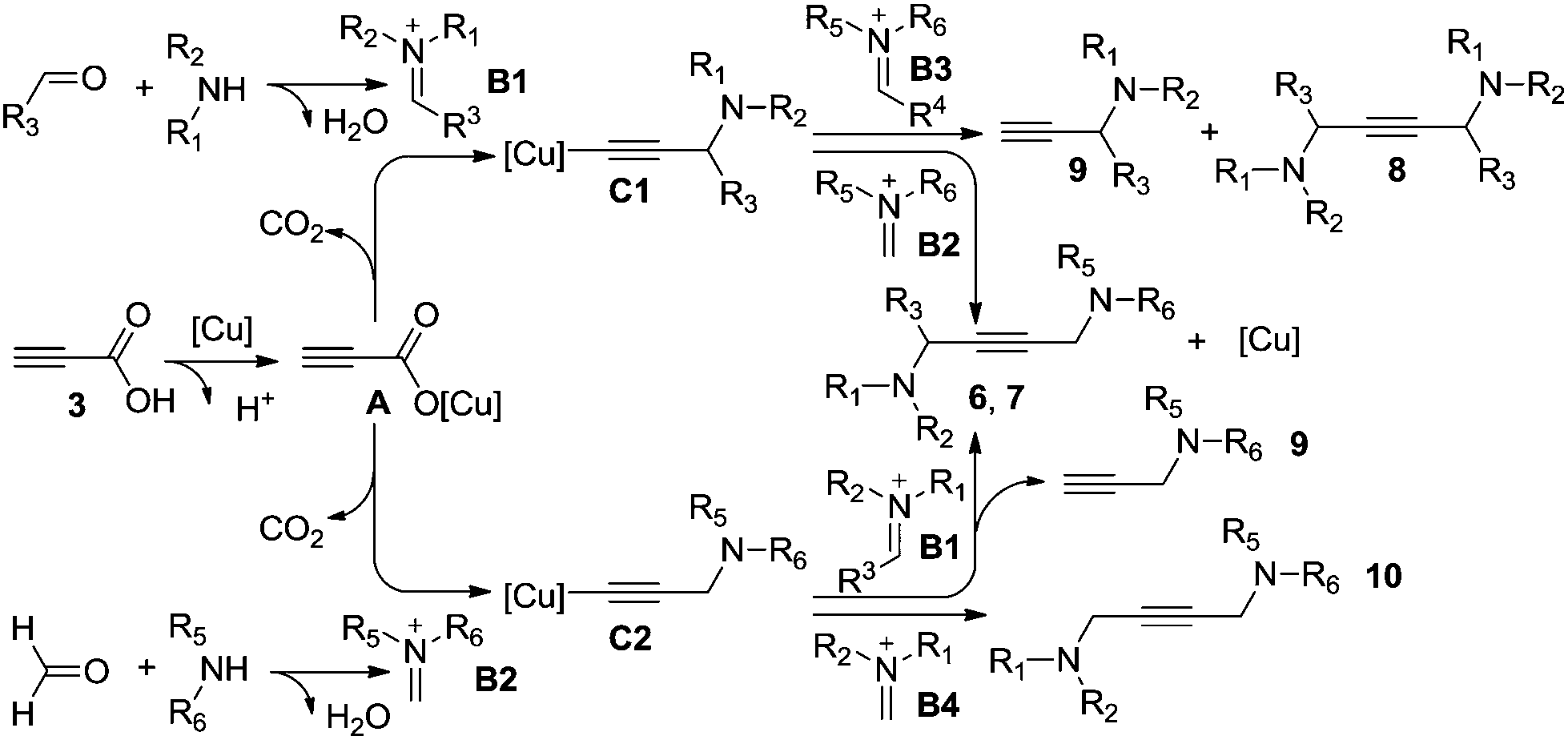 | ||
| Scheme 3 Plausible mechanism. | ||
To summarize, we have developed a novel approach for the unsymmetric construction of 1,4-diamino-2-butynes with good to excellent yields and high chemoselectivities. The concept of steric hindrance is introduced to decarboxylative A3/A3 cross-coupling reaction for the control of chemoselectivity. By simply treating amines and aldehydes with propiolic acid in the presence of CuI/CuCl2, propargylamine–copper intermediates are generated and subsequently coupled with various amines and formaldehyde in situ, to obtain a broad scope of unsymmetric 1,4-diamino-2-butynes. Meanwhile, the reaction is tolerant to oxygen and water, which contributes to the ongoing efforts in developing green chemistry.
Acknowledgements
We are grateful for the generous financial support by the Natural Science Foundation of Shanghai (15ZR1418800), the Shanghai Municipal Education Commission (ZZGCD15028), the Opening Project of Shanghai Key Laboratory of Chemical Biology, the Shanghai University of Engineering Science (nhrc-2015-15, E1-0501-15-0200) and the National Natural Science Foundation of China (21502148).Notes and references
- (a) A. Bisi, S. Gobbi, L. Merolle, G. Farruggia, F. Belluti, A. Rampa, J. Molnar, E. Malucelli and C. Cappadone, Eur. J. Med. Chem., 2015, 92, 471 CrossRef CAS PubMed; (b) N. Sha, K. Kumar, G. Bhargava, K. M. Land, K. H. Chang, R. Arora, S. Sen and V. Kumar, Eur. J. Med. Chem., 2014, 74, 657 CrossRef PubMed; (c) M. N. Bjorn, R. Bjorn and U. Hacksell, J. Med. Chem., 1988, 31, 511 Search PubMed.
- (a) M. Yang, K. Stappert and A. V. Mudring, J. Mater. Chem. C, 2014, 2, 458 RSC; (b) M. Jagoda, S. Warzeska, H. Pritzkow, H. Wadepohl, P. Imhof, J. C. Smith and R. Krämer, J. Am. Chem. Soc., 2005, 127, 15061 CrossRef CAS PubMed.
- (a) C. Koradin, N. Gommermann, K. Polborn and P. Knochel, Chem. – Eur. J., 2003, 9, 2797 CrossRef CAS PubMed; (b) C. M. R. Volla and P. Vogel, Org. Lett., 2009, 11, 1701 CrossRef CAS PubMed; (c) N. D. Doan, J. Q. Zhang, M. Traore, W. Kamdem and W. D. Lubell, J. Pept. Sci., 2015, 21, 387 CrossRef CAS PubMed; (d) M. A. Youngman and S. L. Dax, J. Comb. Chem., 2001, 3, 469 CrossRef CAS PubMed.
- (a) A. Dachs, A. Torrent, A. Roglans, T. Parella, S. Osuna and M. Sola, Chem. – Eur. J., 2009, 15, 5289 CrossRef CAS PubMed; (b) S. A. Shackelford, J. L. Belletire, J. A. Boatz, S. Schneider, A. K. Wheaton, B. A. Wight, H. L. Ammon, D. V. Peryshkov and S. H. Strauss, Org. Lett., 2010, 12, 2714 CrossRef CAS PubMed.
- Y. Kim and H. Nakamura, Chem. – Eur. J., 2011, 17, 12561 CrossRef CAS PubMed.
- Z. W. Lin, D. Y. Yu, Y. N. Sum and Y. G. Zhang, ChemSusChem, 2012, 5, 625 CrossRef CAS PubMed.
- (a) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (b) F. D. Hu, J. H. Yang, Y. Xia, C. Ma, H. P. Xia, Y. Zhang and J. B. Wang, Org. Chem. Front., 2015, 2, 1450 RSC; (c) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef CAS PubMed; (d) C. Liu, H. Zhang, W. Shi and A. W. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed.
- (a) D. Tanaka, S. P. Romeril and A. G. Myers, J. Am. Chem. Soc., 2005, 127, 10323 CrossRef CAS PubMed; (b) J. Tang, A. Biafora and L. J. Gooßen, Angew. Chem., Int. Ed., 2015, 54, 13130 CrossRef CAS PubMed; (c) S. L. Zhang, Y. Fu, R. Shang, Q. X. Guo and L. Liu, J. Am. Chem. Soc., 2010, 132, 638 CrossRef CAS PubMed; (d) J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS PubMed; (e) P. Hu, Y. Shang and W. P. Su, Angew. Chem., Int. Ed., 2012, 51, 5945 CrossRef CAS PubMed; (f) J. Kan, S. J. Huang, J. Lin, M. Zhang and W. P. Su, Angew. Chem., Int. Ed., 2015, 54, 2199 CrossRef CAS PubMed; (g) Y. C. Zhu, X. Y. Li, X. Y. Wang, X. Q. Huang, T. Shen, Y. Q. Zhang, X. Sun, M. C. Zou, S. Song and N. Jiao, Org. Lett., 2015, 17, 4702 CrossRef CAS PubMed; (h) J. M. Miao, P. Fang, S. Jagdeep and H. B. Ge, Org. Chem. Front., 2016, 3, 243 RSC; (i) G. Z. Wang, R. Shang, W. M. Cheng and Y. Fu, Org. Lett., 2015, 17, 4830 CrossRef CAS PubMed.
- For a recent review of the decarboxylation reaction, see: (a) N. Rodriguez and L. J. Gooßen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (b) J. D. Weaver, A. Recio III, A. J. Grenning and J. A. Tung, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (c) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671 RSC; (d) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670 CrossRef CAS; (e) J. Xuan, Z. G. Zhang and W. J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed; (f) C. Shen, P. F. Zhang, Q. Sun, S. Q. Bai, T. S. A. Hor and X. Q. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (g) M. O. Konev and E. R. Jarvo, Angew. Chem., Int. Ed., 2016, 55, 11340 CrossRef CAS PubMed.
- (a) K. Park and S. Lee, RSC Adv., 2013, 3, 14165 RSC; (b) H. D. Feng, D. S. Ermolat'ev, G. H. Song and E. V. Van der Eycken, Adv. Synth. Catal., 2012, 354, 505 CrossRef CAS; (c) H. D. Feng, H. H. Jia and Z. H. Sun, Adv. Synth. Catal., 2015, 357, 2447 CrossRef CAS; (d) H. H. Jia, H. D. Feng and Z. H. Sun, Org. Biomol. Chem., 2015, 13, 8177 RSC; (e) J. Lim, J. Choi, H. S. Kim, I. S. Kim, K. C. Nam, J. Kim and S. Lee, J. Org. Chem., 2016, 81, 303 CrossRef CAS PubMed; (f) D. L. Priebbenow, P. Becker and C. Bolm, Org. Lett., 2013, 15, 6155 CrossRef CAS PubMed; (g) M. Yu, D. L. Pan, W. Jia, W. Chen and N. Jiao, Tetrahedron Lett., 2010, 51, 1287 CrossRef CAS; (h) H. D. Feng, H. H. Jia and Z. H. Sun, J. Org. Chem., 2014, 79, 11812 CrossRef CAS PubMed; (i) S. Y. Li, X. Li, F. Yang and Y. J. Wu, Org. Chem. Front., 2015, 2, 1076 RSC; (j) X. Li, F. Yang, Y. J. Wu and Y. S. Wu, Org. Lett., 2014, 16, 992 CrossRef CAS PubMed.
- (a) K. Park, G. Bae, J. Moon, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2010, 75, 6244 CrossRef CAS PubMed; (b) J. Moon, M. Jeong, H. Nam, J. Ju, J. H. Moon, H. M. Jung and S. Lee, Org. Lett., 2008, 10, 945 CrossRef CAS PubMed; (c) S. Tartaggia, O. D. Lucchi and L. J. Gooßen, Eur. J. Org. Chem., 2012, 1431 CrossRef CAS; (d) X. Li, F. Yang and Y. J. Wu, RSC Adv., 2014, 4, 13738 RSC.
- W. Kim, K. Park, A. Park, J. Choe and S. Lee, Org. Lett., 2013, 15, 1654 CrossRef CAS PubMed.
- H. D. Feng, D. S. Ermolat'ev, G. H. Song and E. V. Van der Eycken, J. Org. Chem., 2012, 77, 5149 CrossRef CAS PubMed.
- For a recent review of the A3-coupling reaction, see: (a) V. A. Peshkov, O. P. Pereshivko and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790 RSC; (b) W. J. Yoo, L. Zhao and C. J. Li, Aldrichimica Acta, 2011, 44, 43 CAS; (c) D. Seidel, Org. Chem. Front., 2014, 1, 426 RSC.
- (a) T. Sugiishi, A. Kimura and H. Nakamura, J. Am. Chem. Soc., 2010, 132, 5332 CrossRef CAS PubMed; (b) Y. L. Shao, F. J. Zhang, J. Zhang and X. G. Zhou, Angew. Chem., Int. Ed., 2016, 55, 11485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00499g |
| This journal is © the Partner Organisations 2017 |