
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced sol–gel synthesis of polymer-supported silsesquioxane membranes†
Hiroki Nagasawa ,
Mai Nishibayashi,
Masakoto Kanezashi,
Tomohisa Yoshioka and
Toshinori Tsuru*
,
Mai Nishibayashi,
Masakoto Kanezashi,
Tomohisa Yoshioka and
Toshinori Tsuru*
Department of Chemical Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashihiroshima, Hiroshima 739-0047, Japan. E-mail: tsuru@hiroshima-u.ac.jp
First published on 10th January 2017
Abstract
A novel approach to the preparation of polymer-supported silsesquioxane membranes is developed via photo-induced sol–gel processing. An organically bridged silsesquioxane thin layer by coating a silsesquioxane sol with a photo-acid generator onto a polymeric SPES/PSF composite nanofiltration membrane followed by irradiation with UV at room temperature. This polymer-supported silsesquioxane membrane derived from 1,2-bis(trimethoxysilyl)ethane displays selective water permeation with a water permeance of 3.0 × 10−6 mol m−2 s−1 Pa−1, and shows a separation factor of 99 in the pervaporation of a 90 wt% isopropanol aqueous solution.
Introduction
Silsesquioxane is an organosilicon compound with a chemical formula [RSiO3/2]n where Si is the element silicon, O is oxygen, and R is either hydrogen or an organic group. Recent reports have shown that organically bridged silsesquioxane is an ideal candidate for the fabrication of microporous membranes with well-controlled properties.1,2 Organically bridged silsesquioxane membranes are mostly prepared via a sol–gel technique that uses bis-silyl-type alkoxides as a precursor.3 In particular, the membranes prepared using 1,2-bis(triethoxysilyl)ethane (BTESE) have been extensively studied over the past few years because the silsesquioxane networks with a C2H4-bridged structure have micropores with the size of 0.5–0.7 nm,4,5 which is suitable for applications in gas separation,6,7 pervaporation,8,9 and reverse osmosis.10In fabricating such silsesquioxane membranes, a composite structure consisting of a separation active layer on a porous support is preferred because it provides good mechanical strength and can greatly reduce the thickness of the separation active layer. Porous ceramic such as α-alumina is the most common material used for such porous supports.1–10 In contrast, little work has been performed on the fabrication of silsesquioxane membranes using polymeric supports, although the use of polymeric supports instead of ceramic ones may offer great potential for cost-savings and large-scale manufacturing. Development of polymer-supported silsesquioxane membranes has long been a challenging task, because the firing step, which is essential in a sol–gel process, is usually done at 200–400 °C. Recently, we developed a new type of layered-hybrid membrane consisting of a BTESE-derived layer on a polymeric support by lowering the firing temperature in order to avoid thermal degradation of the polymeric supports.11–13 The BTESE-derived membrane prepared onto a polymeric nanofiltration membrane at a firing temperature of 120 °C showed a stable water flux of 2.3 kg m−2 h−1 and a separation factor of 2500 for the vapor permeation dehydration of 90 wt% isopropanol–water mixtures at 105 °C.12 However, it is a paradoxical fact that a separation factor tends to decrease with decreasing firing temperature due to the reduced formation of siloxane bonding via thermal condensation of the silanol groups.14 Therefore, achieving higher cross-linking at lower temperature is a key issue for the development of polymer-supported silsesquioxane membranes.
One option to reduce the membrane fabrication temperature is to use a photochemical method such as photo-induced sol–gel processing.15,16 Previously, we proposed photo-induced cationic polymerization for the fabrication of ceramic-supported silsesquioxane membranes.17 To obtain a cross-linked structure at low temperature without firing, we coated silsesquioxane sols onto porous ceramic supports together with a photo-acid generator (PAG), and then irradiated the dried gel with UV light to generate a photo-acid that would induce further polymerization in the coated layer via acid-catalyzed reactions.17 The obtained membrane showed a high separation factor of 525 and a water flux of 3.12 kg m−2 h−1 during the pervaporation of 90 wt% isopropanol–water mixtures at 40 °C.17 As an extension of this achievement, in this work, we applied a photo-induced sol–gel process for fabricating polymer-supported silsesquioxane membranes. The membranes were fabricated by coating PAG-containing sols directly onto polymeric nanofiltration membranes followed by irradiation with UV light. The effect of photochemical processing on the pervaporation performance of the membranes was investigated. To avoid any damage of polymeric supports caused by UV irradiation, we determined the optimal light-to-sample distance for photochemical cross-linking. Furthermore, we examined the effect that plasma-pretreatment of the supports exerted on the pervaporation performance.
Experimental
Sol preparation
Ethylene-bridged bis(alkoxides), 1,2-bis(trimethoxysilyl)ethane (BTMSE) and 1,2-bis(triethoxysilyl)ethane (BTESE), were selected as precursor alkoxides. Polymeric silsesquioxane sols were prepared via acid-catalyzed hydrolysis and polycondensation reactions at 50 °C with water and acetic acid (AcOH) in 2-propanol (IPA). Details of the procedure for sol preparation are described in a previous report.17 The concentration of the sols was 1.0 wt%, and the molar ratios of BTMSE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:AcOH and BTESE:H2O:AcOH for sol preparation were 1:3:0.3 and 1:60:0.3, respectively. The reaction times for BTMSE- and BTESE-derived sols were 1 and 11 h, respectively. After cooling the sols to room temperature, (4-methylphenyl)[4-(2-methylpropyl)phenyl]-iodonium hexafluorophosphate (Irgacure 250, 75% solution in propylene carbonate) was added as a PAG with a molar ratio of BTMSE, or BTESE, to PAG of 1:0.012.
:H2O:AcOH and BTESE:H2O:AcOH for sol preparation were 1:3:0.3 and 1:60:0.3, respectively. The reaction times for BTMSE- and BTESE-derived sols were 1 and 11 h, respectively. After cooling the sols to room temperature, (4-methylphenyl)[4-(2-methylpropyl)phenyl]-iodonium hexafluorophosphate (Irgacure 250, 75% solution in propylene carbonate) was added as a PAG with a molar ratio of BTMSE, or BTESE, to PAG of 1:0.012.
Membrane preparation
The scheme of the membrane fabrication and the reactions in the photo-induced polymerization are described in Fig. 1. A polymeric nanofiltration membrane consisting of an ultrathin sulfonated polyethersulfone (SPES) skin layer on a porous polysulfone (PSF) layer, NTR 7450, was kindly supplied by Nitto Denko, Inc., Japan, and was used as a support for the silsesquioxane membrane preparation. The silsesquioxane sols were coated onto the polymeric supports using a drop-coating method. In the coating procedure, 5 ml of sol was poured onto a support (size: 40 mm × 75 mm) and was allowed to penetrate into the pores of the support surface. The excess solution was drained after 1 min. The supports coated with silsesquioxane sols were dried at room temperature, and were finally irradiated with UV light using a metal-halide lamp (250 W, 250–450 nm) in a quartz cell under a N2 flow. The UV irradiation times were 15 s and 30 s for BTMSE- and BTESE-derived membranes, respectively. The distance from the UV-light source to the samples was set at 200 mm. The UV irradiation conditions described above were decided according to the results of photo-induced reaction analysis of silsesquioxane films and UV tolerance testing of the polymeric supports.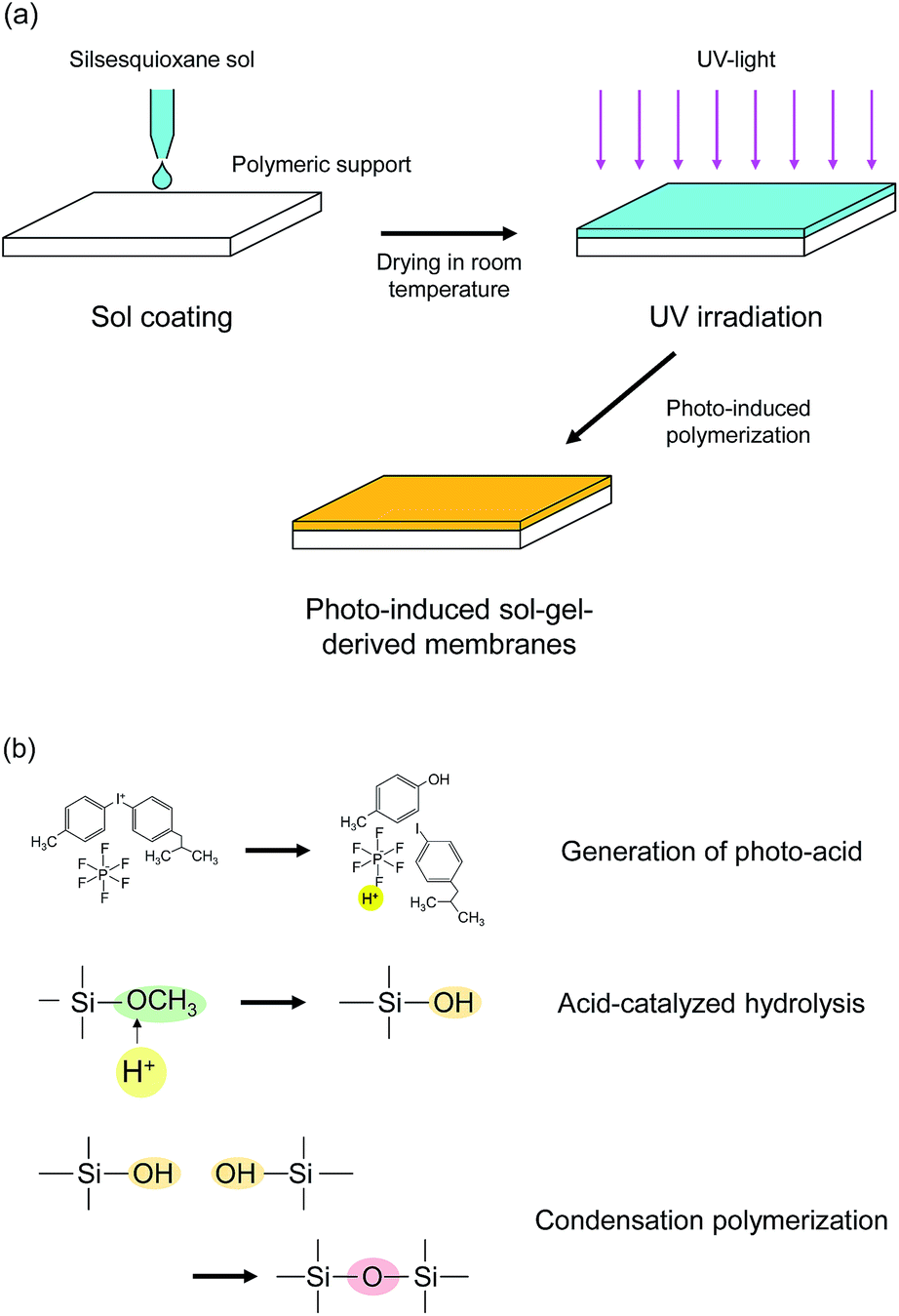 | ||
| Fig. 1 Schematic diagram of (a) membrane preparation procedure and (b) photo-induced polymerization reactions. | ||
Furthermore, a pretreatment of polymeric supports using water vapor plasma was applied to enhance the adhesion between the silsesquioxane layer and the NTR 7450 support. The plasma pretreatment was conducted in a capacitively coupled radio frequency plasma reactor (BPD-1H, Samco, Inc., Japan). A hydrophilic surface was obtained using plasma of a mixture of H2O vapor and Ar. The flow rates of H2O vapor and Ar were both 10 ml min−1. The pressure and RF power input for generating plasma discharge were 15 Pa and 10 W, respectively. To avoid heat damage during the treatment, the temperature of the polymeric supports was maintained at 0 °C. The plasma-treated supports were coated with silsesquioxane sols and irradiated with UV using the same procedure as with the untreated ones.
Characterization
The morphology of the polymer-supported silsesquioxane membranes was examined using field-emission scanning electron microscopy (FE-SEM, S-4800, Hitachi, Japan). The chemical structure of the membranes was evaluated by Fourier transform infrared spectroscopy (FTIR, FT/IR-4100, Jasco, Japan).The progress of the photo-induced reactions was monitored via measurement of the pencil hardness and the FTIR spectra of the silsesquioxane films. The silsesquioxane sols were coated onto silicon wafers or KBr plates via spin-coating, and were irradiated with UV.
The UV tolerance of the polymeric supports was examined in order to determine the optimal UV irradiation conditions for membrane preparation without any damage to the polymeric supports caused by the UV light. The polymeric supports were irradiated with UV at light–substrate distances of 50, 120, and 200 mm. The FTIR spectra of the supports before and after UV irradiation were measured to evaluate the UV tolerance. The changes in the surface morphology were observed by SEM.
The contact angles of the supports were measured to study the effect that plasma pretreatment had on the wettability of the support surface.
Pervaporation measurement
Pervaporation dehydration performance of the membranes was evaluated using a typical experimental apparatus, as shown in Fig. 2. The membrane module had an effective area of 340 mm2 and was placed in a thermostatic chamber at 40 °C. A mixture of 90 wt% IPA and 10 wt% water was circulated through the permeation module using a peristaltic pump. The composition of the feed solution was confirmed by gas chromatography (GC-14B, Shimadzu, Japan) and adjusted to 90 wt% by adding a small amount of water and/or IPA during the test period. The permeate side of the membrane was evacuated at low pressure (<100 Pa), and the permeate vapor was collected with a liquid nitrogen trap to obtain the permeation flux. The composition of the permeate was determined by gas chromatography. The separation factor of the water to IPA, α, was defined as follows.where xwater, xIPA, ywater, and yIPA denote the molar fractions of water and IPA in the feed and the permeate, respectively. The permeance of the i-th component, Pi, was obtained using the following equation.
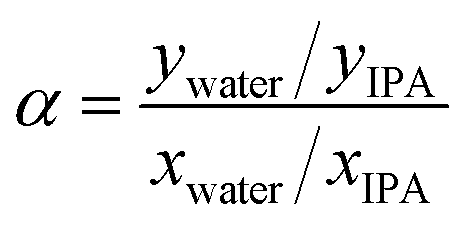
| Pi = Ji/(p1,i − p2,i) |
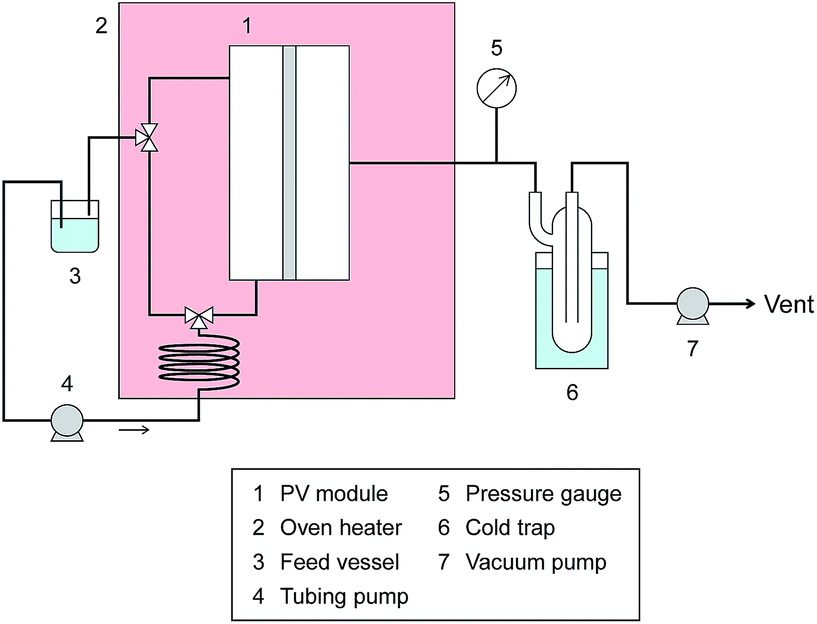 | ||
| Fig. 2 Schematic diagram of the PV equipment. | ||
Results and discussion
Analysis of photo-induced sol–gel reactions
The pencil hardness of the silsesquioxane thin films prepared from BTMSE-derived sols with/without PAG was measured after exposure to UV light for different irradiation times. The films were irradiated using UV light with a distance of the UV-light source to the sample (light-to-sample distance) of 50 mm. As shown in Fig. 3, the hardness of the films with and without PAG was similar before UV irradiation, showing a pencil hardness of lower than 6B. The pencil hardness of the films containing PAG was immediately increased to 2H after UV irradiation of 60 s or more, showing the formation of cross-linked networks, whereas that of the films without PAG remained unchanged. This result indicates that PAG is necessary to initiate the polymerization or cross-linking in BTMES-derived silsesquioxane thin films via UV irradiation.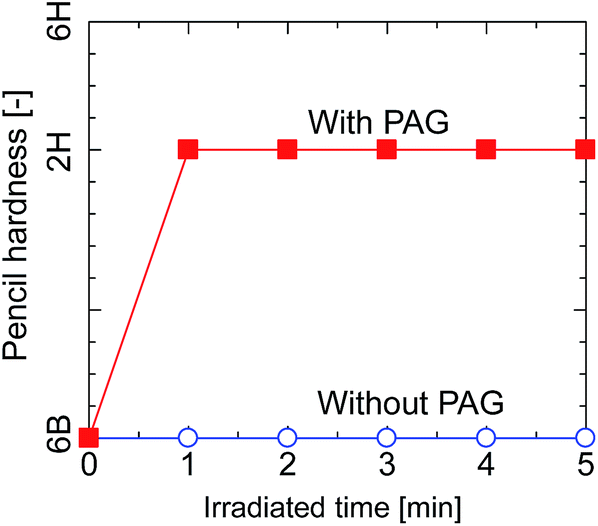 | ||
| Fig. 3 Pencil hardness of BTMSE-derived films with and without PAG as a function of the UV irradiation time. | ||
The FTIR spectra of silsesquioxane films were measured before and after exposure to UV light and are displayed in Fig. 4. The films were prepared using silsesquioxane sols containing PAG. The light-to-sample distance was fixed at 200 mm. The FTIR spectrum of the BTMSE-derived film without UV irradiation is shown in Fig. 4a. The strong band at 1100–1000 cm−1 was characteristic of Si–O–Si stretching, confirming the formation of siloxane bonds. The peaks at 1270 cm−1 were ascribed to the vibration of methylene groups in the bridging units.6 A characteristic peak of methoxy groups appears at 1150 cm−1.18 The peak at 2840 cm−1 is also characteristic of methoxy groups and was ascribed to stretching of the C–H bonds.18 The broad band at 900 cm−1 was ascribed to Si–O stretching in the silanol groups.19 The broad band at 3300 cm−1, the peaks at 3000–2880 cm−1, and the peaks at 820–750 cm−1 were attributed to O–H, C–H, and Si–C structures, respectively.6,20 As shown in Fig. 4b, after exposure to UV light for 120 s, a decline in intensity was observed for the peaks characteristic of methoxy groups at 2840 and 1190 cm−1 after exposure to UV light. Simultaneously, the intensity of the silanol groups at 3300 and 900 cm−1 showed a significant increase after UV irradiation. This indicates the hydrolysis of the methoxy groups during the UV irradiation.
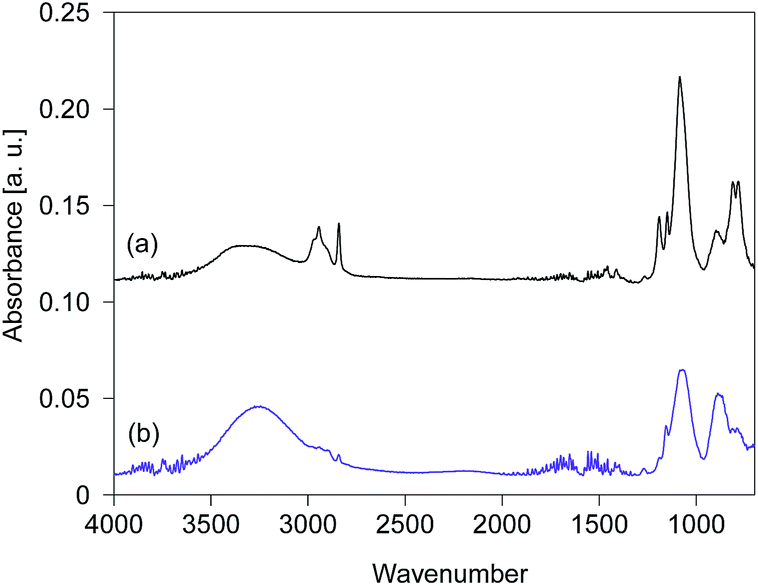 | ||
| Fig. 4 FTIR spectra of BTMSE-derived films with PAG (a) before and (b) after UV irradiation for 120 s. The light-to-sample distance was 200 mm. | ||
The time course changes in the peak areas of methoxy groups at 2840 cm−1 and silanol groups at 900 cm−1 during UV irradiation were measured and are shown in Fig. 5. The peak areas were normalized by the maximum values. The peak areas of the methoxy groups decreased very fast in the first 15 s and approached a steady value, while simultaneously the peak areas of the silanol groups increased markedly, suggesting that the photo-acid catalyzed hydrolysis of methoxy groups was completed within a very short period of UV irradiation.17 After hydrolysis of the methoxy groups, a subsequent condensation of silanol groups occurred quickly to form the cross-linked structure, as confirmed by the pencil hardness measurement. These results indicate that the photo-induced sol–gel processing initiated by the generation of photo-acid can be used to prepare silsesquioxane thin films with hard and crosslinked structures within a very short period of UV irradiation without elevating the processing temperature.
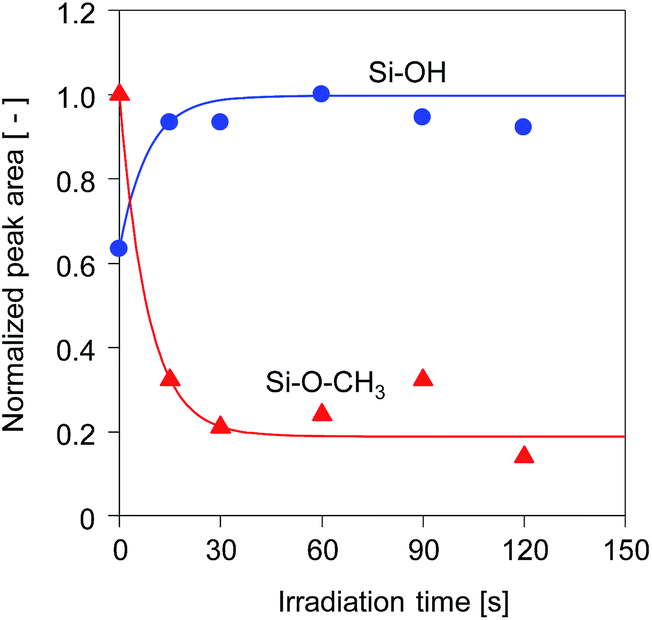 | ||
| Fig. 5 Time course change in the peak areas of methoxy groups at 2840 cm−1 and OH groups at 900 cm−1 for BTMSE-derived films with PAG. The light-to-sample distance was 200 mm. | ||
The stability of SPES/PSF membranes under UV irradiation
In terms of membrane preparation, a higher degree of UV exposure is preferable because it gives a higher degree of reaction in the silsesquioxane layer. On the other hand, it is also important to consider the stability of a polymeric support during exposure to UV irradiation. The effect of UV irradiation on the morphology of the SPES/PSF nanofiltration membrane was investigated by cross-sectional SEM observation and is displayed in Fig. 6. The sample before exposure to UV light had a composite structure, which consisted of a smooth and thin SPES top layer on the porous PFS support. The sample was then irradiated with UV light for 20 min at a distance of 50 mm from the UV-light source. The surface of the sample was obviously damaged after the exposure to UV. The surface became rough, and some visible defects were apparent. | ||
| Fig. 6 Cross-sectional images of a SPES/PSF support (a) before and (b) after UV-irradiation. The distance from the UV-light source to the sample was 50 mm, and the irradiation time was 20 min. | ||
ATR-FTIR spectroscopy was employed to investigate the change in the chemical structure of the SPES/PSF nanofiltration membrane after exposure to UV light. The FTIR absorption spectrum of the sample without UV irradiation is shown in Fig. 7a. The spectrum agreed well with those previously reported for SPES and SPES/PFS membranes.21,22 The peaks at 1584 and 1487 cm−1 were ascribed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of the aromatic ring. The aryl–O–aryl stretching of the aromatic ether appeared at 1241 cm−1. The doublet peaks at 1323 and 1295 cm−1 represent the asymmetric stretching of OSO and the peak at 1151 cm−1 represents the symmetric stretching of OSO. The peak at 1503 cm−1 was ascribed to the stretching mode of the C–C bonds in PSF. The absorption bands of aromatic SO3 groups are characteristic of the topmost SPES layer, but these could not be identified due to extremely thin SPES layer thickness. Fig. 7b shows the spectrum of the sample after UV irradiation for 5 min at a distance of 50 mm from the UV-light source. After exposure to UV light, the absorption peaks, which did not exist in the unirradiated one, appeared at 3200 and 1720 cm−1. The peak at 3200 cm−1 was ascribed to the stretching vibration of hydroxyl groups. The peak at 1720 cm−1 was assigned to the carbonyl groups. The formation of hydroxyl and carbonyl groups was attributed to the UV light-induced degradation of SPES and/or PSF layer via cleavage of the C–S and C–O bonds.23
C stretching vibration of the aromatic ring. The aryl–O–aryl stretching of the aromatic ether appeared at 1241 cm−1. The doublet peaks at 1323 and 1295 cm−1 represent the asymmetric stretching of OSO and the peak at 1151 cm−1 represents the symmetric stretching of OSO. The peak at 1503 cm−1 was ascribed to the stretching mode of the C–C bonds in PSF. The absorption bands of aromatic SO3 groups are characteristic of the topmost SPES layer, but these could not be identified due to extremely thin SPES layer thickness. Fig. 7b shows the spectrum of the sample after UV irradiation for 5 min at a distance of 50 mm from the UV-light source. After exposure to UV light, the absorption peaks, which did not exist in the unirradiated one, appeared at 3200 and 1720 cm−1. The peak at 3200 cm−1 was ascribed to the stretching vibration of hydroxyl groups. The peak at 1720 cm−1 was assigned to the carbonyl groups. The formation of hydroxyl and carbonyl groups was attributed to the UV light-induced degradation of SPES and/or PSF layer via cleavage of the C–S and C–O bonds.23
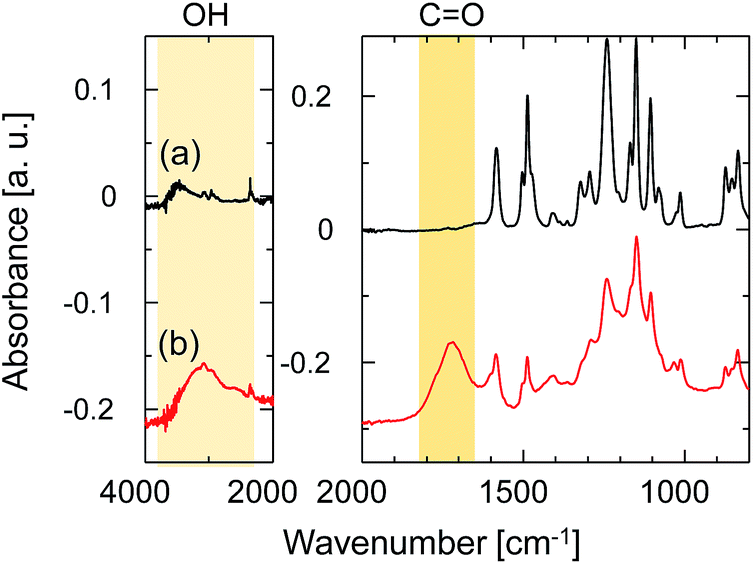 | ||
| Fig. 7 ATR-FTIR spectra of the SPES/PSF support (a) before and (b) after UV-irradiation. The distance from the UV-light source to the sample was 50 mm, and the irradiation time was 20 min. | ||
The ratio of the FTIR absorbance areas of the carbonyl groups at 1720 cm−1 to the CC stretching of the aromatic ring at 1584 cm−1 was plotted as a function of the UV irradiation time in Fig. 8 to determine how the degradation of the membrane had advanced. The absorbance areas for the carbonyl groups and the CC stretching of the aromatic ring represent the amount of degradation products and the original structure of the SPES/PSF membrane at the surface, respectively. Therefore, the area ratio of these two peaks corresponded to the degree of UV light-induced degradation at the surface of the membrane. The light-to-sample distance ranged from 50 to 200 mm. The peak area ratio tended to increase linearly with increases in the irradiation time in all cases. The peak area ratio of the sample exposed to UV at a light-to-sample distance of 50 mm was increased immediately after UV irradiation, which indicated a rapid degradation of the SPES and PSF structures. On the other hand, at a longer light-to-sample distance, the absorption peaks of the product of UV light-induced degradation could not be identified at the initial stage, because the UV light intensity was decreased with distance from the light source to the samples. The peak area ratios of the samples exposed to UV at light-to-sample distances of 120 and 200 mm started to increase after a lag period of 30 and 60 s, respectively. The cross-sectional SEM images of the SPES/PSF support before and after UV irradiation for 30 s with a light-to-sample distance of 200 mm suggested no obvious structural change, as shown in Fig. 9, indicating no degradation in this case.
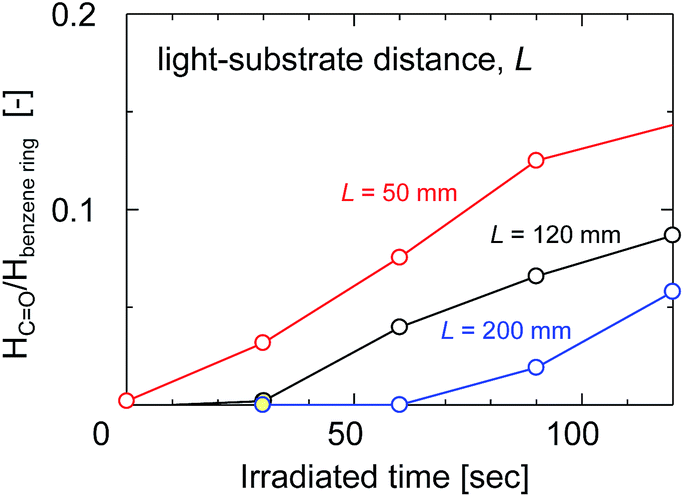 | ||
| Fig. 8 The absorbance peak area ratio of the carbonyl groups at 1750 cm−1 to the aromatic C–H in-plane bend at 1100 cm−1 as a function of irradiation time with different light-to-sample distances. | ||
 | ||
| Fig. 9 Cross-sectional images of the SPES/PSF support (a) before and (b) after UV-irradiation. The light-to-sample distance and the irradiation time were 200 mm and 30 s, respectively. | ||
Pervaporation performance of polymer-supported silsesquioxane membranes
Silsesquioxane membranes were prepared onto SPES/PSF supports via UV-induced polymerization. To prevent damage to the SPES/PSF supports during UV irradiation, the light-to-sample distance for the preparation of BTMSE-derived membranes was set at 200 mm, and the UV irradiation time was limited to 15 s. Fig. 10 shows the time course for water permeance and the separation factor for a polymer-supported BTMSE-derived membrane and SPES/PSF support for the pervaporation of a 90 wt% IPA/water mixture at 40 °C. The BTMSE-derived membrane showed a water permeance of 2.9 × 10−6 mol m−2 s−1 Pa−1, which was similar to that of 2.4 × 10−6 mol m−2 s−1 Pa−1 for the SPES/PSF support. The separation factor was 6.6, which was only slightly higher than the separation factor of 2.3 for the SPES/PSF support. In our previous work, the ceramic-supported BTMSE-derived membrane prepared with the same photo-induced sol–gel process showed a separation factor of several hundreds.17 Therefore, if the BTMSE-derived layer was formed properly, the polymer-supported BTMSE-derived membrane should have had a similar separation performance.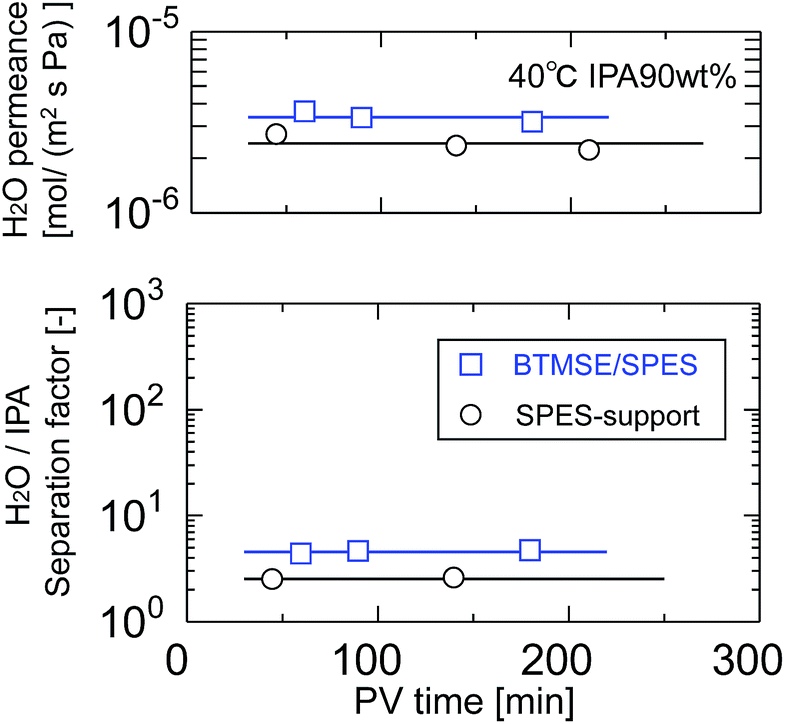 | ||
| Fig. 10 Time course of H2O permeance and the separation factor of a BTMSE-derived photo-induced sol–gel polymer-supported membrane and a SPES/PSF support. | ||
To confirm the formation of a BTMSE-derived silsesquioxane layer on the SPES/PSF support, the ATR-FTIR spectra were measured for the membrane before and after pervaporation measurement, as shown in Fig. 11. For the membrane before pervaporation measurement, an absorbance band corresponding to the stretching vibration of Si–O–Si was observed at 1000–1100 cm−1, together with the peaks that originated from the SPES/PSF support, which indicated that the BTMSE-derived layer had been formed on the SPES surface. However, after pervaporation measurement, the band corresponding to Si–O–Si disappeared. This indicated that the BTMSE-derived layer had been peeled off from the SPES surface during the pervaporation measurement. This result strongly suggested that the adhesion between the BTMSE-derived layer and the SPES support was very low. The adhesion of ceramic coatings onto a polymer substrate, in general, is given by chemical bonds formed through the surface condensation reactions and/or the development of an interlocked structure at the interface through the diffusion of sol particles into the substrate pores.24 However, in photo-induced sol–gel processing, crosslinking within the pores to form an interlocked structure is difficult because the UV light intensity decreases rapidly with depth from the surface, even though sol solutions might have diffused into the substrate pores and formed a gel. Therefore, to enhance the adhesion, the formation of chemical bonds between the coated layer and the surface of the support is important. Chemical bonds at the interface can form via the condensation reaction between the BEMSE-derived gel layer and the SPES surface. However, it is difficult to form such chemical bonds because there is no reaction site such as OH groups on the surface of the original structure of the SPES.
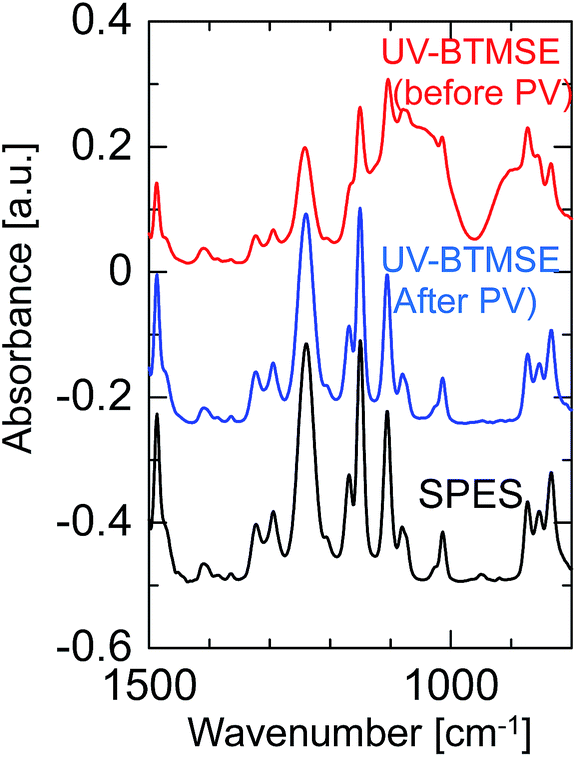 | ||
| Fig. 11 FTIR spectra of a BTMSE-derived photo-induced sol–gel polymer-supported membrane before and after PV operation. | ||
To overcome this adhesion problem, we employed a water-vapor-plasma pretreatment for the NTR support. Water-vapor plasma can form OH groups on the surface of the substrate, that can subsequently react with the OH-groups of the silsesquioxane layer to form a chemically bonded adhesive interface.25 We previously reported a plasma-assisted multi-layer coating for the preparation of silsesquioxane membranes onto the hydrophobic nanoporous intermediate layers, and reported that the gas selectivity of the membranes can be improved by better adhesion between each layer.26 Fig. 12 shows the contact angle of the SPES surface as a function of water-vapor-plasma treatment time. After treatment with the water-vapor plasma, the contact angle decreased from 30° to 9°. The decreased contact angle indicates that the surface of SPES was hydrophilized by reacting with excited water molecules and hydroxyl radicals to form a hydroxylated surface. The contact angle was constant even when the treatment time was extended, suggesting that the surface of the SPES was immediately modified by the water-vapor plasma.
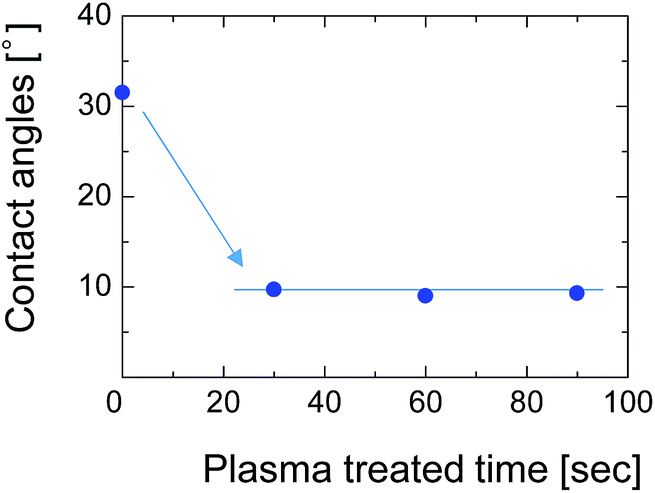 | ||
| Fig. 12 Contact angle of the SPES/PSF support treated with water-vapor plasma. | ||
After pretreatment with water-vapor-plasma, a BTMSE-derived layer was formed on the plasma-pretreated support. Fig. 13 shows the cross-sectional SEM images of the BTMES-derived membrane prepared onto the plasma-pretreated support with the light-to-sample distance of 200 mm and the UV irradiation time of 15 s. The BTMES-derived silsesquioxane layer with a thickness of approximately 200 nm was formed on the SPES/PSF support. Fig. 14 shows the pervaporation performance of BTMSE-derived membranes prepared onto a support with and without water-vapor plasma pretreatment. The membrane formed onto the pretreated support showed a separation factor of 99, which was significantly higher compared with that of the membrane formed onto an untreated support. It should be also noted that the membrane showed a stable performance during the pervaporation measurement. This result clearly demonstrated the effectiveness of water-vapor-plasma pretreatment in enhancing the adhesion between the silsesquioxane layer and the SPES skin layer.
 | ||
| Fig. 13 Cross-sectional image of the BTMSE-derived silsesquioxane membrane prepared on the plasma-pretreated SPES/PSF support. The light-to-sample distance and the irradiation time were 200 mm and 15 s, respectively. | ||
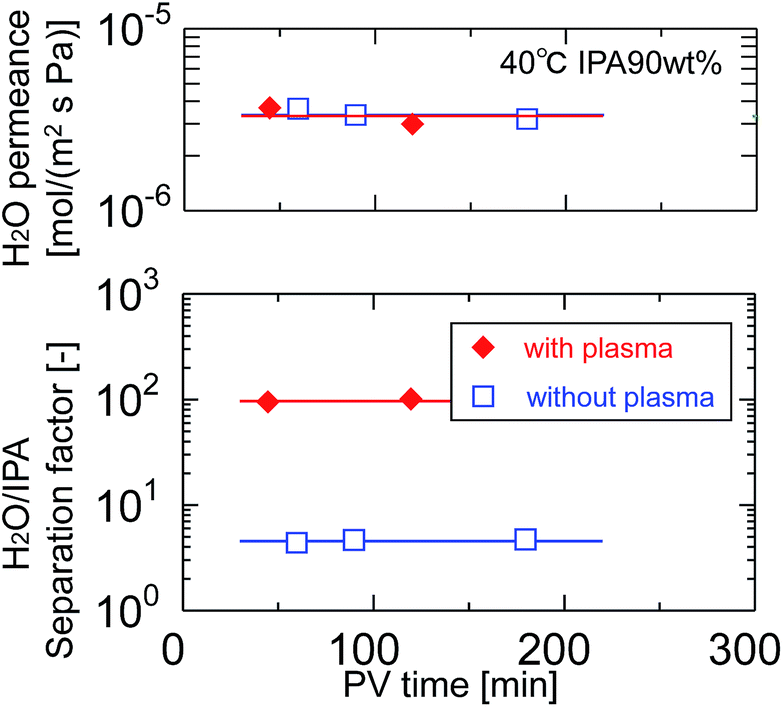 | ||
| Fig. 14 Time course of H2O permeance and separation factor of BTMSE-derived photo-induced sol–gel polymer-supported membranes prepared onto supports with and without water-vapor plasma pretreatment. | ||
We also prepared polymer-supported silsesquioxane membranes with BTESE as a silicon precursor by following the same procedure used for the preparation of BTMSE-derived membranes. The light-to-sample distance and the UV irradiation time for the preparation of BTESE-derived membranes were set at 200 mm and 30 s, respectively. The required UV irradiation time was longer than that for BTMSE-derived membranes due to the lower reactivity of ethoxy groups compared with methoxy groups (ESI-1†). As presented in Table 1, the polymer-supported silsesquioxane membranes with permselectivity in pervaporation can also be prepared by using different precursors via photo-induced sol–gel processing.
| Membrane | Precursor | Pretreatment | H2O permeance [10−6 mol m−2 s−1 Pa−1] | Separation factor [—] |
|---|---|---|---|---|
| SPES/PSF | — | — | 2.3 | 2.3 |
| BTMSE/SPES | BTMSE | — | 2.9 | 6.6 |
| BTMSE/plasma–SPES | BTMSE | Water-vapor-plasma | 3.0 | 99 |
| BTESE/plasma–SPES | BTESE | Water-vapor-plasma | 2.6 | 46 |
The trade-offs between H2O permeance and the separation factor in the pervaporation of the 90 wt% IPA/water mixture for photo-induced sol–gel-derived C2H4-bridged silsesquioxane membranes are displayed in Fig. 15. The membrane formed onto a support with a water-vapor-plasma pretreatment exhibited a higher separation factor compared with those formed onto an untreated support, indicating that pretreatment is a useful strategy for the formation of silsesquioxane membranes on polymeric supports. Photo-induced sol–gel-derived polymer-supported membranes showed only moderate selectivity compared with ceramic-supported photo-induced sol–gel-derived membranes15 probably due to the difference in the UV irradiation time in membrane preparation. Total UV irradiation time for the preparation of a ceramic-supported membrane was 12 min, while that for the polymer-supported membrane was limited to 15 s in order to minimize the damage of a polymeric support. Therefore, the silsesquioxane networks formed on the polymeric supports might have had a looser structure than that of the network structure formed on the ceramic supports. It is noteworthy that, although the separation factor was improved by the silsesquioxane layer formation, the water permeance was unchanged compared with that for SPES/PSF supports. It is surprising that a selective top layer with no or a quite small resistance was obtained. The high level of H2O permeance separation factor can be ascribed to the hydrophilic nature of the photo-induced sol–gel-derived silsesquioxane layers, which has a large number of OH-groups. Although further studies will be needed to promote the separation performance of polymer-supported silsesquioxane membranes prepared via photo induced sol–gel processing, we have successfully demonstrated here a novel strategy for preparing ceramic membranes such as silsesquioxane membranes at low-temperature within extremely sort processing time onto polymeric supports via photo-induced sol–gel processing.
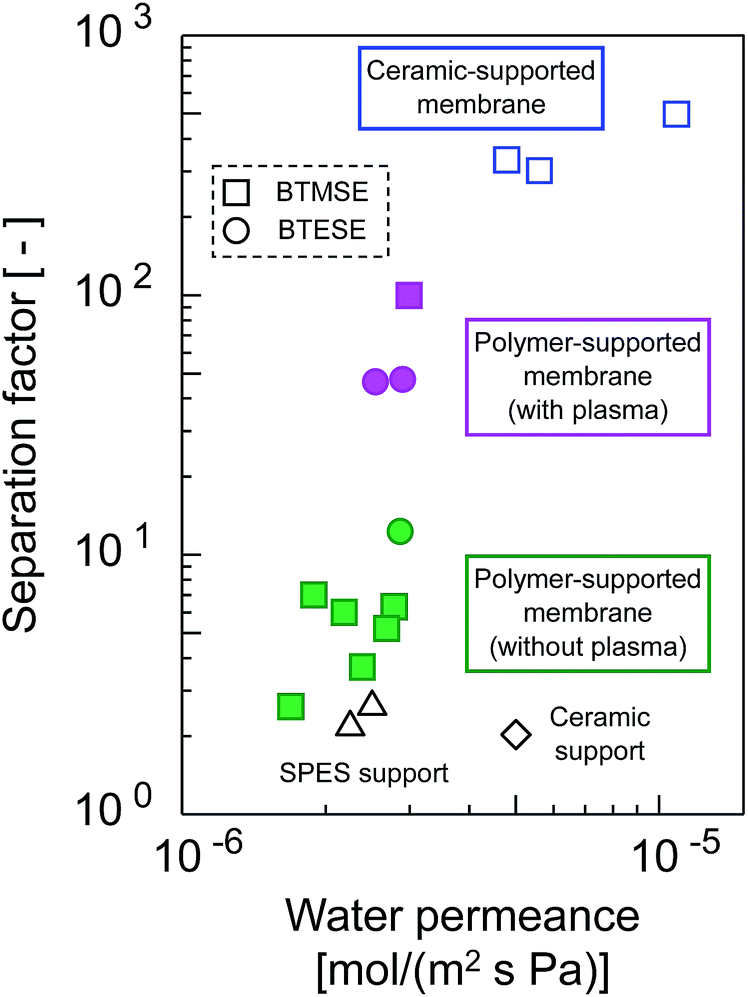 | ||
| Fig. 15 Trade-offs of H2O permeance and the separation factor for photo-induced sol–gel-derived C2H4-bridged silsesquioxane membranes. | ||
Conclusions
Photo-induced sol–gel processing was applied for the preparation of novel polymer-supported silsesquioxane membranes. An organically bridged silsesquioxane thin layer was formed onto a polymeric SPES/PSF composite nanofiltration membrane by coating a silsesquioxane sol with a photo-acid generator followed by UV irradiation at room temperature. The hydrolysis of alkoxy groups in the films catalyzed by UV-generated acids and the subsequent condensation of silanols resulted in the formation of siloxane networks. To avoid photodegradation of the polymeric support surface, the effects of UV-irradiation time and light-to sample distance on the polymeric supports were investigated via FTIR and SEM. The adhesion of the silsesquioxane layer to the polymeric support was low when the support surface was hydrophobic, but it was improved when the support surface was pretreated by water-vapor plasma. The plasma pretreatment was effective for the preparation of a stable silsesquioxane layer that withstood the pervaporation test. The separation properties were investigated by pervaporation of a 90 wt% IPA/water mixture at 40 °C. The polymer-supported silsesquioxane membrane derived from 1,2-bis(trimethoxysilyl)ethane displayed a water flux of 3.0 × 10−6 mol m−2 s−1 Pa−1 and a separation factor of 99.References
- H. L. Castricum, A. Sahm, R. Kreiter, D. H. A. Blank, J. F. Vente and J. E. Ten Elshof, J. Mater. Chem., 2008, 18, 2150–2158 RSC.
- M. Kanezashi, K. Yada, T. Yoshioka and T. Tsuru, J. Am. Chem. Soc., 2009, 131, 414–415 CrossRef CAS PubMed.
- H. L. Castricum, G. G. Paradis, M. C. Mittelmeijer-Hazeleger, R. Kreiter, J. F. Vente and J. E. ten Elshof, Adv. Funct. Mater., 2011, 21, 2319–2323 CrossRef CAS.
- T. Niimi, H. Nagasawa, M. Kanezashi, T. Yoshioka, K. Ito and T. Tsuru, J. Membr. Sci., 2014, 455, 375–383 CrossRef CAS.
- H. Nagasawa, T. Niimi, M. Kanezashi, T. Yoshioka and T. Tsuru, AIChE J., 2014, 60, 4199–4210 CrossRef CAS.
- H. Qi, J. Han and N. P. Xu, J. Membr. Sci., 2011, 382, 231–237 CrossRef CAS.
- H. L. Castricum, H. F. Qureshi, A. Nijmeijer and L. Winnubst, J. Membr. Sci., 2015, 488, 121–128 CrossRef CAS.
- T. Tsuru, T. Shibata, M. Kanezashi and T. Yoshioka, J. Membr. Sci., 2012, 421–422, 25–31 CrossRef CAS.
- H. F. Qureshi, A. Nijmeijer and L. Winnubst, J. Membr. Sci., 2013, 446, 19–25 CrossRef CAS.
- R. Xu, J. Wang, M. Kanezashi, T. Yoshioka and T. Tsuru, Langmuir, 2011, 27, 13996–13999 CrossRef CAS PubMed.
- G. Gong, J. Wang, H. Nagasawa, M. Kanezashi, T. Yoshioka and T. Tsuru, J. Membr. Sci., 2014, 464, 140–148 CrossRef CAS.
- G. Gong, J. Wang, H. Nagasawa, M. Kanezashi, T. Yoshioka and T. Tsuru, J. Membr. Sci., 2014, 472, 19–28 CrossRef CAS.
- G. Gong, H. Nagasawa, M. Kanezashi and T. Tsuru, ACS Appl. Mater. Interfaces, 2016, 8, 1106–11069 Search PubMed.
- J. Wang, M. Kanezashi, T. Yoshioka and T. Tsuru, J. Membr. Sci., 2012, 415–416, 810–815 CrossRef CAS.
- C. Belon, A. Chemtob, C. Croutxe-Barghorn, S. Rigolet, V. Le Houerou and C. Gauthier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4150–4158 CrossRef CAS.
- A. Chemtob, C. Belon, C. Croutxe-Barghorn, J. Brendle, M. Soulard, S. Rigolet, V. L. Houerou and C. Gauthier, New J. Chem., 2010, 34, 1068–1072 RSC.
- M. Nishibayashi, H. Yoshida, M. Uenishi, M. Kanezashi, H. Nagasawa, T. Yoshioka and T. Tsuru, Chem. Commun., 2015, 51, 9932–9935 RSC.
- Y. S. Li, W. Lu, Y. Wang and T. Tran, Spectrochim. Acta, Part A, 2009, 73, 922–928 CrossRef PubMed.
- H. De Paz, A. Chemtob, C. Croutxe-Barghorn, D. Le Nouen and S. Rigolet, J. Phys. Chem. B, 2012, 116, 5260–5268 CrossRef CAS PubMed.
- Y. S. Li, T. Tran, Y. Xu and N. E. Vecchio, Spectrochim. Acta, Part A, 2006, 65, 779–786 CrossRef PubMed.
- L. Puro, M. Manttari, A. Pihlajamaki and M. Nystrom, Chem. Eng. Res. Des., 2006, 84(A2), 87–96 CrossRef CAS.
- C. Klaysom, B. P. Ladewig, G. Q. M. Lu and L. Wang, J. Membr. Sci., 2011, 368, 48–53 CrossRef CAS.
- N. N. Rupoasih, H. Suyanto, M. Sumadiyasa and N. Wendri, Open J. Org. Polym. Mater., 2013, 3, 12–18 CrossRef.
- T. P. Chou and G. Cao, J. Sol–Gel Sci. Technol., 2003, 27, 31–41 CrossRef CAS.
- T. M. Long, S. Prakash, M. A. Shannon and J. S. Moore, Langmuir, 2006, 22, 4104–4109 CrossRef CAS PubMed.
- X. Ren, M. Kanezashi, H. Nagasawa and T. Tsuru, RSC Adv., 2015, 5, 59837–59844 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21161e |
| This journal is © The Royal Society of Chemistry 2017 |