
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquefaction of lignite with a Ru/C catalyst in supercritical ethanol†
Rongrong Miaoa,
Qianqiu Zhanga,
Yuzhen Shia,
Junjie Gua,
Ping Ninga and
Qingqing Guan *ab
*ab
aFaculty of Environmental Science and Engineering, Kunming University of Science and Technology, Kunming 650500, China. E-mail: 15545488@qq.com
bCollaborative Innovation Center of Western Typical Industry Environmental Pollution Control, Kunming University of Science and Technology, Kunming 650500, China
First published on 4th January 2017
Abstract
High moisture content materials present challenges for direct liquefaction. We herein report an effective way to liquefy high moisture content feedstocks in supercritical ethanol with a Ru/C catalyst. The Ru/C catalyst converts some of the ethanol into hydrogen through steam reforming. Additional hydrogen is produced by the water gas shift reaction. During this process, water is converted into gaseous products. Compared to the case with no added catalyst, use of Ru/C suppressed solid residues, improved hydrogen yields, and improved the quality of the oil fraction. Oil quality was improved by enhancing production of long chain alkanes (LCAs), and suppressing production of phenols and polycyclic aromatic hydrocarbons (PAHs). The effects of reaction temperature and time on the product distribution and composition were also investigated. Higher temperatures favored gasification. The maximum oil yield, 6.4 wt%, and minimum solid residue yield, 0.9 wt% (calculated by oil or solid/the mass of ethanol and lignite), were obtained at 400 °C and 60 min reaction time with an ethanol/lignite (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L) ratio of 9:1, and 50 wt% catalyst loading relative to lignite. The dominant compounds in the oil were LCAs, esters, and phenols. The major gaseous products were CH4, H2, and CO.
:L) ratio of 9:1, and 50 wt% catalyst loading relative to lignite. The dominant compounds in the oil were LCAs, esters, and phenols. The major gaseous products were CH4, H2, and CO.
1. Introduction
There is increasing interest in the liquefaction of coal and biomass to produce liquid hydrocarbons,1,2 since liquid fuel is currently considered the best energy carrier for the transportation sector. However, high moisture content materials such as microalgae and lignite pose challenges for direct liquefaction.3–5 A feedstock drying procedure is commonly required, which increases cost dramatically.Liquefaction in high-temperature water (HTW) is one method of avoiding the need for drying. HTW is generally defined as liquid water above 200 °C or at sub/supercritical water (SCW) conditions.6–8 In HTW liquefaction, water serves as both solvent and reactant, and there is no need to remove moisture from the feedstock prior to reaction. Numerous investigations have covered liquefaction of various materials in HTW.7–11 The disadvantages of HTW liquefaction include the need for high temperatures and pressures, and the production of liquid fractions with a relatively high oxygen content.12 In additional, oil/water separation is necessary after HTW liquefaction.
Methods that utilize organic solvents such as tetralin, toluene, or acetone instead of water have also been investigated.13 Among supercritical organic solvents, ethanol may be the most attractive for liquefaction, because it is both effective and renewable.14
Supercritical ethanol exhibits excellent properties for liquefaction. The critical temperature and pressure of ethanol (243.2 °C, 6.38 MPa) are far below that of water. Ethanol can act both as solvent and reactant. Ethanol can generate a H2-rich reaction environment through steam reforming in the presence of water.14 Additionally, ethanol can react with acidic components through esterification.12
Previous studies have reported liquefaction of biomass, biomass-coal mixtures, and upgrading of crude bio-oil in supercritical ethanol. Feedstocks in these studies included lignocellulose,15 lignin,16 cellulose,15,17 sewage sludge,18 and microalgae.19,20 However, most of these feed stocks were pretreated by drying to remove the water.21 Some researchers used mixtures of ethanol and water as solvent. Although synergistic effects were found with an ethanol/water solvent system, post-reaction separation of the water phase was needed,1,22–24 and the separation of water soluble products was cost too much energy. Thus, it is important to seek effective methods of liquefaction in water/ethanol systems that avoid the need for post-reaction water separation and the water soluble products, thereby decreasing operational and capital costs.
Use of ethanol as H2-donor through steam reforming with water could potentially remove water and upgrade oil by hydrogenation. Catalyst are necessary to improve yields. Some catalysts such as KOH, K2CO3, Ca(OH)2, H3PO4, FeCl3, ZnCl2, AlCl3,4 Pd/C, Pt/C, Ru/C,7 Ni/SiO2–Al2O3, and CoMo/γ-Al2O3 have been tested for liquefaction. Corrosion is a major disadvantage of using homogeneous catalysts such ans KOH, K2CO3, Ca(OH)2, H3PO4, FeCl3, ZnCl2, AlCl3.4 Heterogeneous catalysts have gained attention due to their low corrosivity and ease of recovery. Ruthenium (Ru) in particular has proved to be one of the most effective catalysts for gasification. Previous research showed that Ru/C catalysts were highly active in gasification reactions, producing relatively high yields of hydrogen.25,26 It is well known that hydrogen gas has a positive effect on liquefaction in the presence of catalysts such as Pt and Ru. Thus, Ru was the choice catalyst for lignite liquefaction.7,27
Liquefaction of lignite with Ru/C catalyst in supercritical ethanol was tested. Lignite is considered a low rank coal with high moisture content (10–40 wt%), sometimes as high as 75 wt%. There are about 2.6 trillion tons of worldwide lignite reserves. The high volatility and poor thermal stability of lignite make it a less suitable fuel for direct use, but a promising candidate for liquefaction. However, few studies have investigated the catalytic liquefaction of lignite.
To the best of the authors' knowledge, no work has been reported on the liquefaction of high moisture feedstocks with Ru. The current work seeks to fill that gap by systematically investigating lignite liquefaction in the presence of ethanol with Ru catalysis. The effect of each component in the Ru/ethanol/lignite liquefaction system was determined by comparison with a control run lacking that component. The effects of reaction temperature and time on the product distribution were studied for temperatures between 350 and 450 °C, and reaction times between 5 and 120 min. Changes to the properties of the oil were identified and analyzed by gas chromatography/mass spectrometry (GC/MS).
2. Materials and methods
2.1 Materials
The lignite used in this study originated from Zhaotong (Yunnan, China). Table 1 provides elemental and chemical characterization of the lignite. The lignite was not dried before performing the ultimate analysis and the experiments. Prior to use, the lignite samples were sieved to a particle size smaller than 80 mesh (<0.178 mm). The Ru/C catalyst and dichloromethane were purchased from Sigma-Aldrich. The metal loading of Ru on carbon was 5 wt%. The anhydrous alcohol was obtained from FengChuan chemical company. Aside from sieving the lignite, all chemicals were used as received.2.2 Catalyst characterization
BET surface area measurements were carried out on a Tristar II 3020 system. BET characterization results of the Ru/C catalyst are shown in Fig. 1. The N2 adsorption–desorption isotherm is of type I, indicating a microporous material. The BET surface area of Ru/C is 748.50 m2 g−1, and the pore volume is 0.487086 cm3 g−1 with an average pore diameter of 6.2616 nm.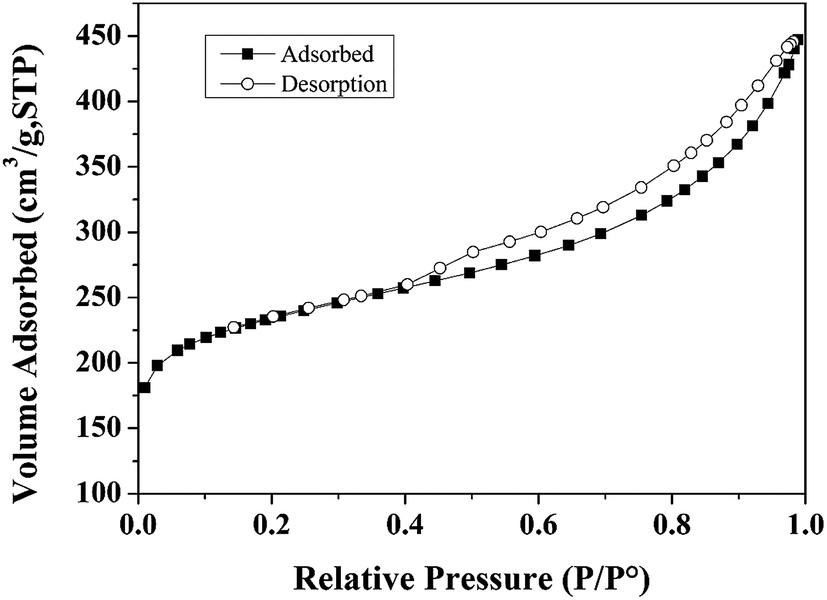 | ||
| Fig. 1 Nitrogen adsorption–desorption isotherms. | ||
X-ray photoelectron spectroscopy (XPS) data were obtained with a ULVAC PHI 5000 Versa Probe-II instrument. The spectra of all the elements in the sample are shown in Fig. 2. Fig. 2a shows the binding energy of C 1s + Ru 3d, and O 1s, indicating the presence of C and O. The spectrum intensity of Ru is somewhat low due to the low concentration of Ru, 5 wt% relative to carbon. As shown in Fig. 2b, the peaks located from 456 eV to 471 eV indicate the presence of Ru, RuCl3, RuO2 and RuOx. Fig. 2c indicates that only a small portion of the oxygen on the catalyst is present as ruthenium oxide.
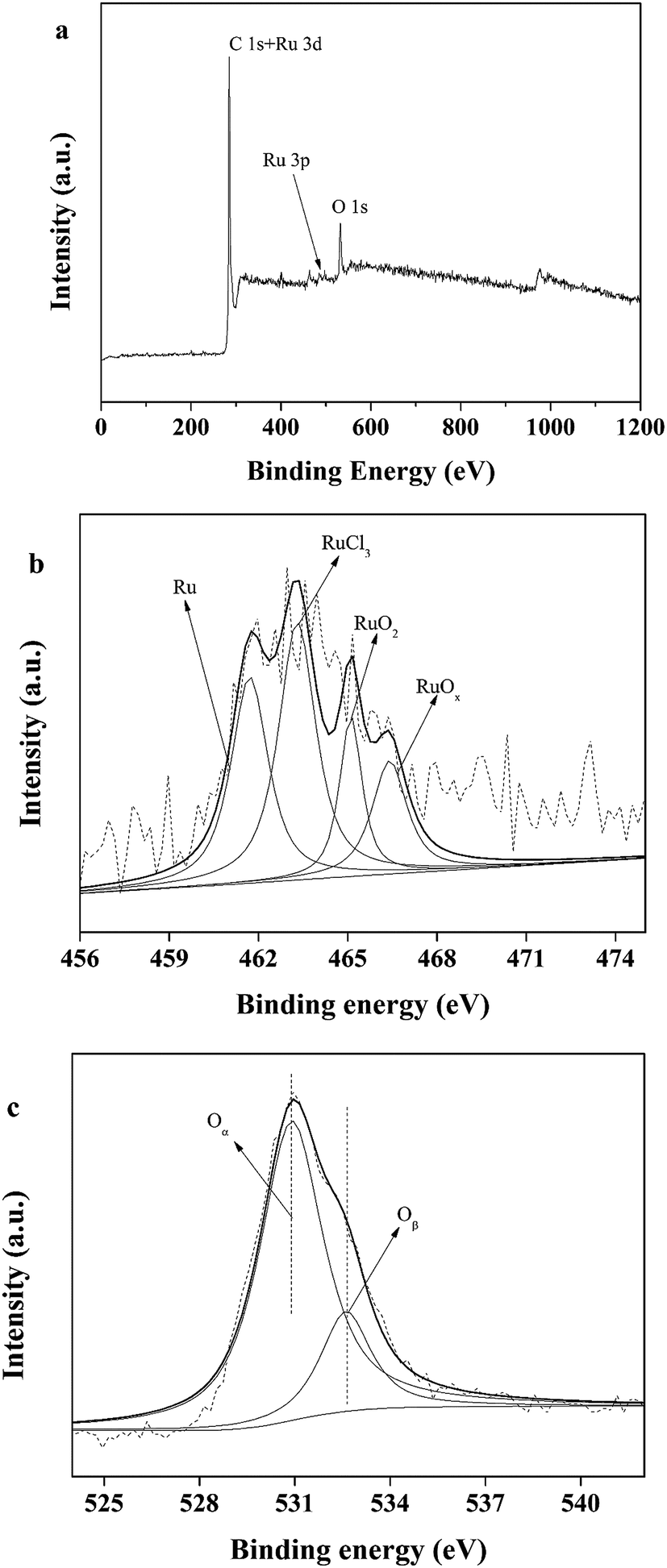 | ||
| Fig. 2 XPS profiles of Ru/C. | ||
2.3 Procedure
316-stainless steel mini-batch (10 mL) reactors, previously described in detail, were employed for liquefaction experiments.28,29 Reactions were initiated by placing the reactors vertically in a Techne Fluidized Sand Bath (model SBL-2).In a typical run, 0.096 g of lignite powder (particle size <0.178 mm), 0.048 g of Ru/C catalyst (50 wt% of lignite), and 0.87 g ethanol were added to the reactor. After the reactor was loaded, the cap assembly was connected and securely tightened to seal the reactor. The air inside the reactor was replaced with helium by repeated cycles of evacuation and charging with He (280 kPa). The 280 kPa of helium that remained in the reactor served as an internal standard for the quantification of gas yields. After helium was loaded, the reactor valve was closed and the reactor assembly was lowered vertically in the preheated Techne Fluidized Sand Bath set to the desired reaction temperature. The temperature was controlled by a Techne TC-8D temperature controller with a precision of ±2 °C. The reactor body was entirely immersed in the heated fluidized bed of aluminum oxide particles, but the valve remained a few inches above the surface, as it could not withstand the high temperatures of the sand bath. The reactor temperature reached the set-point temperature of the sand bath in about 3 min and remained isothermal for the desired reaction time, at which point the reactors were removed from the sand bath. A fan was employed to cool the reactors to room temperature in just a few minutes. Hence, heat-up and cool-down times were usually short compared to the overall reaction times.
After cooling to room temperature, the reactor was connected to a GC gas sampling valve. The reactor valve was opened and gases in the reactor flowed into the GC sample loop. Once gas analysis was complete, residual gases in the reactor were expelled completely. Reactors were opened, and dichloromethane was added to recover the liquid and solid products. To ensure the complete recovery, the reactor was washed three times, each with 9 mL dichloromethane. The collected solid and liquid phases were separated by organic filter head. The separated solid residue and filter head were dried in an oven at 75 °C for 48 h and weighed. The weight of solid residue was calculated by subtracting the weight of the filter head and the catalyst. The dichloromethane and ethanol were removed from the liquid phase using a rotary evaporator under vacuum. The material remaining in the flask after rotary evaporation was the oil.
The gaseous products were analyzed by an Agilent Technologies model 7820A gas chromatograph (GC) equipped with a thermal conductivity detector (GC-TCD) and a 15 ft × 1/8 in. i.d. stainless steel column, packed with 60 × 80 mesh Carboxen 1000 (Supelco). Argon served as the carrier gas. The liquid phase was weighted by an electronic balance AL204 and analyzed by a GC-MS (PE SQ 8T-680) equipped with an Elite-5MS capillary column.
To investigate the effect of temperature and reaction time on oil composition, the oil fractions obtained at different reaction conditions were analyzed by GC-MS. 5 mL of dichloromethane was added in the rotary to dissolved the oil and 5 μL of solvent was injected for each analysis so that spectra could be better compared run to run.
The method adopted in this work as well as in many other studies4,30,31 for the separation and quantification of the oil products causes some errors, since low boiling point components such as propane may be lost during the filtration and rotary evaporation steps. Some results indicated that these lighter materials are present in small quantities, but are difficult to quantify. It also should be noted that the total mass of products varied considerably, mainly because of the behavior of ethanol as both reactant and solvent. The presence of ethanol greatly enhanced apparent gas yields and also affects the yield and composition of the liquefied oil. As both ethanol and lignite participated in the reaction, we used the sum of the mass of ethanol and lignite as the bases for the calculations of oil, solid residue, and gas yields. Further discussion can be found in Section 3. All results reported herein represent mean values for at least two independent trials conducted under nominally identical conditions to determine uncertainty.
Unless otherwise indicated, the yield of each product was calculated as follows:
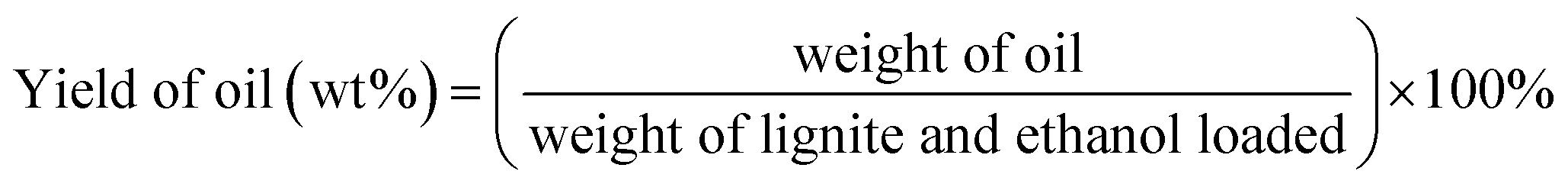
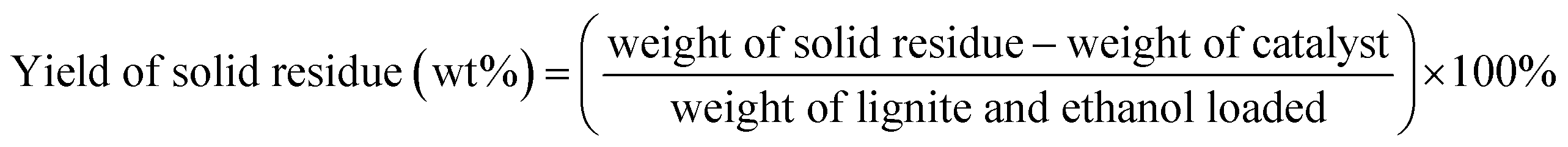
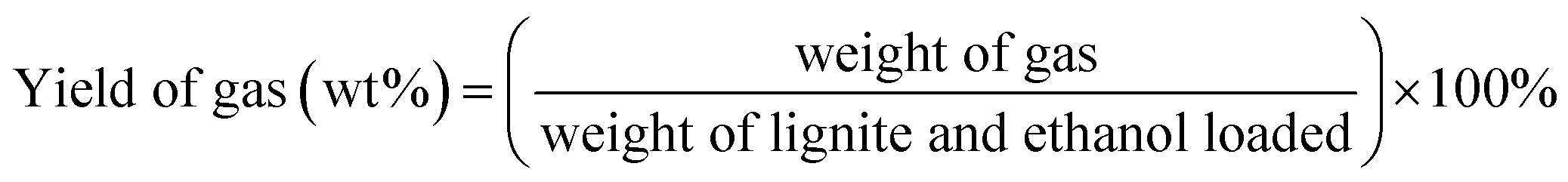
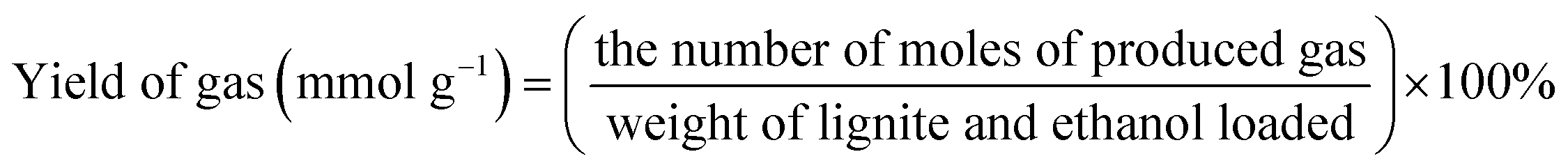
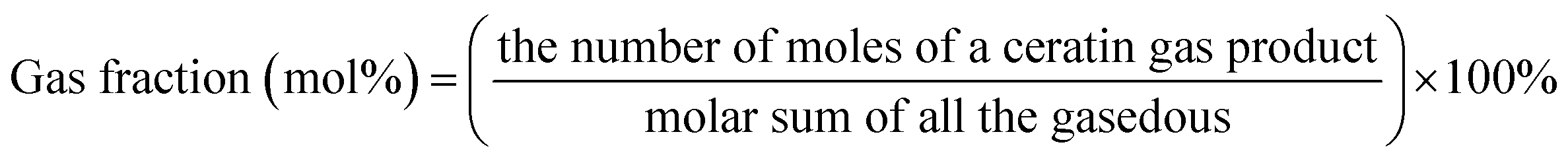
3. Results and discussion
3.1 The effect of Ru/C catalyst
To understand the effect of Ru/C on liquefaction of lignite, control experiments were performed with and without Ru/C. As shown in Fig. 3, gas composition and product yields were dramatically different between the run with Ru/C (C), and without Ru/C (A).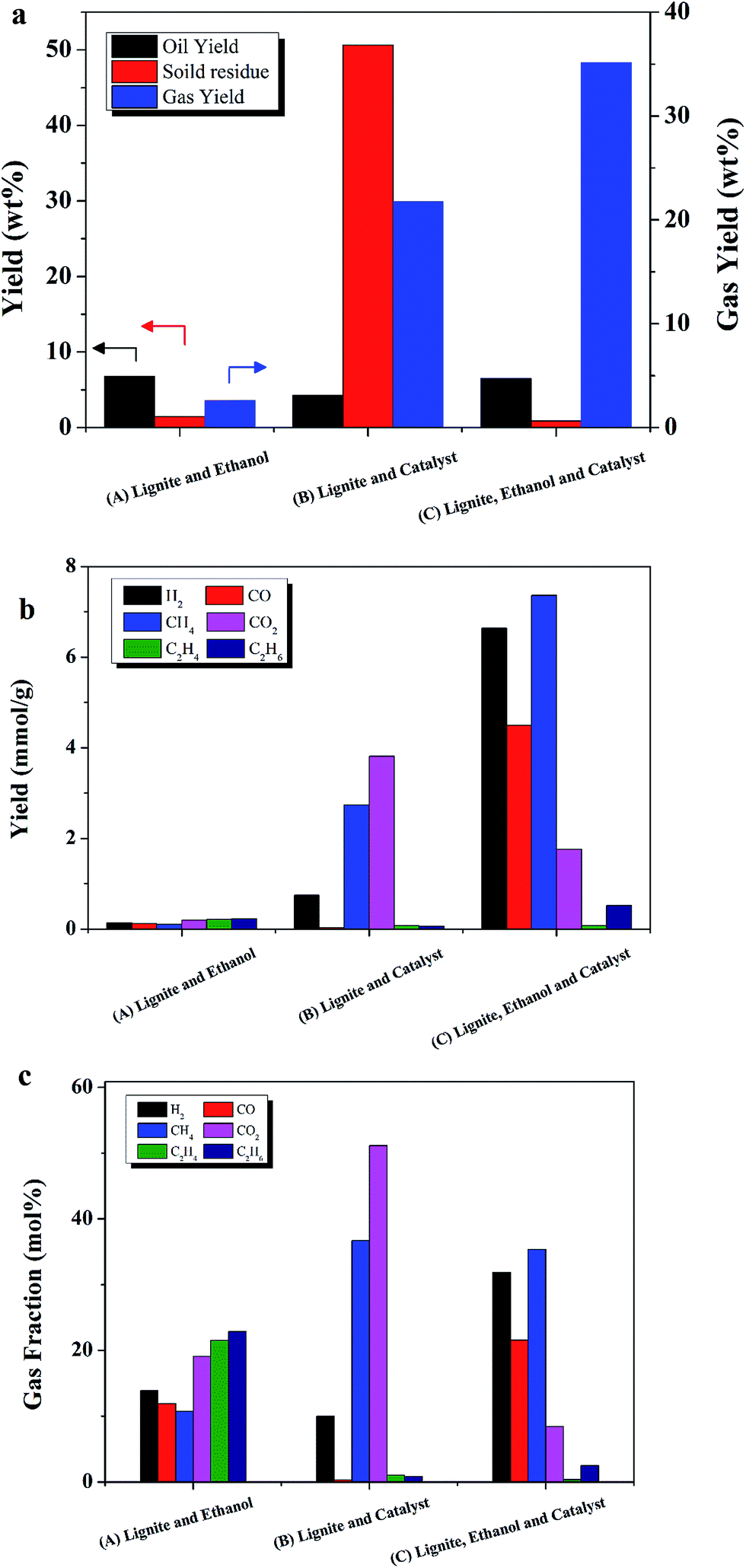 | ||
| Fig. 3 The effect of Ru/C catalyst on (a) wt% product yields by phase, (b) gas component yield (mmol g−1), and (c) mol% gas fraction. All runs were conducted at 400 °C for 60 min with (A) 0.87 g ethanol and 0.096 g lignite; (B) no ethanol, 0.096 g lignite, and 0.048 g Ru/C; (C) 0.87 g ethanol, 0.096 g lignite, and 50 wt% Ru/C. | ||
Fig. 3a shows that gas yield was 35 wt% for sample C, the case with ethanol, lignite, and catalyst. It should be noted that comparing with the mass of lignite loaded, the gas yield was over 100 wt%. It was concluded that ethanol took part in the liquefaction and gasification reactions, leading to large apparent gas yields.
Only 2.6 wt% gas yield was detected when lignite and ethanol (sample A) were added without catalyst, as shown in Fig. 3a and b. The dominant gas species was C2H6, followed by C2H4 and CO2, as shown in Fig. 3c. Without catalyst, formation of gas was inhibited, indicating that ethanol cannot serve as an effective H2-donor without catalyst. 6.7% oil yield was achieved, indicating oil mainly was from lignite. Since the oil yield was 67% as comparing with lignite weight, liquefaction was the dominant process. Supercritical ethanol is an excellent solvent for macromolecules. Since the dielectric constant of ethanol decreases from 25 to below 4 at supercritical conditions, ethanol becomes more hydrophobic and is better able to dissolve hydrocarbons as it approaches its critical point.32 Moreover, ethanol can react with acids to produce esters. Since the lignite contained 36 wt% volatile material and only 7 wt% ash, it was hypothesized that ethanol could increase the oil yield from lignite liquefaction through esterification.
The effect of Ru/C was investigated. It should be noted that the experiments B was performed without ethanol, so the yield was only divided by weight of lignite. When no ethanol was added (sample B), owing to the lack of solvent, the yield of solid residue was 50 wt%, the maximum value in this study. The yield of oil was only 4 wt%. Gas yield was 21 wt%, suggesting that Ru/C significantly increased gas yield even without ethanol. Clearly, Ru/C can enhance the steam reforming reaction between lignite with water.
When ethanol, lignite, and Ru/C were present, sample C, oil yield was 6.4 wt%, which was close to the 6.8 wt% oil yield obtained for sample A, the case with only ethanol and lignite (no catalyst). It should be noted that there was 35 wt% gas yield and only 0.9 wt% solid residues. The gas yield was increased 13 fold compared to sample A (without Ru).
The main gas fractions for sample C, the case with ethanol, lignite, and catalyst, were H2 and CH4, followed by CO, CO2, C2H6, and C2H4. Compared to the case without catalyst, sample A, methane production increased 73 fold and hydrogen production increased more than 48 fold with the presence of Ru/C. Duan et al. reported abundant production of CH4 and CO2 during hydrothermal liquefaction of microalga with Ru/C catalyst, but only 10 mol% yield of H2.7 These results confirm that ethanol serves as a H2-donor in the presence of Ru/C.
To confirm that liquefaction in supercritical ethanol with Ru/C provides an efficient way to remove water from high moisture materials, additional experiments about reaction of water and ethanol with and without Ru/C were carried out. The results show that almost 100% of the water in lignite was converted during the process with Ru/C, meanwhile little hydrogen was detected without Ru/C. Thus, Ru/C catalyst converts some of the ethanol into hydrogen through steam reforming with water (for more details, see the ESI†).
Additionally, oil yield was not increased with Ru/C, but solid residue yield was suppressed, and gas yield increased significantly. Moreover, the quality of the oil was enhanced, as shown in Fig. 4.
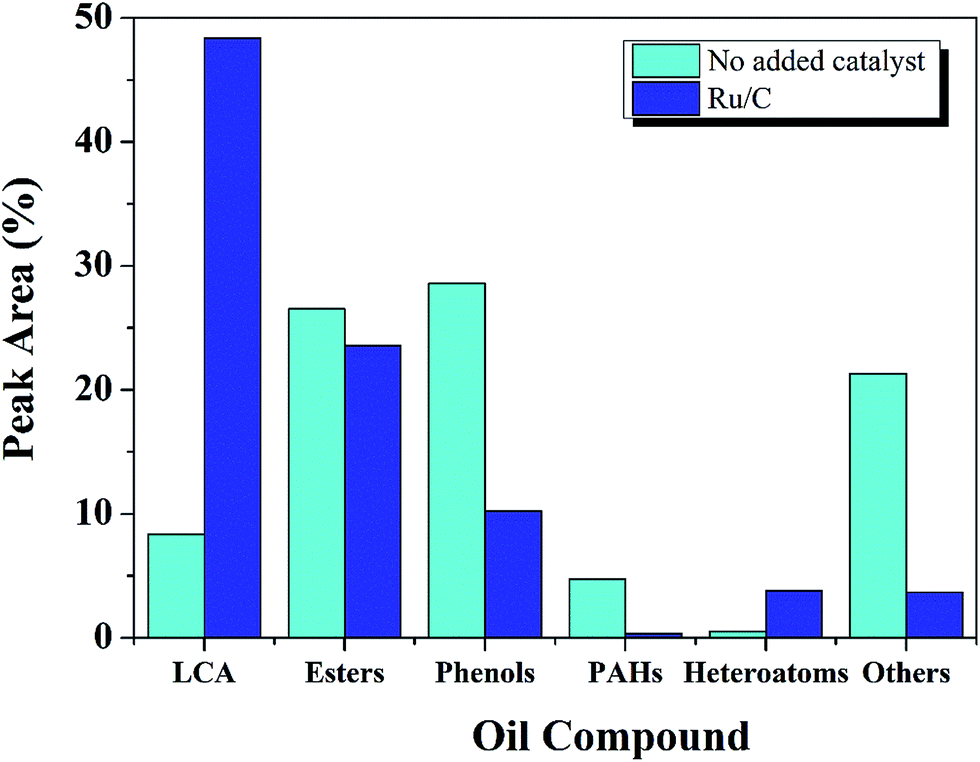 | ||
| Fig. 4 The effect of Ru/C catalyst on oil compound distribution (400 °C, 60 min, L:E ratio of 1:9, and 50 wt% Ru/C relative to lignite). | ||
Compounds within the oil fraction were identified by GC-MS and classified into five groups: phenols, long-chain alkanes (LCAs), esters, polycyclic aromatic hydrocarbons (PAHs), and others. The others including: ketones, aldehydes, ethers, acids, benzenes and alcohols. The total peak areas of the selected major components accounted for 90% of the total chromatogram. Some components contained more than one functional group but were categorized into only one group.
The major category of compounds formed without catalyst was the phenols, which accounted for 28%. This result was expected since the structure of lignite is dominated by aromatic rings. Previous research has shown that lignite has many functional groups, including methyl, ethyl, phenolic hydroxyl, and carboxylic acid groups.33 The complex structure of lignite leads to the large diversity of components within the liquid product.
Without Ru, esters were the second largest compound in the oil, followed by others and LCAs. Esterification between ethanol and hydrocracked reaction intermediates may be responsible for the ester formation.16 The organic oxygen in lignite was reported to be present in the form of alcohol, phenol, carbonyl, carboxyl, and ether groups. The acids likely arose from the oxygen-containing functional groups of lignite.33 Esters were also found by Jade,12 and Tang34 while studying the liquefaction of biomass with supercritical ethanol, confirming esterification between ethanol and acids.
Others mainly include ketones (7%), alcohols (5%), benzene (4%) and ethers (2%). Higher alcohols might arise from the alcoholysis between ethanol and reaction intermediates, a phenomenon observed previously in the liquefaction of lignite with ethanol.33
In comparison with the non-catalytic process, the effect of Ru/C was a 6 fold increase in LCAs, a dramatic decrease in phenols, and a reduction in the others group. These results suggest that the catalyst may promote decarbonylation or decarboxylation of oxygen containing functional groups. Related investigations also reported35–37 that catalysts may reduce O content by converting carboxylic acids into CO233,38,39 and LCAs.
Ru/C may also increase the LCA yield through hydrocracking and hydrogenation pathways. Hydrogen yields may seem low in some cases due to the high activity of Ru/C for methanation, CO2 + 4H2 → CH4 + 2H2O, which consumes hydrogen and produces CH4.
Besides the phenols and others group, heteroatoms and PHAs also decreased 86% and 92%, respectively, with the introduction of Ru catalyst. Since LCAs have a higher heating value, the presence of Ru/C improved the quality of the oil significantly.
In conclusion, supercritical ethanol with Ru catalyst provides an effective reaction environment for the liquefaction of high moisture material. By enhancing the steam reforming reaction, water can be converted into gaseous products efficiently. Although the Ru catalyst decreased the oil yield through hydrocracking and hydrogenation, it also increased gaseous product yields and suppressed the formation of solid residue. The presence of Ru catalyst accelerated hydrogenation reactions, improving the quality of the oil significantly.
3.2 The effect of temperature
As reported by many researchers, temperature can affect both yield and product quality in a liquefaction process.9,13,17,40,41 To determine the effect of temperature, experiments were performed at 350, 400, and 450 °C; with an ethanol/lignite (E/L) ratio of 9:1, 50 wt% Ru/C relative to lignite, and a reaction time of 60 min. The critical temperature of ethanol is 243 °C. Thus, all reactions were performed at supercritical conditions.
Temperature significantly affected the products yields, as shown in Fig. 5. Oil yield increased from 5.1 to 6.4 wt% as temperature increased from 350 to 400 °C, but then decreased to 1.5 wt% at 450 °C. Yield of solid residue tripled from 400 to 450°. This phenomenon may be explained by observations from a related process. In coal hydrocracking, high temperatures, near 350 °C, enhance desirable decomposition reactions leading to more hydrocracking intermediates. Formation of these unstable intermediates results in increased oil and gas production, and reduced yields of solid residue. However, further increases in temperature increase gas formation by accelerating the decomposition and hydrocracking reactions among the intermediates. Further increases in temperatures also promote condensation and re-polymerization reactions to form solid residue. Similar results were reported by Le and Chad,42,43 who found that high temperatures promoted oil yields, but that further increases in temperature resulted in increased formation of solid residue and gas.
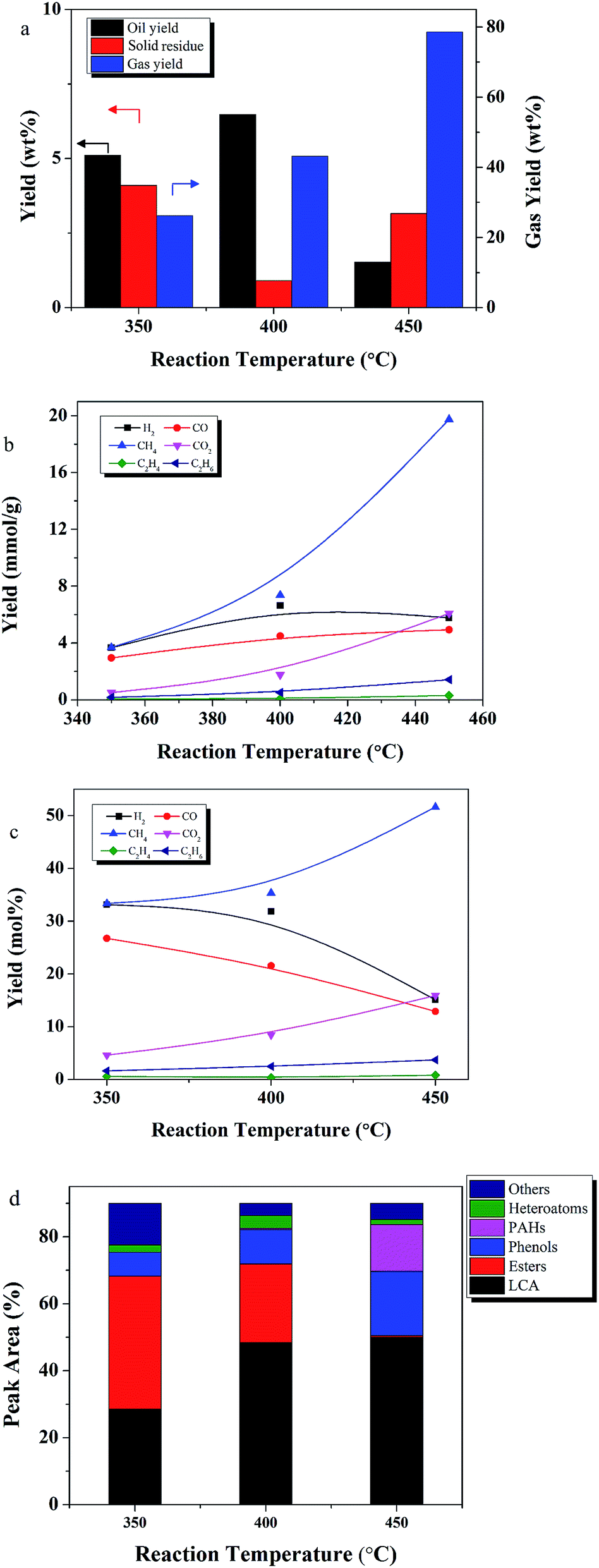 | ||
| Fig. 5 The effect of temperature on (a) yield of each product fraction, (b) yield (mmol g−1) of each gas component, (c) mol% yield of each gas component, and (d) oil product distribution (reaction conditions: 60 min, lignite/ethanol ratio of 1:9, and 50 wt% Ru/C relative to lignite). | ||
Temperature also had a significant influence on gas composition and gas yield. Gas yield increased 4.5 fold as the temperature increased from 350 to 450 °C. The major gas products were CH4, H2, CO, CO2. C2H4, and C2H6 were also detected. Fig. 5c showed that CH4 increased as H2 and CO decreased, indicating that Ru/C has a positive effect on the methanation reaction.7
Esters decreased from 39 to 23% with the increase in temperature from 350 to 400 °C. The esters were likely converted into alkanes, a phenomenon observed previously in the upgrading of extra-heavy crude oil in supercritical ethanol.44–46 When temperature increased from 400 to 450 °C, the esters disappeared, PAHs appeared and were detected at 13%, and phenols doubled. These results are consistent with an enhancement of cyclization reactions and SR formation at high temperatures, since PAHs are considered to be the precursors of SR. Similar results were reported by Zhu et al., who observed that phenolic compounds tend to polymerize into high molecular weight solid products at high temperatures.47
The solid residues were subjected to elemental analysis. Results are shown in Table 2. The carbon content increased significantly compared with the raw material (Table 1), while the oxygen and hydrogen content decreased significantly. These changes may be caused by hydrodeoxygenation and/or deoxygenation processes during liquefaction. The decrease of N content indicated denitrogenation and/or hydrodenitrogenation. H/C and O/C all decreased, while the higher heating value (HHV) increased after liquefaction.
| Experimental conditions | C | H | N | S | O | H/C | O/C | HHV (MJ kg−1) |
|---|---|---|---|---|---|---|---|---|
| 350 °C–60 min | 74.79 | 3.92 | 0.94 | 0.43 | 13.73 | 0.05 | 0.18 | 28.47 |
| 400 °C–60 min | 78.77 | 2.72 | 0.73 | 0.57 | 8.44 | 0.03 | 0.11 | 29.05 |
| 450 °C–60 min | 64.20 | 2.06 | 0.71 | 0.63 | 8.62 | 0.03 | 0.13 | 23.16 |
3.3 The effect of time
The effect of reaction times between 5 and 120 min on the yields of product fractions was investigated at different temperatures from 350 to 450 °C, an E:L ratio of 9:1, and 50 wt% Ru/C relative to lignite.
Fig. 6a shows the effect of different reaction times on the yields of product fractions at 350 °C. An oil yield of 4.1 wt% was obtained in 5 min, which was very close to the volatile material content in the lignite, suggesting that only the readily volatile part of the lignite was liquefied at the low reaction temperature of 350 °C and short reaction time of 5 min. As reaction time increased to 15 min, gas yield doubled and oil yield decreased.
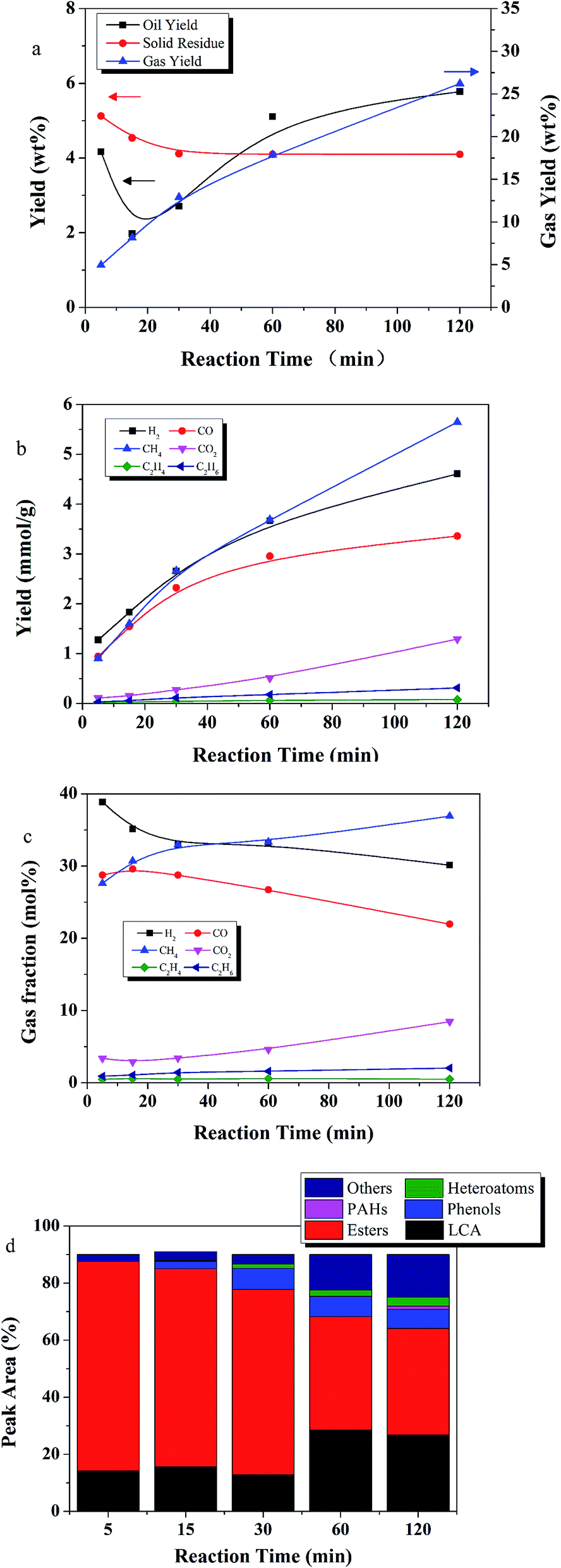 | ||
| Fig. 6 The effect of time on (a) yield of product fractions, (b) gas component yield (mmol g−1), (c) mol% yield of each gas component, and (d) oil product distribution (reaction conditions: 350 °C, E:L ratio of 9:1, 50 wt% catalyst loading relative to lignite). | ||
Fig. 6b and c show detailed information about the gaseous products. The gas composition follows the trend, in order of abundance, CH4 > H2 > CO > CO2 > C2H6 > C2H4. CO and H2 decreased as the CH4 increased, suggesting Ru/C accelerated methanation.
The oil compound distribution is shown in Fig. 6d. At 350 °C, esters were the major compounds, especially at short reaction times. Esters accounted for more than 64% at reactions times of 30 min and shorter. LCAs were the second most predominant species, followed by phenols, others and heteroatoms. No PAHs were detected until 120 min. As reaction time increased, more LCAs and phenols were produced, along with compounds within the other group.
The effects of reaction time at a temperature of 400 °C are depicted in Fig. 7. Oil yield steadily increased from 2.5 to 6.4 wt% as reaction time increased from 5 to 60 min, then leveled off. Solid residue yield decreased as reaction time increased from 5 to 60 min. Gas yield increased with reaction time.
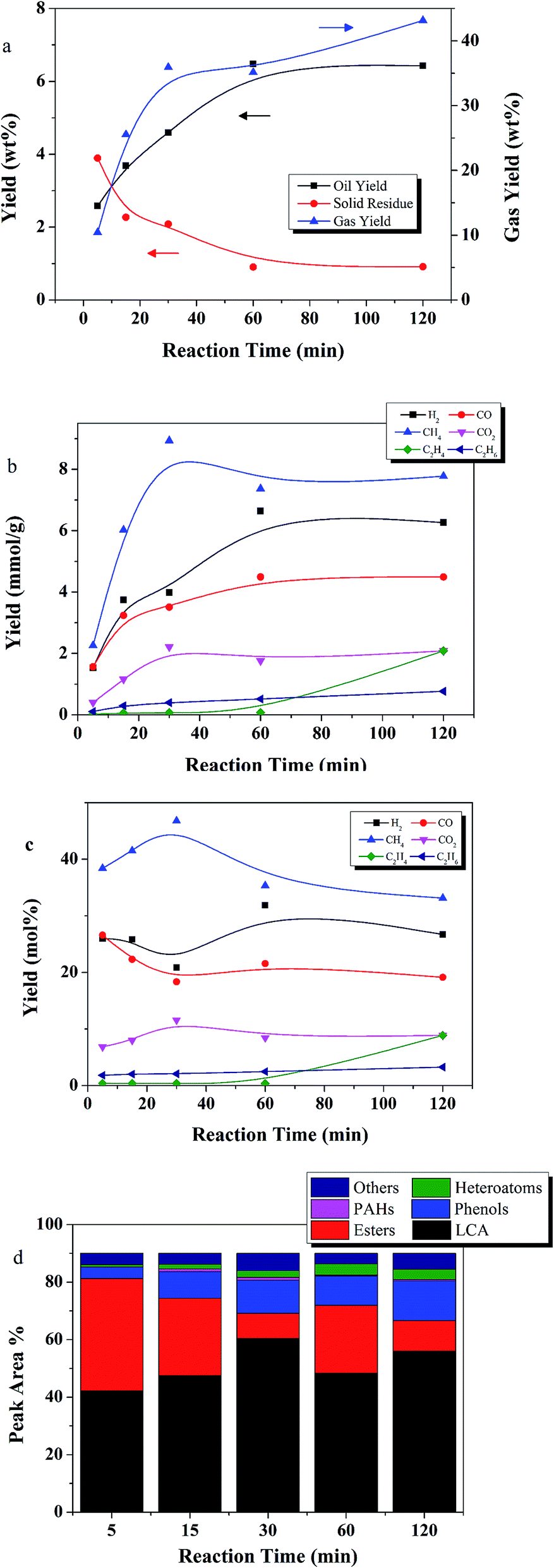 | ||
| Fig. 7 The effect of time on (a) yields of product fractions, (b) gas component yield (mmol g−1), (c) mol% yield of gas components, and (d) oil product distribution (reaction conditions: 400 °C, E:L ratio of 9:1, 50 wt% catalyst loading relative to lignite). | ||
As shown in Fig. 7d, LCAs generally increased with reaction time while yield of esters generally decreased, possibly due to the catalytic effect of Ru/C. Phenols increased from 4% at 5 min to 13% at 120 min. The most abundant compounds were LCAs, accounting for nearly two-thirds of the total area at the optimum reaction time, 60 min.
Thus, increasing reaction temperature from 350 to 400 °C, and lengthening reaction time to 60 min had and overall positive effect on lignite liquefaction with Ru/C.
Fig. 8 shows the temporal variation of products at 450 °C from 5 to 120 min. The gases are the dominant product. Gas yield increased from 32 to 78 wt% as time increased from 5 to 60 min. Thereafter, gas yield leveled off. The SR yield gradually decreased from 2.2 wt% at 5 min, to 1.5 wt% at 60 min, and then leveled off. Thus, gasification was the dominant reaction at 450 °C.
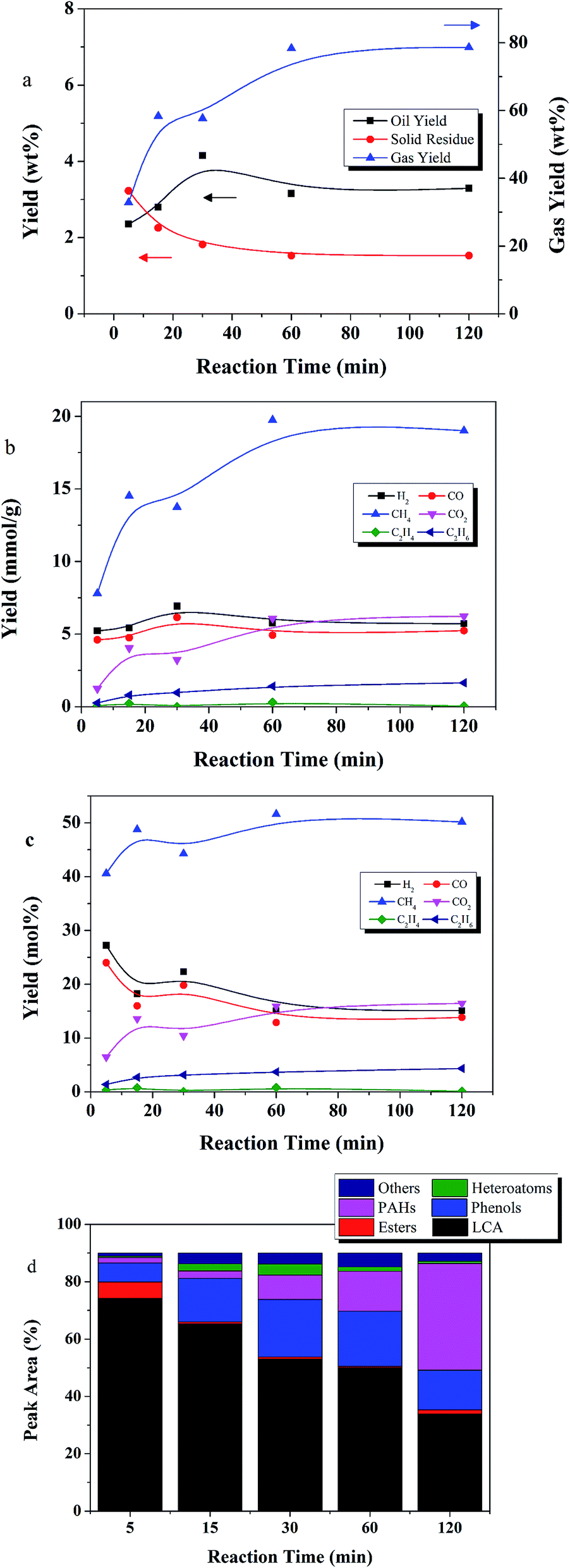 | ||
| Fig. 8 The effect of reaction time on (a) yields of product fractions, (b) gas component yield (mmol g−1), (c) mol% yield of gas components, and (d) oil product distribution (reaction conditions: 450 °C, E:L ratio of 9:1, 50 wt% catalyst loading relative to lignite). | ||
Oil compound distributions were in agreement with this observation. LCAs dominated at 5 min reaction time, accounting for 74% of the peak area, but decreased to 34% peak area at 120 min. From 5 to 120 min, peak area of PAHs increased 20 fold, and peak area of phenols doubled. Guan similarly observed a decrease in LCAs with time while studying the gasification of algae at 500 °C.28 The dominant LCAs at 5 min were C21–C27 alkanes. After 120 min, C12–C18 alkanes became the dominant LCA species, suggesting that LCA bonds were broken as reaction time increased.
Gas phase products CH4, CO2, and C2H6 increased with reaction time. The highest CH4 yield achieved was 19.7 mmol g−1 at 60 min reaction time. After 5 min, only 7.7 mmol g−1 CH4 was produced. CO and H2 achieved high yields of 4.5 and 5.0 mmol g−1, respectively, at 5 min, and changed only slightly thereafter.
Overall, for the high reaction temperature of 450 °C, longer reaction times favored gaseous products and PAH formation, and disfavored LCA yield.
4. Conclusions
Efficient liquefaction of lignite in supercritical ethanol can be achieved with a Ru/C catalyst. Ru/C was effective in utilizing ethanol as an in situ hydrogen supply through steam reforming with water. Additional hydrogen is produced by the water gas shift reaction. During this process, water is converted into gaseous products. The maximum oil yield, 6.4 wt%, and minimum solid residue yield, 0.9 wt%, were obtained at 400 °C and 60 min with an E:L ratio of 9:1, and 50 wt% catalyst loading relative to lignite. The dominant species in the oil were LCAs, esters, and phenols. Gas yield was 20.8 mmol g−1 at these conditions. The major gaseous products were CH4, H2 and CO. Ru/C catalyzed hydrogenation reactions, thereby improving the quality of the oil. Lower or higher temperatures led to reduced oil yields. In general, liquefaction in supercritical ethanol by Ru/C provides an effective way to treat high moisture materials and produce high quality fuels.
Acknowledgements
The authors are grateful for the financial supports from National Natural Science Foundation of China (21307049), Analysis and Measurement Foundation of Kunming University of Science and Technology (2016T20080118) and Collaborative Innovation Center of Western Typical Industry Environmental Pollution Control. The authors thank Dr Natalie Rebacz for helpful suggestions for improving the manuscript.References
- R. Yang, Y. Chen, Y. Wu, D. Hua, M. Yang, C. Li, Z. Chen and J. Liu, Production of liquid fuel via coliquefaction of coal and Dunaliella tertiolecta in a sub-/supercritical water–ethanol system, Energy Fuels, 2013, 27(5), 2619–2627 CrossRef CAS.
- S. Vasireddy, Clean liquid fuels from direct coal liquefaction: Chemistry, catalysis, technological status and challenges, Energy Environ. Sci., 2011, 4(2), 311–345 Search PubMed.
- P. Duan, X. Bai, Y. Xu, A. Zhang, F. Wang, L. Zhang and J. Miao, Non-catalytic hydropyrolysis of microalgae to produce liquid biofuels, Bioresour. Technol., 2013, 136, 626–634 CrossRef CAS PubMed.
- B. Jin, P. Duan, Y. Xu, B. Wang, F. Wang and L. Zhang, Lewis acid-catalyzed in situ transesterification/esterification of microalgae in supercritical ethanol, Bioresour. Technol., 2014, 162, 341–349 CrossRef CAS PubMed.
- X. Li, D. E. Priyanto, R. Ashida and K. Miura, Two-stage conversion of low-rank coal or biomass into liquid fuel under mild conditions, Energy Fuels, 2015, 29(5), 3127–3133 CrossRef CAS.
- T. M. Brown, P. Duan and P. E. Savage, Hydrothermal liquefaction and gasification of Nannochloropsis sp., Energy Fuels, 2010, 24(6), 3639–3646 CrossRef CAS.
- P. Duan and P. E. Savage, Hydrothermal liquefaction of a microalga with heterogeneous catalysts, Ind. Eng. Chem. Res., 2011, 50(1), 52–61 CrossRef CAS.
- P. Duan, Z. Chang, Y. Xu, X. Bai, F. Wang and L. Zhang, Hydrothermal processing of duckweed: effect of reaction conditions on product distribution and composition, Bioresour. Technol., 2013, 135, 710–719 CrossRef CAS PubMed.
- B. Jin, P. Duan, Y. Xu, F. Wang and Y. Fan, Co-liquefaction of micro- and macroalgae in subcritical water, Bioresour. Technol., 2013, 149, 103–110 CrossRef CAS PubMed.
- H. Li, S. Hurley and C. Xu, Liquefactions of peat in supercritical water with a novel iron catalyst, Fuel, 2011, 90(1), 412–420 CrossRef CAS.
- O. N. Fedyaeva and A. A. Vostrikov, Non-isothermal liquefaction of liptobiolith coal in supercritical water flow and effect of zinc additives, J. Supercrit. Fluids, 2013, 83, 86–96 CrossRef CAS.
- J. Chumpoo and P. Prasassarakich, Bio-oil from hydro-liquefaction of bagasse in supercritical ethanol, Energy Fuels, 2010, 24(3), 2071–2077 CrossRef CAS.
- H. Mazaheri, K. T. Lee, S. Bhatia and A. R. Mohamed, Sub/supercritical liquefaction of oil palm fruit press fiber for the production of bio-oil: effect of solvents, Bioresour. Technol., 2010, 101, 7641–7647 CrossRef CAS PubMed.
- F. Mariño, Hydrogen production via catalytic gasification of ethanol. A mechanism proposal over copper–nickel catalysts, Int. J. Hydrogen Energy, 2004, 29(1), 67–71 CrossRef.
- D. Liu, Q. Li, A. Zhao, L. Song, P. Wu and Z. Yan, Hydro-liquefaction of sawdust and its three components in supercritical ethanol with [BMIM]Cl/NiCl2 catalyst, Chem.–Eng. J., 2015, 279, 921–928 CrossRef CAS.
- A. Riaz, C. S. Kim, Y. Kim and J. Kim, High-yield and high-calorific bio-oil production from concentrated sulfuric acid hydrolysis lignin in supercritical ethanol, Fuel, 2016, 172, 238–247 CrossRef CAS.
- S. Brand and J. Kim, Liquefaction of major lignocellulosic biomass constituents in supercritical ethanol, Energy, 2015, 80, 64–74 CrossRef CAS.
- H. Li, X. Yuan, G. Zeng, D. Huang, H. Huang, J. Tong, Q. You, J. Zhang and M. Zhou, The formation of bio-oil from sludge by deoxy-liquefaction in supercritical ethanol, Bioresour. Technol., 2010, 101(8), 2860–2866 CrossRef CAS PubMed.
- Y. Le, Y. Li and P. E. Savage, Near- and supercritical ethanol treatment of biocrude from hydrothermal liquefaction of microalgae, Bioresour. Technol., 2016, 211, 779–782 CrossRef PubMed.
- H. Huang, X. Yuan, G. Zeng, J. Wang, H. Li, C. Zhou, X. Pei, Q. You and L. Chen, Thermochemical liquefaction characteristics of microalgae in sub- and supercritical ethanol, Fuel Process. Technol., 2011, 92(1), 147–153 CrossRef CAS.
- F. Mondragon, G. Quintero, A. Jaramillo, J. Fernandez and P. J. Hall, The catalytic liquefaction of coal in the presence of ethanol, Fuel Process. Technol., 1998, 53(3), 171–181 CrossRef CAS.
- J. Zhang and Y. Zhang, Hydrothermal liquefaction of microalgae in an ethanol–water co-solvent to produce biocrude oil, Energy Fuels, 2014, 28(8), 5178–5183 CrossRef CAS.
- Y. Chen, Y. Wu, P. Zhang, D. Hua, M. Yang, C. Li, Z. Chen and J. Liu, Direct liquefaction of Dunaliella tertiolecta for bio-oil in sub/supercritical ethanol–water, Bioresour. Technol., 2012, 124, 190–198 CrossRef CAS PubMed.
- K. Khampuang, N. Boreriboon and P. Prasassarakich, Alkali catalyzed liquefaction of corncob in supercritical ethanol–water, Biomass Bioenergy, 2015, 83, 460–466 CrossRef CAS.
- J. Yu, X. Lu, Y. Shi, Q. Chen, Q. Guan, P. Ning, S. Tian and J. Gu, Catalytic gasification of lignite in supercritical water with Ru/CeO2–ZrO2, Int. J. Hydrogen Energy, 2016, 41, 4579–4591 CrossRef CAS.
- Q. Guan, X. Huang, J. Liu, J. Gu, R. Miao, Q. Chen and P. Ning, Supercritical water gasification of phenol using a Ru/CeO2 catalyst, Chem.–Eng. J., 2016, 283, 358–365 CrossRef CAS.
- J. A. Onwudili and P. T. Williams, Enhanced methane and hydrogen yields from catalytic supercritical water gasification of pine wood sawdust via pre-processing in subcritical water, RSC Adv., 2013, 3(30), 12432–12442 RSC.
- Q. Guan, P. E. Savage and C. Wei, Gasification of alga Nannochloropsis sp. in supercritical water, J. Supercrit. Fluids, 2012, 61, 139–145 CrossRef CAS.
- Q. Guan, C. Wei and P. E. Savage, Hydrothermal gasification of Nannochloropsis sp. with Ru/C, Energy Fuels, 2012, 26(7), 4575–4582 CrossRef CAS.
- C. Xu and J. Donald, Upgrading peat to gas and liquid fuels in supercritical water with catalysts, Fuel, 2012, 102, 16–25 CrossRef CAS.
- S. N. Cheng, I. D'Cruz, M. C. Wang, M. Leitch and C. B. Xu, Highly efficient liquefaction of woody biomass in hot-compressed alcohol–water co-solvents, Energy Fuels, 2010, 24, 4659–4667 CrossRef CAS.
- P. Duan, B. Jin, Y. Xu and F. Wang, Co-pyrolysis of microalgae and waste rubber tire in supercritical ethanol, Chem.–Eng. J., 2015, 269, 262–271 CrossRef CAS.
- H.-Y. Lu, X.-Y. Wei, R. Yu, Y.-L. Peng, X.-Z. Qi, L.-M. Qie, Q. Wei, J. Lv, Z.-M. Zong, W. Zhao, Y.-P. Zhao, Z.-H. Ni and L. Wu, Sequential thermal dissolution of huolinguole lignite in methanol and ethanol, Energy Fuels, 2011, 25(6), 2741–2745 CrossRef CAS.
- Z. Tang, Y. Zhang and Q. X. Guo, Catalytic hydrocracking of pyrolytic lignin to liquid fuel in supercritical ethanol, Ind. Eng. Chem. Res., 2010, 49(5), 2040–2046 CrossRef CAS.
- K. Charutawai, S. Ngamprasertsith and P. Prasassarakich, Supercritical desulfurization of low rank coal with ethanol/KOH, Fuel Process. Technol., 2003, 84(1–3), 207–216 CrossRef CAS.
- J. W. Chen, C. B. Muchmore and A. C. Kent, Extraction and desulfurization of chemically degraded coal with supercritical fluids. Final report, july 1, 1984-june 30, 1985, 1985 Search PubMed.
- D. Borah and M. K. Baruah, Electron transfer process: 1. Removal of organic sulphur from high sulphur Indian coal, Fuel, 1999, 78(9), 1083–1088 CrossRef CAS.
- J. S. Gethner, Thermal and oxidation chemistry of coal at low temperatures, Fuel, 1985, 64(10), 1443–1446 CrossRef CAS.
- A. M. Vassallo, Y. L. Liu, L. S. K. Pang and M. A. Wilson, Infrared spectroscopy of coal maceral concentrates at elevated temperatures, Fuel, 1991, 70(5), 635–639 CrossRef CAS.
- J. Zhang, W. T. Chen, P. Zhang, Z. Luo and Y. Zhang, Hydrothermal liquefaction of chlorella pyrenoidosa in sub- and supercritical ethanol with heterogeneous catalysts, Bioresour. Technol., 2013, 133, 389–397 CrossRef CAS PubMed.
- P. Duan, B. Jin, Y. Xu, Y. Yang, X. Bai, F. Wang, L. Zhang and J. Miao, Thermo-chemical conversion of Chlorella pyrenoidosa to liquid biofuels, Bioresour. Technol., 2013, 133, 197–205 CrossRef CAS PubMed.
- C. M. Huelsman and P. E. Savage, Intermediates and kinetics for phenol gasification in supercritical water, Phys. Chem. Chem. Phys., 2012, 14(8), 2900–2910 RSC.
- L. Yang, R. Ma, Z. Ma and Y. Li, Catalytic conversion of chlorella pyrenoidosa to biofuels in supercritical alcohols over zeolites, Bioresour. Technol., 2016, 209, 313–317 CrossRef CAS PubMed.
- R. O. Idem, S. P. R. Katikaneni and N. N. Bakhshi, Thermal cracking of canola oil: reaction products in the presence and absence of steam, Energy Fuels, 1996, 10(6), 1150–1162 CrossRef CAS.
- C. Ovalles, E. Filgueiras, A. Morales, C. E. Scott, F. Gonzalez-Gimenez and B. Pierre Embaid, Use of a dispersed iron catalyst for upgrading extra-heavy crude oil using methane as source of hydrogen, Fuel, 2003, 82(8), 887–892 CrossRef CAS.
- T. Yang, Y. Jie, B. Li, X. Kai, Z. Yan and R. Li, Catalytic hydrodeoxygenation of crude bio-oil over an unsupported bimetallic dispersed catalyst in supercritical ethanol, Fuel Process. Technol., 2016, 148, 19–27 CrossRef CAS.
- Z. Zhu, L. Rosendahl, S. S. Toor, D. Yu and G. Chen, Hydrothermal liquefaction of barley straw to bio-crude oil: Effects of reaction temperature and aqueous phase recirculation, Appl. Energy, 2015, 137, 183–192 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21818k |
| This journal is © The Royal Society of Chemistry 2017 |