
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerizable ionic liquids and polymeric ionic liquids: facile synthesis of ionic liquids containing ethylene oxide repeating unit via methanesulfonate and their electrochemical properties†
Borong Wuabcde,
Zhen-Wei Zhanga,
Mu-Hua Huang *a and
Yiyuan Pengd
*a and
Yiyuan Pengd
aSchool of Materials Science and Engineering, Beijing Institute of Technology, Beijing, 100081, China. E-mail: mhhuang@bit.edu.cn
bBeijing Key Laboratory of Environment Science and Engineering, Beijing 100081, China
cCollaborative Innovation Center of Electric Vehicles in Beijing, Beijing 100081, China
dBeijing Higher Institution Engineering Research Center of Power Battery and Chemical Energy Materials, Beijing 100081, China
eKey Laboratory of Small Functional Organic Molecule, Ministry of Education, Jiangxi Normal University, Nanchang 330022, China
First published on 17th January 2017
Abstract
Polymeric ionic liquids have shown great potential in numerous application fields, and an easy access to polymerizable monomer is the key to the polymerized ionic liquids. The polymerizable monomer is usually synthesized via the quaternization of a corresponding bromide, which is usually prepared from bromination of the corresponding alcohol but with a low yield. In this study, a polymerizable monomer was prepared by quaternization of a mesylate, derived from the corresponding alcohol, followed by an ion exchange reaction. The polymerizable ILs M1–M4 showed ionic conductivity as high as 10−3 S cm−1, and the oxygen effect was observed with the lithium salt. Polymeric ionic liquids P1 and P2 were prepared via radical polymerization on M1 and M2, and the ionic conductivity of polymeric ionic liquid P2 was as high as 2.2 × 10−4 S cm−1 with electrochemical stability exceeding 7 V (vs. Li/Li+).
Introduction
With the fast development of electric vehicles and mobile electric devices, rechargeable batteries with high performance are highly required. Lithium ion batteries (LIBs) have been improved rapidly and possess high energy density and excellent cyclic stability. However, several safety problems have been encountered and numerous accidents have occurred due to LIBs. To solve the safety issue of LIBs, electrolytes are the main components that could be improved, which function as the charge carriers between electrodes. Traditionally, organic liquid electrolytes, such as carbonate ester, were widely used,1 which have drawbacks such as volatility, leakage, flammability, and toxicity. Thus, the synthesis of solvent-free electrolytes has attracted the attention from materials scientists.Solid polymer electrolyte (SPE)2,3 is a popular concept used by people to reconstruct old electronic device systems. Polyethylene oxide (PEO)-based SPE has particularly been paid attention to for application in LIBs. Pure PEO has poor ionic conductivity, which hardly meets the requirement for LIBs, and the addition of carbonate ester makes the leakage of carbonate ester a possibility. Ionic liquid has numerous advantages:4 low vapor pressure, non-flammability, and good ion conduction, and these properties make it a better candidate for application in LIBs.
Solid or quasi-solid electrolytes,5 such as polymer electrolytes, usually have good mechanical stability and simple processing. Among them, polymeric ionic liquids (PILs)6–8 are the most promising solid electrolyte candidates, since they combine the advantages of ionic liquids and polymers. Plenty of pioneering studies have been directed at obtaining high ionic conductivity PILs.3,9–26 Since the first PIL synthesized by the Ohno group,27 several attempts have been made to investigate the tether effect28,29 between the charge center and backbone on the ionic conductivity improvement of the PILs. The introduction of oxygen atom in the spacer was found to decrease the ionic conductivity of PILs30,31 since the oxygen trapped the mobile cation was different from that in the traditional SPE. The Vidal group researched the nitrogen element in the spacer and an ionic conductivity as high as 6.5 × 10−4 S cm−1 at 20 °C was achieved.32 From the reported study, it was strongly suggested that the tether effect played an important role in determining the ionic conductivity. However, a systematic study on the tether effect as well as the heteroatom effect on ionic conductivity of ILs as well as PILs was not available.
Moreover, an easy access to polymerizable ILs is highly required in order to accelerate the development of high performance PILs suitable for battery applications. In this sense, the synthetic method towards ionic liquid monomers is very important as well as is study on the structure–property relationship of ILs and PILs. In this study, the design and synthesis of polymerizable ionic liquid monomer M1 to M4 as well as PILs P1 and P2 is presented (Fig. 1) towards developing a highly efficient synthetic procedure. In addition, the ionic conductivity of the synthesized ILs and PILs was investigated.
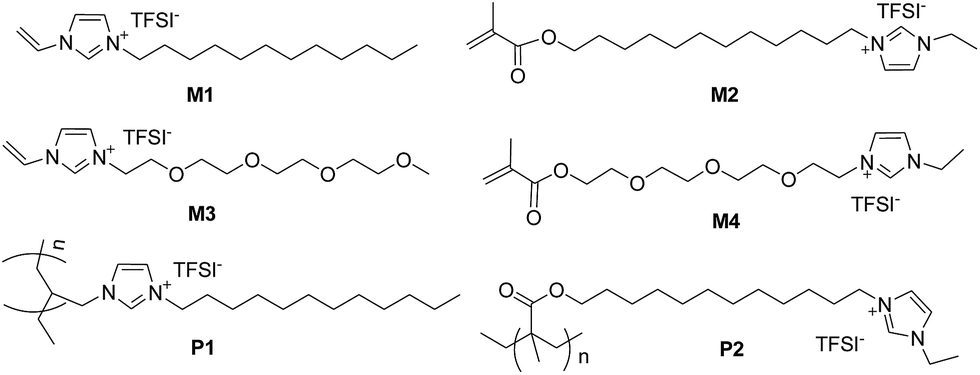 | ||
| Fig. 1 Structure of some polymerizable ionic liquids and polymeric ionic liquids in this study. | ||
Results and discussion
Synthesis and characterization of polymerizable ionic liquid monomer M1 to M4
Our study initiated with the synthesis of polymerizable ionic liquid M1. According to the reported procedure, we tried the quaternization reaction between bromide 1 and N-vinylimidazole 2 in organic solvents, such as methanol and chloroform, and the imidazolium bromide was obtained as a precipitate from acetonitrile. In order to make the synthetic procedure greener and with higher economic efficiency, the solvent-free quaternization reaction was carried out at 80 °C. The reaction was effective and gave the target product in 96% isolated yield, which was transferred into polymerizable ionic liquid M1 by exchange with LiTFSI in 90% isolated yield (Scheme 1).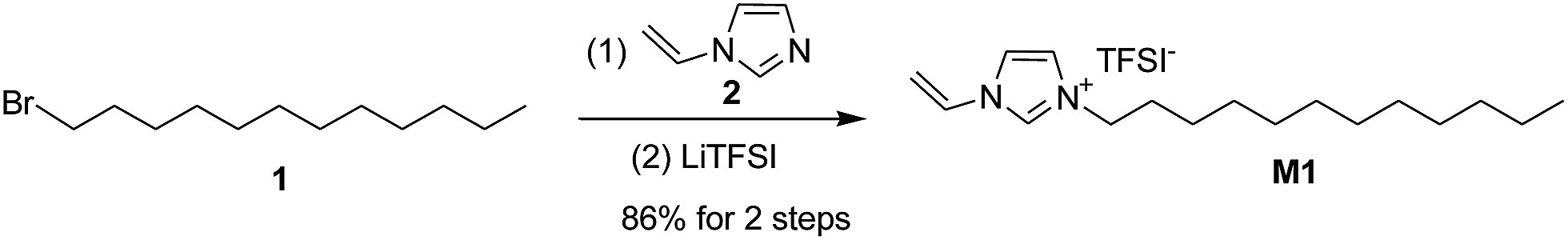 | ||
| Scheme 1 Synthesis of ionic liquid M1 via alkylbromide 1. | ||
The chemical structure of M1 was determined by examining its 1H-NMR spectrum (Fig. 2), which was very similar to that of its corresponding bromide (Fig. S1†).
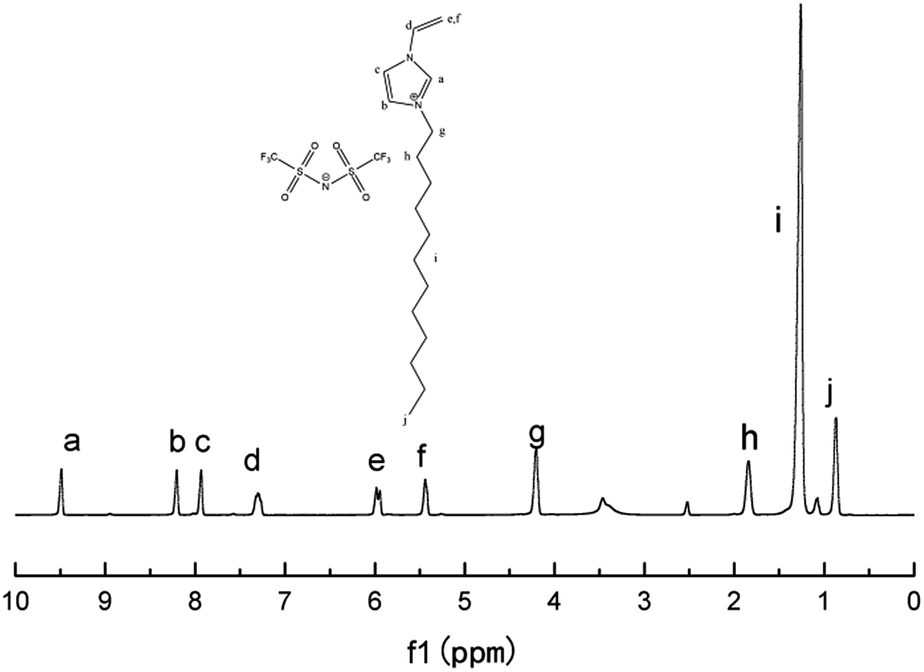 | ||
| Fig. 2 1H-NMR spectrum of ionic liquid monomer M1 in DMSO-D6. | ||
The structure was further confirmed by high-resolution mass spectroscopy (Fig. 3 and 4).
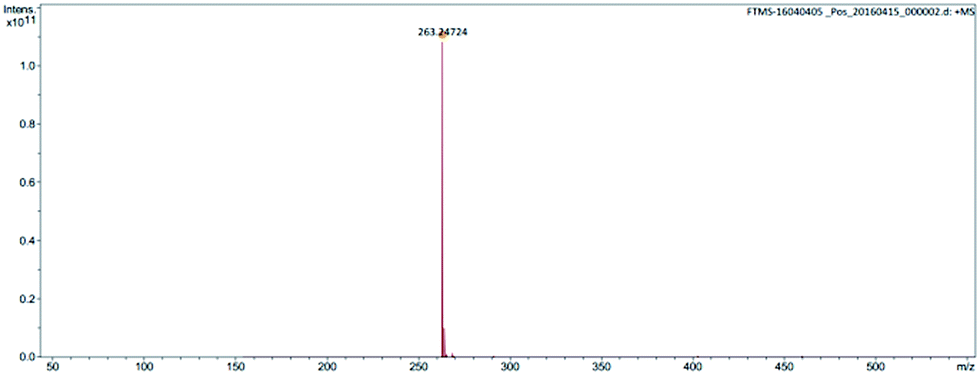 | ||
| Fig. 3 Mass spectrum of cation of ionic liquid monomer M1. | ||
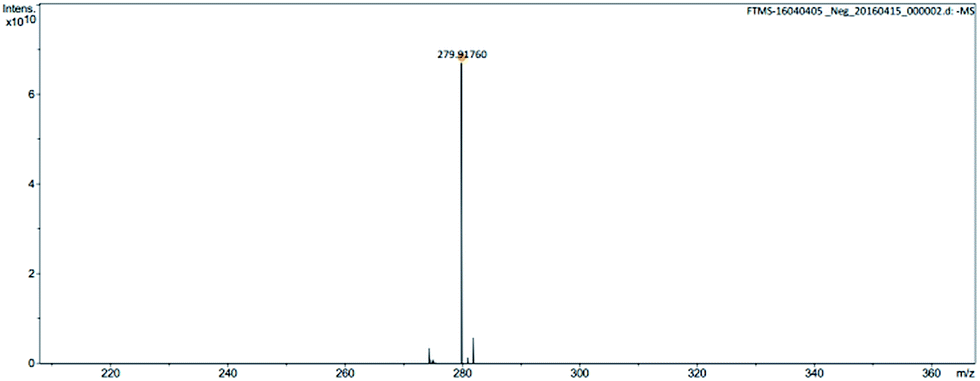 | ||
| Fig. 4 Mass spectrum of anion of ionic liquid monomer M1. | ||
With M1 prepared, we moved to the synthesis of M2 using dibromide 3 (Scheme 2).
 | ||
| Scheme 2 Synthesis of ionic liquid M2 via alkylbromide 3. | ||
The commercially available dibromide 3 was monosubstituted by methacrylic acid in the presence of Cs2CO3. It was found that the monoester reaction outcome strongly depended on the reaction conditions. Using excess dibromide and adding acid into the dibromide solution, monoester 4 was isolated in 45% yield after flash column chromatography. Bromide 4 was transferred into polymerizable ionic liquid M2 in 95% yield by treating 4 with N-ethylimidazole, 5 followed by ion exchange with LiTFSI.
The chemical structure of M2 was determined by examining its 1H-NMR spectrum (Fig. 5), which was very similar to that of its corresponding bromide (Fig. S2†). This was further confirmed via high-resolution mass spectroscopy (Fig. 6).
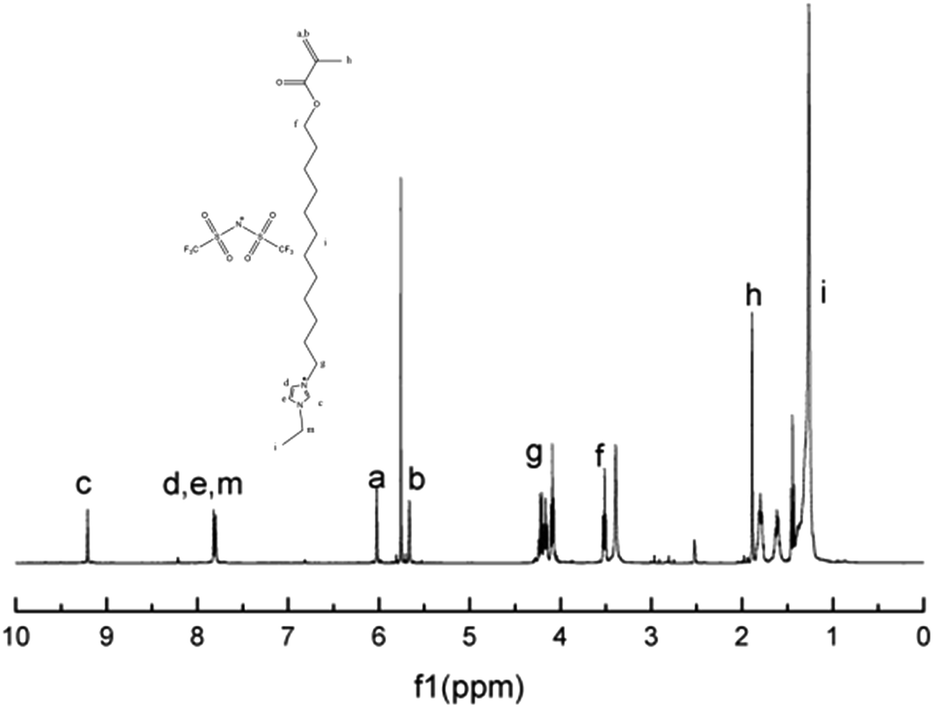 | ||
| Fig. 5 1H-NMR spectrum of ionic liquid monomer M2. | ||
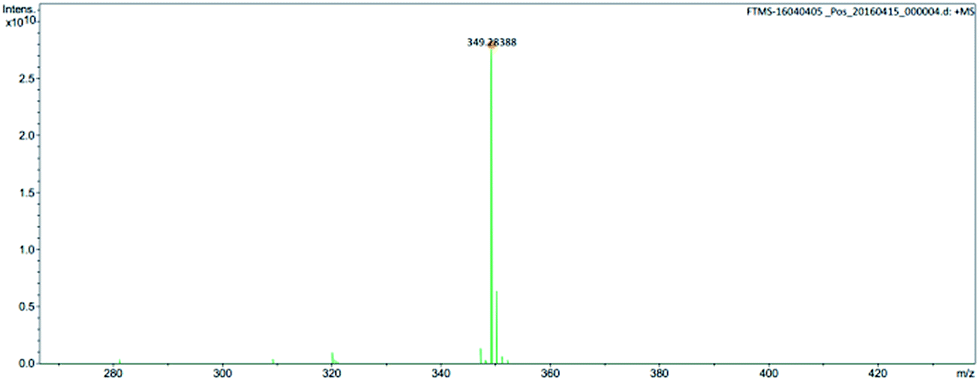 | ||
| Fig. 6 Mass spectrum of anion of ionic liquid monomer M2. | ||
With the experience of quaternization reaction and ion exchange reaction obtained from the synthesis of M1 and M2, we began with the synthesis of M3 and M4 following the same procedure. However, the bromination of alcohol 6 did not work using either HBr or PBr3. As an alternative, alcohol 6 was transformed into mesylate 7 (Fig. S3†) in 99% isolated yield by treating 6 with methanesulfonyl chloride (MsCl) in the presence of Et3N. To our delight, the quaternization reaction between mesylate 7 and N-vinylimidazole 2 worked nicely to give imidazolium mesylate 8 in 97% yield, which was transferred into M3 in 100% yield (Scheme 3).
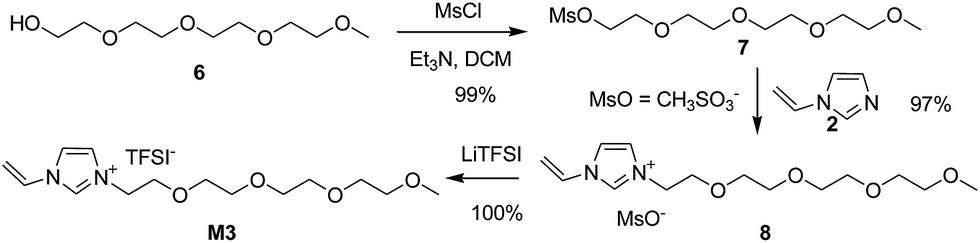 | ||
| Scheme 3 Synthesis of ionic liquid M3 via mesylate. | ||
The chemical structure of M3 was determined by examining its 1H-NMR spectrum (Fig. 7).
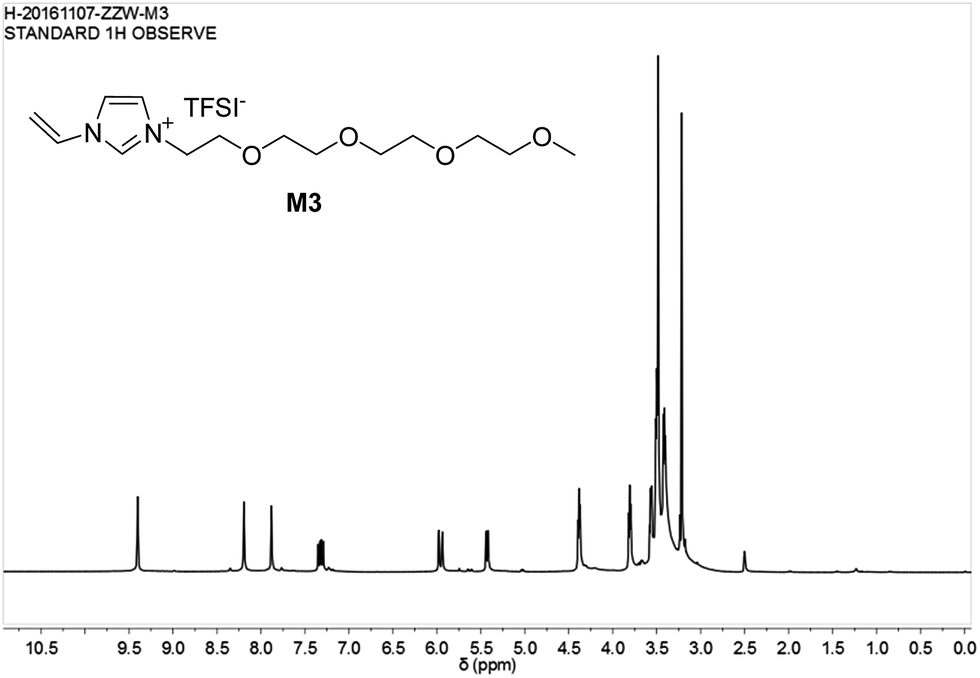 | ||
| Fig. 7 1H-NMR spectrum of ionic liquid monomer M3. | ||
It was further confirmed by high-resolution mass spectroscopy of M3 (Fig. S6†).
Encouraged by success on the synthesis of M3, we moved to the synthesis of M4 starting from diol 9. Diol 9 was transformed into dimesylate 10 (Fig. S4†) in 99% isolated yield by treating 6 with methanesulfonyl chloride (MsCl) in the presence of Et3N. The monosubstitution of dimesylate 10 was achieved using methacrylic acid in basic conditions to give monoester 11 (Fig. S5†) in 45% isolated yield. Finally, ionic liquid M4 was obtained in 94% yield by the quaternization reaction between mesylate 11 and N-ethylimidazole 5, followed by ion exchange reaction with LiTFSI (Scheme 4).
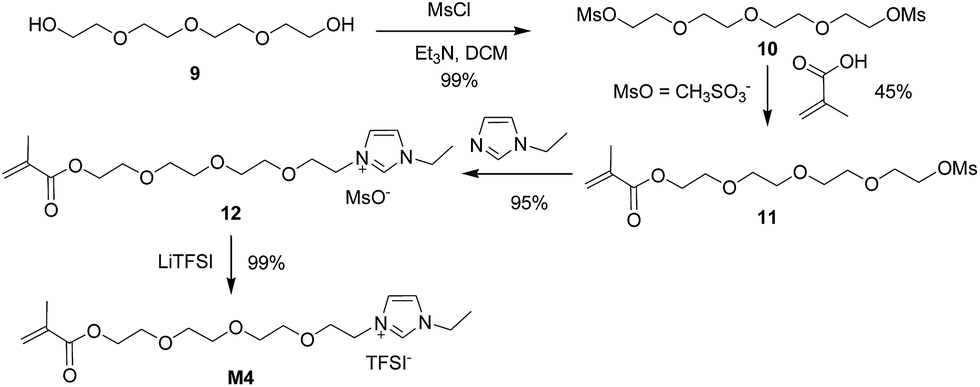 | ||
| Scheme 4 Synthesis of ionic liquid M4 via mesylate. | ||
The mesylate anion of IL 12 was transferred into TFSI− by exchange with LiTFSI in water, and M4 was obtained. This was confirmed via the 1H NMR spectrum of M4 (Fig. 8).
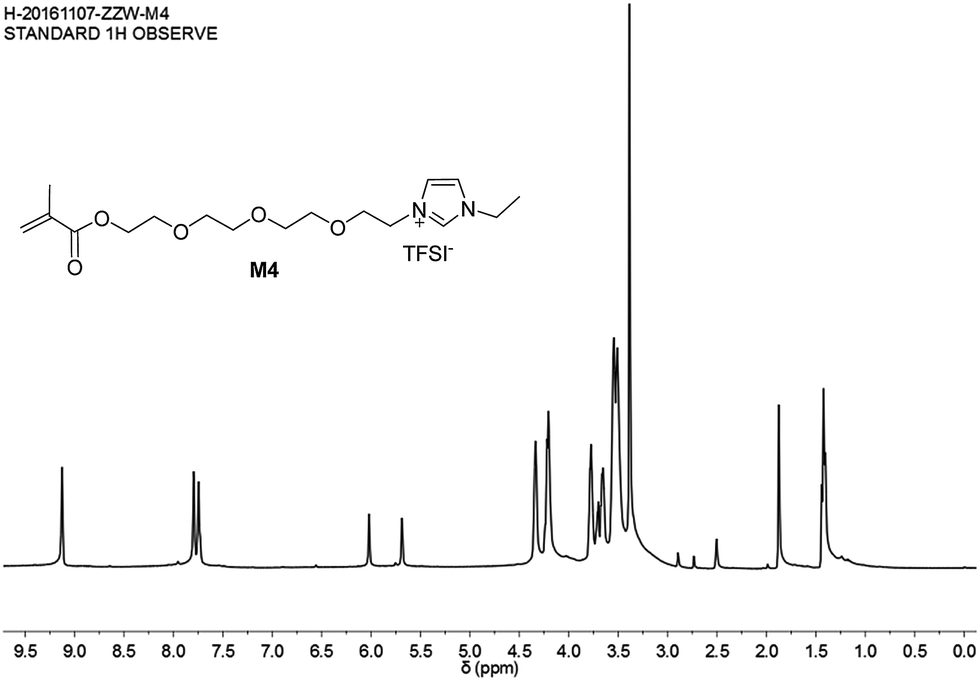 | ||
| Fig. 8 1H-NMR spectrum of ionic liquid monomer M4. | ||
Synthesis of polymeric ionic liquid P1 and P2
With polymerizable ionic liquid monomer M1 and M2 in hand, we conducted the radical polymerization reaction. M1 was first polymerized in the presence of AIBN in DMF with heating to give P1 as a rubber-like solid; its 1H-NMR spectrum showed the complete disappearance of vinyl group between 5–6 ppm (Fig. 9).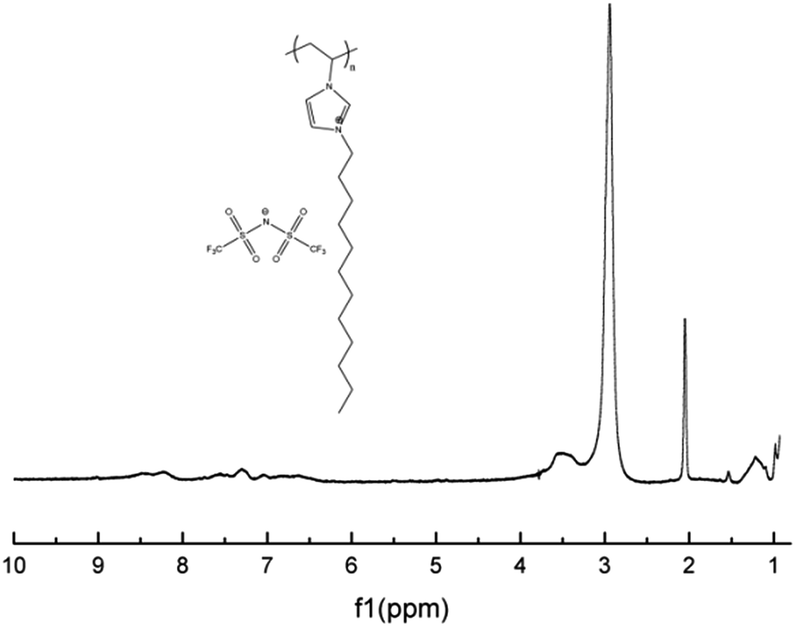 | ||
| Fig. 9 1H-NMR spectrum of polymeric ionic liquid P1. | ||
Similarly, M2 was polymerized to give P2 with AIBN as the initiator. The structure of P2 was characterized by examining its 1H-NMR spectrum (Fig. 10).
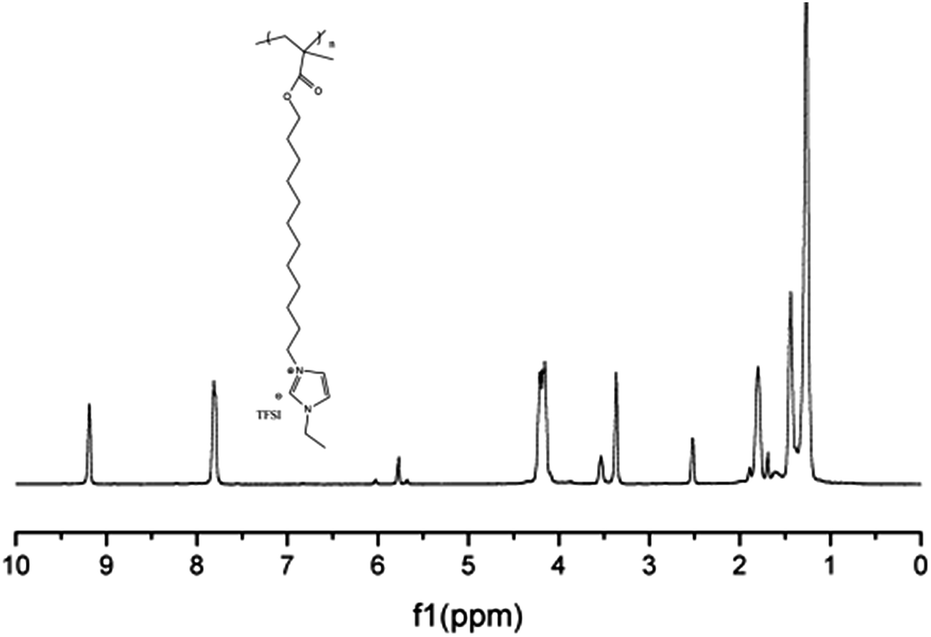 | ||
| Fig. 10 1H-NMR spectrum of polymeric ionic liquid P2. | ||
Molecular weight determinations were attempted using GPC, but unfortunately no peak could be observed despite several attempts. As an alternative, we carried out the molecular weight measurement via 1H-NMR observations according to known methods,33 which gave molecular weights of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 588 g mol−1 for P1 and 15782 g mol−1 for P2.
588 g mol−1 for P1 and 15782 g mol−1 for P2.
Ionic conductivity of ionic liquid monomer M1 to M4 and polymer P1 and P2
The ionic conductivity of pure ionic liquids M1 to M4 was measured to be 1.36 × 10−3 S cm−1, 1.3 × 10−3 S cm−1, 1.79 × 10−3 S cm−1 and 1.89 × 10−3 S cm−1, respectively, at 30 °C.The ionic conductivity of P1 and P2 was dependent on the temperature, as shown in Fig. 11.
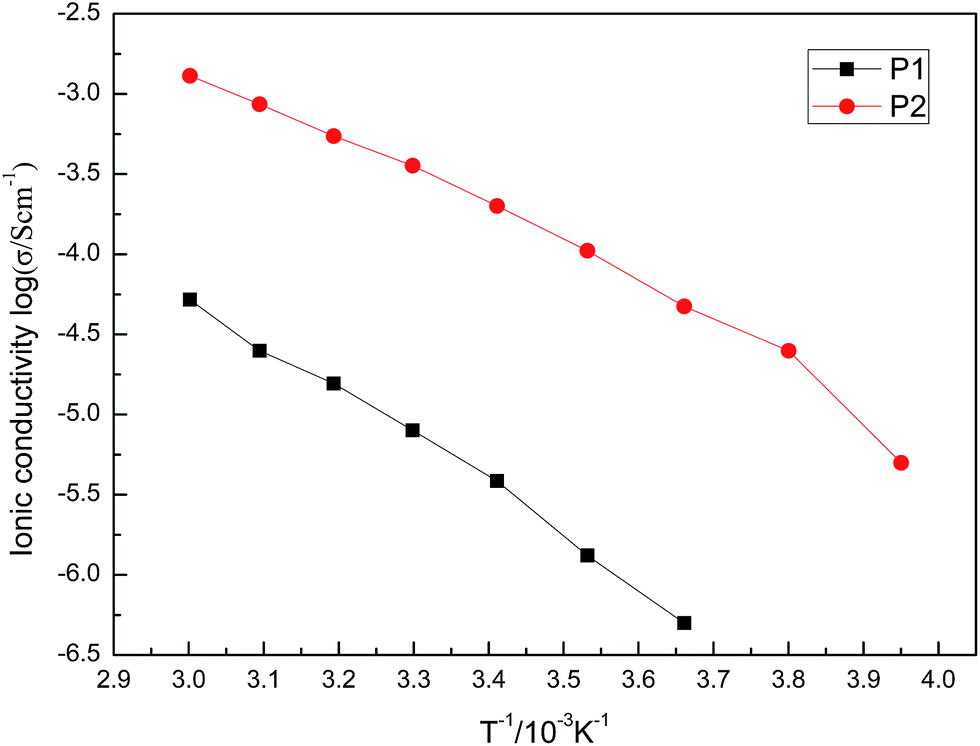 | ||
| Fig. 11 Temperature dependence of polymer P1 and P2. | ||
It was shown that P1 and P2 have higher ionic conductivity at higher temperatures, which arises from the movement improvement of the polymer chains and free volume. It also showed a nearly linear relationship between logσ and T. The activation energies of P1 and P2 were calculated to be 58.58 kJ mol−1 and 48.73 kJ mol−1, respectively. The ionic conductivity of P1 and P2 was measured to be 1.1 × 10−6 S cm−1 and 2.2 × 10−4 S cm−1, respectively, at 30 °C. Ionic conductivity of P1 decreased to one thousandth of M1 after polymerization of M1, which was observed for most of the PILs. However, P2 decreased to one tenth of M2 after M2 polymerization. This 100-fold difference of ionic conductivity definitely comes from the structural differences between P1 and P2. The long alkyl chain between the polymer backbone and charge centre keeps the TSFI− ions moving more freely compared with the case charge centre attached to the polymer backbone. Essentially, the large difference of ionic conductivity also corresponds to the big difference of Tg between P1 and P2, at −22 °C vs. −67 °C.
Lithium salt concentration dependence of M1 to M4 and P1 to P2
In order to evaluate the potential of ILs and PILs in LIBs, the interaction between ILs (PILs) and lithium salt was investigated.As shown in Fig. 12, with the increase of the ILs M1–M4 content, the ionic conductivity of the LiTFSI and ILs composites went up. Pure LiTFSI had ionic conductivity that was too low to be measured. When 94 wt% M1 was added into LiTFSI, the ionic conductivity increased to 8 × 10−4 S cm−1 at 30 °C; similarly, 9.6 × 10−4 S cm−1 for M2, 1.17 × 10−3 S cm−1 for M3, and 1.43 × 10−3 S cm−1 for M4 was observed. Clearly, oxygen-containing ionic liquids M3 and M4 showed higher ionic conductivity than all-carbon chain ionic liquids M1 and M2. This may be the result of dissociation ability of the ethylene oxide chain with the lithium salt. M2 had higher ionic conductivity than M1 and M4 higher than M3 under the same conditions. This may be caused by the longer chain of M2 and M4 than that of M1 and M3 as well as the plasticizing effect of the ester moiety.
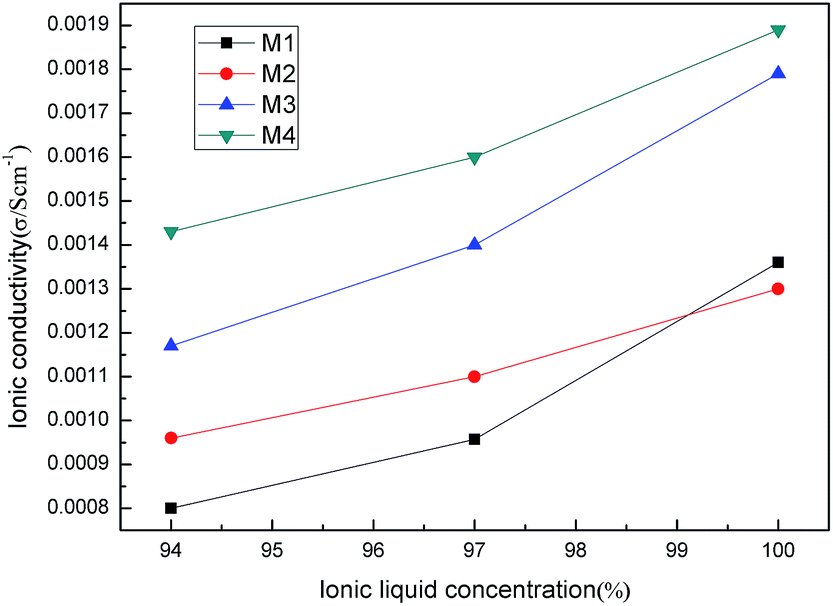 | ||
| Fig. 12 IL helps lithium salt ionic conductivity at 30 °C. | ||
In the case of PILs, P2 was investigated towards LiTFSI due to its high ionic conductivity. When LiTFSI was introduced into P2 during the polymerization process, the ionic conductivity of the composite decreased. The addition of LiTFSI to P2 may make a dimer of TFSI− formed, which will have bigger ion volume corresponding to the poor mobility, and thus provides lower ionic conductivity than pure P2. Higher Li-salt concentration resulted in higher dimer concentration in the system and lower ionic conductivity (Fig. 13).
 | ||
| Fig. 13 Lithium salt concentration dependence of P2. | ||
Electrochemical stability
For further application of P2 as solid electrolyte, it is very important to know its electrochemical stability. Generally, the point corresponding to the irreversible change is defined to be the decomposition voltage, which, in other words, means the stability boundary or so-called window.As shown in Fig. 14, non-oxidizable electrochemical stability of P2 exceeded 7 V (vs. Li/Li+), whereas anti-reductive electrochemical stability was about 2.5 V. This demonstrated high potential for applications in high-voltage systems.
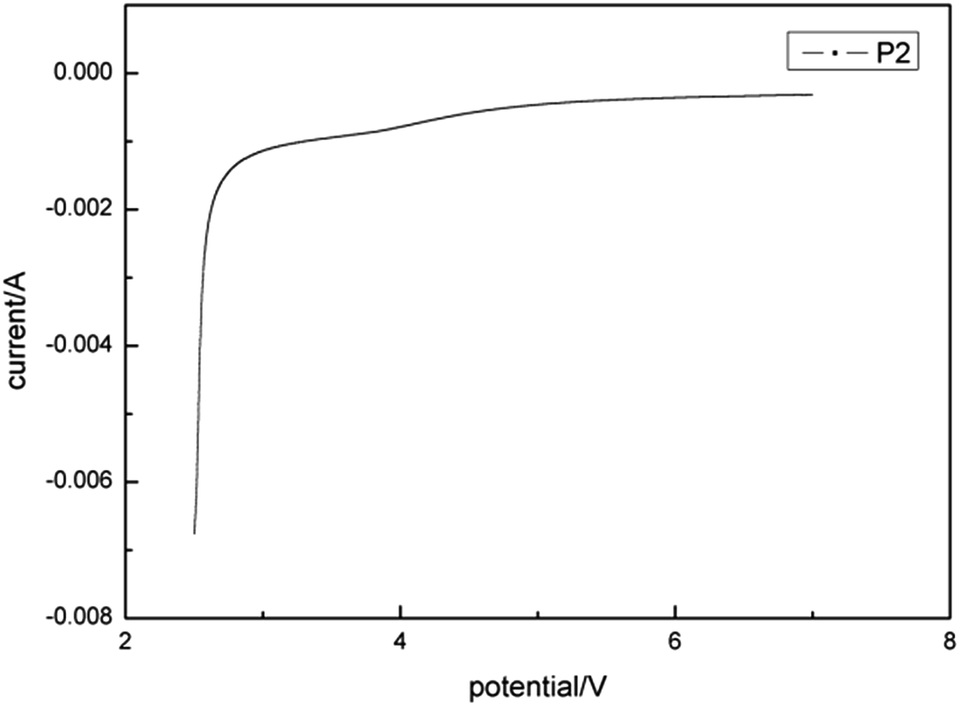 | ||
| Fig. 14 Electrochemical stability of P2. | ||
Thermal stability
An ideal solid electrolyte for lithium batteries will have high thermal stability. The decomposition temperature of P1 and P2 was measured by TGA (Fig. S7†).P1 lost 5% weight at 369 °C, whereas P2 at 234 °C, but P2 has higher maximum decomposed temperature, and decomposed more completely than P1.
Experimental
Materials
1-Vinyl-1H-imidazole, 1-bromoundecane, azodiisobutyronitrile (AIBN), 1,12-dibromo dodecane, N-ethylimidazole, 2,5,8,11-tetraoxatridecan-13-ol, methanesulfonyl chloride, 2,2′-((oxybis(ethane-2,1-diyl))bis(oxy))diethanol, cesium carbonate, methanesulfonyl chloride, lithiumbis((trifluoromethyl)sulfonyl)-amide (LiTFSI) and methanol were purchased from Tianjin Baiheng technology Co., Ltd. Dichloromethane, trichloromethane, and ethylacetate were purchased from Beijing Chemical Works. Anhydrous ether, and N,N-dimethylformamide were purchased from Beijing Tong Guang Fine Chemical Co., Ltd. All reagents were used as received.Characterisation method
The 1H-NMR spectrum of sample dissolved in either CDCl3 or DMSO-D6 was recorded on a Varian Mercury plus 400 M nuclear magnetic resonance spectrometer. The molecular weight of ionic liquid monomers was obtained through the Fourier transform ion cyclotron resonance mass spectrometer, a Bruker APEX IV. The ionic conductivity was measured as follows:| σ = d/(R×A) |
Synthetic procedures
1H NMR (400 MHz, DMSO-D6): δ 0.85 (s, 3H), 1.23 (m, 18H), 1.82 (s, 2H), 4.20 (s, 2H), 5.42 (d, J = 6.3 Hz, 1H), 5.97 (d, J = 15.5 Hz, 1H), 7.32 (m, 1H), 7.96 (s, 1H), 8.23 (s, 1H), 9.59 (d, J = 7.2 Hz, 1H).
To a suspension of imidazolium bromide (2 g, 5.84 mmol) in deionized water (30 ml), LiTFSI (1.68 g, 5.84 mmol) in one portion was added, and the mixture was shaken vigorously in a separation funnel, followed by addition of dichloromethane into the abovementioned mixture. After being shaken 3–4 times, the lower layer was collected. The organics were dried with anhydrous sodium sulfate, filtered, and concentrated to give a light yellow oil (2.85 g, 5.256 mmol, 90%).
1H NMR (400 MHz, DMSO-D6): δ = 0.85 (s, 3H), 1.23 (m, 18H), 1.82 (s, 2H), 4.20 (s, 2H), 5.42 (d, J = 6.3 Hz, 1H), 5.97 (d, J = 15.5 Hz, 1H), 7.32 (m, 1H), 7.96 (s, 1H), 8.23 (s, 1H), 9.59 (d, J = 7.2 Hz, 1H).
1H NMR (400 MHz, DMSO-D6): δ = 1.33 (m, 20H), 1.95 (s, 3H), 3.58 (t, J1 = 6.72 Hz, J2 = 6.71 Hz, 2H), 4.15 (t, J1 = 6.5 Hz, J2 = 6.6 Hz, 2H), 5.73 (d, J = 1.40 Hz, 1H), 6.08 (s, 1H).
1H NMR (400 MHz, DMSO-D6): δ = 1.23 (m, 23H), 1.86 (s, 3H), 3.50 (s, 2H), 4.19 (dd, J1 = 6.6 Hz, J2 = 13.9 Hz, 2H), 5.66 (s, 1H), 5.99 (s, 1H), 7.85 (s, 3H), 9.34 (s, 1H).
To a suspension of the above imidazolium bromide (1 g, 2.912 mmol) in deionized water (30 ml) was added LiTFSI (0.842 g, 2.912 mmol) in one portion, and the mixture was shaken vigorously in a separation funnel followed by addition of dichloromethane into the above mixture. After being shaken 3–4 times, the down layer was collected. The organics were dried with anhydrous sodium sulfate, filtered, and concentrated to give a light yellow oil (1.50 g, 2.76 mmol, 95%).
1H NMR (400 MHz, DMSO-D6): δ = 1.23 (m, 23H), 1.86 (s, 3H), 3.50 (s, 2H), 4.19 (dd, J1 = 6.6 Hz, J2 = 13.9 Hz, 2H), 5.66 (s, 1H), 5.99 (s, 1H), 7.85 (s, 3H), 9.34 (s, 1H).
1H NMR (400 MHz, DMSO-D6): δ = 3.03 (s, 3H), 3.33 (s, 3H), 3.50 (m, 2H), 3.61 (m, 10H), 3.74 (m, 2H), 4.34 (m, 2H).
To the mesylate 7 (1 g, 3.492 mmol) was added 1-vinyl-1H-imidazole (0.34 ml, 3.492 mmol), the resulting mixture was stirred at 100 °C for 10 h. The resulting reaction mixture was washed with ethyl acetate to give the imidazolium mesylate 8 as an oil (1.288 g, 3.387 mmol, 97%).
1H NMR (400 MHz, DMSO-D6): δ = 3.00 (s, 3H), 3.55 (s, 3H), 3.75 (m, 2H), 3.85 (m, 10H), 4.11 (t, J1 = 4.3 Hz, J2 = 4.5 Hz, 2H), 4.80 (t, J1 = 4.2 Hz, J2 = 4.4 Hz, 2H), 5.60 (dd, J1 = 8.6 Hz, J2 = 2.5 Hz, 1H), 6.10 (dd, J1 = 15.6 Hz, J2 = 2.5 Hz, 1H), 7.5 (m, 1H), 7.9 (s, 1H), 8.0 (s, 1H), 10.3 (s, 1H).
To a suspension of mesylate 8 (1 g, 2.630 mmol) in deionized water (30 ml) was added LiTFSI (0.755 g, 2.630 mmol, 1 eq.), and the mixture was shaken vigorously in a separation funnel followed by dichloromethane addition into the above mixture. After being shaken 3–4 times, the bottom layer was collected. The organics were dried with anhydrous sodium sulfate, filtered, and concentrated to give M3 as a light yellow oil (1.487 g, 2.630 mmol, 100%).
1H NMR (400 MHz, DMSO-D6): δ 3.24 (s, 3H), 3.44 (dd, J1 = 5.7 Hz, J2 = 3.4 Hz, 2H), 3.52 (dd, J1 = 6.7 Hz, J2 = 4.1 Hz, 10H), 3.83 (m, 2H), 4.4 (m, 2H), 5.45 (dd, J1 = 8.7 Hz, J2 = 2.3 Hz, 1H), 6.0 (dd, J1 = 15.6 Hz, J2 = 2.4 Hz, 1H), 7.3 (dd, J1 = 15.6 Hz, J2 = 8.7 Hz, 1H), 7.9 (s, 1H), 8.2 (s, 1H), 9.4 (s, 1H).
1H NMR (400 MHz, DMSO-D6): δ 3.07 (d, J = 0.8 Hz, 3H), 3.65 (m, 8H), 3.76 (m, 4H), 4.37 (dd, J1 = 3.70 Hz, J2 = 4.70 Hz, 4H).
To a solution of dimesylate 10 (1 g, 2.853 mmol, 2 equiv.) in DMF (30 ml) was added methacrylic acid (0.13 ml, 1.427 mmol, 1 equiv.), cesium carbonate (0.465 g, 1.427 mmol, 1 equiv.) and hydroquinone (1 wt% of methacrylic acid). The resulting reaction mixture was stirred at 32 °C for 48 h to give a clear solution. The solvents were removed under reduced pressure, and the residue was purified by flash column chromatography to give the title monoester 11 as a yellow oil (0.437 g, 1.283 mmol, yield 45%).
1H NMR (400 MHz, DMSO-D6): δ 1.95 (m, 3H), 3.08 (d, 3H), 3.65 (m, 6H), 3.76 (m, 4H), 4.30 (m, 2H), 4.38 (m, 4H), 5.58 (m, 1H), 6.13 (dd, J1 = 0.93 Hz, J2 = 1.44 Hz, 1H).
To mesylate 11 (1 g, 2.936 mmol) was added 1-ethyl-1H-imidazole (0.33 ml, 2.936 mmol), and the resulting mixture was stirred at 100 °C for 10 h. The resulting reaction mixture was washed with ethyl acetate to give imidazolium mesylate 12 as an oil (1.217 g, 2.789 mmol, 95%).
1H NMR (400 MHz, DMSO-D6): δ 1.57 (m, 3H), 1.94 (s, 3H), 2.75 (s, 3H), 3.64 (m, 12H), 4.32 (m, 4H), 4.52 (m, 2H), 5.59 (s, 1H), 6.11 (s, 1H), 7.34 (s, 1H), 7.65 (s, 1H), 9.77 (s, 1H).
To a suspension of imidazolium mesylate 12 (1 g, 2.291 mmol) in deionized water (30 ml) was added LiTFSI (0.658 g, 2.291 mmol 1 eq.), and the mixture was shaken vigorously in a separation funnel, followed by addition of dichloromethane into the abovementioned mixture. After being shaken 3–4 times, the bottom layer was collected. The organics were dried with anhydrous sodium sulfate, filtered, and concentrated to give M3 as a light yellow oil (1.351 g, 2.176 mmol, 95%).
1H NMR (400 MHz, DMSO-D6): δ 1.44 (t, J1 = 7.2 Hz, J2 = 6.2 Hz, 3H), 1.89 (s, 3H), 3.63 (m, 12H), 4.23 (d, J = 5.9 Hz, 4H), 4.35 (m, 2H), 5.70 (s, 1H), 6.04 (s, 1H), 7.76 (s, 1H), 7.81 (s, 1H), 9.14 (s, 1H).
1H NMR (400 MHz, DMSO-D6): δ 0.80 (s, 3H), 1.23 (m, 18H), 1.82 (s, 2H), 4.20 (s, 2H), 7.32 (m, 1H), 7.96 (s, 1H), 8.23 (s, 1H), 9.59 (d, J = 7.2 Hz, 1H).
1H NMR (400 MHz, DMSO-D6): δ = 1.24 (m, 23H), 1.87 (s, 3H), 3.51 (s, 2H), 4.19 (dd, J1 = 6.6 Hz, J2 = 13.9 Hz, 2H), 7.79 (s, 3H), 9.17 (s, 1H).
Conclusion
Ionic liquid monomer M1 to M4 and polymeric ionic liquid P1 to P2 were designed and synthesized in high yield. It is worth noting that the mesylate of ethylene oxide-containing alcohol could be synthesized in perfect yield and it could be transferred into imidazolium mesylate with high efficiency. M1 to M4 had ionic conductivity around 1–2 × 10−3 S cm−1. P2 showed ionic conductivity of 2.2 × 10−4 S cm−1, which was 200 times that of P1 due to the long alkyl chain tether separated polymer backbone and charge centre. Further systematic study of electrochemical properties of M1 to M4 and their corresponding PILs is ongoing and will be reported in due course.Acknowledgements
The authors thank the National Natural Science Foundation of China (No. 21202008), Beijing Natural Science Foundation (No. 2162039), Jiangxi Provincial Department of Science and Technology (No. 20133BBE50014) and the Science Foundation of the Education Department of Jiangxi province (No. KJLD13020) for generous support.Notes and references
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS PubMed.
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed.
- A. S. Shaplov, R. Marcilla and D. Mecerreyes, Electrochim. Acta, 2015, 175, 18–34 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- X. Chen, J. Zhao, J. Zhang, L. Qiu, D. Xu, H. Zhang, X. Han, B. Sun, G. Fu, Y. Zhang and F. Yan, J. Mater. Chem., 2012, 22, 18018–18024 RSC.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- J. Yuan and M. Antonietti, Polymer, 2011, 52, 1469–1482 CrossRef CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- N. Nishimura and H. Ohno, Polymer, 2014, 55, 3289–3297 CrossRef CAS.
- Z. Jia, W. Yuan, C. Sheng, H. Zhao, H. Hu and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1339–1350 CrossRef CAS.
- Y. S. Vygodskii, A. S. Shaplov, E. I. Lozinskaya, K. A. Lyssenko, D. G. Golovanov, I. A. Malyshkina, N. D. Gavrilova and M. R. Buchmeiser, Macromol. Chem. Phys., 2008, 209, 40–51 CrossRef CAS.
- A. S. Shaplov, L. Goujon, F. Vidal, E. I. Lozinskaya, F. Meyer, I. A. Malyshkina, C. Chevrot, D. Teyssie, I. L. Odinets and Y. S. Vygodskii, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4245–4266 CrossRef CAS.
- A. S. Shaplov, P. S. Vlasov, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. M. Pavlov, C. Wandrey, A. Bhide, M. Schonhoff and Y. S. Vygodskii, Macromolecules, 2011, 44, 9792–9803 CrossRef CAS.
- A. S. Shaplov, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, P. Aubert, O. V. Okatova, G. M. Pavlov, L. I. Komarova, C. Wandrey and Y. S. Vygodskii, Electrochim. Acta, 2011, 57, 74–90 CrossRef CAS.
- M. Dobbelin, I. Azcune, M. Bedu, A. Ruiz De Luzuriaga, A. Genua, V. Jovanovski, G. Cabanero and I. Odriozola, Chem. Mater., 2012, 24, 1583–1590 CrossRef CAS.
- A. S. Shaplov, D. O. Ponkratov, P. Aubert, E. I. Lozinskaya, C. Plesse, A. Maziz, P. S. Vlasov, F. Vidal and Y. S. Vygodskii, Polymer, 2014, 55, 3385–3396 CrossRef CAS.
- Z. Jia, W. Yuan, H. Zhao, H. Hu and G. L. Baker, RSC Adv., 2014, 4, 41087–41098 RSC.
- H. Hu, W. Yuan, L. Lu, H. Zhao, Z. Jia and G. L. Baker, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2104–2110 CrossRef CAS.
- A. S. Shaplov, D. O. Ponkratov, P. S. Vlasov, E. I. Lozinskaya, L. V. Gumileva, C. Surcin, M. Morcrette, M. Armand, P. Aubert, F. Vidal and Y. S. Vygodskii, J. Mater. Chem. A, 2015, 3, 2188–2198 CAS.
- M. A. Aboudzadeh, A. S. Shaplov, G. Hernandez, P. S. Vlasov, E. I. Lozinskaya, C. Pozo-Gonzalo, M. Forsyth, Y. S. Vygodskii and D. Mecerreyes, J. Mater. Chem. A, 2015, 3, 2338–2343 CAS.
- M. A. Aboudzadeh, H. Zhu, C. Pozo-Gonzalo, A. S. Shaplov, D. Mecerreyes and M. Forsyth, Electrochim. Acta, 2015, 175, 74–79 CrossRef CAS.
- A. S. Shaplov, E. I. Lozinskaya, P. S. Vlasov, S. M. Morozova, D. Y. Antonov, P. Aubert, M. Armand and Y. S. Vygodskii, Electrochim. Acta, 2015, 175, 254–260 CrossRef CAS.
- G. Colliat-Dangus, M. M. Obadia, Y. S. Vygodskii, A. Serghei, A. S. Shaplov and E. Drockenmuller, Polym. Chem., 2015, 6, 4299–4308 RSC.
- L. Porcarelli, J. R. Nair, C. Gerbaldi, A. S. Shaplov, Y. S. Vygodskii, M. Salsamendi and D. Mecerreyes, ACS Appl. Mater. Interfaces, 2016, 8, 10350–10359 CAS.
- Y. V. Nelyubina, A. S. Shaplov, E. I. Lozinskaya, M. I. Buzin and Y. S. Vygodskii, J. Am. Chem. Soc., 2016, 138, 10076–10079 CrossRef CAS PubMed.
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, ACS Energy Lett., 2016, 1, 678–682 CrossRef CAS.
- H. Ohno and K. Ito, Chem. Lett., 1998, 751–752 CrossRef CAS.
- M. D. Green and T. E. Long, Polym. Rev., 2009, 49, 291–314 CrossRef CAS.
- H. Ohno, Macromol. Symp., 2007, 249–250, 551–556 CrossRef.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2004, 50, 255–261 CrossRef CAS.
- W. Ogihara, S. Washiro, H. Nakajima and H. Ohno, Electrochim. Acta, 2006, 51, 2614–2619 CrossRef CAS.
- J. Juger, F. Meyer, F. Vidal, C. Chevrot and D. Teyssie, Tetrahedron Lett., 2009, 50, 128–131 CrossRef CAS.
- H. He, M. Zhong, B. Adzima, D. Luebke, H. Nulwala and K. Matyjaszewski, J. Am. Chem. Soc., 2013, 135, 4227–4230 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26459j |
| This journal is © The Royal Society of Chemistry 2017 |