
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an indirect competitive enzyme-linked immunosorbent assay for screening ethopabate residue in chicken muscle and liver†
Zhaozhou Li *,
Yao Wang,
Daomin Li,
Xiujin Chen,
Zhili Li,
Hongli Gao,
Li Cao,
Songbiao Li and
Yuze Hou
*,
Yao Wang,
Daomin Li,
Xiujin Chen,
Zhili Li,
Hongli Gao,
Li Cao,
Songbiao Li and
Yuze Hou
Henan Engineering Laboratory of Livestock Disease Diagnosing and Food Safety Testing, College of Food and Bioengineering, Henan University of Science and Technology, No. 263 Kaiyuan Avenue, Luolong District, Luoyang 471023, China. E-mail: ilizhaozhou@126.com; Fax: +86 379 64282342; Tel: +86 379 64282342
First published on 20th July 2017
Abstract
Ethopabate (ETP) is a coccidiostat that is frequently used to prevent and treat coccidiosis in chickens. Illegal use and abuse of ETP can lead to drug residues in edible animal tissues, which present a potential health risk to consumers. To rapidly monitor ETP residues, a monoclonal antibody-based indirect competitive enzyme-linked immunosorbent assay (ic-ELISA) has been developed that has simple sample preparation and clean-up. After immunogen preparation, inoculation and cell fusion, a monoclonal antibody (4G7) was obtained with the lgG2 isotype. The 4G7 antibody had the ability to specifically recognize ETP with an IC50 value of 0.66 μg L−1, it was found to have weak and insignificant cross-reactivity for some structure-related analogs. With the optimized ic-ELISA protocol, the detection limits of ETP were calculated as 0.21 μg kg−1 and 0.34 μg kg−1 in chicken muscle and liver samples, respectively. The recoveries ranged from 85.4% to 98.4% with a coefficient of variation of less than 15%. Furthermore, good correlations between the results of ic-ELISA and high-performance liquid chromatography demonstrated the reliability of the developed ic-ELISA. This proposed method is a rapid, sensitive and useful tool that offers a cost-effective alternative to current published procedures without any concession in the ability to detect ETP residues in edible animal tissues.
1. Introduction
Coccidiosis is a severe parasitic disease caused by a protozoa that is found in the intestinal epithelium. It is especially likely to occur in warm and humid environments. It not only causes high mortality rates but also leads to bloody diarrhea, reduction in feed and water intake, emaciation, poor weight gain and poor egg production.1 Ethopabate (ETP), methyl-4-acetamido-2-ethoxybenzoate, is a coccidiostat frequently used to prevent an otherwise costly outbreak of coccidiosis in chicken. It has a synergistic effect with some anticoccidial drugs.2 The use of ETP in the chicken industry may result in ETP residues in chicken products if adequate withdrawal time for the animal has not been observed or if ETP has been improperly administered. The residue presents a potential health risk to consumers. Therefore, The US Code of Federal Regulations has established maximum residual limits (MRLs) for ETP to be 500 μg kg−1 in chicken muscle and 1500 μg kg−1 in chicken liver. It is not allowed to be present in eggs because giving it to laying hens is prohibited. The European Union (EU) has banned the administration of ETP via animal feed to prevent and treat coccidiosis.3Residue monitoring is an effective way for assurance of the food safety and wholesomeness. Numerous methods, including spectrofluorimetric assays,2 spectrofluorimetric methods,4 gas chromatography,5 high-performance liquid chromatography (HPLC) with UV detection6 or by fluorimetry,7 liquid chromatography coupled to triple quadrupole mass spectrometry8–10 or time-of-flight-mass spectrometry,11 have been previously used for ETP residue analysis. Although these approaches are sensitive and reliable, these methods often require extensive sample pretreatments and sophisticated laboratory equipment with highly trained operators. These approaches are not suitable for a high sampling frequency or a rapid assessment of results. Therefore, there has been an urgent need for a rapid, specific and high-throughput screening method to monitor ETP residue in edible animal tissues.
Currently, an indirect competitive enzyme-linked immunosorbent assay (ic-ELISA) has been shown to be a highly sensitive, low-cost and rapid tool for high-throughput screening for residues in animal tissues. It is also the most popular bioassay for residue analysis. Specificity will be gained by using an antibody for capturing and detecting the target molecule. So far, the antibody-based ic-ELISA is generally believed to be a highly sensitive, specific and reliable method compared to detection based on other recognition elements, such as molecular imprinting polymers and aptamers.12,13 To the best of our knowledge, no studies have been published regarding the preparation of a monoclonal antibody against ETP or the development of an immunoassay for simultaneous screening of ETP residues in edible animal tissues.
The purpose of this study was to develop and validate an ic-ELISA method for rapid determination of ETP residues in chicken muscle and liver. Methyl-4-amino-2-ethoxybenzoate (MAE) was selected as a hapten and further conjugated with carrier proteins to prepare a mono-clonal antibody (mAb) against ETP. Then, the mAb was produced through a normal protocol for mice immunization and cell fusion. The most sensitive hapten and mAb were used to establish an ic-ELISA with high sensitivity, specificity and low cost. Under optimum conditions, the ic-ELISA was used for ETP analysis in real samples as well as validated by high-performance liquid chromatography (HPLC) method. The proposed method not only broadens the applicability of an antibody-based ELISA method but also presents an alternative approach for simultaneous screening ETP residues in the foods of animal origin.
2. Materials and methods
2.1. Reagents and instruments
The ETP standard was purchased from Hubei Widely Chemical Technology Co., Ltd, China. Methyl-4-amino-2-ethoxybenzoate (MAE) was received from Hangzhou Sage Chemical Co., Ltd. Methyl-4-acetamidobenzoate (MAB) was from Beijing Realchem Technology Co., Ltd. Methyl-4-acetamido-2-methoxybenzoate (MAMB) was from Nanjing Chemlin Chemical Co., Ltd. Methyl-4-amino-2-methoxybenzoate (MAM), and 4-aminobenzoic acid methyl ester (AAME) were from TCI (Shanghai) Development Co., Ltd. Methanol, acetonitrile, acetic acid, 25% glutaric dialdehyde and hydrochloric acid were obtained from Sinopharm Chemical Reagent Co., Ltd. Bovine serum albumin (BSA), ovalbumin (OVA), Freund's adjuvants (complete and incomplete), polyethylene glycol 1500 (PEG 1500, 50%), 3,3′,5,5′-tetramethylbenzidine (TMB), horseradish peroxidase labeled goat anti-rabbit IgG (HRP-IgG), culture media RPMI-1640, hypoxanthine aminopterin thymidine (HAT) and hypoxanthine thymidine (HT) were purchased form Sigma-Aldrich (St. Louis, MO). All other chemicals and organic solvents were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China) and were of analytical grade.The SP2/0 mouse plasmacytoma cell line came from Shanghai Suer Biological Technology Co., LTD (China). Fetal calf serum was purchased from Hangzhou Sijiqing Biological Engineering Materials Co., Ltd. (Hangzhou, China). Water used in these experiments was filtered from a Millipore Milli-Q purification system.
The determination of ETP, MAE, MAB, MAMB, MAM and AAME, whose chemical structures are shown in Fig. S1,† was carried out on a Varian liquid chromatographic system (Varian Company, USA) equipped with a Prostar 210 pump, a LC workstation version 6.41 system software and a Prostar 325 UV-Vis detector. Chromatographic separation was performed on a C18 reversed-phase column (5 μm, 4.6 mm × 250 mm Zorbax SB, Agilent Corporation, USA). The mobile phase consisted of 35% water and 65% methanol (by volume). Effluents were monitored at a wavelength of 268 nm. The flow rate was 1 mL min−1, the injection volume was 10 μL, and the column temperature was maintained at room temperature.12,14
2.2. Buffers and solutions
The following solutions were used in the experiments. The incubation solution was 0.01 mol L−1 phosphate-buffered saline (PBS) buffer (pH 7.2). The coating solution was 0.05 mol L−1 carbonate buffer (pH 9.6). The washing solution was 0.01 mol L−1 PBS buffer containing 0.1% Tween-20 (PBST) with a pH value of 7.2. The blocking solution was 0.01 mol L−1 PBS buffer with 1% (w/v) OVA and 4% (w/v) cane sugar as well as a pH value of 7.2. TMB substrate solution was prepared by adding 3.3 mg TMB in 250 μL dimethyl sulfoxide to 25 mL of citrate-phosphate buffer containing 3.25 μL of a 30% H2O2 solution. The stopping reagent was selected as 2 mol L−1 H2SO4 solution.2.3. Preparation of hapten–protein conjugates
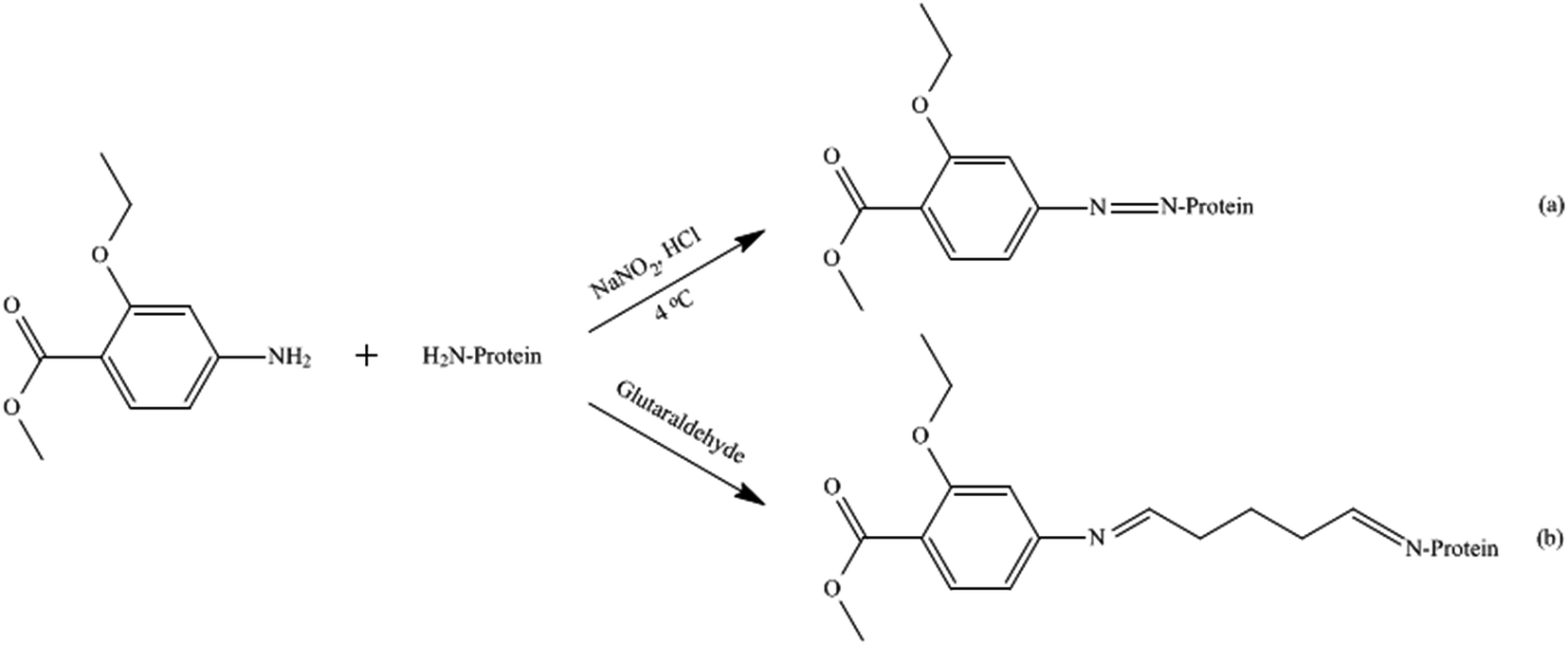 | ||
| Fig. 1 Synthetic process of MAE conjugates by diazotization (a) and glutaraldehyde (b) methods. | ||
After centrifugation for 15 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g to remove the precipitate, the hapten density (number of hapten molecules per molecule of carrier protein) of the conjugate was estimated directly by using a double-beam spectrophotometer (Agilent 8453, USA). Furthermore, the resulting conjugate solution was lyophilized with a freeze-dryer (Labconco, Kansas, MO, USA) and stored at −20 °C.
000g to remove the precipitate, the hapten density (number of hapten molecules per molecule of carrier protein) of the conjugate was estimated directly by using a double-beam spectrophotometer (Agilent 8453, USA). Furthermore, the resulting conjugate solution was lyophilized with a freeze-dryer (Labconco, Kansas, MO, USA) and stored at −20 °C.
2.4. Generation of monoclonal antibody against ethopabate
Animal experiments were performed in compliance with the laws and guidelines of the Chinese specifications and regulations and approved by the Animal Ethics Committee of the Henan University of Science and Technology.18,19 Female BALB/c mice (8–10 weeks old) were purchase from the Henan Laboratory Animal Center (Zhengzhou, China). They were respectively immunized through intraperitoneal injections with MAE–BSA(DIA) and MAE–BSA(GA) conjugates. The first injection consisted of each immunogen emulsified with Freund's complete adjuvant at a dosage of 100 μg for each mouse. Subsequently, the mice were administered boost immunizations biweekly with the same dosage of the immunogen emulsified in Freund's incomplete adjuvant. After the fifth immunization, tail bleeding was performed and then the obtained antiserum was determined. The mouse exhibiting the highest antibody titer was sacrificed after the last immunization, and the splenocytes were fused with SP2/0 myeloma cells.20 Furthermore, the cells were selected using the selection medium after the fusion. With a noncompetitive indirect ELISA method, the growing hybridoma cells were screened for antibody production using the coating antigen MAE–OVA with 100 μL for each well. By using the limiting dilution method, the positive clones were cloned using single cells for multiple rounds until monoclones were obtained. The selected positive monoclone with highly specific recognition of ETP was used for ascites production. Subsequently, ascites fluid containing monospecific antibodies was collected, centrifuged (2000g, 5 min), and then purified with ammonium sulfate precipitation and DEAE iron-exchanged chromatography. The purified monoantibody was finally separated into 500 μL aliquots and stored at −20 °C.2.5. Indirect ELISA and ic-ELISA
For conjugate-coated indirect competitive assays, ELISA plates were coated with 100 μL per well of MAE–OVA solution in the coating buffer through overnight incubation at 4 °C. Coated plates were washed three times with PBS containing 0.1% Tween-20 (PBST) incubated with 200 μL of 1% OVA in PBS at 37 °C for 0.5 h and washed with PBST again. Next, 100 μL of antibody were added to each well. Followed by incubation at 37 °C for 0.5 h, the plates were washed as described and then incubated with 100 μL of HRP-lgG (1:5000) at 37 °C for 0.5 h. After washing the plates with PBST, 100 μL of substrate solution were added to each well and incubated for 15 min at room temperature in the dark. The enzymatic reaction was stopped with 2 mol L−1 H2SO4 solution. The absorbance was immediately read at 450 nm on a 550 plate reader (Bio-Rad, Hercules, California, USA).21
The protocol used for the ic-ELISA was similar to a previously described protocol.22 The plates were coated with 100 μL of the coating conjugates. Coated plates were washed and blocked as described above. Afterwards, 50 μL per well of the antibody plus 50 μL per well of analyte in PBS or sample were added. The plates were then incubated, washed and measured with a microplate reader at 450 nm to obtain the absorbency (B) of every well. The competitive inhibitory curves were developed by plotting absorbance against the logarithm of analyte concentrations and were fitted to a four-parameter logistic equation: Y = (A − B)/[1 − (X/C)D] + B, where A is the maximum absorbance in the absence of ETP, B is the minimum absorbance at infinite concentration, C is the concentration producing 50% of the maximal absorbance, D is the slope at the inflection point of the sigmoid curve, X is the concentration of hapten, and Y is the OD value. From the equation, IC50 values (concentrations at which the binding of the antibody to the coating antigen are inhibited by 50%) were determined.23
2.6. Sample preparation
The chicken muscle or liver samples were purchased from Dazhang supermarket (Luoyang, China). Firstly, they were cut into pieces and homogenized. Two grams of each sample were weighed in a 50 mL disposable plastic centrifuge tube. Twenty milliliters of acetonitrile and 2 g anhydrous Na2SO4 were added. After 5 min of vortex mixing, the mixtures were centrifuged at 4000g for 10 min. The resulting supernatants were transferred into new tubes and the same extraction procedure was repeated. Subsequently, the supernatant extracts were combined and evaporated at 35 °C under vacuum. The dry residue was reconstituted by 2 mL of 5% methanol–water and transferred into a 5 mL centrifuge tube. Followed by the addition of 2 mL hexane, fat in the supernatant liquid was removed after a 10 min vortex mix. The residual layer was up to 2 mL with 5% methanol–water and was analyzed by the ic-ELISA method. The concentration of ETP in the real sample was calculated with the standard curve obtained with the standard substance.122.7. Validation of the ic-ELISA
Immunoassay validation was performed using 20 different frozen tissue samples, which were purchased from Dazhang supermarket (Luoyang, China) and had previously been proved by high-performance liquid chromatography (HPLC) to be free of ETP. Each tissue sample was analyzed using the ELISA method to calculate the limit of detection (LOD) and demonstrate the range of blank matrix effects. The determination of LOD was based on 20 blank samples accepting no false positive rates, with an average value plus triple standard deviation (SD). The limit of quantification (LOQ) was calculated as the blank value plus ten times its SD. According to the European commission decision 2002/657/EC24 and the European guidelines for the validation of the screening method,25 the decision limit and detection capability were assessed by CCα and CCβ. CCα was determined by analyzing 60 blank samples per matrix fortified with the analyte at the permitted limit (MRL). The decision limit CCα (α = 5%) was calculated as the concentration at the MRL plus 1.64 times the corresponding standard deviation. The detection capability CCβ (β = 5%) was expressed as the value of the decision limit plus 1.64 times the corresponding standard deviation.The accuracy and precision of the ELISA method were represented by coefficient of variation (CV) and recovery, respectively. CV was calculated by the analysis of 5 spiked duplicate blanks at levels of 0.5, 1 and 2 times the MRL. The recovery (%) was calculated using the following equation: (concentration measured/concentration spiked) × 100. Furthermore, each concentration level was detected 5 repetitions in a time span of 2 months.
2.8. Comparison of the ic-ELISA with HPLC
To assess the qualitative and quantitative accuracy of the ic-ELISA method, a comparison between the ic-ELISA and HPLC was performed using blank and naturally contaminated chicken samples. Nine natural contaminated chicken muscle or liver samples were respectively screened from Dazhang supermarket (Luoyang, China). Furthermore, the samples were subjected to the ic-ELISA procedure and HPLC analysis according to the procedure described in the section of 2.1. A one-dimensional linear regression model was applied to assess the correlation of the two detection methods.2.9. Theoretical calculations
Descriptive statistical parameters, such as the mean value, SD, and CV, were calculated. Statistical analysis of the data were carried out by using Origin 8.5. Molecular modeling was performed using Gaussian 09 and GaussView 5.0.3. Results and discussion
3.1. Hapten design
Hapten design and synthesis is a key step in the development of a rapid immunoassay method. Our intended approach to hapten design was the preparation of an antibody against ETP. It is reported that the chemical structure and electronic distribution of the target analyte play important roles in the formation of high-quality antibodies.26 Therefore, molecular modeling was carried out by using Gaussian 09 and GaussView 5.0. The DFT B3LYP method was used for energy calculation (basis set at 6-31G). The atomic charges (NBO charges) and electrostatic potentials of the molecules were calculated using Gaussian 09 and GaussView 5.0. Fig. 2 shows the electrostatic potential contours of ETP and its structural analogs with the minimum energy conformations. It is demonstrated that the electrostatic potential contour of MAE–GA conjugate is closest to that of ETP. The molecular charge distribution of these compounds is shown in Table 1. In terms of electrostatic potentials and molecular charges, MAE–GA mimicked the target analyte ETP more authentically than other two haptens. With the hapten of MAE, the obtained antibody exhibits limited cross-reactivity (CR) with MAM and AAME. Therefore, it can be deduced that the atomic charge at C2 plays an important role and substituent in this position has the predominant effect on antibody binding. Furthermore, with MAM or AAME as the hapten, the resulting antibodies might be specific only for MAM or AAME and might not be specific for ETP and MAE. Due to that possibility, MAE was selected as the hapten due to its similarity in spatial structure and charge distribution. | ||
| Fig. 2 The electrostatic potential contours of ETP (a), MAE–GA (b), MAM–GA (c) and AAME–GA (d). | ||
| Structure | Molecular charge distribution | |||||
|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | C5 | C6 | |
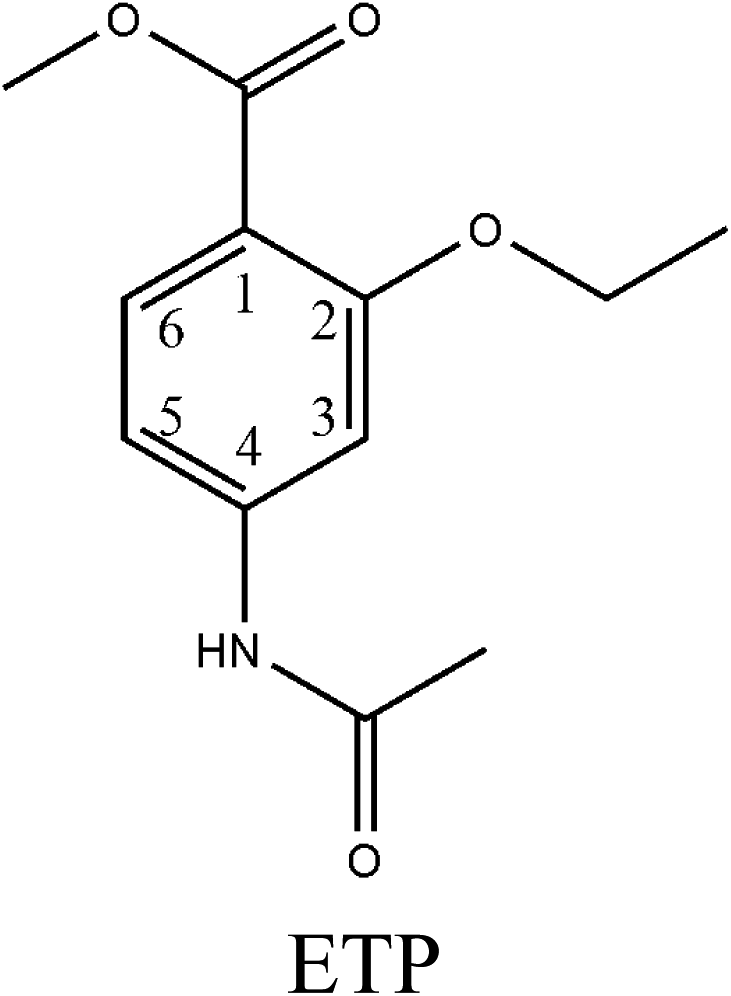 |
−0.226 | 0.322 | −0.297 | 0.171 | −0.271 | −0.178 |
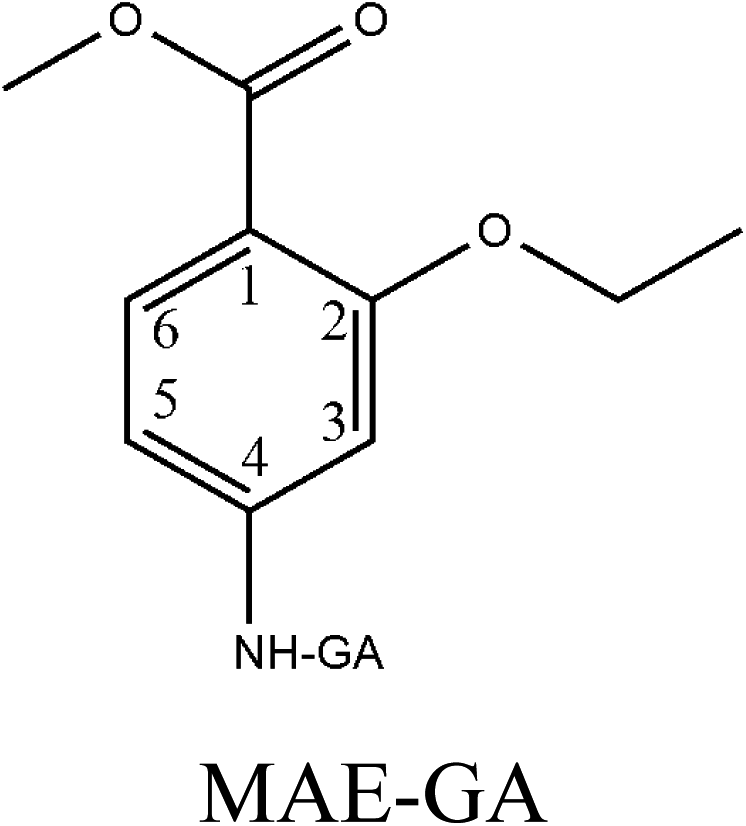 |
−0.229 | 0.318 | −0.295 | 0.174 | −0.273 | −0.181 |
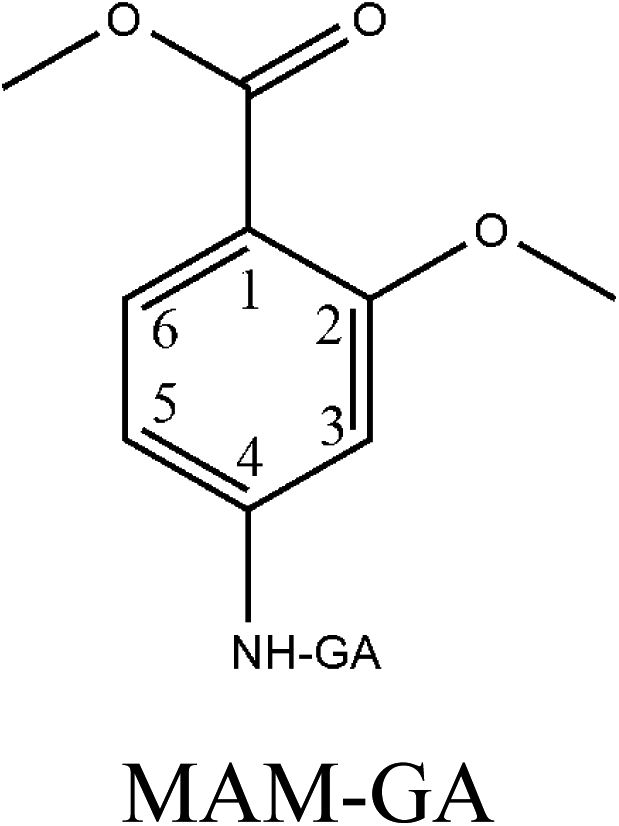 |
−0.21 | 0.358 | −0.35 | 0.191 | −0.286 | −0.163 |
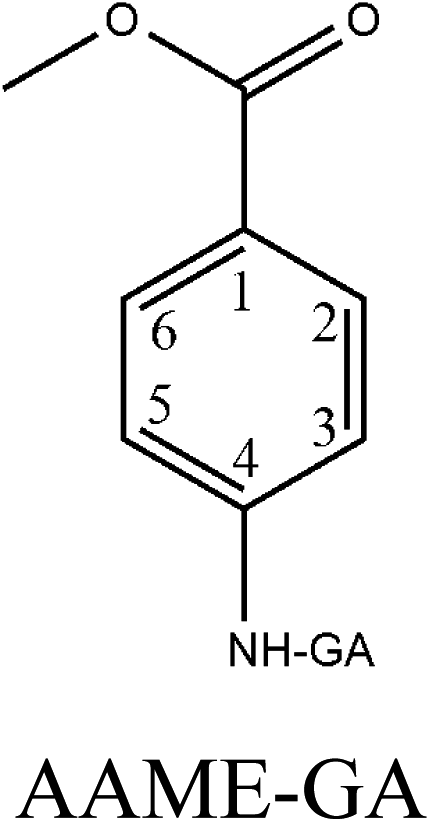 |
−0.178 | −0.18 | −0.272 | 0.17 | −0.266 | −0.179 |
The molecular modeling has borne out further evidence for the optimal hapten design criteria. For producing a highly specific antibody, it is essential to prepare a near-perfect mimic of the target analyte. The hapten should contain a handle terminated with a functional group capable of covalent bonding to the carrier protein. As the hapten, it should be identical to the target molecule in structure, geometry, electronic and hydrophobic properties.27 In general, a linear saturated aliphatic bridge with 3 to 6 carbon atoms are preferred for minimum physico-chemical and antigenic interferences over the hapten or the immune system, respectively. The linking spacer should be better placed at distal positions from characteristic chemical moieties. On the other hand, it will be less favorable for antibody affinity and specific recognition when the binding positions nest to highly antigenic groups or central location with respect to the molecular longitudinal axis.28 For coupling to carrier proteins, amino group is the preferred moiety because it is compatible to a variety of reaction conditions, and it is readily activated to react with carboxyl groups, which are usually abundant in carrier proteins. After coupling with the spacer arm at right positions of the target molecule, the electronic properties will undergo a minimal change and there may also be a slight alteration in geometry. However, severe molecular charge changes that will destabilize the resulting bioconjugate and affect the immunogenic properties. Therefore, it is recommended that low or moderate hapten densities are in favor of high-affinity antibody production due to the relatively strong interactions with T- and B-cell antigen receptors.29
3.2. Estimation of hapten–protein ratios
For developing an ELISA method to detect ETP, the most important thing is to prepare an appropriate immunogen with which immunizing animals to obtain the highly specific antibody toward the target compound. Verification of the conjugate synthesis was performed on the double-beam spectrophotometer (Agilent 8453, USA). The hapten/protein ratio was estimated according to the ultraviolet absorbance spectra of haptens, carrier proteins, and conjugates as follows: [εconjugation − εprotein]/εhapten, where ε is the absorbance coefficient of analyte. The incorporation rates of MAE–BSA(DIA), MAE–OVA(DIA), MAE–BSA(GA) and MAE–OVA(GA) were calculated as 24.7, 17.5, 27.3 and 16.4, respectively. This proved that the conjugates were prepared successfully.The hapten density is a very important parameter which generally defines the quality and quantity of antibody produced. A higher density could increase the antibody titer with lower affinity. In contrast, a lower density induces a slower immune response with higher affinity antibodies. It is reported that high antibody titers with moderate antibody affinities are usually obtained with a hapten density of 15 to 30 molecules per carrier protein. Furthermore, the formations of hapten–protein conjugates are not always reproducible, even under the best experimental conditions. These changes will produce inconsistent hapten–protein stoichiometries resulting large variation in the preparation of desired antibodies. Therefore, careful selection of hapten density for hapten–protein conjugation is very important for antibody generation.30
3.3. Antibody production and characterization
According to the bleeding serum characterization results, the spleen cells from the immunized mice were used for the fusion experiment. After several cell fusions and cultures, the hybridoma cells 4A3, 3B2, 2F3, 5F7, 6D9 and 4G7, which were rescreened and subcloned three times. The titers of monoclonal antibodies (mAbs) were determined by the indirect ELISA (Table 2). Among them, 4G7 showed the highest titer and best sensitivity toward ETP. Therefore, it was selected to produce the mAb. The subclass of the mAb 4G7 was identified as the IgG2 isotype possessing a kappa light chain by a mouse monoclonal isotyping kit.| Hybridoma cells | B/B0 values | |||||
|---|---|---|---|---|---|---|
| MAE–BSA(GA) | MAE–BSA(DIA) | |||||
| 4A3 | 5F7 | 4G7 | 3B2 | 2F3 | 6D9 | |
| Titer (1:X × 103) |
16 | 32 | 256 | 8 | 32 | 128 |
| ETP (500 μg L−1) | 0.27 | 0.13 | 0.04 | 0.34 | 0.12 | 0.07 |
| MAE (500 μg L−1) | 0.11 | 0.06 | 0.03 | 0.14 | 0.05 | 0.05 |
The cross-reactivity (CR) of the mAb toward the structural relative compounds was also assessed. Four related analogs (MAM, MAMB, AAME and MAB) were selected for CR testing. The CR values were calculated with the following formula: CR = (IC50 of ETP/IC50 of competitor) × 100%. Standard curves were prepared with up to 2 μmol L−1 of each compound in PBS and measured with the proposed ELISA. Due to the low water solubility of the studied compounds, higher concentrations could not be studied. Some inhibition was observed with non-target compounds, and the CR values were below 2%. The CR values for MAM, MAMB, AAME and MAB were 0.42%, 0.67%, 1.24% and 1.37%, respectively. This result could be interpreted from a comparison of the chemical structures of these compounds.
3.4. The standard curve for the ic-ELISA
The optimum conditions for the ic-ELISA were determined as a coating antigen (MAE–OVA) concentration of 0.2 μg mL−1 and a 1:2.56 × 105 ratio of antibody (4G7) dilution. These conditions were optimum for developing a linear response curve and were kept constant for the rest of the experiment. As shown in Fig. 3, the standard curve that was based on the ETP matrix calibration ranges from 0.1 μg L−1 to 30.0 μg L−1. The IC50 value of this method for ETP was 0.66 μg L−1.
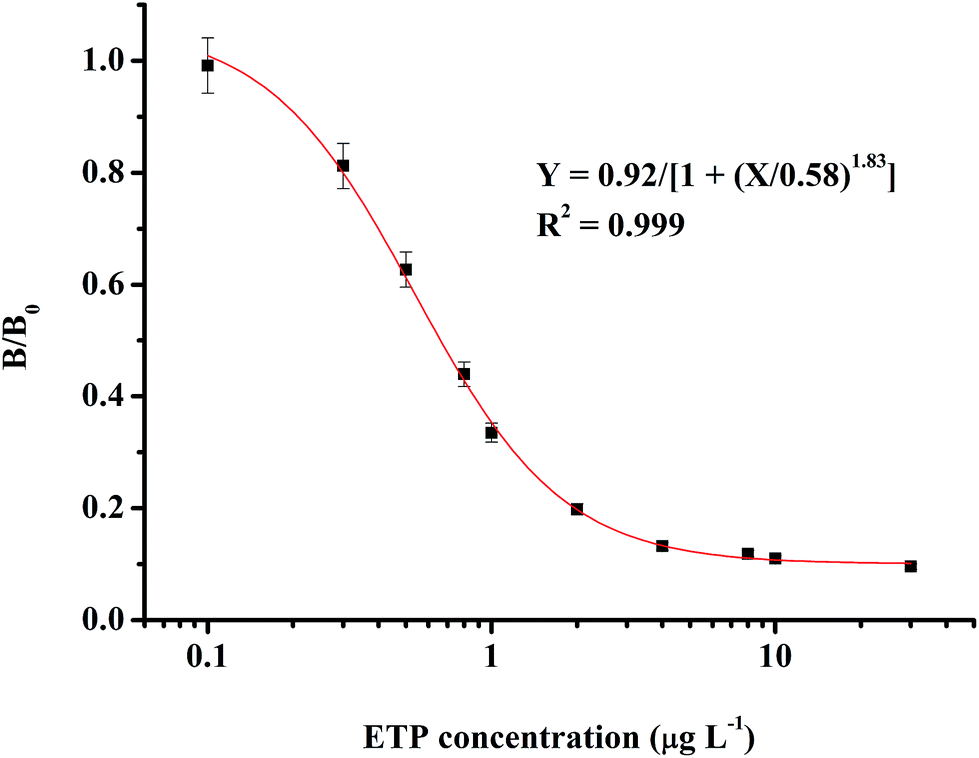 | ||
| Fig. 3 Standard curve for detecting EPT by the ic-ELISA. | ||
3.5. Validation of the ic-ELISA method
The assay validation was carried out by investigating the LOD, LOQ, CCα, CCβ, accuracy, repeatability and reproducibility. Based on the sample preparation procedure, the LOD, LOQ, CCα and CCβ values were calculated as 0.21 μg kg−1, 0.32 μg kg−1, 135.23 μg kg−1 and 144.71 μg kg−1 in the chicken muscle samples, respectively. For the chicken liver samples, the LOD, LOQ, CCα and CCβ values were determined as 0.34 μg kg−1, 0.47 μg kg−1, 183.12 μg kg−1 and 194.71 μg kg−1, respectively.To evaluate the accuracy, repeatability and reproducibility of the ELISA method, a spiked recovery test was conducted at levels of 0.5, 1 and 2 times the MRL. The mean recovery and coefficient of variance (CV) were calculated by repeated analysis (n = 5) of the spiked samples and comparison of the intra- and inter-assay CVs. As shown in Table 3, the recoveries and CVs of the above samples spiked with ETP are listed. The ETP recoveries were in the range of 86.9–95.9%. The CVs were less than 4.4%. According to the standards of the European Commission (2002),24 when the mass fractions are greater than 10 μg kg−1, the mean recovery should be in the range from 80% to 110%, and the intra- and inter-assay CVs should be no more than 15%. Therefore, the accuracy and precision are acceptable. Furthermore, this ELISA method has good repeatability and reproducibility.
| Sample | Spiked ETP (μg kg−1) | Intra-assay | Inter-assay | ||||
|---|---|---|---|---|---|---|---|
| Mean ± SD (μg kg−1) | Recovery (%) | CV (%) | Mean ± SD (μg kg−1) | Recovery (%) | CV (%) | ||
| Muscle | 250 | 217.3 ± 9.6 | 86.9 | 4.4 | 222.3 ± 9.6 | 88.9 | 4.3 |
| 500 | 454.0 ± 14.1 | 90.8 | 3.1 | 459.0 ± 13.3 | 91.8 | 2.9 | |
| 1000 | 926.0 ± 21.3 | 92.6 | 2.3 | 947.0 ± 16.1 | 94.7 | 1.7 | |
| Liver | 750 | 663.8 ± 27.9 | 88.5 | 4.2 | 681.8 ± 20.5 | 90.9 | 3.0 |
| 1500 | 1384.5 ± 42.9 | 92.3 | 3.1 | 1407.0 ± 22.5 | 93.8 | 1.6 | |
| 3000 | 2826.0 ± 59.3 | 94.2 | 2.1 | 2877.0 ± 31.6 | 95.9 | 1.1 | |
3.6. Comparison of the ELISA and HPLC analysis
Eighteen naturally contaminated chicken samples were used for the final validation of this ELISA method. Among them, lower concentrations were found in 9 muscle samples than those of 9 liver samples. After simultaneously detecting the ETP concentration with both the proposed ic-ELISA and HPLC methods, the results measured with the ELISA correlated well with measurements from the instrumental method (shown in Fig. 4(a) and (b)). The correlation coefficient values were calculated as 0.997 and 0.996 for muscle and liver samples, respectively. It has been suggested that the present ic-ELISA method is reliable for the trace analysis of ETP residues in chicken muscle and liver.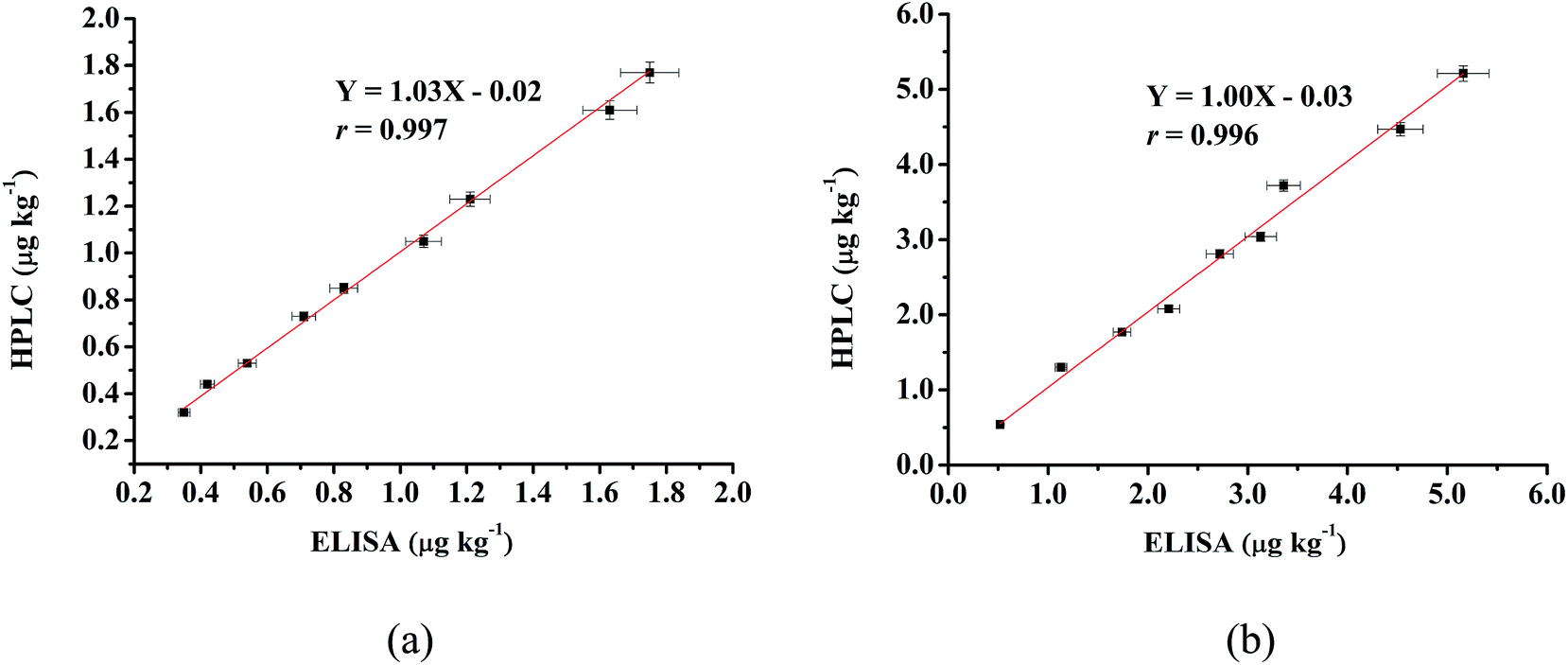 | ||
| Fig. 4 Correlation between the ELISA and HPLC results of naturally contaminated muscle (a) and liver (b) samples. | ||
3.7. Matrix effect and elimination
The matrix effect is one of the common challenges of an immunoassay in the residue analysis. ELISA methods often have a high potential for nonspecific binding between antibodies and non-target analytes and are prone to matrix effect. In the samples or sample extracts, structural analogs might affect the specific recognition of antibodies. The resulting matrix interference could reduce the analytical sensitivity and cause false positives by lowering color development. This occurs when either the enzyme activity is inhibited by the presence of inferences in the sample extracts, the interaction between the analyte and the antibody is hindered, or both of them has occurred concurrently in the immunoassay.31The matrix effects can be reduced by dilution of sample extract or special cleanup procedures using solid phase extraction or addition of heavy metal slats for precipitation of certain interferences. It is generally acknowledged that the advantages of the immunoassay are ease of use, flexibility, high throughput and low cost. Therefore, sample cleanup procedure should be kept as simple as possible. Dilution is a commonly used procedure to reduce the interferences from matrices, however, it also reduce the quantifiable sensitivity of the assay.32 After dilution, this established ELISA method still maintains enough sensitivity. If the dilution factor is too great, the dilution error will be found in the analysis. Dilution is not the only way for the elimination of matrix effects. Many sample preparation protocols, such as centrifugation, filtration and other pretreatments, can be used to incorporate the sample dilution for the removal of matrix interferences.32
4. Conclusions
The present study firstly described the generation and characterization of a specific monoclonal antibody against ETP. Based on this excellent antibody, a sensitive ic-ELISA method coupled with a simple procedure for sample preparation was developed. The described methodology showed excellent analytical parameters, with detection limits of 0.21 μg kg−1 and 0.34 μg kg−1 in chicken muscle and liver samples, respectively. The recoveries ranged from 85.4% to 98.4% with coefficients of variation less than 15%. It was found that this new method ensured high-throughput screening, reduced the time and solvent required in the sample pretreatment and met the requirements for ETP residue analysis. Both the prepared monoclonal antibody and the proposed ic-ELISA method have been shown to be reliable tools that can be applied for the measurement of ETP residues in edible animal tissues and products in surveillance programs.Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. U1504330), Young Academic Leaders Training Program of Henan University of Science and Technology, Program for Tackling Key Problems in Science and Technology of Henan Province, China (No. 162102310417), Key Science and Technology Program of Educational Commission of Henan Province, China (No. 14B550015), Program for Innovative Research Team in Science and Technology in Henan University of Science and Technology and Doctoral Research Initiation Grant from Henan University of Science and Technology (No. 13480030).References
- J. Chico, A. Rubies, F. Centrich, R. Companyo, M. D. Prat and M. Granados, Anal. Bioanal. Chem., 2013, 405, 4777–4786 CrossRef CAS PubMed.
- J. J. Nasr and S. Shalan, Luminescence, 2014, 29, 1188–1193 CrossRef CAS PubMed.
- Global MRL Database, http://vetdrugs.globalmrl.com/, accessed 2016.7.30.
- A. M. El-Kosasy, L. A. Hussein, N. Magdy and M. M. Abbas, Spectrochim. Acta, Part A, 2015, 150, 430–439 CrossRef CAS PubMed.
- R. L. Smallidge Jr, J.–Assoc. Off. Anal. Chem., 1978, 61, 561–563 CAS.
- R. H. M. M. Granja, A. M. Montes Nino, R. A. M. Zucchetti, R. E. Montes Nino and A. G. Salerno, J. AOAC Int., 2008, 91, 1483–1487 CAS.
- J. J. Nasr, S. Shalan and F. Belal, Food Anal. Methods, 2013, 6, 1522–1528 CrossRef.
- K. Pietruk, M. Olejnik, P. Jedziniak and T. Szprengier-Juszkiewicz, J. Pharm. Biomed. Anal., 2015, 112, 50–59 CrossRef CAS PubMed.
- L. Clarke, M. Moloney, J. O'Mahony, R. O'Kennedy and M. Danaher, Food Addit. Contam., Part A: Chem., Anal., Control, Exposure Risk Assess., 2013, 30, 958–969 CrossRef CAS PubMed.
- M. Cronly, P. Behan, B. Foley, E. Malone, S. Earley, M. Gallagher, P. Shearan and L. Regan, J. Pharm. Biomed. Anal., 2010, 53, 929–938 CrossRef CAS PubMed.
- J. L. Matus and J. O. Boison, Drug Test. Anal., 2016, 8, 465–476 CrossRef CAS PubMed.
- Z. Li, Z. Li, D. Li, H. Gao, X. Chen, L. Cao, Y. Hou and S. Li, Anal. Methods, 2015, 7, 9295–9303 RSC.
- Z. Li, C. Qin, D. Li, Y. Hou, S. Li and J. Sun, J. Pharm. Biomed. Anal., 2014, 98, 210–220 CrossRef CAS PubMed.
- V. Hormazabal and M. Yndestad, J. Liq. Chromatogr. Relat. Technol., 1996, 19, 2517–2525 CrossRef CAS.
- G. Degand, A. Bernes-Duyckaerts and G. Maghuin-Rogister, J. Agric. Food Chem., 1992, 40, 70–75 CrossRef CAS.
- F. Xu, W. Jiang, J. Zhou, K. Wen, Z. Wang, H. Jiang and S. Ding, J. Agric. Food Chem., 2014, 62, 3108–3113 CrossRef CAS PubMed.
- M. C. Zhang, Y. Wang, X. N. Yu, Y. R. Hu and S. H. Liu, Food Agric. Immunol., 2014, 25, 229–242 CrossRef CAS.
- Decree No. 588 of the State Council of the People's Republic of China, 2011.
- Professional Standard No. SN/T 3986-2014 of the General Administration of quality supervision, inspection and Quarantine of the people's Republic of China, 2015.
- G. Kohler and C. Milstein, Nature, 1975, 256, 495–497 CrossRef CAS PubMed.
- F. A. Esteve-Turrillas, J. Parra, A. Abad-Fuentes, C. Agullo, A. Abad-Somovilla and J. V. Mercader, Anal. Chim. Acta, 2010, 682, 93–103 CrossRef CAS PubMed.
- J. V. Mercader, J. Parra, F. A. Esteve-Turrillas, C. Agullo, A. Abad-Somovilla and A. Abad-Fuentes, Food Control, 2012, 26, 162–168 CrossRef CAS.
- K. Hu, X. Huang, Y. Jiang, W. Fang and X. Yang, Aquaculture, 2010, 310, 8–12 CrossRef CAS.
- European Union, Off. J. Eur. Communities: Legis., 2002, 221, 8–9 Search PubMed.
- European Commission, Guideline for the validation of screening methods (initial validation and transfer), 2010, pp. 1–18 Search PubMed.
- M. Franek, I. Diblikova, I. Cernoch, M. Vass and K. Hruska, Anal. Chem., 2006, 78, 1559–1567 CrossRef CAS PubMed.
- M. H. Goodrow and B. D. Hammock, Anal. Chim. Acta, 1998, 376, 83–92 CrossRef CAS.
- E. Ceballos-Alcantarilla, C. Agullo, A. Abad-Fuentes, A. Abad-Somovilla and J. V. Mercader, RSC Adv., 2015, 5, 51337–51341 RSC.
- D. L. Brandon and R. G. Binder, Food Agric. Immunol., 2006, 17, 53–61 CrossRef CAS.
- K. V. Singh, J. Kaur, G. C. Varshney, M. Raje and C. R. Suri, Bioconjugate Chem., 2004, 15, 168–173 CrossRef CAS PubMed.
- N. A. Lee, S. Wang, R. D. Allan and I. R. Kennedy, J. Agric. Food Chem., 2004, 52, 2746–2755 CrossRef CAS PubMed.
- Y. Song, Y. Ge, Y. Zhang, B. Liu, Y. Lu, T. Dong and S. Wang, Anal. Bioanal. Chem., 2009, 393, 2001–2008 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra20736g |
| This journal is © The Royal Society of Chemistry 2017 |