
A practical protocol for the synthesis of bibenzyls via C(sp3)–H activation of methyl arenes under metal-free conditions†
Promod
Kumar
,
Tirumaleswararao
Guntreddi
,
Rahul
Singh
and
Krishna Nand
Singh
*
Department of Chemistry (Centre of Advanced Study), Institute of Science, Banaras Hindu University, Varanasi 221005, India. E-mail: knsingh@bhu.ac.in; knsinghbhu@yahoo.co.in
First published on 28th October 2016
Abstract
A variety of bibenzyl derivatives have been synthesized with excellent atom economy via C(sp3)–H–C(sp3)–H coupling of readily available methyl arenes using K2S2O8 under metal-free and environmentally benign conditions.
Introduction
Transition metal-catalyzed C–C bond formation is one of the most important transformations in organic synthesis.1 During the past few years, C–H activation reactions have attracted a great deal of attention for the preparation of valuable compounds.2 Most recently, the versatility of transition metal catalysts has allowed the discovery of a variety of new strategies in activating both Csp2–H and Csp3–H bonds for carbon–carbon bond creation.3 Yet, only a few methods have been developed for Csp3–HCsp3–H bond formation because of the low reactivity of Csp3–H bonds.4 The direct coupling of two Csp3–H bonds to generate a C–C bond is quite demanding and challenging because of its high atom economy. Methyl arenes are inexpensive and abundant organic molecules, and the methods for their direct/selective functionalization are highly tempting. Although adequate attention has been paid to the Csp3–H activation of methyl arenes,5 many of the reactions described so far invariably employ expensive/toxic transition metals and severely suffer from the excessive use of methyl arenes and oxidants.The bibenzyl motif is generally found in many natural products and exhibits potential biological and agricultural activities.6 Some of the bibenzyl derivatives are used as starting materials for the synthesis of highly useful drug molecules.7 Traditional methods for the synthesis of bibenzyls involve reduction of stilbene/diphenylacetylene derivatives.8 In recent times, bibenzyl derivatives have been synthesized by using homocoupling of benzyl halides,9 benzylmagnesium halides,10 and phenylacetic acids11 (Scheme 1). However, these methods require pre-functionalization of the starting materials as well as the use of expensive transition metal catalysts. Therefore, a metal-free and mild protocol for the synthesis of bibenzyl derivatives employing economical and readily available starting materials is highly demanding.
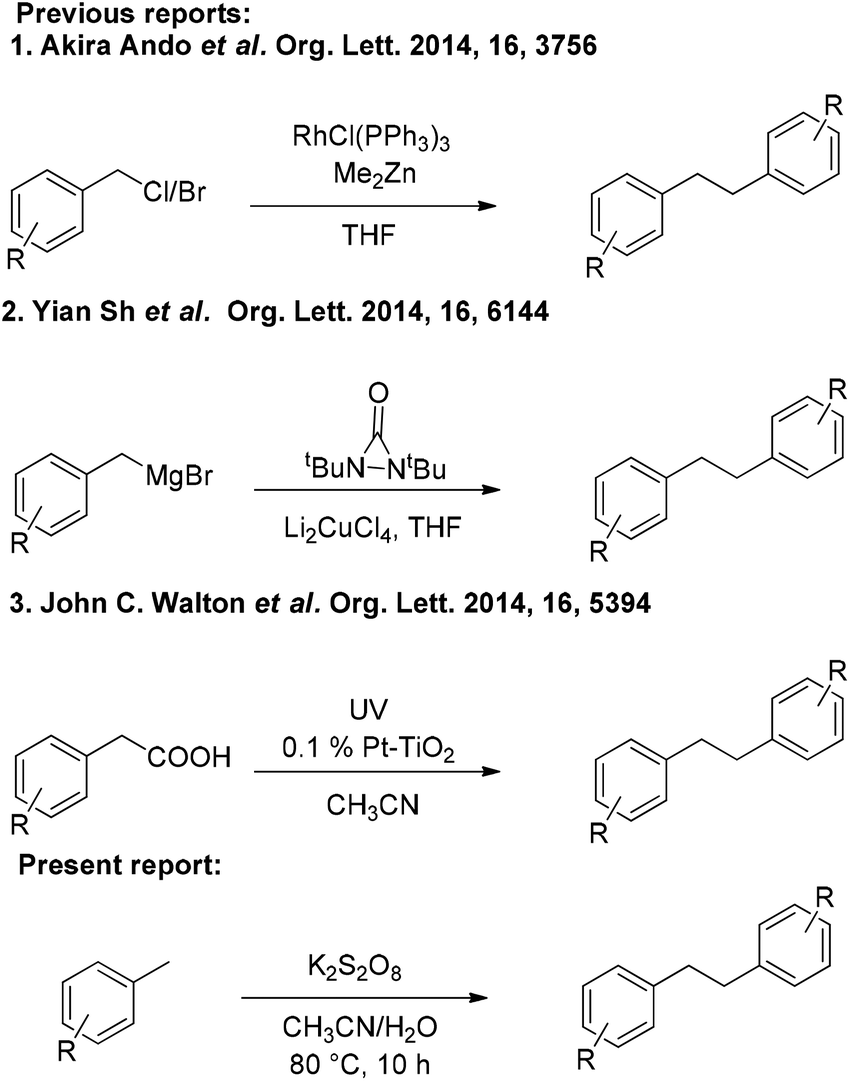 | ||
| Scheme 1 Previous and present reports. | ||
In view of the above and as a part of our ongoing interest in developing new protocols involving C–H activation,12 and others,13 we disclose herein an efficient K2S2O8 mediated homocoupling of methyl arenes for the synthesis of bibenzyl derivatives under metal-free conditions (Scheme 1).
Results and discussion
In order to optimize the reaction conditions, a model reaction employing p-xylene as a substrate was thoroughly investigated by varying different parameters such as the catalyst, oxidant, solvent(s) and temperature, and the outcome is given in Table 1.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol %) | Oxidant | Solvent/solvents (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
Temp (°C) | Yieldb (%) |
| a Reaction conditions: 1b (1.0 mmol), catalyst (20 mol%), oxidant (2.0 mmol), solvent (2 ml), 10 h. b Isolated yield. | |||||
| 1 | AgNO3 (20) | K2S2O8 | CH3CN/H2O | 60 | 56 |
| 2 | AgNO3 (20) | K2S2O8 | CH3CN/H2O | 70 | 68 |
| 3 | AgNO3 (20) | K2S2O8 | CH3CN/H2O | 80 | 75 |
| 4 | AgNO3 (20) | K2S2O8 | CH3CN/H2O | 90 | 73 |
| 5 | — | K2S2O8 | CH3CN/H2O | 80 | 76 |
| 6 | — | K2S2O8 | DCE/H2O | 80 | 0 |
| 7 | — | K2S2O8 | DMSO/H2O | 80 | 0 |
| 8 | — | K2S2O8 | DMF/H2O | 80 | 0 |
| 9 | — | K2S2O8 | DMA/H2O | 80 | 0 |
| 10 | — | K2S2O8 | NMP/H2O | 80 | 0 |
| 11 | — | Na2S2O8 | CH3CN/H2O | 80 | 68 |
| 12 | — | (NH4)2S2O8 | CH3CN/H2O | 80 | 61 |
| 13 | — | TBHP | CH3CN/H2O | 80 | 0 |
| 14 | — | DTBP | CH3CN/H2O | 80 | 0 |
| 15 | (NH4)2Ce(NO3)6 | CH3CN/H2O | 80 | 0 | |
| 16 | K2S2O8 | CH3CN | 80 | 0 | |
| 17 | K2S2O8 | H2O | 80 | 0 | |
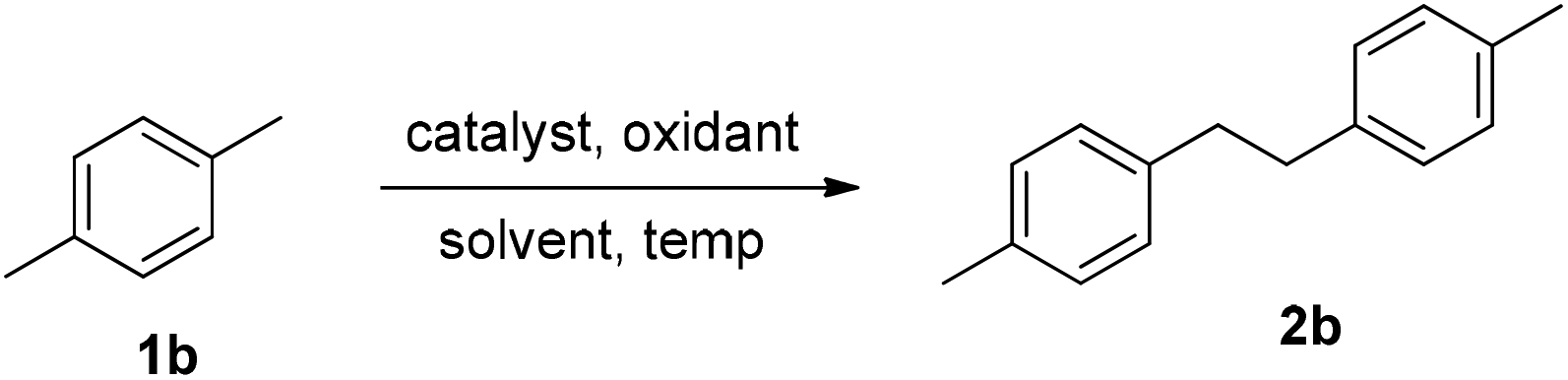
To our outmost delight, when the reaction was conducted using AgNO3 (20 mol%) as the catalyst and K2S2O8 (2 equiv.) as the oxidant in CH3CN/H2O (1:1) at 60 °C for 10 hours, the homocoupled product 1,2-di-p-tolylethane (2b) was formed in 56% yield (Table 1, entry 1). Increasing the reaction temperature to 70 °C and 80 °C increased the product yields to 68% and 75% respectively (entries 2 & 3). Further increase in the temperature to 90 °C, however, did not improve the product yield again (entry 4). Notably, when the reaction was carried out in the absence of AgNO3 at 80 °C, the product 2b was obtained in 76% yield (entry 5). Afterwards, the effect of different solvent systems such as DCE/H2O, DMSO/H2O, DMF/H2O, DMA/H2O and NMP/H2O was studied to improve the product yield, which remained entirely futile (entries 6–10). Switching the oxidant to Na2S2O8 and (NH4)2S2O8 also did not augment the product yield (entries 11 & 12). Surprisingly the use of oxidants like TBHP, DTBP and (NH4)2Ce(NO3)6 remained completely ineffective (entries 13–15). The reaction also remained worthless, when conducted using solitary solvents like CH3CN and H2O (entries 16 & 17). It unequivocally reveals that the choice of the oxidant and biphasic solvent system is crucial for the success of the reaction.
With the optimized reaction conditions in hand (entry 5), the scope and limitations of the homocoupling of various methylarenes involving the C(sp3)–H bond were subsequently explored in detail (Scheme 2).
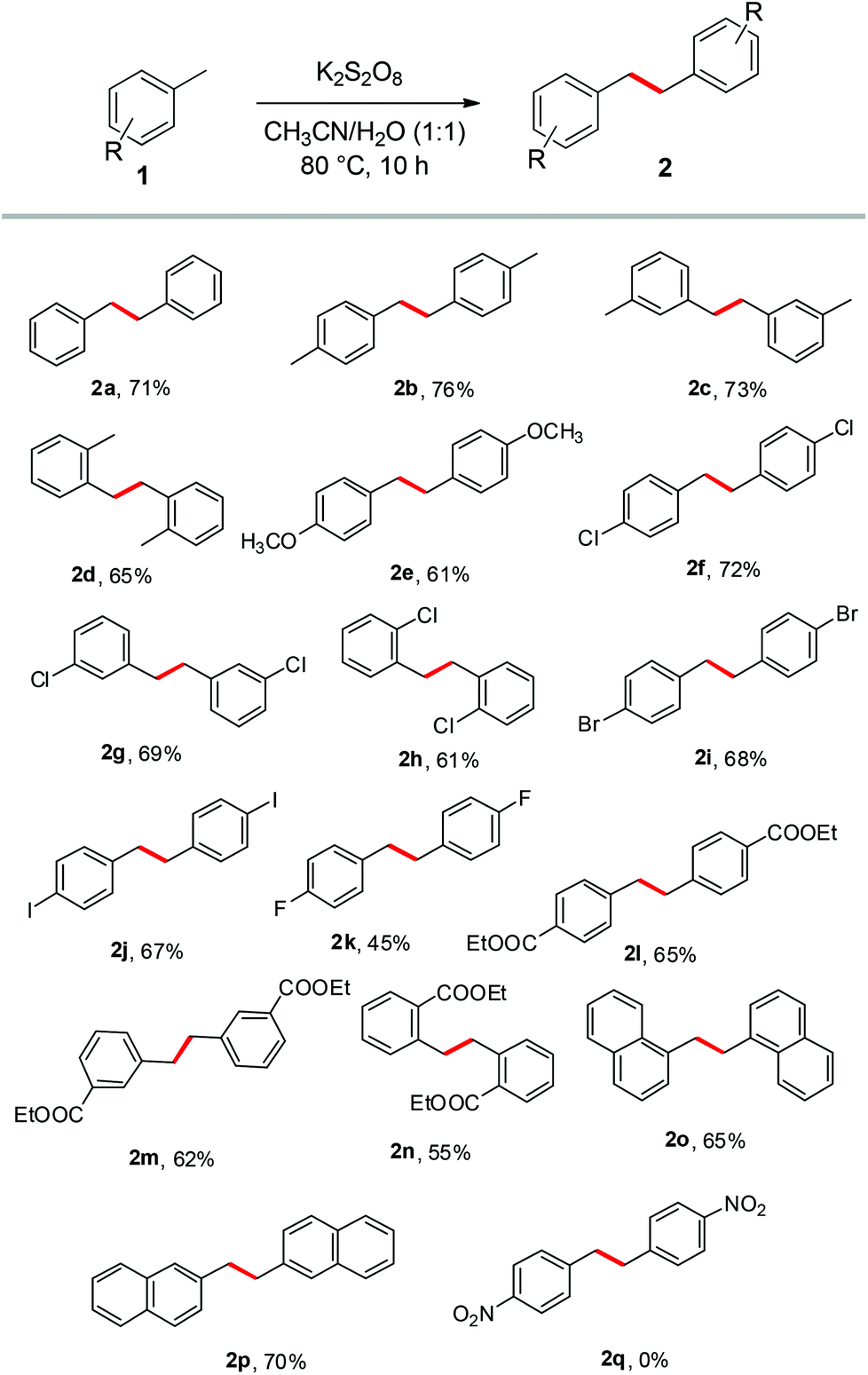 | ||
| Scheme 2 The scope and versatility of the homocoupling of methylarenes. Conditions: Methylarene 1 (1.0 mmol), K2S2O8 (2.0 mmol), solvents (2 ml), 10 h. | ||
Evidently, toluene and its derivatives like m-xylene, o-xylene, p-methoxytoluene, p-chlorotoluene, m-chlorotoluene, o-chlorotoluene, p-bromotoluene, p-iodotoluene and p-fluorotoluene were readily transformed into their corresponding bibenzyls 2a–k in reasonably high yields. Dimethyl benzenes such as p-xylene, o-xylene and m-xylene surprisingly underwent dimerization involving only one methyl group to afford their corresponding bibenzyls 2b–d, with the other methyl group remaining intact. Toluene derivatives containing electron withdrawing groups such as ethyl-4-methylbenzoate, ethyl-3-methylbenzoate and ethyl-2-methylbenzoate also endured the reaction smoothly to give the desired products 2l–n in good yields. o-Substituted toluene derivatives offered lower yields compared to m- and p-substituted toluenes, perhaps due to the steric hindrance. Bicyclic methylarenes such as 1-methylnaphthalene and 2-methylnaphthalene also worked well leading to the formation of the products 2o & 2p in high yields. The results showed a wide scope and good tolerance of functional groups for the reaction, although 4-nitrotoluene and 4-methylphenol noticeably did not undergo the reaction at all under the established conditions.
Ultimately, it was thought worthwhile to extend the applicability of the reaction for cross coupling of two different methyl arenes. As a result, a representative reaction involving toluene (1a) and 1-methylnaphthalene (1o) was carried out under the established conditions by using different molar ratios of the reactants. But despite all our efforts, we could not exceed the yield of the cross coupled product 3a above 25%, which was invariably accompanied by the formation of the homocoupled products 2a and 2o (Scheme 3). A number of other cross-coupling reactions including two electronically different methyl arenes (e.g. toluene and chlorotoluene) have also been carried out under the established conditions. But we could not observe any isolable cross-coupled product. Furthermore, an intramolecular cyclization coupling using p-xylene and m-xylene was also tried by varying their molar ratios to afford a cyclophane type product, but the reaction did not proceed beyond the formation of 2b and 2c respectively, even at higher temperatures and longer reaction times. An effort was also made to cyclize the isolated products 2b and 2c under the standard conditions, but the reaction could not succeed and the starting reactants 2b and 2c remained as it is.
 | ||
| Scheme 3 Cross coupling reaction between 1a and 1o. Conditions: 1a (1.0 mmol), 1o (1.0 mmol), K2S2O8 (4.0 mmol), solvent (4 ml), CH3CN/H2O (1:1), 80 °C, 10 h. | ||
In order to gain an insight into the mechanistic pathway, a typical reaction as shown in Scheme 4 was carried out in the presence of a radical scavenger TEMPO, which quenched the reaction thereby revealing the involvement of a radical mechanism.
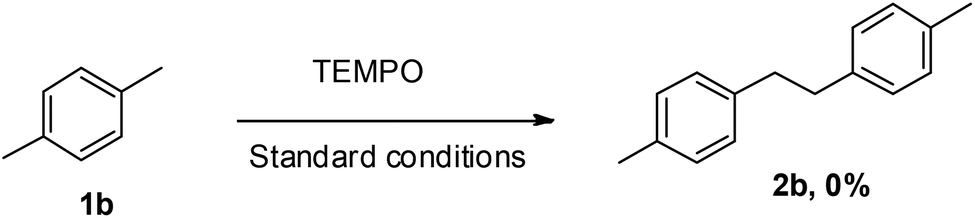 | ||
| Scheme 4 Radical trapping experiment. | ||
Based on the above observations, a plausible mechanism is outlined in Fig. 1, which is initiated with the thermal decomposition of K2S2O8 to form the sulfate radical (SO4−˙). The reaction of the sulfate radical with methylarene 1 gives rise to a benzyl radical, which then dimerizes to afford the bibenzyl derivatives 2.
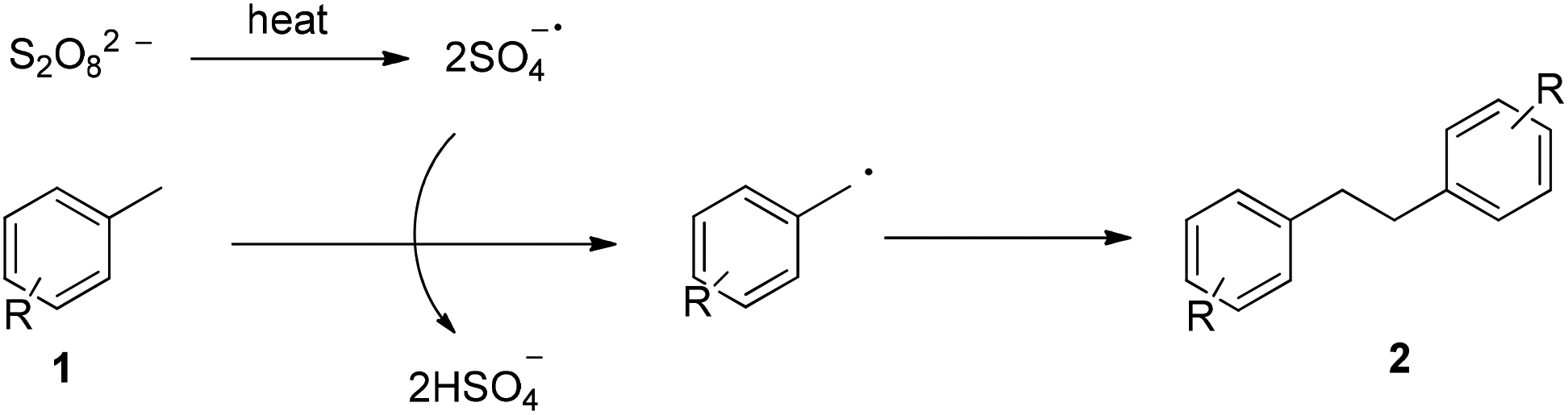 | ||
| Fig. 1 Plausible mechanism. | ||
Conclusions
In summary, a novel and efficient K2S2O8 mediated C(sp3–H) C(sp3–H) homocoupling of inexpensive methyl arenes has been developed in aqueous solution to afford bibenzyls. In addition to the broad substrate scope and functional group compatibility, the transformation is simple and metal-free. In view of its operational simplicity, easy availability of starting materials and mild reaction conditions, this method should find important applications in organic synthesis.Acknowledgements
P. K. thanks UGC, New Delhi for the award of a DSK-Postdoctoral Fellowship with UGC Award Letter No. F.4-2/2006 (BSR)/CH/13-14/0165.Notes and references
- (a) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed; (b) N. Miyaura, Cross-Coupling Reactions, Springer, Berlin, 2002 Search PubMed; (c) A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed.
- (a) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC; (d) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (f) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (g) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (h) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (i) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed.
- (a) S. Guin, S. K. Rout, A. Banerjee, S. Nandi and B. K. Patel, Org. Lett., 2012, 14, 5294 CrossRef CAS PubMed; (b) Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (c) F. Xiong, C. Qian, D. Lin, W. Zeng and X. Lu, Org. Lett., 2013, 15, 5444 CrossRef CAS PubMed; (d) Y.-F. Liang, X. Li, X. Wang, Y. Yan, P. Feng and N. Jiao, ACS Catal., 2015, 5, 1956 CrossRef CAS; (e) Y. Aihara, M. Tobisu, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2014, 136, 15509 CrossRef CAS PubMed; (f) H. Yang, P. Sun, Y. Zhu, H. Yan, L. Lu, X. Qu, T. Li and J. Mao, Chem. Commun., 2012, 48, 7847 RSC; (g) M.-B. Zhou, C.-Y. Wang, R.-J. Song, Y. Liu, W.-T. Wei and J.-H. Li, Chem. Commun., 2013, 49, 10817 RSC; (h) Z. Li, Y. Zhang, L. Zhang and Z.-Q. Liu, Org. Lett., 2014, 16, 382 CrossRef CAS PubMed; (i) S.-L. Zhou, L.-N. Guo, S. Wang and X.-H. Duan, Chem. Commun., 2014, 50, 3589 RSC; (j) G. Qin, X. Chen, L. Yang and H. Huang, ACS Catal., 2015, 5, 2882 CrossRef CAS; (k) J. M. Curto and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 18 CrossRef CAS PubMed.
- (a) S. Pan, J. Liu, Y. Li and Z. Li, Chin. Sci. Bull., 2012, 57, 2382 CrossRef CAS; (b) K. Yang and Q. Song, Org. Lett., 2015, 17, 548 CrossRef CAS PubMed; (c) F.-F. Wang, C.-P. Luo, G. Deng and L. Yang, Green Chem., 2014, 16, 2428 RSC; (d) Z.-Q. Zhu, P. Bai and Z.-Z. Huang, Org. Lett., 2014, 16, 4881 CrossRef CAS PubMed.
- R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062 RSC.
- (a) X. Zhang, J. K. Xu, J. Wang, N. L. Wang, H. Kurihara, S. Kitanaka and X. J. Yao, Nat. Prod., 2007, 70, 24 CrossRef CAS PubMed; (b) Y. Hernandez-Romero, L. Acevedo, L. Sanchez Mde, W. T. Shier, H. K. Abbas and R. Mata, J. Agric. Food Chem., 2005, 53, 6276 CrossRef CAS PubMed; (c) G. Cioffi, P. Montoro, O. L. De Ugaz, A. Vassallo, L. Severino, C. Pizza and N. De Tommasi, Molecules, 2011, 16, 2527 CrossRef CAS PubMed; (d) G. Du, Y. Q. Shen, L. Y. Yang, L. D. Shu, M. L. Wen and Q. F. Hu, Chem. Nat. Compd., 2014, 49, 1019 CrossRef CAS; (e) Y. Li, F. Zhang, Z. H. Wu, K. W. Zeng, C. Zhang, H. W. Jin, M. B. Zhao, Y. Jiang and J. Li, Fitoterapia, 2015, 102, 120 CrossRef CAS PubMed; (f) X.-M. Zhou, C.-J. Zheng, L.-S. Gan, G.-Y. Chen, X.-P. Zhang, X.-P. Song, G.-N. Li and C.-G. Sun, J. Nat. Prod., 2016, 79, 1791 CrossRef CAS PubMed.
- (a) A. Hofer, G. Kovacs, A. Zappatini, M. Leuenberger, M. A. Hediger and M. Lochner, Bioorg. Med. Chem., 2013, 21, 3202 CrossRef CAS PubMed; (b) P. Wyatt and A. Hudson, Org. Biomol. Chem., 2004, 2, 1528 RSC; (c) M. B. Nodwell, A. Bagai, S. D. Halperin, R. E. Martin, H. Knust and R. Britton, Chem. Commun., 2015, 51, 11783 RSC; (d) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS PubMed; (e) D. Shen, C. Miao, S. Wang, C. Xia and W. Sun, Org. Lett., 2014, 16, 1108 CrossRef CAS PubMed; (f) E. Galán, M. L. Perrin, M. Lutz, H. S. J. van der Zant, F. C. Grozema and R. Eelkema, Org. Biomol. Chem., 2016, 14, 2439 RSC.
- (a) Y. Motoyama, M. Takasaki, S.-H. Yoon, I. Mochida and H. Nagashima, Org. Lett., 2009, 11, 5042 CrossRef CAS PubMed; (b) D. Guin, B. Baruwati and S. V. Manorama, Org. Lett., 2007, 9, 1419 CrossRef CAS PubMed; (c) P. H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747 RSC; (d) Y. J. Kim, S. M. Kim, E. J. Cho, H. Hosono and J. W. Yang, Chem. Sci., 2015, 6, 3577 RSC.
- (a) K. Sato, Y. Inoue, T. Mori, A. Sakaue, A. Tarui, M. Omote, I. Kumadaki and A. Ando, Org. Lett., 2014, 16, 3756 CrossRef CAS PubMed; (b) D. Toummini, F. Ouazzani and M. Taillefer, Org. Lett., 2013, 15, 4690 CrossRef CAS PubMed; (c) A. E. Lanterna, A. Elhage and J. C. Scaiano, Catal. Sci. Technol., 2015, 5, 4336 RSC; (d) M. R. Prinsell, D. A. Everson and D. J. Weix, Chem. Commun., 2010, 46, 5743 RSC; (e) S. M. Goldup, D. A. Leigh, R. T. McBurney, P. R. McGonigal and A. Plant, Chem. Sci., 2010, 1, 383 RSC.
- Y. Zhu, T. Xiong, W. Han and Y. Shi, Org. Lett., 2014, 16, 6144 CrossRef CAS PubMed.
- D. W. Manley and J. C. Walton, Org. Lett., 2014, 16, 5394 CrossRef CAS PubMed.
- (a) R. Vanjari, T. Guntreddi and K. N. Singh, Org. Lett., 2013, 15, 4908 CrossRef CAS PubMed; (b) R. Vanjari, T. Guntreddi and K. N. Singh, Green Chem., 2014, 16, 351 RSC; (c) R. Vanjari, T. Guntreddi and K. N. Singh, Chem. – Asian. J., 2016, 11, 696 CrossRef CAS PubMed; (d) R. Vanjari, T. Guntreddi, S. Kumar and K. N. Singh, Chem. Commun., 2015, 51, 366 RSC; (e) S. Kumar, R. Vanjari, T. Guntreddi and K. N. Singh, RSC Adv., 2015, 5, 9920 RSC.
- (a) T. Guntreddi, R. Vanjari and K. N. Singh, Org. Lett., 2014, 16, 3624 CrossRef CAS PubMed; (b) T. Guntreddi, R. Vanjari and K. N. Singh, Org. Lett., 2015, 17, 976 CrossRef CAS PubMed; (c) R. Singh, B. K. Allam, N. Singh, K. Kumari, S. K. Singh and K. N. Singh, Org. Lett., 2015, 17, 2656 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00529b |
| This journal is © the Partner Organisations 2017 |