
A NMR method for relative stereochemical assignments of the tricyclic core of cephalosporolides, penisporolides and related synthetic analogues†
Jian
Wang
a and
Rongbiao
Tong
 *ab
*ab
aDepartment of Chemistry, Hong Kong University of Science and Technology, Clearwater Bay, Kowloon, Hong Kong, China. E-mail: rtong@ust.hk
bHKUST Shenzhen Research Institute, Shenzhen, 518057, China
First published on 26th October 2016
Abstract
The misassignments of the relative configuration of natural products such as cephalosporolides H and I and penisporolide B were frequently detected by synthetic studies (total synthesis) with considerable effort and cost. Reported herein is the development of a NMR analysis method that can be applied to reliably discriminate the four possible diastereomers of the tricyclic core of cephalosporolides and penisporolides using only proton and/or carbon NMR data without relying on sophisticated 2D NMR spectral analysis. The effectiveness of this NMR method was examined with 28 synthetic compounds, leading to detection and/or correction of 11 misassignments.
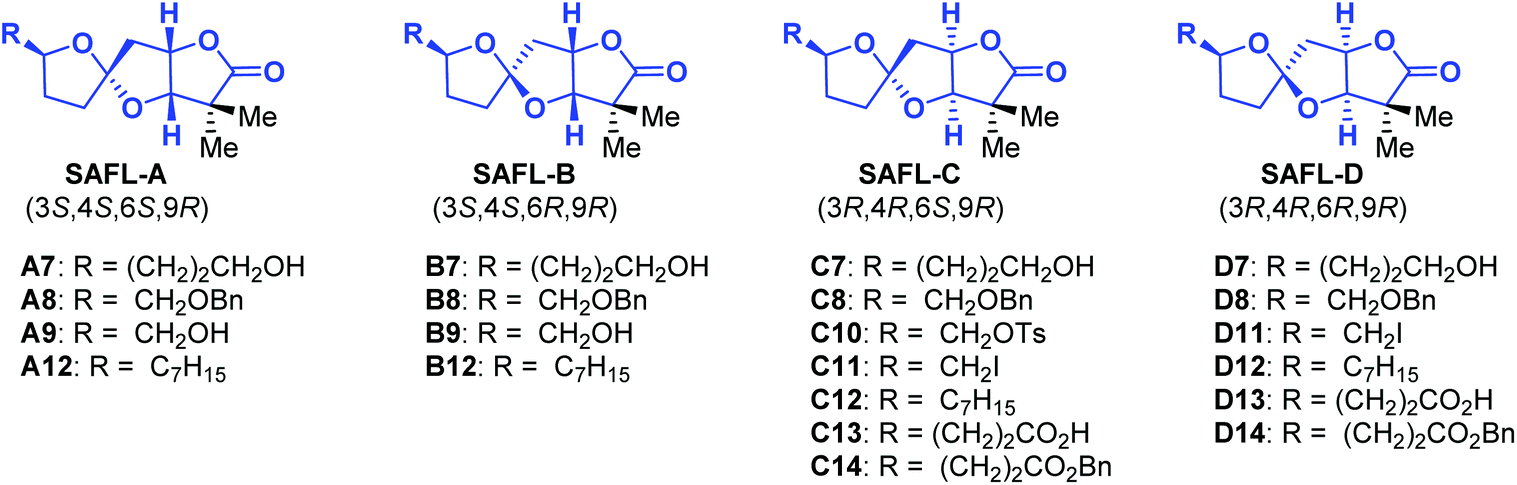
Natural products have played highly significant and multifaceted roles in chemistry. They not only are the most productive source for the development of new drugs1 but also greatly inspire chemists to develop new synthetic strategies and methodologies by challenging the state-of-the-art chemical synthesis.2 Their molecular structures including the relative and absolute configurations are of paramount importance to these roles, in particular, to total synthesis and drug discovery. However, many natural products were reported with wrong structural assignments if suitable single crystals were unavailable for X-ray diffraction analysis.3 There are few general methods available for structural corrections and confirmation besides NMR analysis4 including computational NMR methods and total synthesis. Although unambiguous, total synthesis usually requires tremendous synthetic efforts with considerable cost and time.5 For example, recent synthetic studies6 including one from our group consistently revealed that the structures for cephalosporolides H and I7 and penisporolide B8 were incorrect and that the relative configuration of the tricyclic [5,5]-spiroacetal-cis-fused-γ-lactone (SAFL) should be revised (Fig. 1). In light of the potential biological activity of this family of compounds and new members of SAFL-containing natural products probably being isolated in the future, we are interested in developing a reliable NMR analysis method (similar to the Kishi NMR database4e,f or Rychnovsky acetonide 13C-NMR analysis4g) to determine the relative configuration of the SAFL core without synthetic studies. Prior observation of the spiro-differentiating spin–spin coupling (SSC) pattern first by Ramana9 and then by Britton10 and Brimble6f further inspired us to undertake a comprehensive analysis of the NMR data of the SAFLs reported in the literature and our laboratory with the expectation of stereochemical discrimination of all four possible diastereomers using only the NMR analysis method. Note: since the γ-lactone of the SAFLs is always cis-fused to the spiroacetal with the same configuration (3R,4R or 3S,4S), there are only four diastereomers derived from the four chiral centers (C3, C4, C6 and C9), namely, SAFL-A with the (3S*,4S*,6S*,9R*) relative configuration, (3S*,4S*,6R*,9R*) SAFL-B, (3R*,4R*,6S*,9R*) SAFL-C, and (3R*,4R*,6R*,9R*) SAFL-D (Fig. 1).
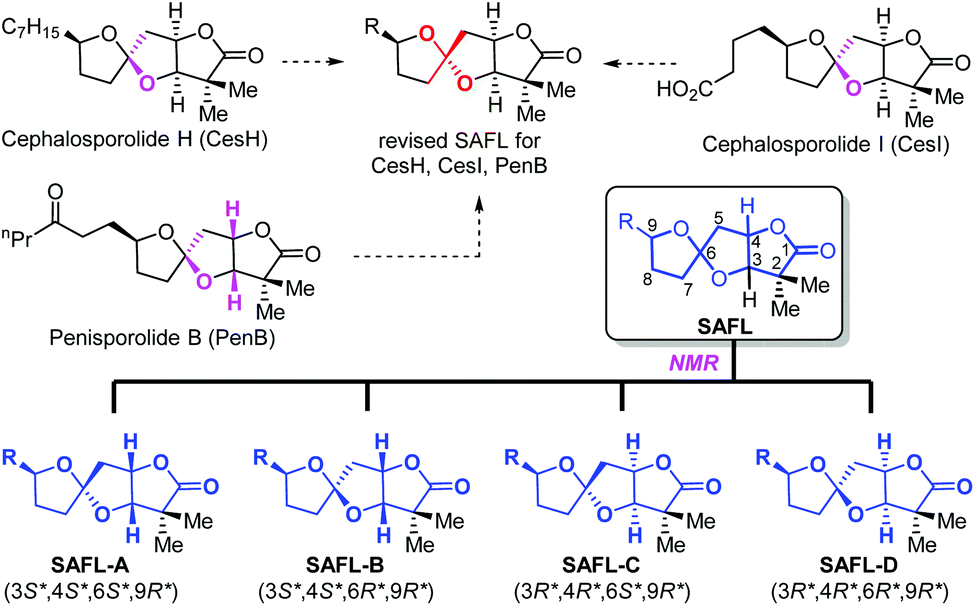 | ||
| Fig. 1 Structural revision of the tricyclic core of cephalosporolides H and I and penisporolides B and their four possible diastereomers. | ||
Our studies started with a comprehensive analysis of the 1H-NMR spectra derived from four synthetic diastereomers SAFL-A1, -B1, -C1, and -D1, whose relative configurations have been unambiguously determined by the combination of NMR spectroscopic (including 2D NMR) and X-ray diffraction analysis.6e After the careful comparative analysis of the 1H-NMR spectra11 (Fig. 2), the spin–spin coupling pattern and chemical shifts of H4, H5 and H8 were identified to be useful for distinguishing these four diastereomers. The first obvious difference was the SSC pattern of H4: triplet (t) at 5.08–5.10 ppm versus doublet of doublet of doublet (ddd) at 5.02 ppm, which is consistent with the observation in the related spiroketal compounds reported by Ramana,9 Britton10 and Brimble.6f This distinct H4 SSC with slightly different chemical shifts differentiated SAFL-A1/D1 from SAFL-B1/C1, corresponding to the stereochemical discrimination of the spiroketal chiral centers (3S*,4S*,6S* vs. 3S*,4S*,6R*), irrespective of the C9 relative configuration. The different SSC patterns of these two pair diastereomers are explained by their slightly different conformations resulting from opposite stereochemistry at the spiroketal carbon, as illustrated by X-ray diffraction analysis of SAFL-B1 and -D1 [Fig. 3(a) and (b)].
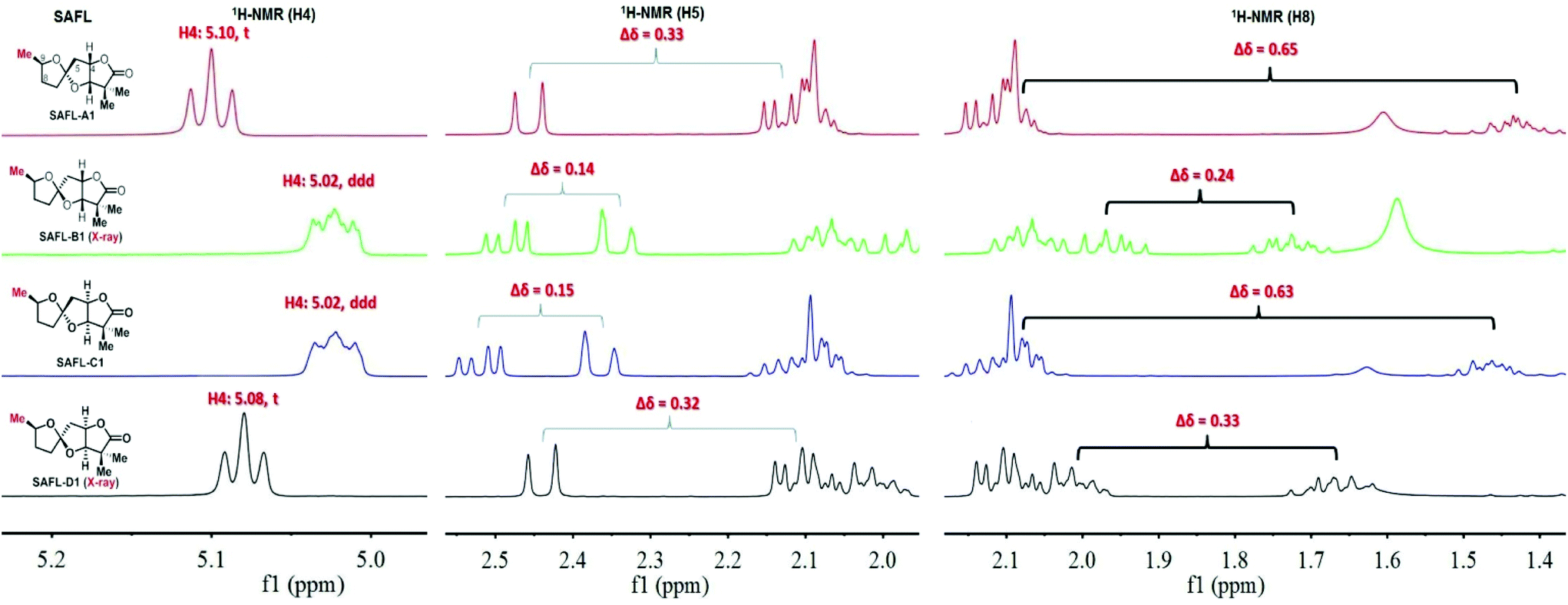 | ||
| Fig. 2 Truncated 1H-NMR spectra of SAFL-A1-D1. Note: full NMR spectra are available in the ESI of ref. 6e. | ||
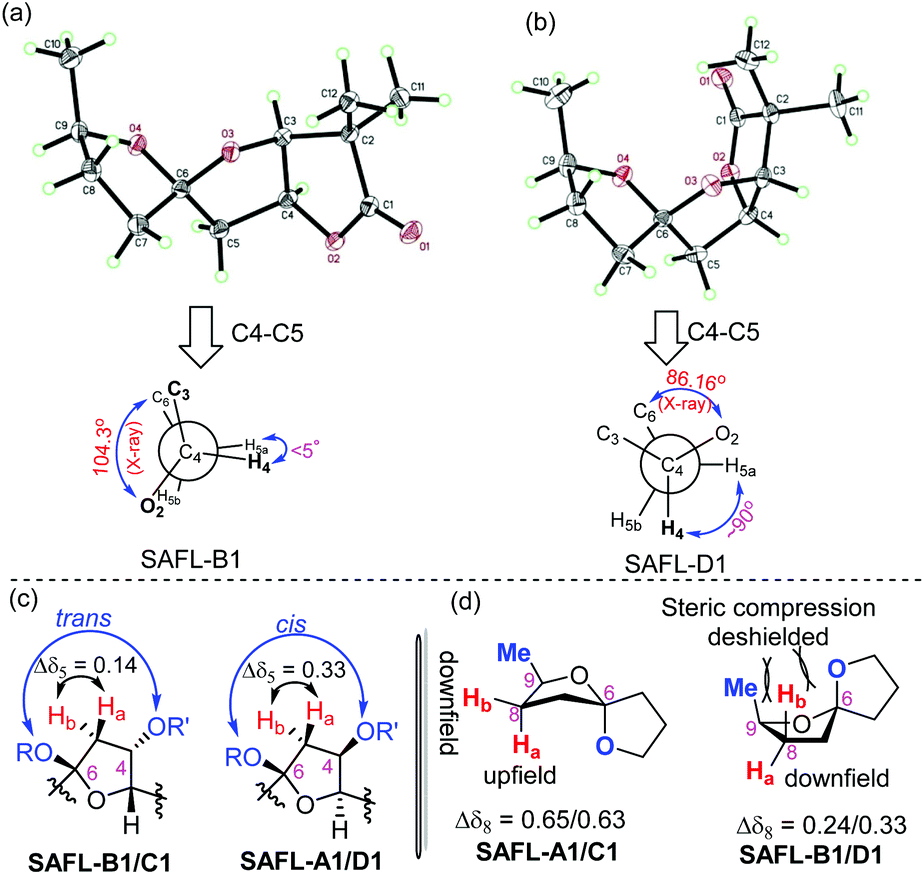 | ||
| Fig. 3 (a) Thermal ellipsoid diagram of SAFL-B1 and Newman projection along C4–C5. (b) Thermal ellipsoid diagram of SAFL-D1 and Newman projection along C4–C5. (c) Chemical difference of H5 in the SAFLs. (d) Chemical difference of H8 in the SAFLs. | ||
The U-shaped conformation of SAFL-D1 [Fig. 3(b)] has a ∼90° dihedral angle of H4 and H5a (H5a is trans-oriented to H4, 86.16° is the torsion angle of O2–C4–C5–C6 in the X-ray data6e), resulting in no spin–spin coupling between H4 and H5a according to the Bothner-By12 approximation of the Karplus correlation13 between 3J-coupling constants and dihedral angles and coincidentally identical 3J coupling constants of H4–H3 and H4–H5b. The V-shaped SAFL-B1 [Fig. 3(a)] reveals an unusual eclipsed conformation along C4–C5 with the <5° dihedral angle of H4–C4–C5–H5a (104.3° is the torsion angle of O2–C4–C5–C6 in the X-ray data6e), which according to the Bothner-By equation explains the ddd SSC of H4 with three different neighboring protons (H5a, H5b and H3). This stereochemical discrimination of the spiroketal chiral center through the H4 SSC pattern could be further consolidated by the significant difference of the two H5 chemical shifts: Δδ5 (δ5a–δ5b) 0.32–0.33 ppm for SAFL-A1/D1 and 0.14–0.15 ppm for SAFL-B1/C1 (Fig. 2). This chemical shift difference may also arise from different conformations revealed by the X-ray diffraction analysis. The fully eclipsed conformation along C4–C5 (B ring) in SAFL-B1 (and probably SAFL-C1) might cause orbital distortions14 of H5 protons or steric compression15 and therefore affect the chemical shifts [H5a (dd) is shifted downfield to 2.5 ppm], while the conformation along C4–C5 in SAFL-D1 (probably and SAFL-A1) is between the staggered and eclipsed conformations, which explains the H5b (dd) proton downfield at ∼2.1 ppm. An alternative rationalization of this difference might be that the diamagnetic anisotropic chemical shift effect of a CH2 is consistently larger when flanking substituents are cis to each other16 [Fig. 3(c)]: cis-4,6-dioxy substitution giving rise to the large Δδ5 (δ5a–δ5b = 0.32–0.33 ppm, SAFL-A1/D1); the trans-4,6-dioxy substitution resulting in the small Δδ5 (δ5b–δ5a = 0.14–0.15 ppm, SAFL-B1/C1).17 A combination of the H4 SSC pattern and H5 chemical shift difference could provide a reliable way to determine the relative configuration of C6 and C3/4 and distinguish SAFL-A1/D1 from SAFL-B1/C1. To differentiate SAFL-A1 from SAFL-D1 (and SAFL-B1 from SAFL-C1) for determination of the relative configuration of C9 and C6, the chemical shifts of H8 were found to be the only useful resonances with a considerable difference. Δδ8 (δ8a–δ8b) 0.65 ppm was observed in SAFL-A1 and only Δδ8 0.33 ppm in SAFL-D1. Similar Δδ8 difference was found between SAFL-B1 (Δδ8 0.24 ppm) and SAFL-C1 (Δδ8 0.63 ppm). However, the rationalization of this chemical shift difference (Δδ8) is much more difficult than the case of Δδ5 because the different C6 substitution is three bonds away from the H8 nuclei. After reviewing the X-ray ORTEP diagrams of SAFL-B1/D1 using the commercial software Mercury, we suspect that the effect of steric compression18 on SAFL-B1/D1 [Fig. 3(d)] might shift downfield of the pseudo-axial H8b, leading to the smaller Δδ8 value (note: the axial protons are upfield of the equatorial ones in most substituted cyclohexanes with the Δδ (δe–δa) value of ∼0.5 ppm. At this stage, the four SAFL diastereomers could be discriminated by the proton NMR analysis and the relative configuration of an unknown tricyclic SAFL may be assigned correctly and readily.
To minimize the errors arising from different machines and sample preparation and generalize this NMR method for different SAFLs, the value of Δδ5 and Δδ8 is further averaged among six different SAFLs with the same relative configuration but different C9 substituents, which had been prepared in our laboratory (Table 1). In general, the results are highly consistent with those observed in the SAFLs with the C9 methyl group. For example, the average of Δδ5 and Δδ8 in the six SAFL-A series is 0.33 ppm and 0.62 ppm with standard deviation (SD) of 0.009 and 0.018, respectively, which suggests the homogeneity of the data and the least influence of the C9 substitution on Δδ5 and Δδ8. Therefore, the average value of Δδ5 and Δδ8 is suggested to be used in the subsequent stereochemical assignments of each type of SAFL as follows. SAFL-A: Δδ5 (ppm) 0.33, Δδ8 (ppm) 0.62; SAFL-B: Δδ5 (ppm) 0.14, Δδ8 (ppm) 0.22; SAFL-C: Δδ5 (ppm) 0.15, Δδ8 (ppm) 0.60; SAFL-D: Δδ5 (ppm) 0.32, Δδ8 (ppm) 0.32.
|
|
||||
|---|---|---|---|---|
| H# | SAFL-A (1–6) | SAFL-B (1–6) | SAFL-C (1–6) | SAFL-D (1–6) |
| Note: Avg = average; SD = standard deviation. | ||||
| H5 (Δδ5) | 0.33, 0.34 | 0.14, 0.15 | 0.15, 0.16 | 0.32, 0.32 |
| 0.34, 0.33 | 0.15, 0.14 | 0.15, 0.15 | 0.33, 0.32 | |
| 0.32, 0.32 | 0.15, 0.13 | 0.15, 0.13 | 0.32, 0.31 | |
| Avg | 0.33 | 0.14 | 0.15 | 0.32 |
| SD | 0.009 | 0.008 | 0.010 | 0.006 |
| H8 (Δδ8) | 0.65, 0.63 | 0.24, 0.22 | 0.63, 0.62 | 0.33, 0.34 |
| 0.60, 0.63 | 0.21, 0.21 | 0.59, 0.58 | 0.32, 0.31 | |
| 0.61, 0.62 | 0.22, 0.18 | 0.57, 0.58 | 0.31, 0.30 | |
| Avg | 0.62 | 0.22 | 0.60 | 0.32 |
| SD | 0.018 | 0.020 | 0.024 | 0.015 |
One challenge in this 1H-NMR analysis is to correctly assign the resonances to H5 and H8, which are usually in the upfield region (1.0–2.5 ppm) and frequently overlapping with other alkyl proton signals. This problem could be solved by an extensive 2D NMR analysis. For example, 1H–1H COSY can correlate the adjacent three-bond protons (e.g., H3–H4–H5, H7–H8–H9) and HMQC (or HSQC) can be used to identify the methylene resonances at C5 and C8. However, the chemical shifts of H5 and H8 might be erroneously reported or swapped with other signals without the authentic 2D NMR spectra, which would prevent the use of this proton NMR analysis method as an effective tool to stereochemically discriminate the SAFLs. In order to address this potential problem, we performed an extensive analysis of the 13C-NMR data derived from the 24 SAFLs that were used in the proton NMR analysis in Table 1. First of all, the carbon resonances that shift most over different types of SAFLs but least within the same type of SAFL should be identified. The value of standard deviation (SD) for each carbon resonance was compared within that of the same type and among the different types of SAFLs for identification of such stereochemistry-responsive carbon(s). The average chemical shifts and the SD values for each carbon within the same type of SAFL are tabulated in Table 2.19 Apparently, the chemical shifts of C3, C7 and C9 vary significantly over different types of SAFLs with the large SD values of 0.996, 1.118 and 0.760, respectively. This suggests that the chemical shifts derived from these carbons may be useful to distinguish the four types (relative configuration) of SAFLs. Secondly, the small SD values within the same type of SAFL were observed in C1 (0.06–0.25), C2 (0.13–0.21), C3 (0.04–0.09), C5 (0.06–0.18), C6 (0.06–0.12), and C7 (0.13–0.22), which suggests that the substituent at C9 has less influence on the chemical shift of these carbons. Taking all these observations together, we concluded the resonances of C3 and C7 were characteristic for each type of SAFL and could be used for stereochemical assignment of an unknown SAFL.20 In order to quantify the assessment of each carbon chemical shift for the stereochemical discrimination in this analysis process, we proposed and defined the σ value for each carbon (Table 2). The σ value of each carbon can be obtained by the division of the SD value of different types of SAFLs by the average of all SD values that are obtained within the same type of SAFL. For example, σ(C3) = 0.996/[(0.04 + 0.09 + 0.04 + 0.04)/4] = 0.996/0.0525 = 19.0. The carbon with larger σ value is better for the stereochemical discrimination. As shown in Table 2, the σ value of C3 (19.0) and C7 (6.30) is larger than that of others, which indicates that C3 and C7 are better representatives for the stereochemical discrimination of SAFLs. Therefore, the relative configuration of SAFL-A could be confirmed by δC3 (ppm) 86.7 and δC7 (ppm) 34.4. Analogously, SAFL-B could be established by δC3 (ppm) 85.1 and δC7 (ppm) 37.0; SAFL-C by δC3 (ppm) 85.2 and δC7 (ppm) 35.6; SAFL-D by δC3 (ppm) 87.1 and δC7 (ppm) 36.3. It is recognized that a single carbon, C3 or C7, could not unambiguously distinguish the four diastereomers and the combination of these two carbon chemical shifts are essential. However, if the spiroketal chiral center is involved, the difference of either C3 or C7 is significant.
| C# | SAFL-A (SD) | SAFL-B (SD) | SAFL-C (SD) | SAFL-D (SD) | SD | σ |
|---|---|---|---|---|---|---|
| 1 | 180.93 (0.25) | 180.94 (0.08) | 180.87 (0.06) | 180.77 (0.19) | 0.078 | 0.54 |
| 2 | 44.38 (0.21) | 44.50 (0.16) | 44.50 (0.15) | 44.61 (0.13) | 0.094 | 0.58 |
| 3 | 86.67 (0.04) | 85.09 (0.09) | 85.24 (0.04) | 87.06 (0.04) | 0.996 | 19.0 |
| 4 | 79.97 (0.14) | 80.43 (0.20) | 80.49 (0.07) | 80.92 (0.83) | 0.389 | 1.25 |
| 5 | 41.77 (0.06) | 41.83 (0.07) | 42.29 (0.18) | 41.62 (0.10) | 0.289 | 2.82 |
| 6 | 115.03 (0.12) | 115.35 (0.11) | 115.55 (0.06) | 115.17 (0.08) | 0.225 | 2.43 |
| 7 | 34.40 (0.16) | 37.04 (0.22) | 35.62 (0.13) | 36.27 (0.20) | 1.118 | 6.30 |
| 8 | 30.22 (0.59) | 30.90 (0.74) | 30.29 (0.80) | 31.12 (0.62) | 0.446 | 0.65 |
| 9 | 78.39(1.55) | 78.03 (4.94) | 77.73 (1.48) | 79.47 (0.87) | 0.760 | 0.34 |
At this stage, the NMR analysis method for stereochemical assignments of the four diastereomeric SAFLs was established and therefore the decision tree21 is summarized in Fig. 4. Although the 13C-NMR method is independent of the 1H-NMR analysis, the combination of 1H- and 13C-NMR analysis usually leads to a quick and conclusive determination of the relative configuration of an unknown SAFL. Practically, the following steps are suggested for an unknown SAFL: (i) SSC of H4 (downfield, t vs. ddd); (ii) δC3 (85–87 ppm); (iii) δC7 (34–37 ppm); (iv) Δδ5 (0.14 vs. 0.33 ppm); (v) Δδ8 (0.22, 0.32, or ∼0.60 ppm). This order of steps does not correspond to the order of priority or importance, but it facilitates a quick determination of the relative configuration of an unknown SAFL because the information of the first three steps can be easily extracted from the spectra without ambiguity.
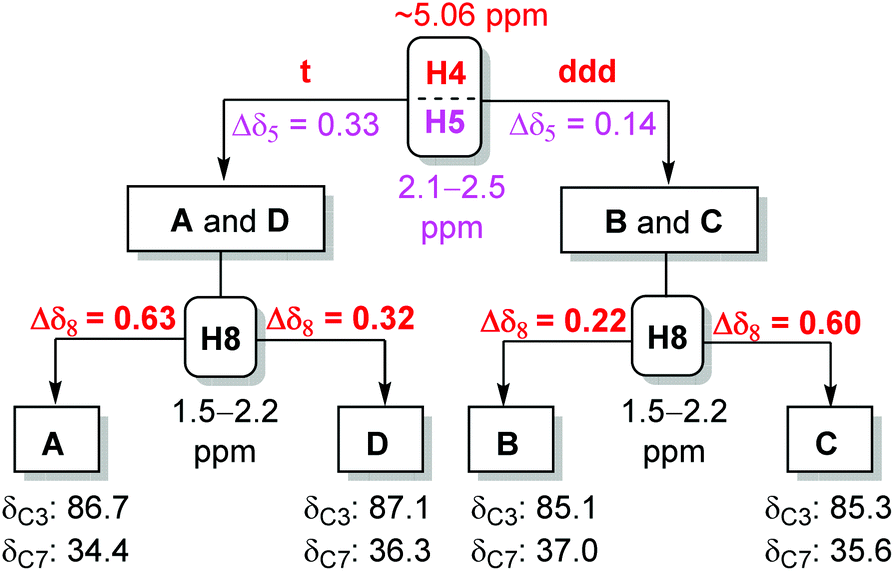 | ||
| Fig. 4 Decision tree used for stereochemical assignments of SAFLs by NMR. | ||
To showcase the application of this NMR method, the reported NMR data of penisporolide A8 was analyzed (Fig. 5). The H4 SSC of penisporolide A was reported as a multiplet at 5.01 ppm. It was more likely to be ddd since in some cases the ddd pattern was not well resolved for clear interpretation. The Δδ5 (0.17 ppm) and the chemical shifts of C3 (85.2 ppm) and C7 (35.5 ppm) were well consistent with that of SAFL-C, although the Δδ8 (0.05 ppm) value was not matching at all. After re-examination of original assignments, it was thought that the resonance (1.55 ppm) assigned to H12 might be derived from H8b, which would result in the matching Δδ8 (0.60 ppm) value as SAFL-C. If the structural revision was true, this NMR analysis could serve as an effective tool to detect possible misassignments of NMR resonances.
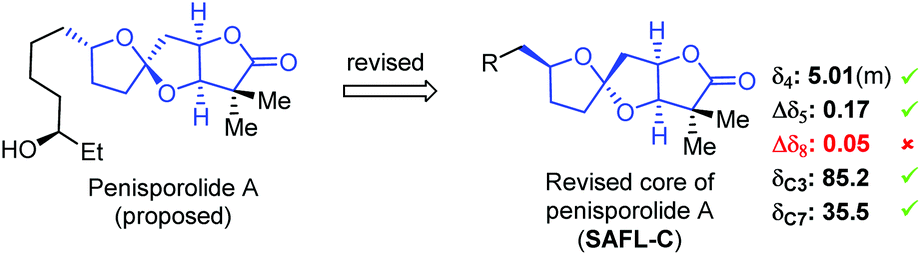 | ||
| Fig. 5 Possible stereochemical revision of the SAFL core of penisporolide A by the NMR analysis method. | ||
To further demonstrate the predictive ability of this NMR analysis method, the assignments of the relative configuration of 19 SAFL-containing compounds reported in the literature were re-examined and the results are presented in Table 3. Most NMR resonances of these compounds had been correctly assigned, which supports the effectiveness of our NMR method. However, several wrong assignments of carbon and proton resonances (carbon 3: B8, B9, C8, and D8; carbon 7: D12; and proton 8: A8, A9, C8 and D8) could be detected and corrected by our NMR analysis method, in support of the predictive ability of our method irrespective of the substitution at C9. The detection and predictive ability of this NMR method would be of high importance to further/future research on this family of natural/non-natural products.
|
|
|||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A7 | A8 | A9 | A12 | B7 | B8 | B9 | B12 | C7 | C8 | C10 | C11 | C12 | C13 | C14 | D7 | D8 | D11 | D12 | D13 | D14 | |
| Complete NMR data are available in the ESI of ref. 6a–f.a Wrong assignment of C3 or C7.b Wrong assignment of H8. ✗ Δδ8 (0.2–0.4 ppm) does not match. | |||||||||||||||||||||
| SSC | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| δ C3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ✓a | ✓ | ✓ | ✓a | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ✓ | ✓ | ✓ | ✓ |
| δ C7 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ✓ | ✓ |
| Δδ5 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Δδ8 | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓b | ✓ | ✓ | ✓ | ✓ |
| Ref. | 6e | 6f | 6f | 6a | 6e | 6f | 6f | 6a | 6e | 6f | 6f | 6e | 6d,e | 6c,d | 6d | 6e | 6f | 6d | 6a–d | 6c,d | 6d |
It is recognized that some natural products contain a similar tricyclic core structure with different substitutions at C2. It would be interesting to attest this NMR method with these types of SAFL-containing compounds (Table 4). Remarkably, our 1H-NMR analysis method was applicable to these SAFL natural products such as cephalosporolides E (1) and F (2),22ent-ascospiroketal B (7),23 and some synthetic compounds (3–6 and 8),6c,9 although the 13C-NMR analysis fails in most cases due to the considerable substituent effects on C3 and/or C7.24 In addition, the wrong assignments of the relative configuration of spiroketal centers of 3 and 4 were easily detected. It is expected that similar NMR analysis methodology will be developed for stereochemical examinations/assignments of other types of compounds.

Conclusions
In summary, a reliable NMR method was established to discriminate the relative configuration of the four possible diastereomers of SAFLs, a tricyclic core structure found in a small family of natural products. The effectiveness of this NMR analysis method was demonstrated by 28 SAFL compounds reported in the literature. This NMR method can also serve as a detection tool for stereochemistry and resonance misassignments as exemplified by the possible stereochemical revision of penisporolide A and compounds 3 and 4 and by the wrong assignments of C3 and/or C7 of 8 synthetic SAFLs (SAFL-B8, -B9, -C8, -D8, and -D12). It is expected that this NMR analysis will provide guidance for stereochemical assignments of new SAFL compounds isolated or synthesized in the future and inspire the development of similar NMR methods for determination of the relative configuration of other types of compounds.Acknowledgements
This research was financially supported by HKUST and the Research Grant Council of Hong Kong (ECS 605912, GRF 605113, and GRF 16305314) and partially supported by the NSFC (Project No. 21472160). We are very grateful to Xin Miao (HKUST) for some preliminary NMR analysis.Notes and references
- For selected reviews, see: (a) A. L. Harvey, Drug Discovery Today, 2008, 13, 894–901 CrossRef CAS PubMed; (b) E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discovery Today, 2016, 21, 204–207 CrossRef CAS PubMed; (c) D. J. Newman and G. M. Cragg, J. Nat. Prod., 2012, 75, 311–335 CrossRef CAS PubMed; (d) G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS PubMed.
- For selected reviews on representative examples, see: (a) M. Büschleb, S. Dorich, S. Hanessian, D. Tao, K. B. Schenthal and L. E. Overman, Angew. Chem., Int. Ed., 2016, 55, 4156–4186 CrossRef PubMed; (b) K. C. Nicolaou, D. Vourloumis, N. Winssinger and P. S. Baran, Angew. Chem., Int. Ed., 2000, 39, 44–122 CrossRef CAS; (c) A. M. Armaly, Y. C. DePorre, E. J. Groso, P. S. Riehl and C. S. Schindler, Chem. Rev., 2015, 115, 9232–9276 CrossRef CAS PubMed; (d) C. Cordier, D. Morton, S. Murrison, A. Nelson and C. O'Leary-Steele, Nat. Prod. Rep., 2008, 25, 719–737 RSC; (e) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323–332 CrossRef CAS PubMed.
- (a) K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 CrossRef CAS PubMed; (b) H.-D. Yoo, S.-J. Nam, Y.-W. Chin and M.-S. Kim, Arch. Pharmacal Res., 2016, 39, 143–153 CrossRef CAS PubMed.
- NMR analysis is the most studied method. For excellent reviews, see: (a) J. M. Seco, E. Quiñoá and R. Riguera, Chem. Rev., 2004, 104, 17–118 CrossRef CAS; (b) G. Bifulco, P. Dambruoso, L. Gomez-Paloma and R. Riccio, Chem. Rev., 2007, 107, 3744–3779 CrossRef CAS PubMed; (c) M. Elyashberg, A. J. Williams and K. Blinov, Nat. Prod. Rep., 2010, 27, 1296–1328 RSC; (d) M. W. Lodewyk, M. R. Siebert and D. J. Tantillo, Chem. Rev., 2011, 112, 1839–1862 CrossRef PubMed. For selected examples, see: (e) J. Lee, Y. Kobayashi, K. Tezuka and Y. Kishi, Org. Lett., 1999, 1, 2181–2184 CrossRef CAS PubMed; (f) S. Higashibayashi, W. Czechtizky, Y. Kobayashi and Y. Kishi, J. Am. Chem. Soc., 2003, 125, 14379–14393 CrossRef CAS PubMed; (g) S. D. Rychnovsky, B. Rogers and G. Yang, J. Org. Chem., 1993, 58, 3511–3515 CrossRef CAS; (h) S. D. Rychnovsky, Org. Lett., 2006, 8, 2895–2898 CrossRef CAS PubMed; (i) W. R. Roush, T. D. Bannister, M. D. Wendt, M. S. VanNieuwenhze, D. J. Gustin, G. J. Dilley, G. C. Lane, K. A. Scheidt and W. J. Smith, J. Org. Chem., 2002, 67, 4284–4289 CrossRef CAS PubMed; (j) S. G. Smith and J. M. Goodman, J. Org. Chem., 2009, 74, 4597–4607 CrossRef CAS PubMed; (k) F. López-Vallejo, M. Fragoso-Serrano, G. A. Suárez-Ortiz, A. C. Hernández-Rojas, C. M. Cerda-García-Rojas and R. Pereda-Miranda, J. Org. Chem., 2011, 76, 6057–6066 CrossRef PubMed; (l) P. H. Willoughby, M. J. Jansma and T. R. Hoye, Nat. Protocols, 2014, 9, 643–660 CrossRef CAS PubMed , and references therein.
- For reviews covering incorrectly assigned structures of natural products and their structural revisions by total synthesis, see: (a) M. E. Maier, Nat. Prod. Rep., 2009, 26, 1105–1124 RSC; (b) T. L. Suyama, W. H. Gerwick and K. L. McPhail, Bioorg. Med. Chem., 2011, 19, 6675–6701 CrossRef CAS PubMed. For recent examples of structural revisions by total synthesis from our group, see: (c) L. Song, K.-H. Lee, Z. Lin and R. Tong, J. Org. Chem., 2014, 79, 1493–1497 CrossRef CAS PubMed; (d) L. Zhu, Y. Liu, R. Ma and R. Tong, Angew. Chem., Int. Ed., 2015, 54, 627–632 CAS; (e) L. Zhu and R. Tong, Synlett, 2015, 1643–1648 CAS; (f) L. Zhu and R. Tong, Org. Lett., 2015, 17, 1966–1969 CrossRef CAS PubMed.
- (a) S. F. Tlais and G. B. Dudley, Org. Lett., 2010, 12, 4698–4701 CrossRef CAS PubMed; (b) S. F. Tlais and G. B. Dudley, Beilstein J. Org. Chem., 2012, 8, 1287–1292 CrossRef CAS PubMed; (c) R. A. Fernandes and M. B. Halle, Asian J. Org. Chem., 2013, 2, 593–599 CrossRef CAS; (d) J. Li, C. Zhao, J. Liu and Y. Du, Tetrahedron, 2015, 71, 3885–3889 CrossRef CAS; (e) J. Wang and R. Tong, J. Org. Chem., 2016, 81, 4325–4339 CrossRef CAS PubMed. Brimble achieved the synthesis of the tricyclic SAFL core of CesH, see: (f) O. C. Finch, D. P. Furkert and M. A. Brimble, Tetrahedron, 2014, 70, 590–596 CrossRef CAS.
- X. Li, Y. H. Yao, Y. A. Zheng, I. Sattler and W. H. Lin, Arch. Pharmacal Res., 2007, 30, 812–815 CrossRef CAS.
- X. Li, I. Sattler and W. H. Lin, J. Antibiot., 2007, 60, 191–195 CrossRef CAS PubMed.
- C. V. Ramana, S. B. Suryawanshi and R. G. Gonnade, J. Org. Chem., 2009, 74, 2842–2845 CrossRef CAS PubMed.
- S. Chang and R. Britton, Org. Lett., 2012, 14, 5844–5847 CrossRef CAS PubMed.
- All NMR data used in this article were collected in CDCl3.
- A. A. Bothner-By, in Advances in Magnetic Resonance, ed. J. S. Waugh, Academic Press, New York, 1965, vol. 1, pp. 195–613 Search PubMed.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- For related cases with geometry and orbital distortions that cause chemical shifts, see: (a) F. M. Beringer, L. L. Chang, A. N. Fenster and R. R. Rossi, Tetrahedron, 1969, 25, 4339–4345 CrossRef CAS; (b) D. J. Sardella and E. Boger, Magn. Reson. Chem., 1989, 27, 13–20 CrossRef CAS.
- M. A. Cooper and S. L. Manatt, J. Am. Chem. Soc., 1970, 92, 4646–4652 CrossRef CAS.
- For similar effects in tetrahydrofurans, see: (a) D. R. Williams, Y. Harigaya, J. L. Moore and A. D'sa, J. Am. Chem. Soc., 1984, 106, 2641–2644 CrossRef CAS; (b) E. D. Mihelich and G. A. Hite, J. Am. Chem. Soc., 1992, 114, 7318–7319 CrossRef CAS. For similar effects in γ-lactones, see: (c) S. A. M. T. Hussain, W. D. Ollis, C. Smith and J. F. Stoddart, J. Chem. Soc., Perkin Trans. 1, 1975, 1480–1492 RSC.
- The upfield shift of cis substituents compared to trans is often observed in a series of succinic anhydrides, see: L. E. Erickson, J. Am. Chem. Soc., 1965, 87, 1867–1875 CrossRef CAS.
- For the effect of steric compression on chemical shifts in cage-like molecules, see: S. Winstein, P. Carter, F. A. L. Anet and A. J. R. Bourn, J. Am. Chem. Soc., 1965, 87, 5247–5249 CrossRef CAS . Also see ref. 15.
- See the ESI.†.
- The characteristic deviation of the chemical shifts of the C7 in the 13C-NMR might be due to the shielding γ/δ-effect caused by hydrogen–hydrogen gauche interactions, especially in these SAFLs where C7 is oriented inside the concave face of the cis-fused-γ-lactone core, see: D. M. Grant and B. V. Cheney, J. Am. Chem. Soc., 1967, 89, 5315–5318 CrossRef CAS.
- For the use of decision trees in NMR, see: F. Qiu, A. Imai, J. B. McAlpine, D. C. Lankin, I. Burton, T. Karakach, N. R. Farnsworth, S.-N. Chen and G. F. Pauli, J. Nat. Prod., 2012, 75, 432–443 CrossRef CAS PubMed.
- For isolation of cephalosporolides E and F: (a) M. J. Ackland, J. R. Hanson, P. B. Hitchcock and A. H. Ratcliffe, J. Chem. Soc., Perkin Trans. 1, 1985, 843–847 RSC; (b) A. Farooq, J. Gordon, J. R. Hanson and J. A. Takahashi, Phytochemistry, 1995, 38, 557–558 CrossRef CAS; (c) V. Rukachaisirikul, S. Pramjit, C. Pakawatchai, M. Isaka and S. Supothina, J. Nat. Prod., 2004, 67, 1953–1955 CrossRef CAS PubMed; (d) J. L. Oller-López, M. Iranzo, S. Mormeneo, E. Oliver, J. M. Cuerva and J. E. Oltra, Org. Biomol. Chem., 2005, 3, 1172–1173 RSC; (e) H. M. T. B. Herath, M. Jacob, A. D. Wilson, H. K. Abbas and N. P. D. Nanayakkara, Nat. Prod. Res., 2013, 27, 1562–1568 CrossRef CAS PubMed.
- J. Wang and R. Tong, Org. Lett., 2016, 18, 1936–1939 CrossRef CAS PubMed.
- For substituent effects on carbon NMR chemical shifts, see: (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS. It was reported that carbon NMR chemical shifts data are less discriminating than those of proton ones, see: (b) M. G. Chini, R. Riccio and G. Bifulco, Eur. J. Org. Chem., 2015, 1320–1324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Ten tables showing NMR data of 24 compounds. See DOI: 10.1039/c6qo00556j |
| This journal is © the Partner Organisations 2017 |