
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free one-pot oxidation of ethylarenes for the preparation of α-ketoamides under mild conditions†
Fuyan Liua,
Kuan Zhanga,
Yanfeng Liua,
Shan Chen*b,
Yiping Chena,
Dela Zhanga,
Chunfu Lin ab and
Bo Wang*ab
ab and
Bo Wang*ab
aState Key Lab of Marine Resource Utilisation in South China Sea, Hainan University, Haikou 570228, PR China. E-mail: wangbo@hainu.edu.cn
bInstitute of Chemical Engineering, College of Materials and Chemical Engineering, Hainan University, Haikou 570228, PR China
First published on 23rd January 2017
Abstract
Here we developed a highly efficient solvent-free, one-pot procedure for synthesizing α-ketoamides from ethylarenes and amines, by oxidizing a C–H bond sp3 center. A copper catalyst was employed, and the reactions proceeded smoothly at ambient temperatures. Most of the tested ethylarenes and amines were successfully converted to their corresponding α-ketoamides in moderate to excellent yields of up to 93% with three equivalents of the oxidant tert-butyl hydroperoxide.
Introduction
α-Ketoamides are key structural motifs in a variety of natural products, biologically important compounds, and pharmaceuticals, such as the immunosuppressant drug FK-506, rapamycin, serine, cysteine proteases and other enzymes, and epoxide hydrolase inhibitors.1 All of these compounds, as well as the HIV replication inhibitors complestatin and chloropeptin I,2 include α-ketoamide frameworks. Furthermore, α-ketoamides are also used as valuable precursors and synthetic intermediates in synthetic organic chemistry, for instance in the synthesis of medicinally useful compounds tetrasubstituted 2-oxazolidin-4-one and 2-oxindoles.3Because of the significance of the α-ketoamide scaffold, it is of great interest and importance to develop efficient methods to synthesize the many types of α-ketoamides that are in demand. Over the past decades, several approaches have been developed for synthesizing α-ketoamides.4–15 Most of these procedures involved the direct amidation of α-keto acids and α-keto acyl halides,4 but in most conditions hazardous reagents were usually involved and harsh conditions were required. Direct oxidations of α-hydroxyamides and α-aminoamides have also been used to successfully produce the corresponding α-ketoamides.5 Recently, one-pot procedures for synthesizing α-ketoamides have attracted the focus of researchers worldwide. Jiao and his co-workers introduced a one-pot procedure for synthesizing α-ketoamides by using a copper catalyst to cleave Csp3–H, Csp2–H, and N–H bonds of aryl acetaldehyde and aniline reactants.6 Some recently developed one-pot approaches have included the direct oxidative amidation of aryl methyl ketones,7c nBu4NI-catalyzed multiple sp3 C–H bond oxidation of ethylarenes and sequential coupling with dialkylformamides,8 I2/IBX-catalyzed oxidative amidation of terminal alkenes with amines in dimethyl sulfoxide (DMSO),9a oxidative amidation of terminal alkynes catalyzed by copper(II) triflate,10a iron-2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO)-catalyzed oxidative amidation of α-keto alcohols,11a copper-catalyzed oxidative amidation of 1-arylethanols with amines,12b amidation of halogen benzenes promoted by palladium nanoparticles,13a DMSO-promoted oxidative amidation of α-ketoaldehydes,14c and tert-butyl hydroperoxide/I2 (TBHP/I2)-promoted tandem reactions of β-diketones with amines.15 Most of these methods, however, suffer from the need to carry out extra steps involving the starting materials before the reactions for making the α-ketoamides. They also suffer from low yields, harsh conditions and toxic reagents, and the need to use large amounts of oxidants that are considered to not be environmentally benign.
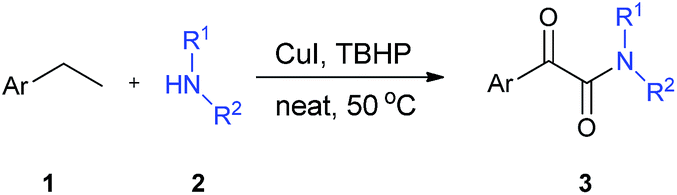
Herein, we present a novel solvent-free and one-pot procedure for the synthesis of α-ketoamides starting from commercially available ethylarenes and amines. In this approach, we used CuI as the catalyst and TBHP as the oxidant. No solvent was required, and the reactions proceeded under mild conditions (Scheme 1).
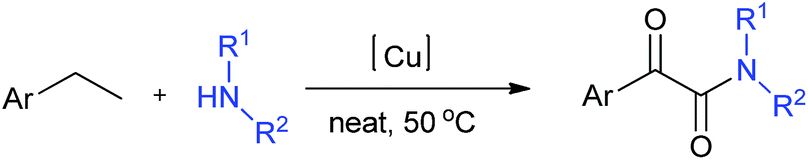 | ||
| Scheme 1 Oxidative synthesis of α-ketoamides from ethylarenes and amines. | ||
Results and discussion
Initial investigations for suitable reaction conditions were performed on a 5 mL scale by employing ethylbenzene (1a) and morpholine (2a) as the model substrates. Conditions such as the identities of the catalyst, oxidants, and solvents, as well as the amounts of loaded catalyst and oxidant, were evaluated, and the results are summarized in Table 1. Carrying out the reaction at 50 °C in acetonitrile in the presence of the oxidant tert-butyl hydroperoxide (TBHP) with copper iodide (CuI) as the catalyst afforded the desired product 3aa in a 63% yield (entry 1, Table 1). CuI was shown to be the most suitable catalyst of all the catalysts tested since this 63% yield of 3aa was better than the yields afforded by the other tested catalysts (CuCl2, Cu2O, CuBr and CuBr2) (see entries 1–5, Table 1). Therefore, in the subsequent investigations, all reactions were performed with CuI as the catalyst.
|
|||||
|---|---|---|---|---|---|
| Entry | Catal. | Oxidant | Solvent | Temp. (°C) | Yieldd (%) |
| a Conditions: 1a (1.0 mmol), 2a (6.0 mmol), catalyst (20 mol%), and oxidant (3 equiv.) under an air atmosphere for 24 h, n.d., not detected; unless otherwise stated, all reaction were performed with 3 equiv. of oxidant.b TBHP: tert-butyl hydroperoxide (TBHP), 70% in water.c Hydrogen peroxide, 30% in water.d Unless otherwise stated, all yields were determined by HPLC analysis.e CuI (10 mol%).f CuI (30 mol%).g Yields obtained from the reaction using 1 equivalent of TBHP.h Yields obtained from the reaction using 2 equivalents of TBHP.i Yields obtained from the reaction using 4 equivalents of TBHP. | |||||
| 1 | CuI | TBHPb | MeCN | 50 | 63 |
| 2 | CuCl2 | TBHP | MeCN | 50 | n.d. |
| 3 | Cu2O | TBHP | MeCN | 50 | n.d. |
| 4 | CuBr | TBHP | MeCN | 50 | 36 |
| 5 | CuBr2 | TBHP | MeCN | 50 | <5 |
| 6 | CuI | TBHP | THF | 50 | 6 |
| 7 | CuI | TBHP | Dioxane | 50 | <5 |
| 8 | CuI | TBHP | MeOH | 50 | 12 |
| 9 | CuI | TBHP | Toluene | 50 | 64 |
| 10 | CuI | TBHP | Neat | 50 | 92 |
| 11 | CuI | DTBP | Neat | 50 | n.d. |
| 12 | CuI | H2O2c | Neat | 50 | n.d. |
| 13 | CuI | DDQ | Neat | 50 | n.d. |
| 14 | CuI | TEMPO | Neat | 50 | n.d. |
| 15 | CuI | TBHP | Neat | 30 | 80 |
| 16 | CuI | TBHP | Neat | 70 | 88 |
| 17 | CuI | TBHP | Neat | 50 | 79e |
| 18 | CuI | TBHP | Neat | 50 | 90f |
| 19 | CuI | TBHP | Neat | 50 | 56g |
| 20 | CuI | TBHP | Neat | 50 | 78h |
| 21 | CuI | TBHP | Neat | 50 | 90i |
The effects of the identity and presence of the solvent on the yields were also evaluated for the purpose of finding suitable conditions. Organic solvents such as tetrahydrofuran (THF), MeCN, 1,4-dioxane, and MeOH were tested at 50 °C. Of all the solvents evaluated, toluene was found to be the best one, having afforded the desired product 3aa with a yield of 64% (entry 9, Table 1), and was thus considered suitable for further investigations. Nevertheless, for the purpose of adhering to at least some of the principles of green chemistry, reactions without organic solvent and with TBHP as the oxidant were also investigated. To our delight, when using three equivalents of TBHP in the absence of organic solvent, great enhancements in the yields of product 3aa were achieved (entry 1, 15–21, Table 1), with yields of 88% for the reaction carried out at 70 °C (entry 16, Table 1), and 92% at 50 °C (entry 10, Table 1). Therefore, in the subsequent investigations, the reactions were performed without organic solvent.
A variety of oxidants, such as TBHP, DTBP, H2O2, DDQ, and TEMPO, and their loadings were also tested to find optimum conditions for the designed reaction. As shown in Table 1, TBHP performed the best of all the oxidants tested. As indicated above, the desired product 3aa was obtained with yields of up to 92% (entry 10, Table 1) when using three equivalents of this oxidant. Using more TBHP (four equivalents) or less of this oxidant (one and two equivalents) resulted in somewhat lower yields. Perhaps, more by-products were generated during the reaction with the larger loading of TBHP, and the oxidation actions were not great enough to convert substrate to product when less of the oxidant was loaded (entry 21, Table 1).
Reaction temperatures were also evaluated to find suitable conditions for the reaction. As shown in Table 1, a temperature of 50 °C was best of all the temperatures evaluated (entry 10, Table 1), with lower and higher temperatures (30 °C and 70 °C) not leading to higher yields of the product 3aa. Therefore, 50 °C was chosen as the suitable reaction temperature for the subsequent reactions.
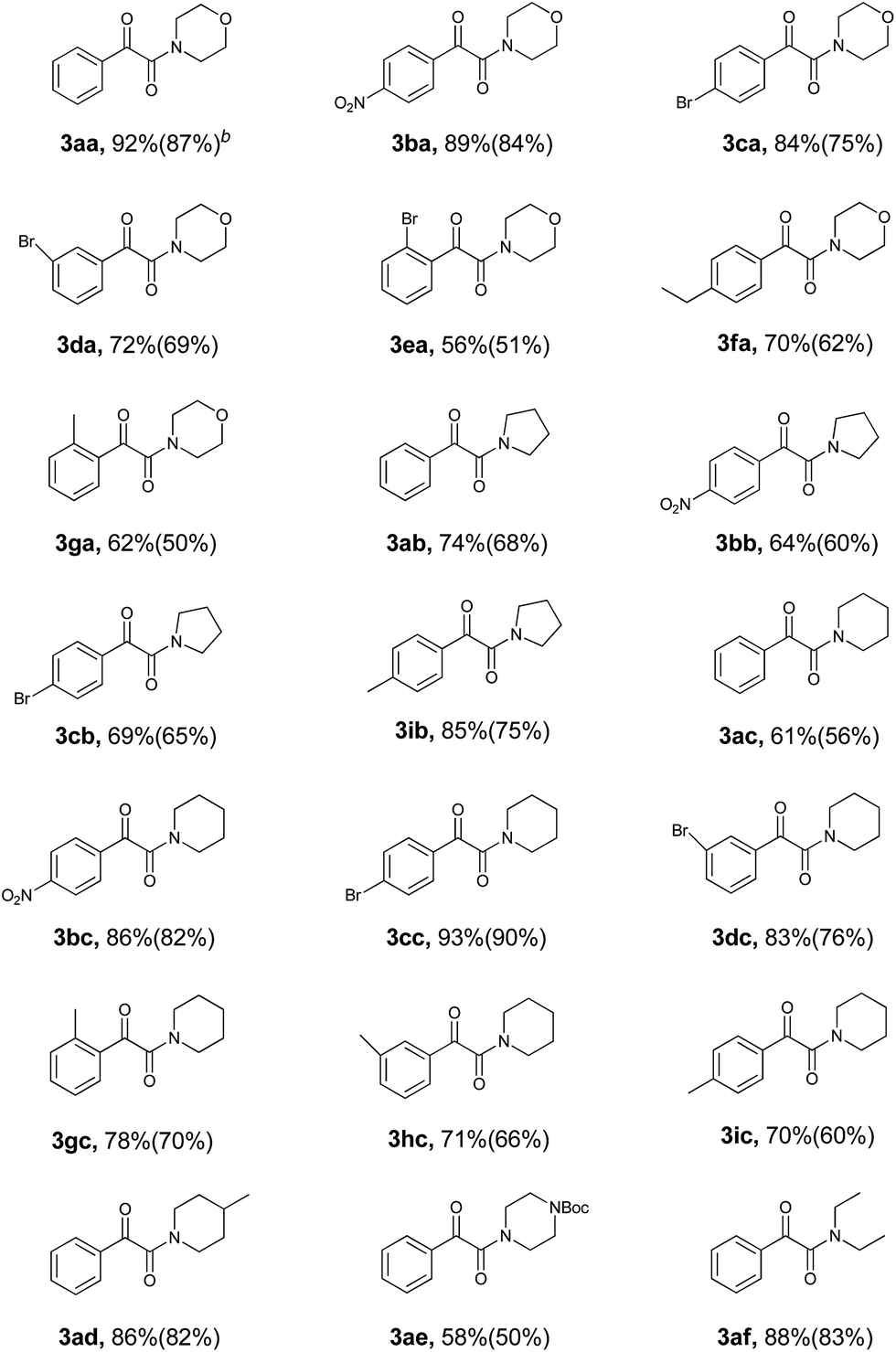
With all of the optimized conditions in hand, a wide range of ethylarenes and amines were then subjected to this reaction to explore the scope of substrates. The results of this investigation are summarized in Table 2. All reactions proceeded well in the absence of solvent at ambient temperature. Yields of product appeared to benefit from the presence of electron-withdrawn substituent(s) on the substrate. For example, the reaction of an ethylarene substrate bearing a nitro group on the ring resulted in the desired product with an excellent yield of 89% (3ba, Table 2). In contrast, the presence instead of an electron-donating ethyl group at the same position resulted in a poorer yield of 70% (3fa, Table 2). Substituent groups on the substrate could therefore either positively or negatively affect product yield, and possibly the reaction rate as well. Note that the reaction of substrates with more than one ethyl group, for example, p-diethylbenzene, with amines resulted in monooxidated amination products in relatively poor yields, possibly due to more by-products having formed.
Based on some related publications,16 a possible mechanism was proposed, as shown in Scheme 2. According to this mechanism, CuI catalyzed the breaking up of the oxidant TBHP into a t-butoxyl radical and a hydroxyl anion (eqn (1) and (2)),17 with the t-butoxyl radical in the following step releasing one hydrogen from the C–H bond of ethylarene on the benzylic ring 1 to form the peroxide 5, which further decomposed to acetophenone 6 with the assistance of TBHP.18 Then, according to the proposed mechanism, acetophenone 6 reacted with amine 2 to form the enamine 7,19 which then further reacted with Cu(II) and t-butoxyl radicals from CuI and TBHP17 to form aminodioxetane 8. Then, according to the mechanism, cleavage of the O–O bond of 8 produced the aryl glyoxal 9,20 which reacted with the introduced amine 2 to give the intermediate 10, and then the reaction of 10 with the t-butoxyl radical from THBP yielded compound 11 and finally the α-ketoamide 3 product.
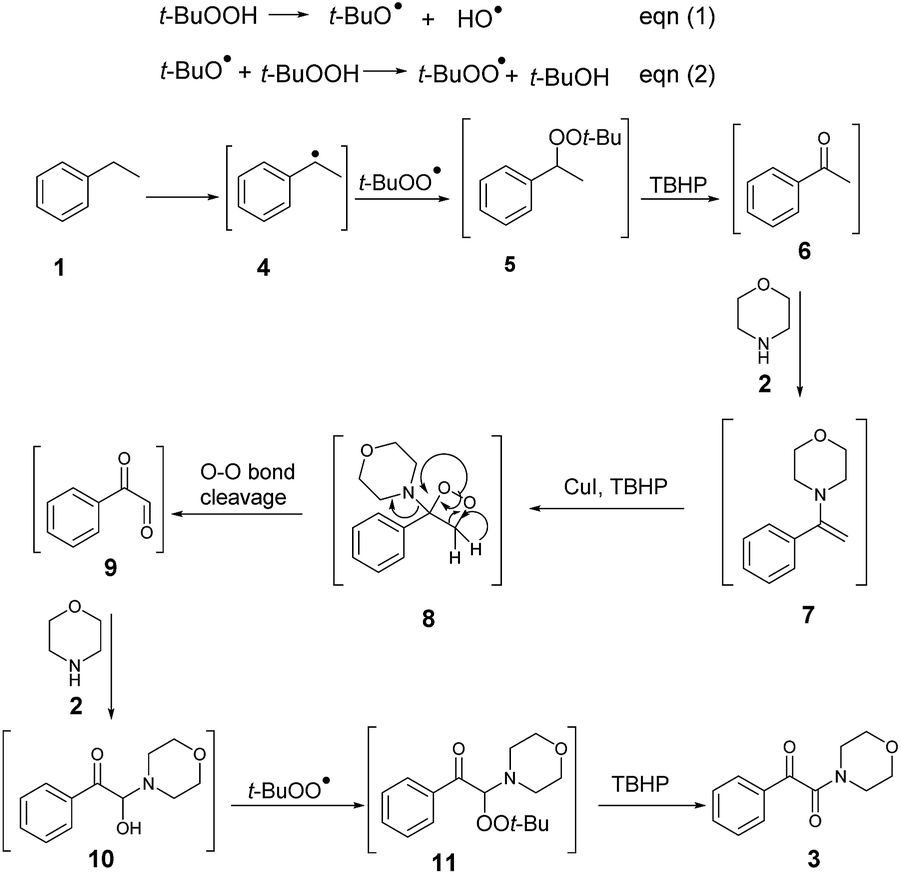 | ||
| Scheme 2 Possible reaction pathway. | ||
To verify the mechanism proposed above, several control experiments were carried out, as are shown in Scheme 3. When the reaction was carried out in the presence of a radical scavenger, for instance TEMPO, the oxidative coupling reaction did not work (a),21 indicating that this conversion likely involved radical intermediates. When acetophenone was used instead of ethylbenzene to react with morpholine, an excellent yield of the product 3 was achieved (b). The reaction of experiment (c) was carried out by employing phenylacetaldehyde instead of ethylbenzene, and this reaction produced the desired compound in very low yield. These examples showed that the oxidation started from the benzylic C–H bond of ethylarene, and not from the methyl group.
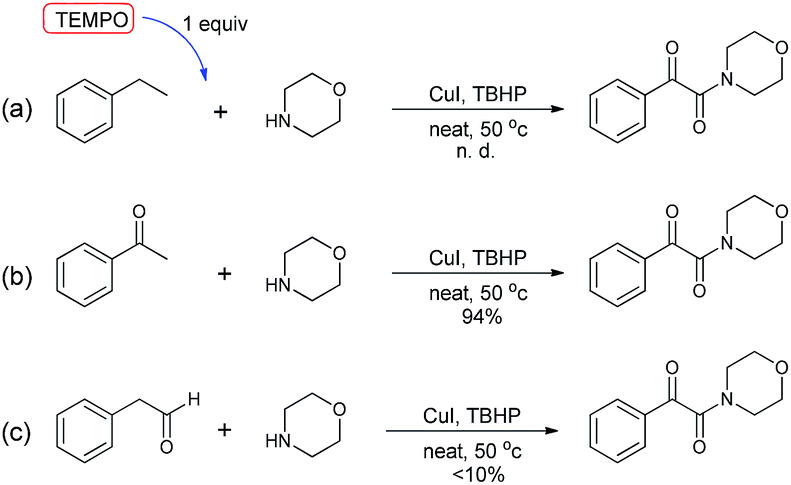 | ||
| Scheme 3 Control experiments to test the proposed mechanism. | ||
Conclusions
In summary, we have developed an efficient one-pot, solvent-free procedure for the synthesis of α-ketoamides through a CuI-catalyzed sp3 C–H bond coupling process aided by aerobic oxidation using ethylarenes and amines. No harsh conditions were needed for this reaction. When using only three equivalents of TBHP, the desired α-ketoamide products were afforded with moderate to excellent yields of up to 93%. These results showed the promise of using this alternative and novel pathway to synthesize α-ketoamides and to enrich the synthetic library for α-ketoamide preparations. This procedure can also be considered to adhere to some of the principles of green chemistry, as the reactions occurred without solvent. Overall, we believe this procedure to have potential in large-scale preparations of α-ketoamides, especially for industrial purposes.Acknowledgements
Financial support from the National Natural Science Foundation of China (21502036), Innovative research team project of Hainan Natural Science Foundation (2016CXTD006), Hainan Provincial Natural Science Foundation (213010) and Hainan University (kyqd1408) are gratefully acknowledged.Notes and references
- (a) M. Hagihara and S. L. Schreiber, J. Am. Chem. Soc., 1992, 114, 6570 CrossRef CAS; (b) Z. Li, A. C. Ortega-Vilain, G. S. Patil, D. L. Chu, J. E. Foreman, D. D. Eveleth and J. C. Powers, J. Med. Chem., 1996, 39, 4089 CrossRef CAS PubMed; (c) B. E. Maryanoff, M. N. Greco, H. C. Zhang, P. Andrade-Gordon, J. A. Kauffman, K. C. Nicolaou, A. Liu and P. H. Brungs, J. Am. Chem. Soc., 1995, 117, 1225 CrossRef CAS; (d) N. Fusetani, S. Matsunaga, H. Matsumoto and Y. Takebayashi, J. Am. Chem. Soc., 1990, 112, 7053 CrossRef CAS; (e) S. Álvarez, R. Álvarez, H. Khanwalkar, P. Germain, G. Lemaire, F. Rodríguez-Barrios, H. Gronemeyer and Á. R. de Lera, Bioorg. Med. Chem., 2009, 17, 4345 CrossRef PubMed; (f) M. L. Stein, H. Cui, P. Beck, C. Dubiella, C. Voss, A. Krüger, B. Schmidt and M. Groll, Angew. Chem., Int. Ed., 2014, 53, 1679 CrossRef CAS PubMed; (g) Y. H. Chen, Y. H. Zhang, H. J. Zhang, D. Z. Liu, M. Gu, J. Y. Li, F. Wu, X. Z. Zhu, J. Li and F. J. Nan, J. Med. Chem., 2006, 49, 1613 CrossRef CAS PubMed; (h) S. L. Harbeson, S. M. Abelleira, A. Akiyama, R. Barrett, R. M. Carroll, J. A. Straub, J. N. Tkacz, C. Wu and G. F. Musso, J. Med. Chem., 1994, 37, 2918 CrossRef CAS PubMed; (i) R. E. Babine and S. L. Bender, Chem. Rev., 1997, 97, 1359 CrossRef CAS PubMed; (j) B. E. Bierer, P. K. Somers, T. J. Wandless, S. J. Burakoff and S. L. Schreiber, Science, 1990, 250, 556 CrossRef CAS PubMed.
- (a) Z. H. Wang, M. Bois-Choussy, Y. X. Jia and J. P. Zhu, Angew. Chem., Int. Ed., 2010, 49, 2018 CrossRef CAS PubMed; (b) H. Deng, J. K. Jung, T. Liu, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 9032 CrossRef CAS PubMed.
- (a) L. Yin, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 7620 CrossRef CAS PubMed; (b) L. Yang, D. X. Wang, Z. T. Huang and M. X. Wang, J. Am. Chem. Soc., 2009, 131, 10390 CrossRef CAS PubMed; (c) D. Tomita, K. Yamatsugu, M. Kanai and M. Shibasak, J. Am. Chem. Soc., 2009, 131, 6946 CrossRef CAS PubMed; (d) J. L. Jesuraj and J. Sivaguru, Chem. Commun., 2010, 46, 4791 RSC.
- (a) D. K. Li, M. Wang, J. Liu, Q. Zhao and L. Wang, Chem. Commun., 2013, 49, 3640 RSC; (b) H. Wang, L. N. Guo and X. H. Duan, Org. Biomol. Chem., 2013, 11, 4573 RSC; (c) F. Heaney, J. Fenlon, P. McArdleb and D. Cunningham, Org. Biomol. Chem., 2003, 1, 1122 RSC; (d) R. P. Singh and J. M. Shreeve, J. Org. Chem., 2003, 68, 6063 CrossRef CAS PubMed.
- (a) L. Banfi, G. Guanti and R. Riva, Chem. Commun., 2000, 985 RSC; (b) T. D. Ocain and D. H. Rich, J. Med. Chem., 1992, 35, 451 CrossRef CAS PubMed; (c) J. E. Semple, T. D. Owens, K. Nguyen and O. E. Levy, Org. Lett., 2000, 2, 2769 CrossRef CAS PubMed; (d) Z. Yang, Z. Zhang, N. A. Meanwell, J. F. Kadow and T. Wang, Org. Lett., 2002, 4, 1103 CrossRef CAS PubMed; (e) T. F. Buckley and H. Rapoport, J. Am. Chem. Soc., 1982, 104, 4446 CrossRef CAS.
- C. Zhang, Z. Xu, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2011, 50, 11088 CrossRef CAS PubMed.
- (a) S. Nekkanti, K. Veeramani, N. P. Kumar and N. Shankaraiaha, Green Chem., 2016, 18, 3439 RSC; (b) N. Mupparapu, R. A. Vishwakarma and Q. N. Ahmed, Tetrahedron, 2015, 71, 3417 CrossRef CAS; (c) R. K. Sharma, S. Sharma, G. Gaba and S. Dutta, J. Mater. Sci., 2016, 51, 2121 CrossRef CAS; (d) M. Lamani and K. R. Prabhu, Chem.–Eur. J., 2012, 18, 4638 CrossRef PubMed; (e) W. Wei, Y. Shao, H. Y. Hu, F. Zhang, C. Zhang, Y. Xu and X. B. Wan, J. Org. Chem., 2012, 77, 7157 CrossRef CAS PubMed.
- B. Du, B. Jin and P. Sun, Org. Biomol. Chem., 2014, 12, 4586 CAS.
- (a) S. Dutta, S. S. Kotha and G. Sekar, RSC Adv., 2015, 5, 47265 RSC; (b) R. Deshidi, S. Devari and B. A. Shah, Eur. J. Org. Chem., 2015, 2015, 1428 CrossRef CAS.
- (a) M. Kumar, S. Devari, A. Kumar, S. Sultan, O. Ahmed, M. Rizvi and B. A. Shah, Asian J. Org. Chem., 2015, 4, 438 CrossRef CAS; (b) R. Deshidi, M. Kumar, S. Devari and B. A. Shah, Chem. Commun., 2014, 50, 9533 RSC; (c) C. Zhang and N. Jiao, J. Am. Chem. Soc., 2010, 132, 28 CrossRef CAS PubMed.
- (a) S. S. Kotha, S. Chandrasekar, S. Sahu and G. Sekar, Eur. J. Org. Chem., 2014, 7451 CrossRef CAS; (b) S. S. Kotha and G. Sekar, Tetrahedron Lett., 2015, 56, 6323 CrossRef CAS.
- (a) C. K. Liu, Z. Fang, Z. Yang, Q. W. Li, S. Y. Guo and K. Guo, RSC Adv., 2016, 6, 25167 RSC; (b) N. Sharma, S. S. Kotha, N. Lahiri and G. Sekar, Synthesis, 2015, 47, 726 CrossRef CAS.
- (a) H. Du, Q. Ruan, M. Qi and W. Han, J. Org. Chem., 2015, 80, 7816 CrossRef CAS PubMed; (b) Y. Wang, X. Yang, C. Zhang, J. Yu, J. Liu and C. Xia, Adv. Synth. Catal., 2014, 356, 2539 CrossRef CAS; (c) C. Zhang, J. Liu and C. Xia, Org. Biomol. Chem., 2014, 12, 9702 RSC; (d) J. Liu, R. Zhang, S. Wang, W. Sun and C. Xia, Org. Lett., 2009, 11, 1321 CrossRef CAS PubMed.
- (a) S. Battula, A. Kumar, A. P. Gupta and Q. N. Ahmed, Org. Lett., 2015, 17, 5562 CrossRef CAS PubMed; (b) T. Truong, G. H. Dang, N. V. Nam, N. T. Truong, D. T. Le and N. T. S. Phan, J. Mol. Catal. A: Chem., 2015, 409, 110 CrossRef CAS; (c) N. Mupparapu, S. Khan, S. Battula, M. Kushwaha, A. P. Gupta, Q. N. Ahmed and R. A. Vishwakarma, Org. Lett., 2014, 16, 1152 CrossRef CAS PubMed.
- X. B. Zhang, M. Wang, Y. C. Zhang and L. Wang, RSC Adv., 2013, 3, 1311 RSC.
- (a) F. T. Du and J. X. Ji, Chem. Sci., 2012, 3, 460 RSC; (b) X. B. Zhang and L. Wang, Green Chem., 2012, 14, 2141 RSC; (c) Q. Zhao, T. Miao, X. B. Zhang, W. Zhou and L. Wang, Org. Biomol. Chem., 2013, 11, 1867 RSC; (d) H. P. Kalmode, S. K. Vadagaonkar and A. C. Chaskar, RSC Adv., 2014, 4, 60316 RSC; (e) C. K. Liu, Z. Yang, S. Y. Guo, Y. Zeng, N. Zhu, X. Li, Z. Fang and K. Guo, Org. Biomol. Chem., 2016, 14, 8570 RSC.
- E. Boess, C. Schmitz and M. Klussmann, J. Am. Chem. Soc., 2012, 134, 5317 CrossRef CAS PubMed.
- (a) F. Napoly, R. Kieffer, L. Jean-Gérard, C. Goux-Henry, M. Draye and B. Andrioletti, Tetrahedron Lett., 2015, 56, 2517 CrossRef CAS; (b) Y. Xu, Z. P. Yang, J. T. Hu and J. Yan, Synthesis, 2013, 45, 370 CrossRef CAS; (c) J. Zhang, Z. Wang, Y. Wang, C. Wan, X. Zheng and Z. Wang, Green Chem., 2009, 11, 1973 RSC.
- G. Stork, R. Terrell and J. Szmuszkovicz, J. Am. Chem. Soc., 1954, 76, 2029 CrossRef CAS.
- (a) H. H. Wasserman and S. Terao, Tetrahedron Lett., 1975, 16, 1735 CrossRef; (b) K. Kaneda, T. Itoh, N. Kii, K. Jitsukawa and S. Teranishi, J. Mol. Catal., 1982, 15, 349–365 CrossRef CAS.
- (a) Y. Shao, Z. Wu, C. Miao and L. Liu, J. Organomet. Chem., 2014, 767, 60 CrossRef CAS; (b) J. J. Warren and J. M. Mayer, J. Am. Chem. Soc., 2010, 132, 7784 CrossRef CAS PubMed; (c) C. W. Chan, Z. Zhou, A. S. C. Chan and W. Y. Yu, Org. Lett., 2010, 12, 3926 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26679g |
| This journal is © The Royal Society of Chemistry 2017 |