DOI:
10.1039/C6RA24662A
(Review Article)
RSC Adv., 2017,
7, 28313-28349
Small hybrid heteroaromatics: resourceful biological tools in cancer research
Received
3rd October 2016
, Accepted 17th April 2017
First published on 30th May 2017
Abstract
Nowadays, hybrid drugs containing two or more covalently linked known potential pharmacophores are designed to simultaneously modulate multiple targets of multifactorial diseases to overcome the side effects associated with a single drug. In this review, an overview of the design strategies employed by various scientists over the past 20 years has been presented. The overview includes the synthesis of different chemical structure-based anticancer hybrids using molecular hybridization techniques. To tackle one of the world's most devastating diseases such as cancer, researchers have exploited the molecular hybridization (MH) technique to synthesize different anticancer hybrids, which include hybrids based on azole, camptothecin, chalcone, pyrrolobenzodiazepine (PBD), coumarin, colchicine, platinum, and some miscellaneous structures. The selection of two or more moieties for generating the hybrid drug is generally aided by the observed (or anticipated) synergistic or additive pharmacological activities of each single moiety. This eventually leads to the identification of novel and better active chemical entities with a superior profile as compared to the parent moieties. In addition to the design strategies, this review also highlights the structure–activity relationship (SAR), mechanism of action, and key features of the synthesized anticancer hybrids.
 Vikrant Abbot | Vikrant Abbot obtained his Bachelor's degree in Pharmacy from the Punjab Technical University in 2009. He started his professional career working as a Production Chemist at Comed Pharmaceuticals Ltd. Baddi for 2.5 years. Thereafter, he completed his Master's degree in Medicinal Chemistry from JUIT, Waknaghat in 2014. Then, he joined academics and is currently working as an Assistant Professor at ISF College of Pharmacy, Moga, Punjab. His present research interests include design and synthesis of small molecules with anticancer and antimicrobial profiles. |
 Poonam Sharma | Poonam Sharma obtained her Master's in Philosophy in 2003 with a gold medal and completed her PhD in chemistry in 2006 from Himachal Pradesh University (India). She was awarded a UGC project fellowship for her PhD research. She was also awarded the Fast Track Young Scientist research project by DST in 2010. Presently, she is working as an Assistant Professor at the Jaypee University of Information Technology, Waknaghat, Solan (India). Her research revolves around physicochemical drug interactions, thermodynamics, heterocyclic bioactive analogs and topical drug delivery. |
 Saurabh Dhiman | Saurabh Dhiman was born in 1990 in Himachal Pradesh, India. He obtained his Bachelor's degree from JUIT, Waknaghat in 2012. He continued his postgraduate studies with specialization in Medicinal Chemistry at the same university and worked in the field of hybrid compounds as anticancer and antimicrobial agents. |
 Malleshappa N. Noolvi | Dr. Malleshappa Noolvi obtained his B. Pharmacy degree from SETs College of Pharmacy, Karnataka in 1997. He completed his M. Pharmacy from Government College of Pharmacy, Bangalore in 1999. He was awarded PhD in Medicinal Chemistry from the Rajiv Gandhi University of Health and Sciences, Bangalore, India. He has twenty-seven international research publications. He has successfully completed various research projects granted by the Department of Science and Technology, New Delhi. Currently, he is working as a Professor at the Shree Dhanvantary Pharmacy College and Shree Dhanvantary Pharmaceutical and Analytical Research Center, KIM, Surat, Gujarat. |
 Harun M. Patel | Harun M. Patel is currently associated with the department of Pharmaceutical Chemistry, R.C. Patel Institute of Pharmaceutical Education and Research, Shirpur Maharashtra, India. He has outstanding research and academic experience. He has more than 70 publications in reputed peer reviewed international journals. His key research interests are working in the field of QSAR, docking study, virtual screening, synthesis of novel anticancer, anti-tubercular and antiepileptic drugs. He has two patents and three international books to his credit. He is a well recognized teacher in medicinal and pharmaceutical chemistry and is well known for his innovative teaching skills. |
 Varun Bhardwaj | Dr Varun Bhardwaj received his PhD in Pharmacy in 2014 under the supervision of Dr Poonam Sharma and Dr S. Chauhan from Jaypee University of Information Technology. He received his Master of Pharmacy degree under the supervision of Prof. Malleshappa N. Noolvi. He worked under two DST funded projects, with 25 publications in international peer reviewed journals. His research interest includes design and synthesis of bioactive heterocycles, physicochemical interactions, and nano/microparticulates, topical formulations especially against microbial infections and drug safety/efficacy profile. |
1. Introduction
Cancer is a cell cycle disease characterized by rapid, uncontrolled, and pathological proliferation of abnormal cells. It is one of the most formidable diseases in the world.1,2 The most frequent forms of cancer are lung cancer, breast cancer, colorectal cancer, stomach cancer, and prostate cancer; among these, prostate cancer (male) and breast cancer (female) represent 29% of all cancer cases.3 Over the past few years, various techniques such as surgery, chemotherapy, immunotherapy, and radiotherapy have been employed to cure cancer. Currently, cancer therapy involves pharmacological interventions targeted at some pathogenic pathway, mostly a particular biological pathway.4 With these advancements in the field of cancer therapy, problems still persist with the available anticancer drugs. Most of the drugs are incapable of overcoming the resistance mechanism involved in the primary and secondary cancer cells and are also unable to differentiate between normal cells and neoplastic cells. This led to the general belief that the drugs simultaneously modulating more than one target would have better therapeutic potential as compared to single target drugs.5 Thus, to simultaneously modulate more than one target and combat different types of cancer, multi-target drugs were generated by combining two or more known pharmacophores to form a new single drug (hybrid drug). A hybrid molecule may be defined as a combination of two different and independently acting compounds that are covalently linked to form one hybrid compound, which leads to a pharmacological potency greater than the sum of each individual moiety's potencies. This approach of developing multi-target drugs has been termed as molecular hybridization (MH). The active compounds or pharmacophoric units are recognized and derived from known bioactive molecules.6 Hybrid anticancer drugs have remarkable advantages over the conventional anticancer drugs because of their improved pharmacodynamic profile. This new method is highly suitable for developing more potent molecules to simultaneously target more than one pathogenic mechanism.7 Molecular hybridization not only enhances the activity or selectivity but can also reduce adverse drug effects.8,9 Molecular hybridization is believed to be analogous to combination therapy with the exception that two drugs are covalently linked and available as a single entity.4
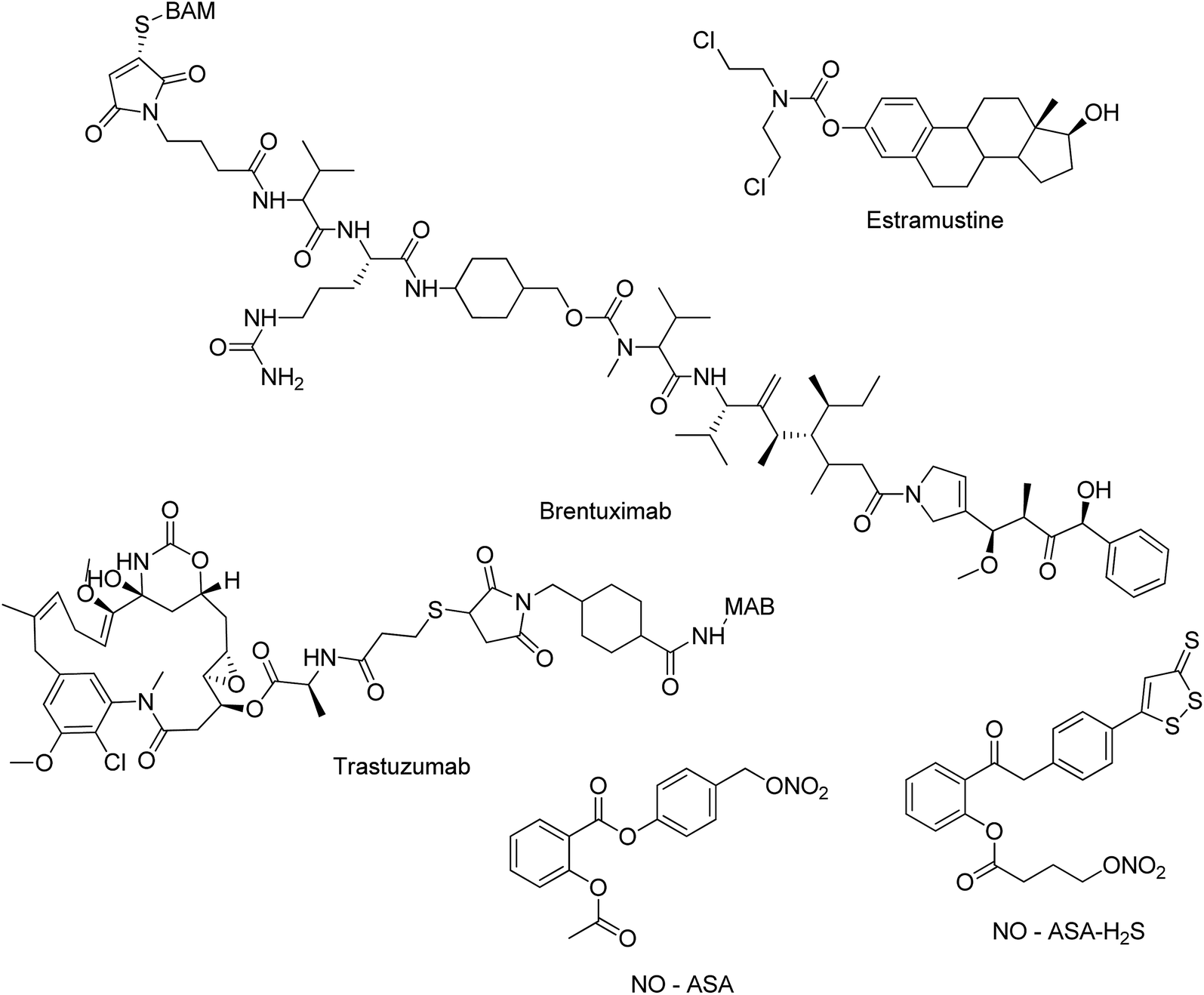
The molecular hybridization strategy has been globally employed by scientists to concomitantly address more than one biological target in cancer treatment that has enabled some anticancer hybrids to reach clinical trials. For example, estramustine hybrid of estradiol and nitrogen mustard is under clinical trial for various solid cancers. Brentuximab (Adcetris®), which is an antibody–drug conjugate, is under clinical trials since 2010. Some other examples include linkage of trastuzumab emtansine-antibody trastuzumab (Herceptin) to the cytotoxic agent mertansine (DM1), under clinical trial in women with advanced HER2 positive breast cancer (EMILIA), and development of NO–Aspirin hybrid (NO–ASA) as a chemopreventive agent. NO–ASA–H2S hybrid seemed to be a promising molecule to be developed as a drug since it inhibited the growth of 11 different types of human cancer cells in culture (Fig. 1).10 Along with molecular hybridization, fragment-based drug discovery also plays an important role in developing hybrid molecules. Using this approach, small chemical fragments are identified and combined together to produce a lead molecule having higher affinity.11
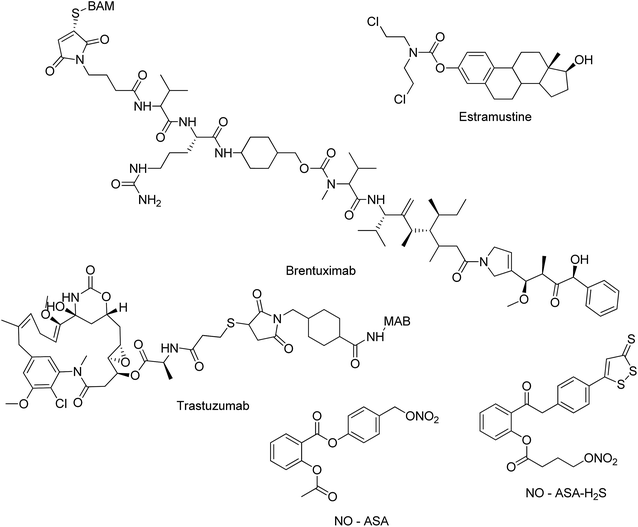 |
| | Fig. 1 Molecules in clinical trials. | |
In this review, various strategies and approaches that have been employed to design hybrid molecules have been discussed. In addition, the structure–activity relationship, mode of action, and key features of the molecules with an impact on biological evaluation have also been highlighted. The major hybrid molecules discussed herein include azoles, camptothecins, chalcones, pyrrolobenzodiazepines (PBDs), coumarins, colchicines, platinum-based hybrids, and some miscellaneous hybrid molecules.
2. Azole-based hybrids
Azole is a class of five-membered nitrogen ring compounds containing at least one non-carbon atom, i.e. nitrogen, along with sulphur or oxygen. Diverse azole-based hybrid molecules have been developed with potential anticancer activity and discussed below:
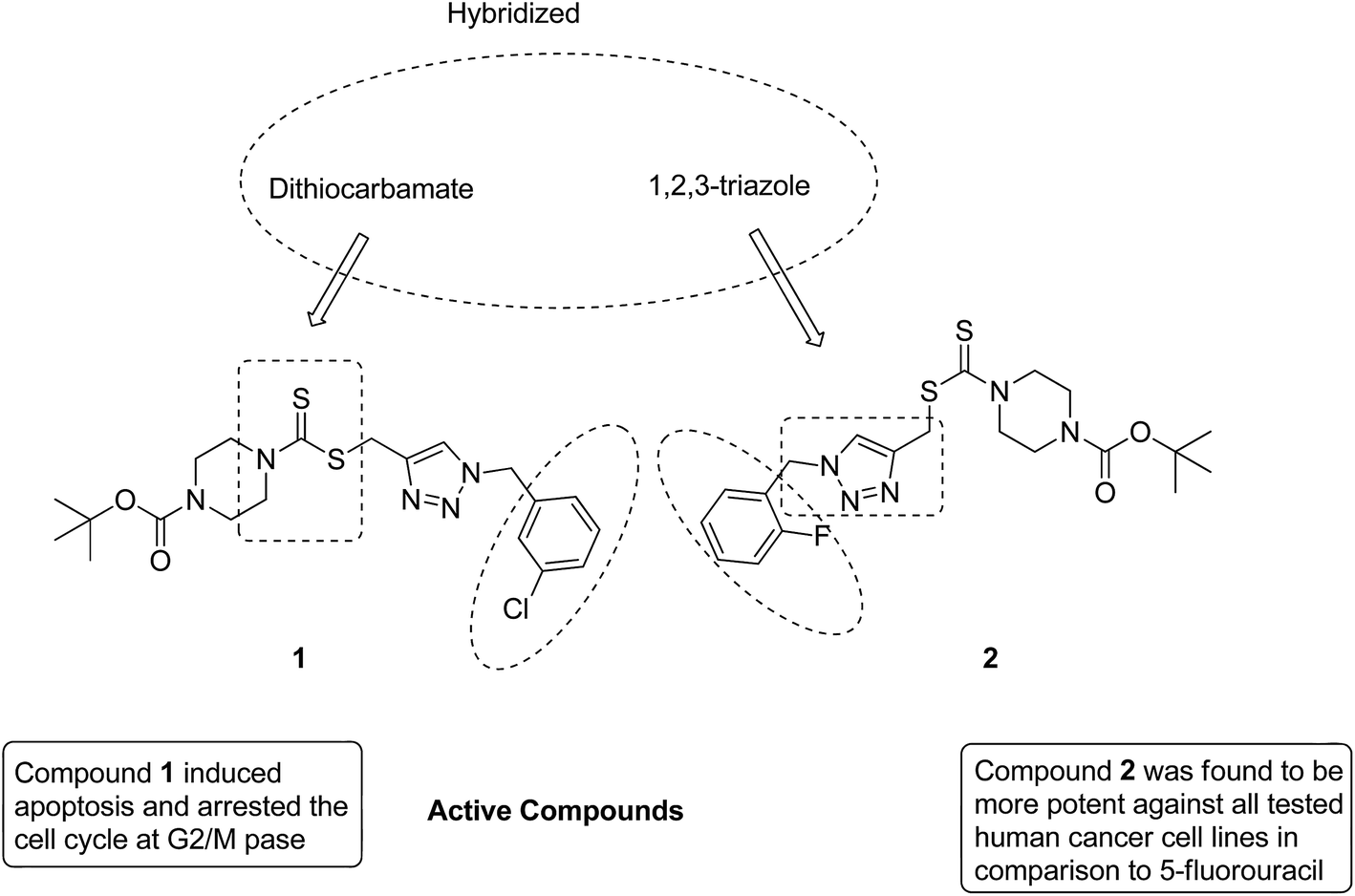
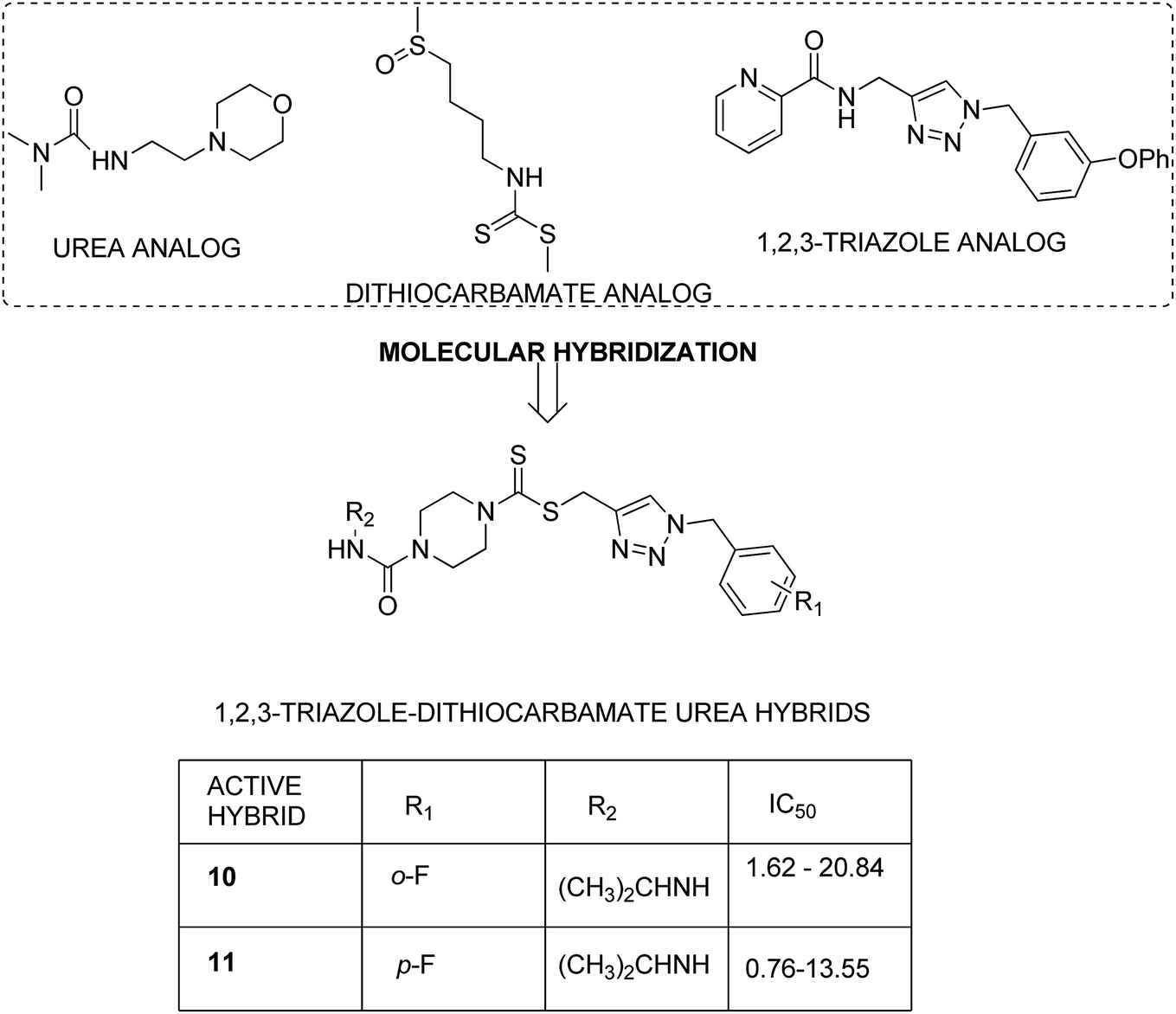
After identifying the importance of 1,2,3-triazole compounds using click chemistry, Duan et al. synthesized novel 1,2,3-triazole–dithiocarbamate hybrids12 and evaluated them against four human tumor cell lines: MGC-803, MCF-7, PC-3, and EC-109. Among the synthesized hybrids, compounds 1 and 2 exhibited broad-spectrum anticancer activity with the IC50 values ranging from 0.73 to 11.63 and 0.49 to 12.45 μM, respectively. These compounds were found to inhibit the proliferation of MGC-803 cells via programmed cell death and imposing the cell cycle at the G2/M phase (Fig. 2).
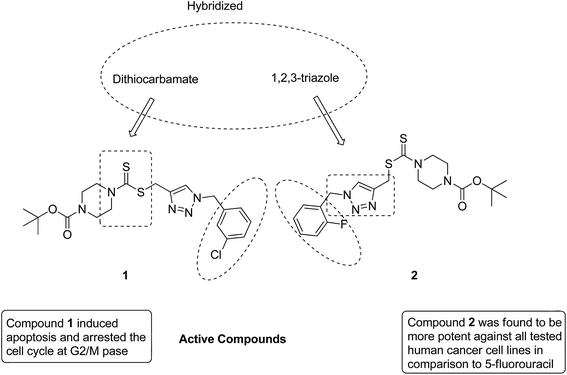 |
| | Fig. 2 Novel hybrids of 1,2,3-triazole-dithiocarbamate. | |
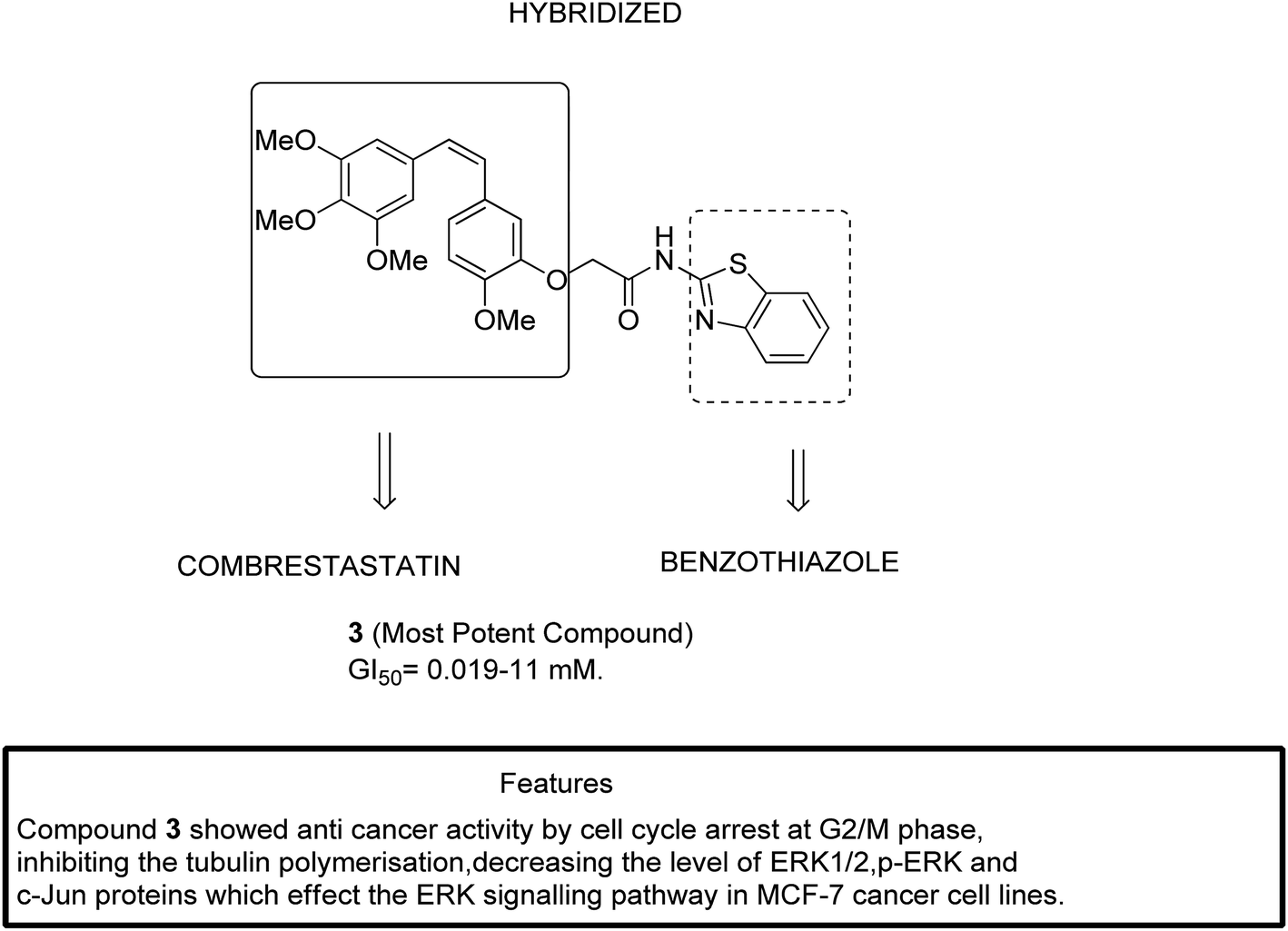
The two major subfamilies extracellular signal-regulated kinase (ERK) and c-Jun of mitogen-activated protein kinases (MAPKs) are the key molecules for regulating breast cancer cell lines. The signaling pathways mediated by the combretastatin scaffold can be used to generate agents that are useful in the treatment of breast cancer. This information led Kamal et al.13 to synthesize combretastatin–amidobenzothiazole hybrid molecules with potential anticancer activity. Among the synthesized hybrids, compound 3 showed significant activity against a breast cancer cell line (MCF-7). Structure–activity relationship (SAR) studies further revealed that compound 3 had no substitution in the benzene ring of benzothiazole, which resulted in its higher activity with the GI50 value ranging from 0.019 to 11 μM (Fig. 3).
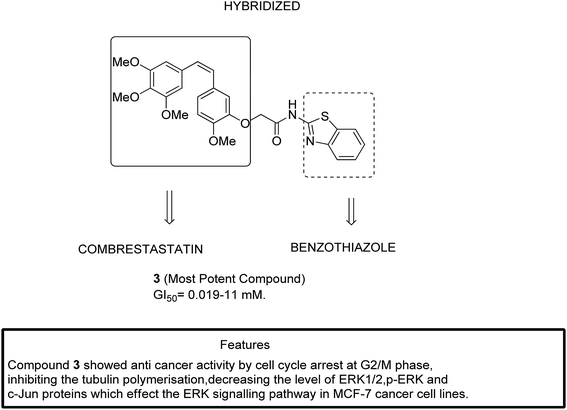 |
| | Fig. 3 Hybrids of combretastatin-amidobenzothiazole. | |
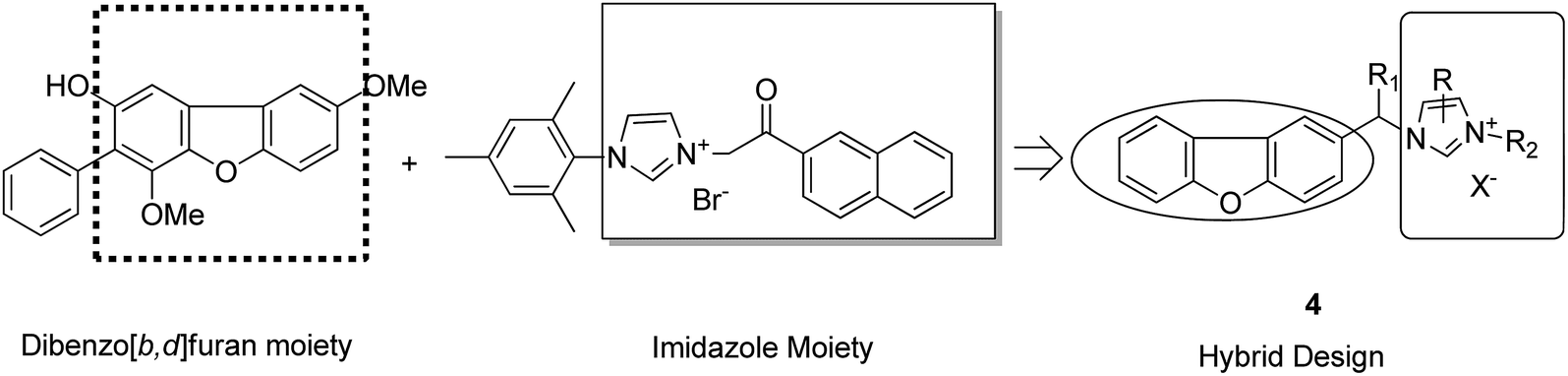
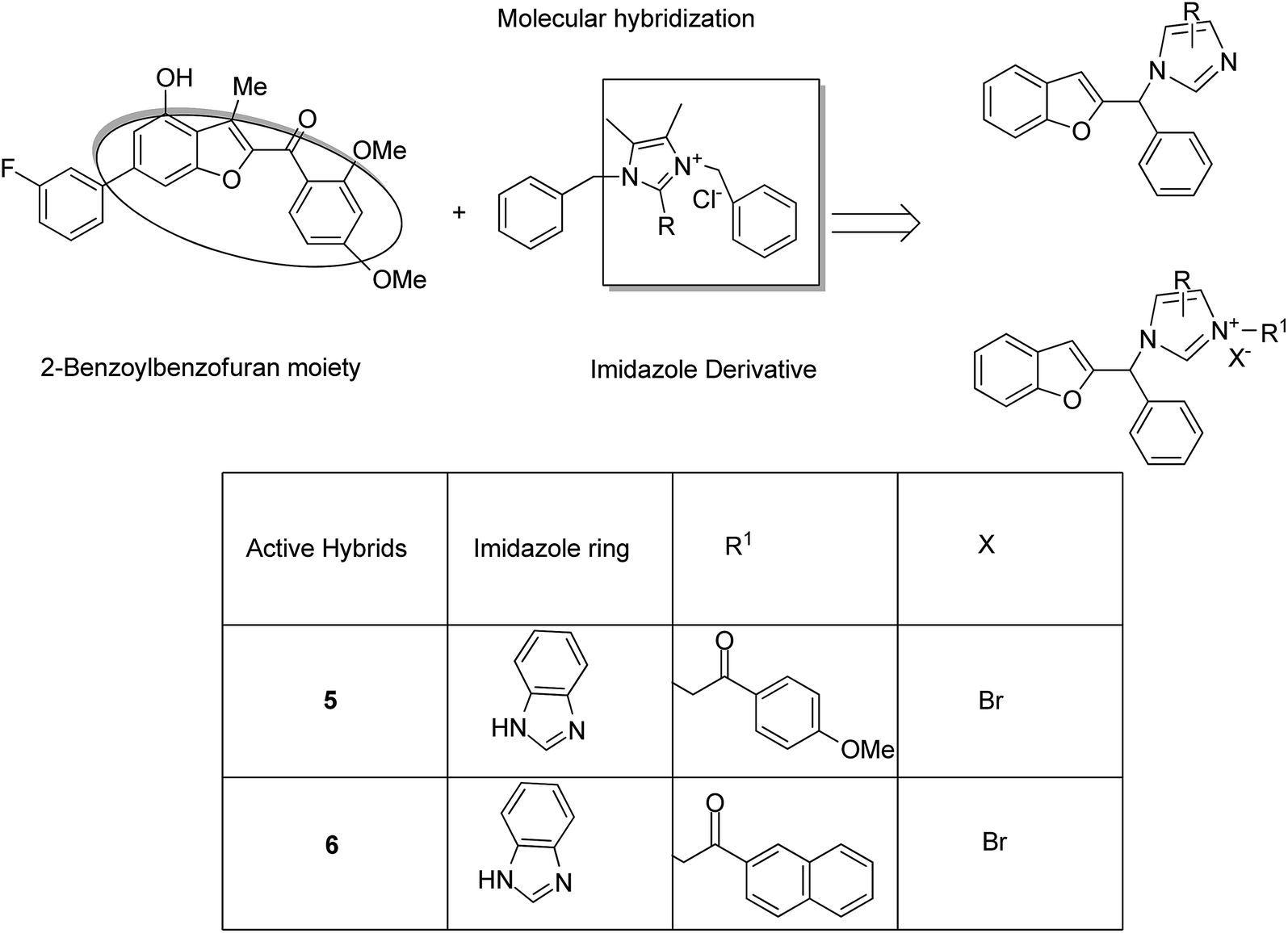
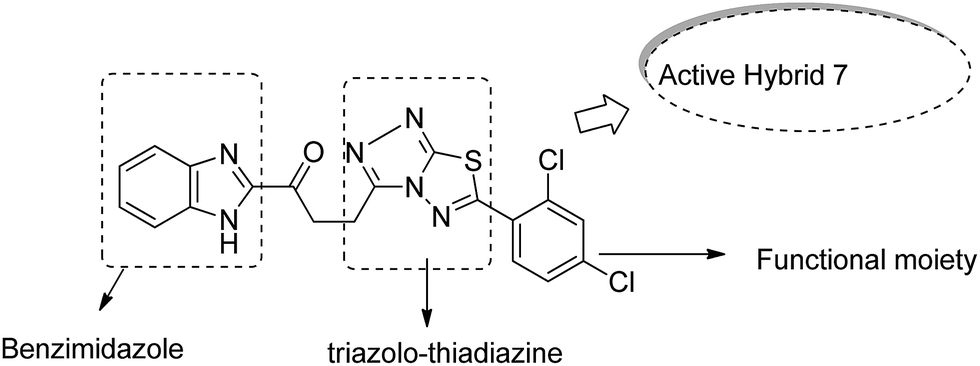
Imidazolium salt hybrids can induce G1 phase cycle cell arrest and cause apoptosis of tumor cells. The success of these hybrid molecules directed Liu et al. to synthesize dibenzo[b,d] furan–imidazole hybrid compounds; moreover, these compounds were assessed against human tumor cell lines.14 Among the synthesized compounds, compound 4 showed significant activity against all human tumor cell lines. SAR suggested that the linking of naphthylacyl or 4-methoxyphenacyl group at the third position of the imidazole ring resulted in alteration of its cytotoxic activity (Fig. 4). Considering the anticancer activities of natural and synthetic 2-benzoylbenzofuran derivatives and the potent cytotoxic activities of imidazole derivatives, Wang et al.15 synthesized novel hybrids of 2-benzoylbenzofuran and imidazole analogs and evaluated their anticancer activity in vitro. Compound 5 and 6 showed the most significant activity against five strains of human cell lines, in particular, against the breast carcinoma (MCF) and myeloid liver carcinoma (SMMC-7721) cell lines. SAR showed that in compounds 5 and 6, substitution of imidazole ring at the third position by 4-methoxyphenacyl resulted in significant biological activity (Fig. 5). Benzimidazoles are known to have diverse biological activities and are of great significance. Various benzimiadazole-linked triazolo-thiadiazole and triazolo-thiadiazines derivatives were synthesized and evaluated against the NCI 60 cell lines panel at the National Cancer Institute, USA. Among the synthesized compounds, 7 showed remarkable cytotoxicity against the MG-MID tumor cell lines. Furthermore, SAR studies have revealed that the presence of chloro group (electron withdrawing) on the para-position and meta-position of a phenyl ring played a significant role in enhancing the biological profile of the compound. The structure of the compound has been presented in Fig. 6.16
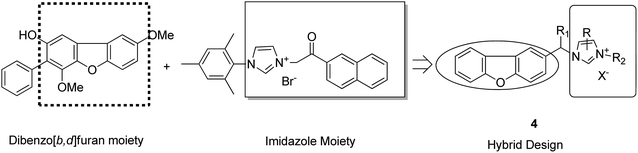 |
| | Fig. 4 Dibenzo[b,d] furan-imidazole hybrids. | |
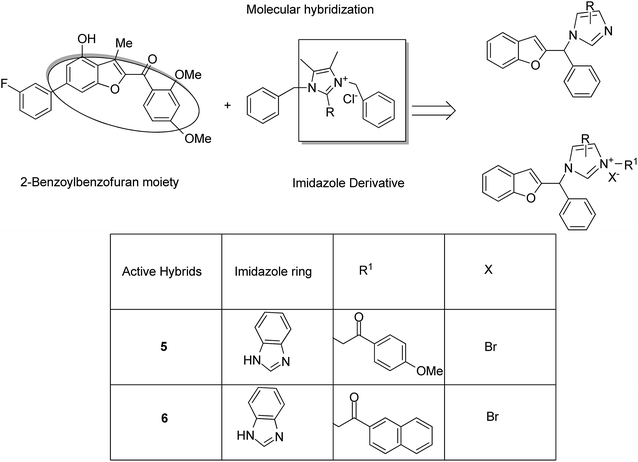 |
| | Fig. 5 Imidazole scaffold-based 2-benzoylbenzofuran hybrids. | |
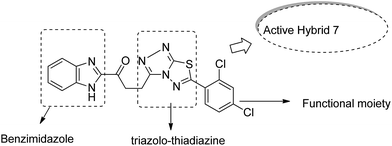 |
| | Fig. 6 Benzimidazole linked with triazolo-thiadiazoles and triazolo-thiadiazines. | |
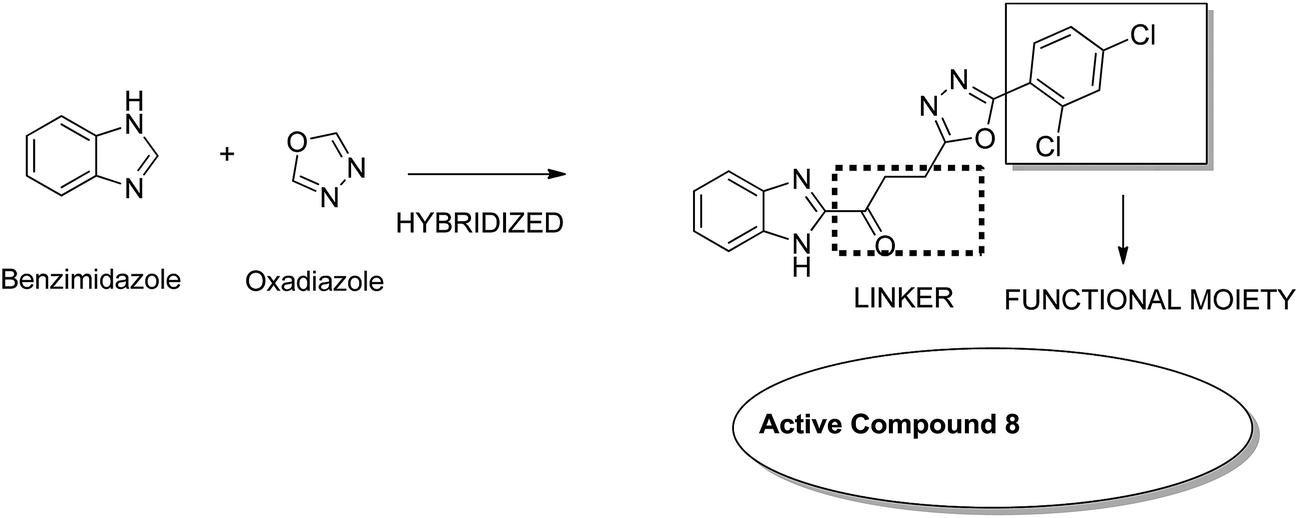
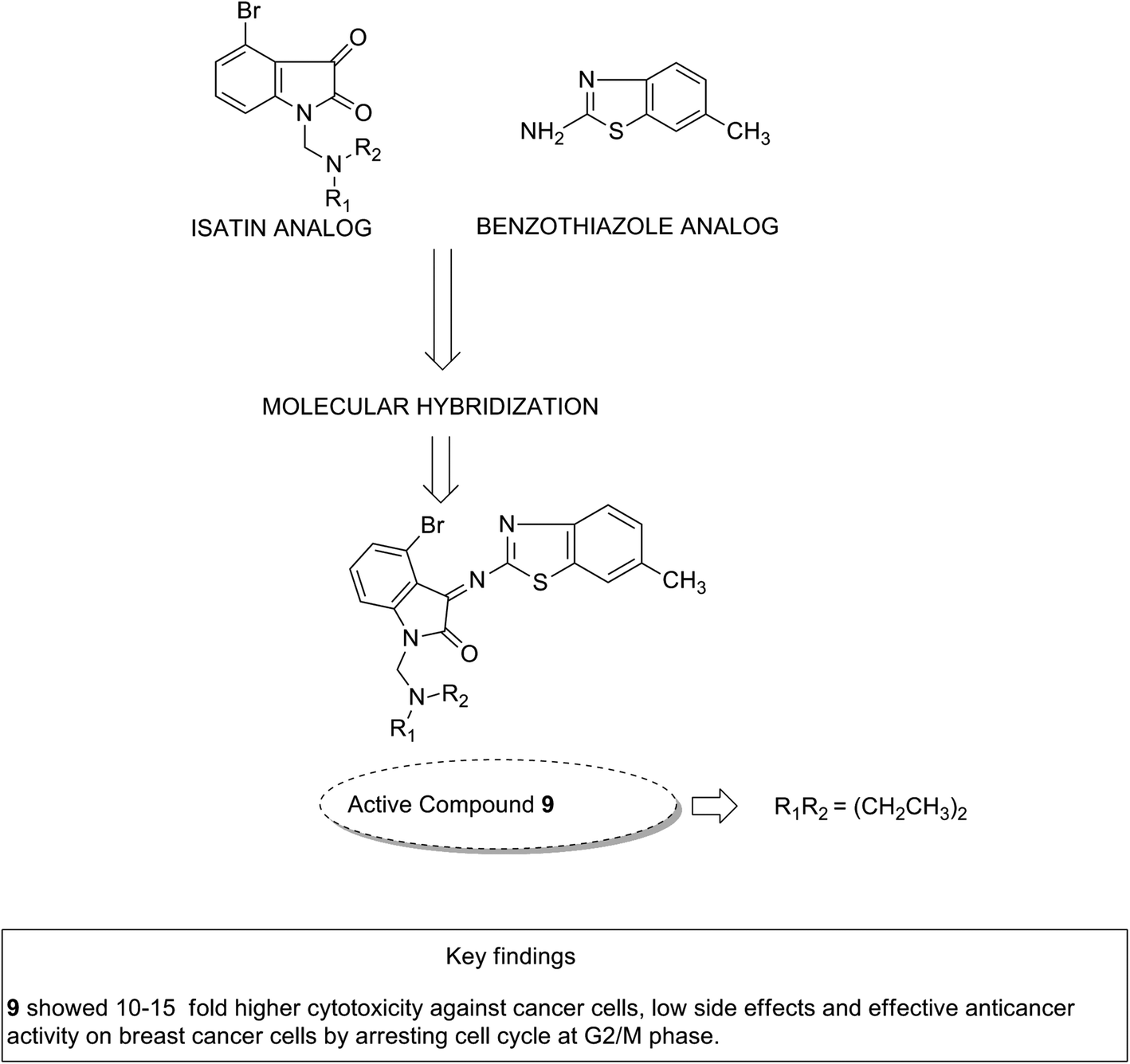
Considering the diverse biological activities of 1,3,4-oxidiazole derivatives, Rashid et al. designed novel oxadiazole nucleus-linked benzimidazole hybrids using a microwave irradiation method. Compound 8 (Fig. 7) was the most active compound with high growth inhibition against the full NCI cell lines panel. According to the SAR studies, the presence of NH2, 2-chloro, and 2,4-dichloro (electron-donating groups) on the phenyl ring at the 5 position of oxadiazole was responsible for the anticancer activity.17 Another study was based on the observations that isatin (1H-indole-2,3-dione) had interesting pharmacological profiles and toleration in human subjects. Solomon et al. fused isatin with benzothiazole and evaluated its activity in breast cancer cell lines. Compound 9 (Fig. 8) showed a better effect with an improved safety profile. These effects were attributed to the arrest in the cell cycle at the G2/M phase.18
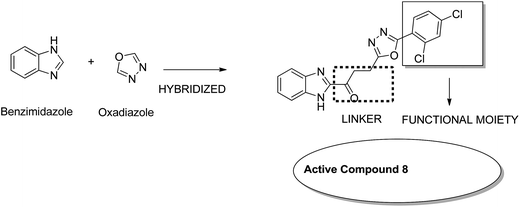 |
| | Fig. 7 Benzimidazoles with oxadiazole nucleus hybrids. | |
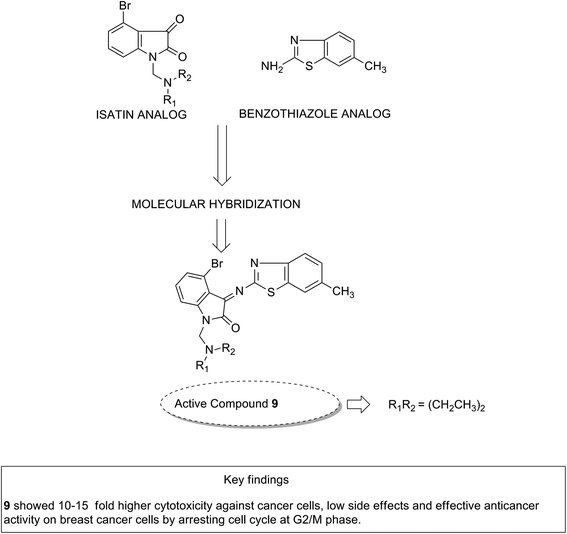 |
| | Fig. 8 Isatin–benzothiazole hybrids. | |
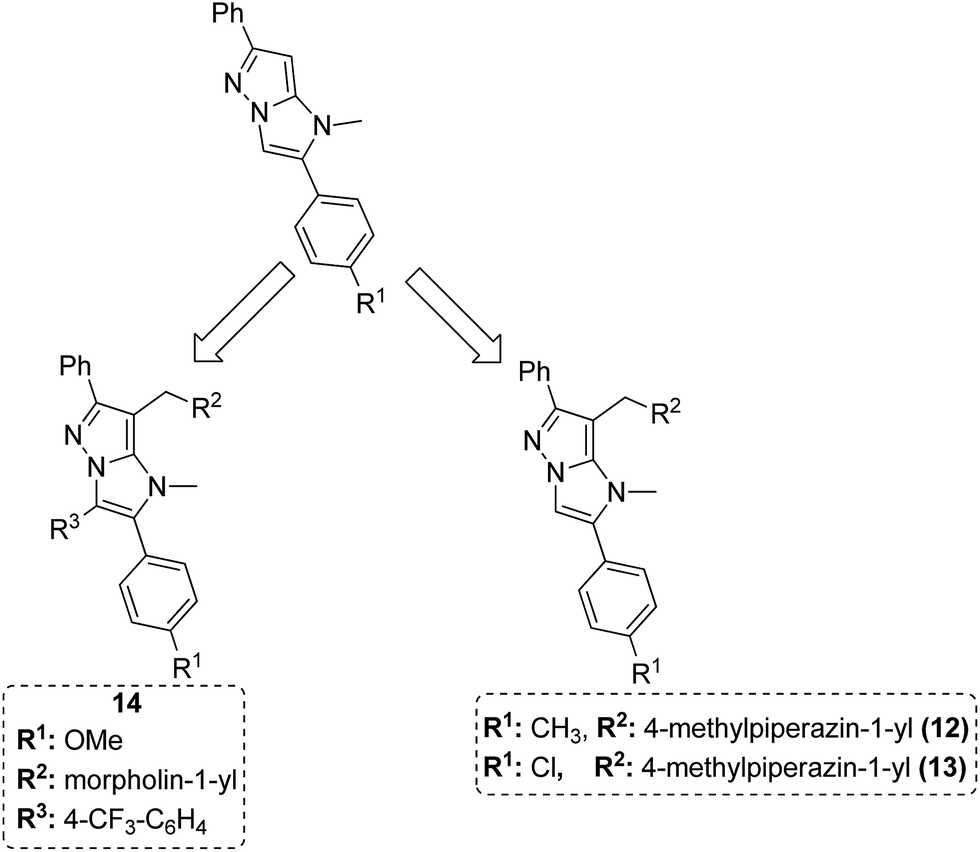
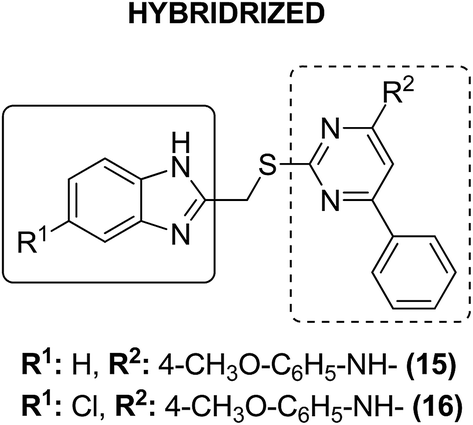
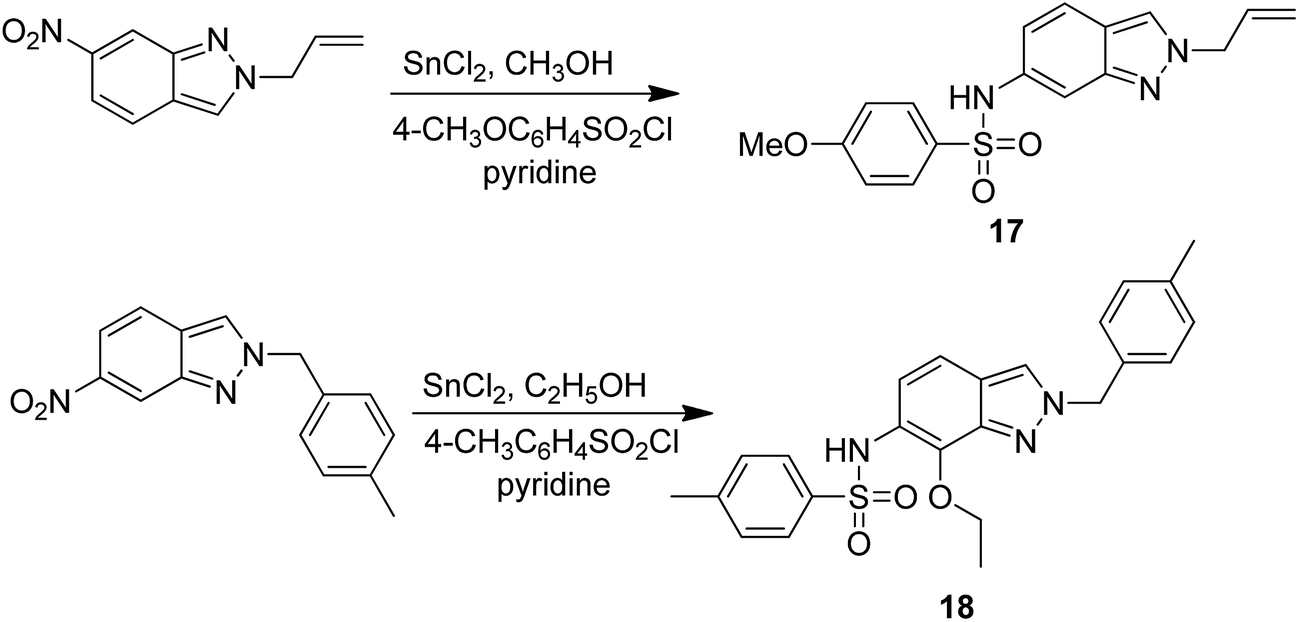
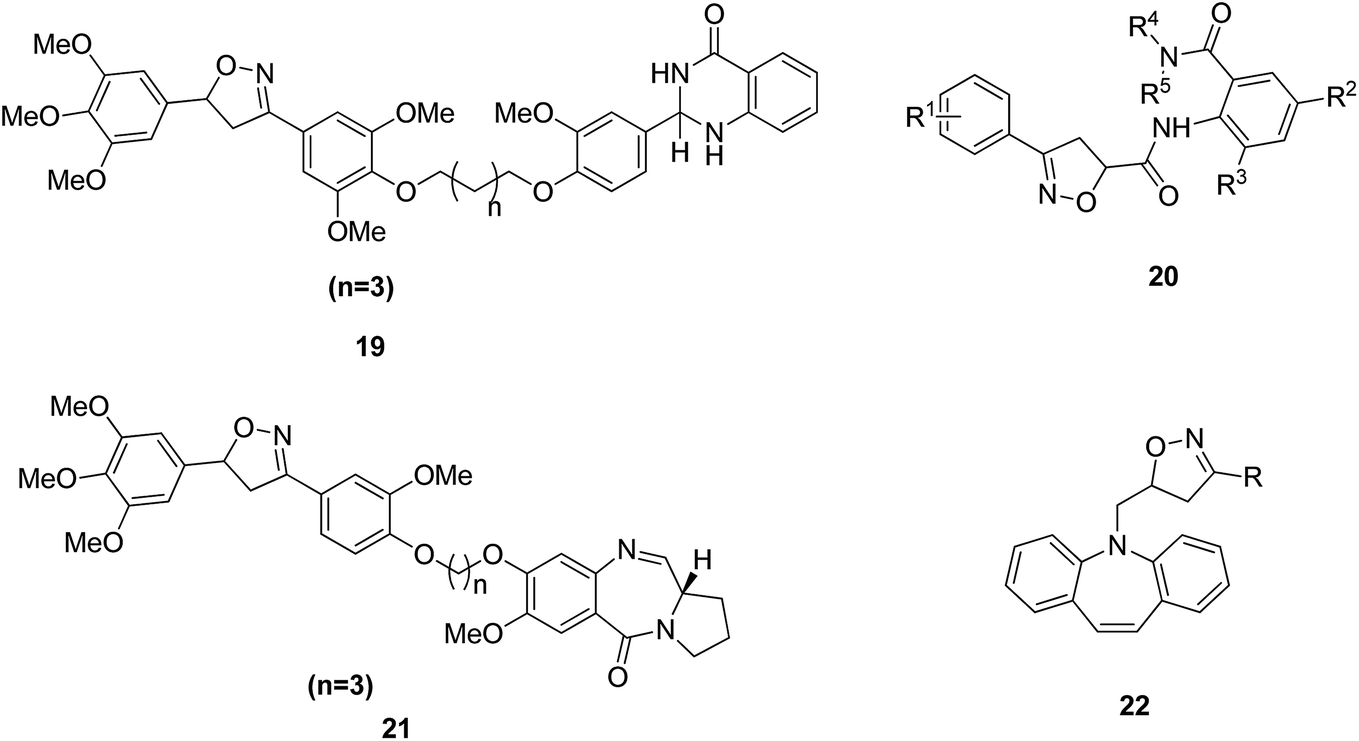
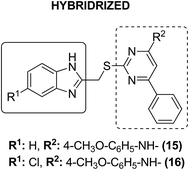
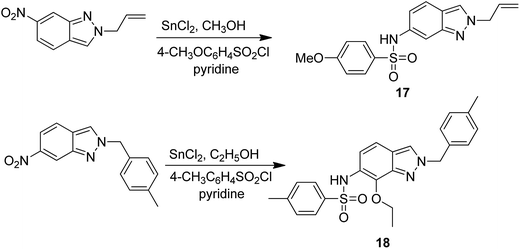
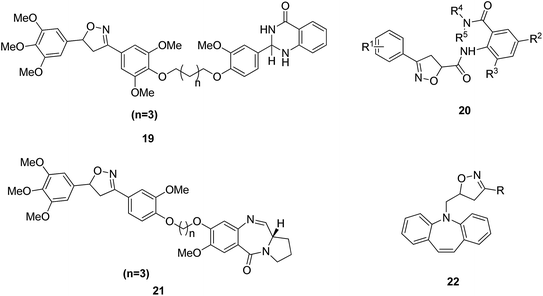
Dithiocarbamates are considered as a privileged scaffold for drug discovery with a wide range of biological activities. Duan et al.19 designed novel hybrids of 1,2,3-triazole dithiocarbamate-urea and evaluated them against human cancer cell lines. The IC50 value for hybrids 10 and 11 (Fig. 9) ranged from 1.62 to 20.84 and 0.76 to 13.55 μM, respectively. Another study was focused on developing the three series of novel imidazo [1,2-b] pyrazole type derivatives: C-2/C-3/C-6 tri (hetero) arylated, C-2/C-3/C-6/C-7 trisubstituted, and C-2/C-6/C-7 trisubstituted imidazo [1,2-b] pyrazoles. Among the total synthesized compounds, 12, 13, and 14 displayed IC50 values less than 10 μM in a 6-cell line panel. The SAR analysis revealed that C-7 amino-methylated compounds such as morpholine or N-methylpiperazine as a 6-membered cycle, are promising candidates for cancer treatment. The presence of a fourth substituent in the C-2/C-3/C-6/C-7 tetra-substituted imidazo [1,2-b] pyrazoles did not produce any significant impact, except for compound 14.20 The chemical structure has been presented in Fig. 10. One more study suggested the impact of the nature of substitution on pyrimidine–benzimidazole hybrid molecules. The hybrid molecules were synthesized and evaluated for anticancer activity against 4 human cancer cell lines. Compounds 15 and 16 (Fig. 11) showed potent anticancer activity against MCF-7, MGC-803, EC-9706, and SMMC-7721 cell lines with the IC50 values ranging from 2.03 to 10.55 μM and 1.06 to 12.89 μM, respectively. The results showed that compound 16 inhibited the proliferation of MGC-803 cancer cells by causing cell death and arresting the cell cycle at the G2/M phase.21 Computational assistance with a novel strategy of preparing some N-[6-indazolyl] aryl-sulfonamides and N-[alkoxy-6-indazolyl] arylsulfonamides has been reported.22 The major steps involved the reduction of 2-alkyl-6-nitroindazoles with tin(II) chloride using various alcohols, following the coupling of the corresponding amine with arylsulfonyl chlorides in pyridine. Compounds 17 and 18 showed significant anticancer activity against A2780 (ovarian carcinoma) and A549 (lung adenocarcinoma) human tumor cell lines, with the IC50 values ranging from 4.21 to 18.6 μM. Furthermore, the results of immunofluorescence microscopy demonstrated that both compounds disrupted the microtubule network similar to the anti-microtubule agent vincristine, verifying the arrest at the G2/M phase of the cell cycle. The molecular docking studies helped in the identification of the paclitaxel binding site as the binding pocket for compound 18, whereas compound 17 was predicted to bind at an unidentified binding site at the α/β subunits interface (Fig. 12). Kaur et al. developed a class of isoxazoline derivatives as anticancer agents.23 Some important synthetic Δ2-isoxazoline scaffolds discussed were 3,5-diaryl-isoxazoline-linked 2,3-dihydroquinazoline hybrids 19, arylisoxazoline-containing anthranilic diamides 20, 3,5-diaryl-isoxazoline-linked pyrrolo[2,1-c][1,4] benzodiazepine (PBD) conjugates 21, and dibenzo [b,f] azepine-tethered isoxazoline derivatives 22 (Fig. 13).
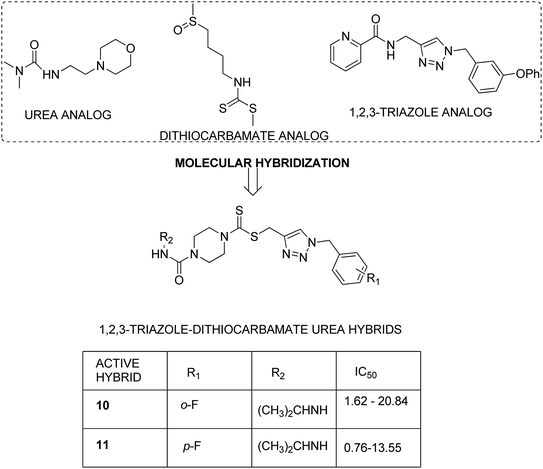 |
| | Fig. 9 1,2,3-Triazole-dithiocarbamate-urea hybrids. | |
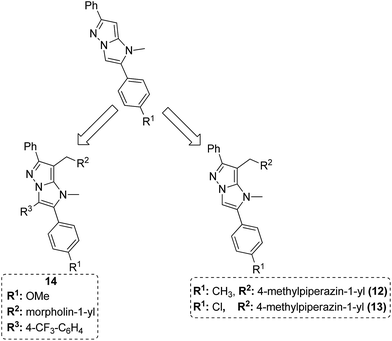 |
| | Fig. 10 Imidazo [1,2-b] pyrazoles. | |
 |
| | Fig. 11 Novel pyrimidine–benzimidazole hybrids. | |
 |
| | Fig. 12 Substituted indazole derivatives. | |
 |
| | Fig. 13 Isoxazoline-containing natural products. | |
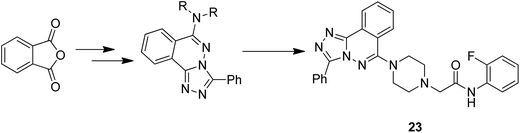
Moreover, two series of novel 1,2,4-triazolo [3,4-a] phthalazine derivatives were synthesized and evaluated for their anticancer activity in the following human cancer cell lines: MGC-803, EC-9706, HeLa, and MCF-7.24 When compared to the standard drug fluorouracil, compound 23 exhibited significantly high anticancer activity, with the IC50 values ranging from 2.0 to 4.5 μM. It was revealed from the flow cytometry analysis that compound 23 induced early cell apoptosis and cell cycle arrest at the G2/M phase. The phenyl group at the C-3 position of the compound was most active and showed better cytotoxic activity (Fig. 14). In another study, the novel hybrid molecules of 1,2-furanonaphthoquinones and 1,2,3-1H-triazoles were synthesized and evaluated for their anticancer activity against four different leukemia cells: MOLT-4, CEM, K562, and KG1. Compounds 24 and 25 showed higher cytotoxic potential against all the tested leukemia cell lines as compared to the classical anticancer compounds such as doxorubicin and cisplatin (Fig. 15).25 Moreover, the modified Pictet-Spengler reaction and click chemistry were used to synthesize a novel series of N-benzenesulfonyl-1,2,3,4-tetrahydroisoquinolines.26 Compounds 26 and 27 were found to be the most potent anticancer agents and selected for molecular docking against the plausible target site AKRIC3. Compound 26 was the most potent compound against the cancer cell lines HuCCA-1, whereas the p-tolyl analog 27 displayed the most potent inhibitory activity against HepG2 cells. The potent and selective action of these compounds was because of the direct junction of their substituents at the R1 and R2 positions. The potent activity of compound 27 against HepG2 cells is due to the presence of the hydrophobic group and it binds to the target site of action via the hydrophobic region (Fig. 16).
 |
| | Fig. 14 Novel 1,2,4-triazolo [3,4-a] phthalazine derivatives. | |
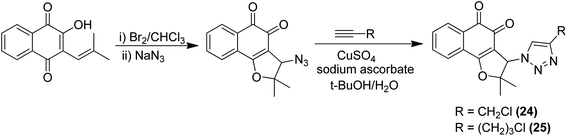 |
| | Fig. 15 1,2-Furanonaphthoquinones and 1,2,3-1H-triazole hybrids. | |
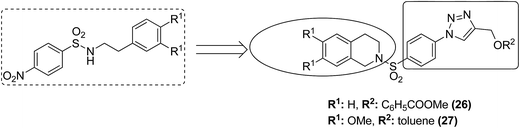 |
| | Fig. 16 Novel N-benzenesulphonyl-1,2,3,4-tetrahydroisoquinoline-based triazoles. | |
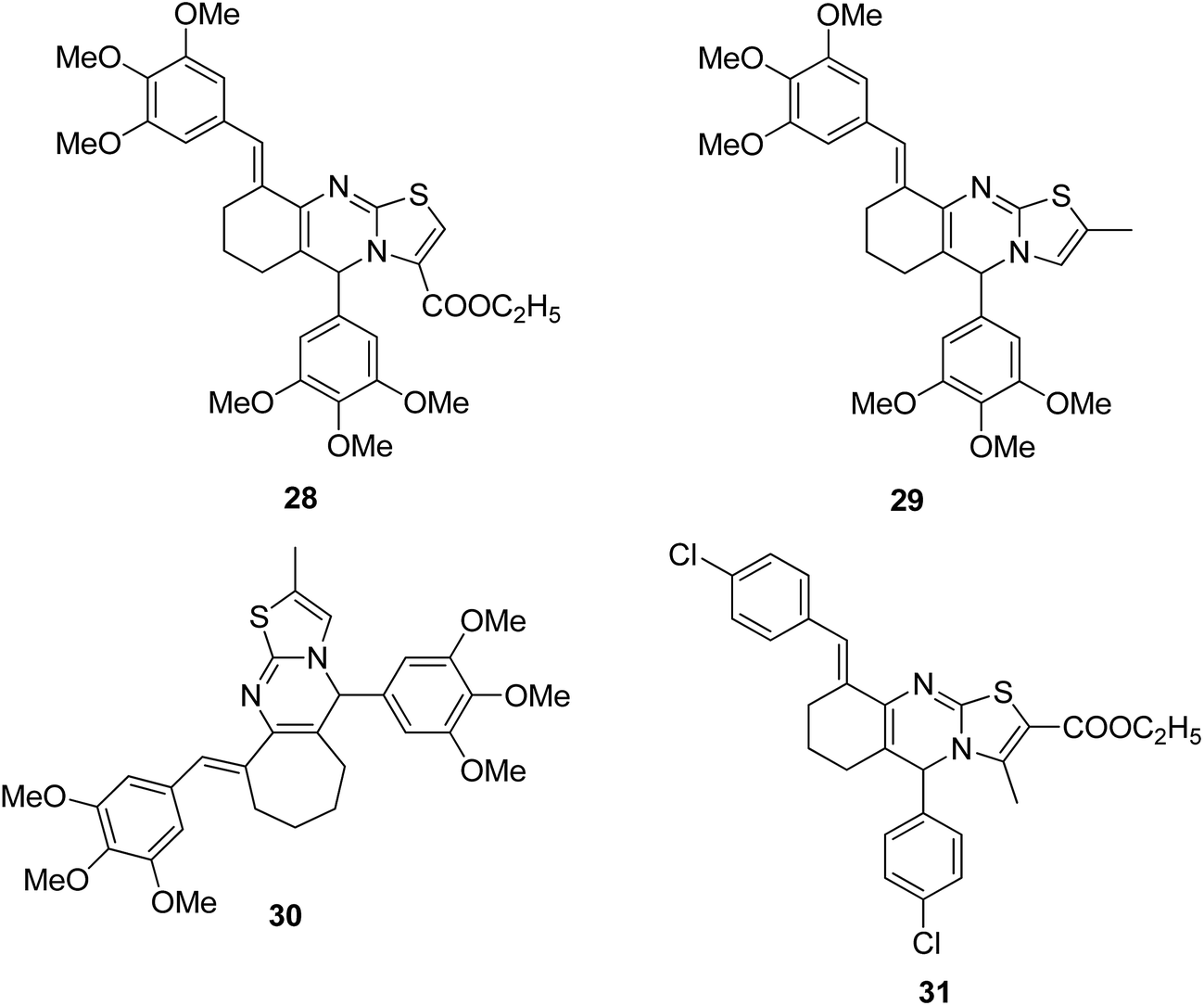
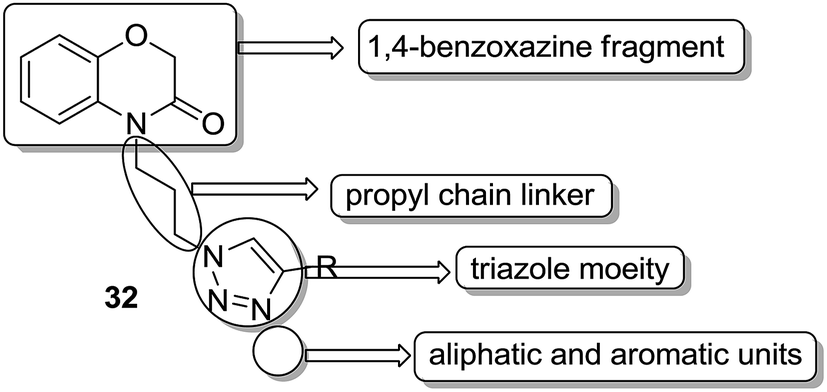
A novel series of hybrid compounds were synthesized using thiazolo[2,3-b]quinazoline and thiazolo[3,2-a]pyrimidine analogs.27 Amongst the synthesized compounds, 28, 29, 30, and 31 showed significant anticancer activity when tested against 60 cancer cell lines at the National Cancer Institute, with the GI50 MG-MID values of 2.4, 1.5, 11.2, and 3.1 μM, respectively (Fig. 17). A different study involved the synthesis of a series of 1,4-benzoxazine-1,2,3-triazole hybrids through a click chemistry approach, and the novel molecules were tested for in vitro cytotoxicity against human HeLa, MIAPACA, MDA-MB-231, and IMR32 cell lines. Compound 32 showed significant activity against MDA-MB-231 and IMR32 cell lines. It was the first study reported on the synthesis and in vitro antiproliferative evaluation of 1,2,3-triazole-1,4-benzoxazine hybrids (Fig. 18).28
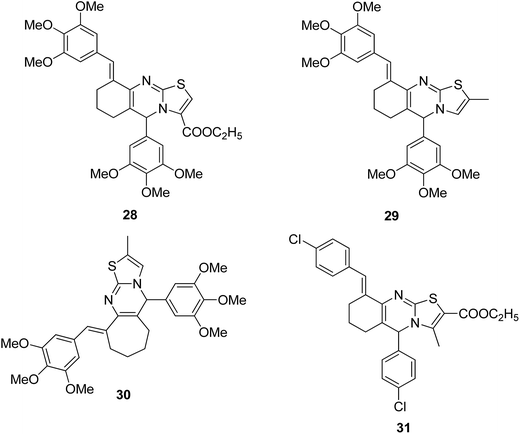 |
| | Fig. 17 Thiazolo[2,3-b]quinazoline and thiazolo[3,2-a]pyrimidine analogs. | |
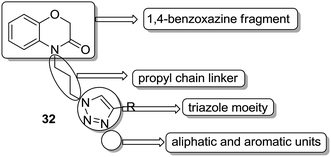 |
| | Fig. 18 1,4-Benzoxazinone-[1,2,3]triazole hybrids. | |
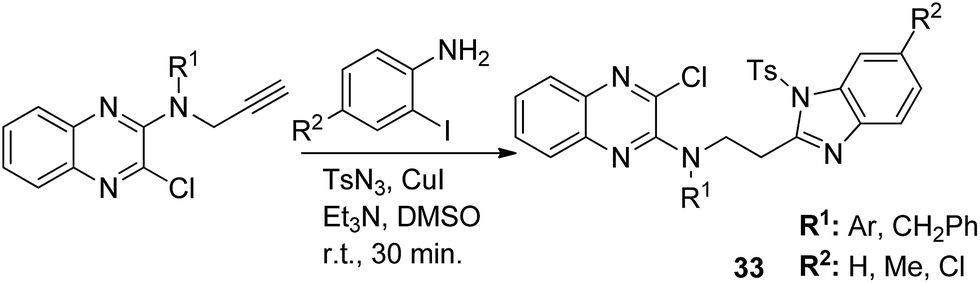
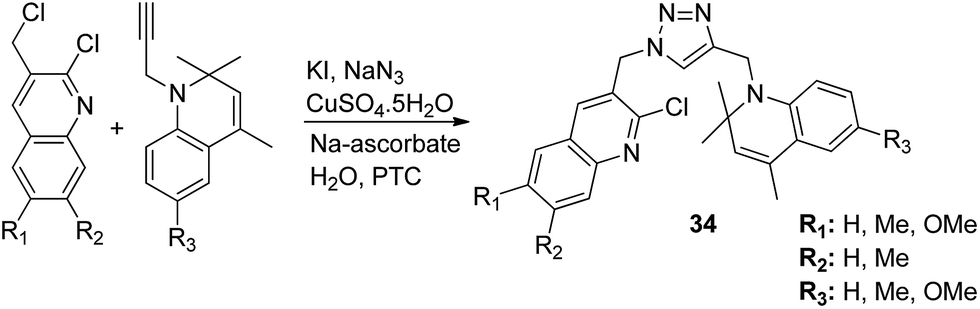
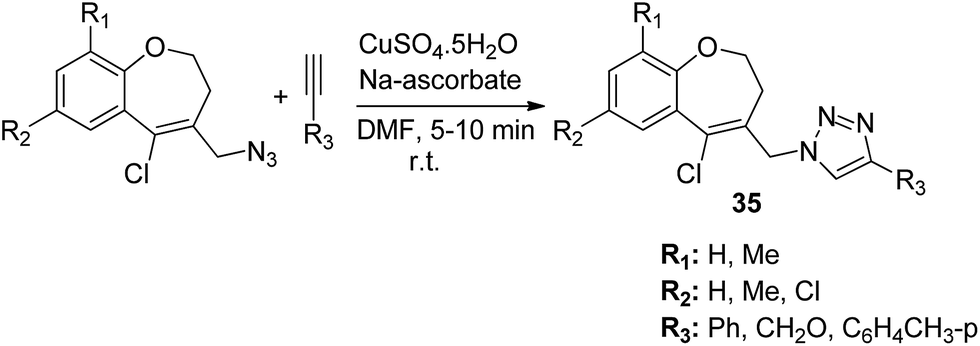
The novel hybrid molecules were synthesized via the multicomponent reaction of N-(prop-2-ynyl)-quinoxalin-2-amine derivatives with 2-iodoanilines and tosyl azide. The solvent system used was 10 mol% copper iodide and triethylamine in DMSO. The products 33 were achieved in good yields within 30 minutes without the use of a ligand and lateral addition of catalyst (Fig. 19). The anticancer activity studies revealed that these compounds induced significant apoptosis in zebrafish embryos.29 In another study, green chemistry was applied to link the three pharmacophoric groups (quinoline, triazole, and dihydroquinoline) to form a novel series of hybrid molecules. The process involved one-pot sequential azidation and copper-catalyzed azide–alkyne cycloaddition (CuAAC) in water under mild conditions. The substitution was performed at the 1 and 4 position of 1,2,3-triazoles having quinolinyl methylene at N-1 and 1,2-dihydroquinolinyl methylene at the C-4 position. The synthesized compounds 34 were evaluated for anticancer activity against various cell lines and found to possess significant activity against lung cancer cells (Fig. 20).30 Kuntala et al. synthesized novel benzoxepine-1,2,3-triazole hybrids via the application of the CuAAC approach. This rapid click reaction was performed by treating 7,9-disubstituted(Z)-4-(azidomethyl)-5-chloro-2,3-dihydrobenzo[b]oxepine with terminal alkynes at room temperature using DMF as the solvent. The synthesized compounds 35 were found to be cytotoxic against lung and colon cancer cell lines (Fig. 21).31
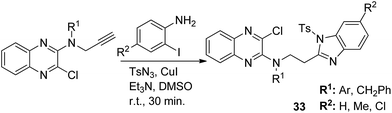 |
| | Fig. 19 Synthesis of quinoxaline–benzimidazole hybrids via a ligand-free MCR. | |
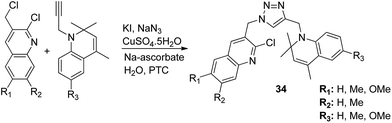 |
| | Fig. 20 Synthesis of a new hybrid template by linking quinoline, triazole, and dihydroquinoline pharmacophoric groups. | |
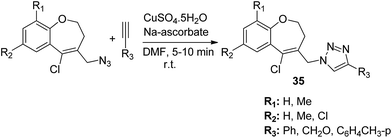 |
| | Fig. 21 Synthesis of novel benzoxepine-1,2,3-triazole hybrids. | |
3. Camptothecin-based hybrids
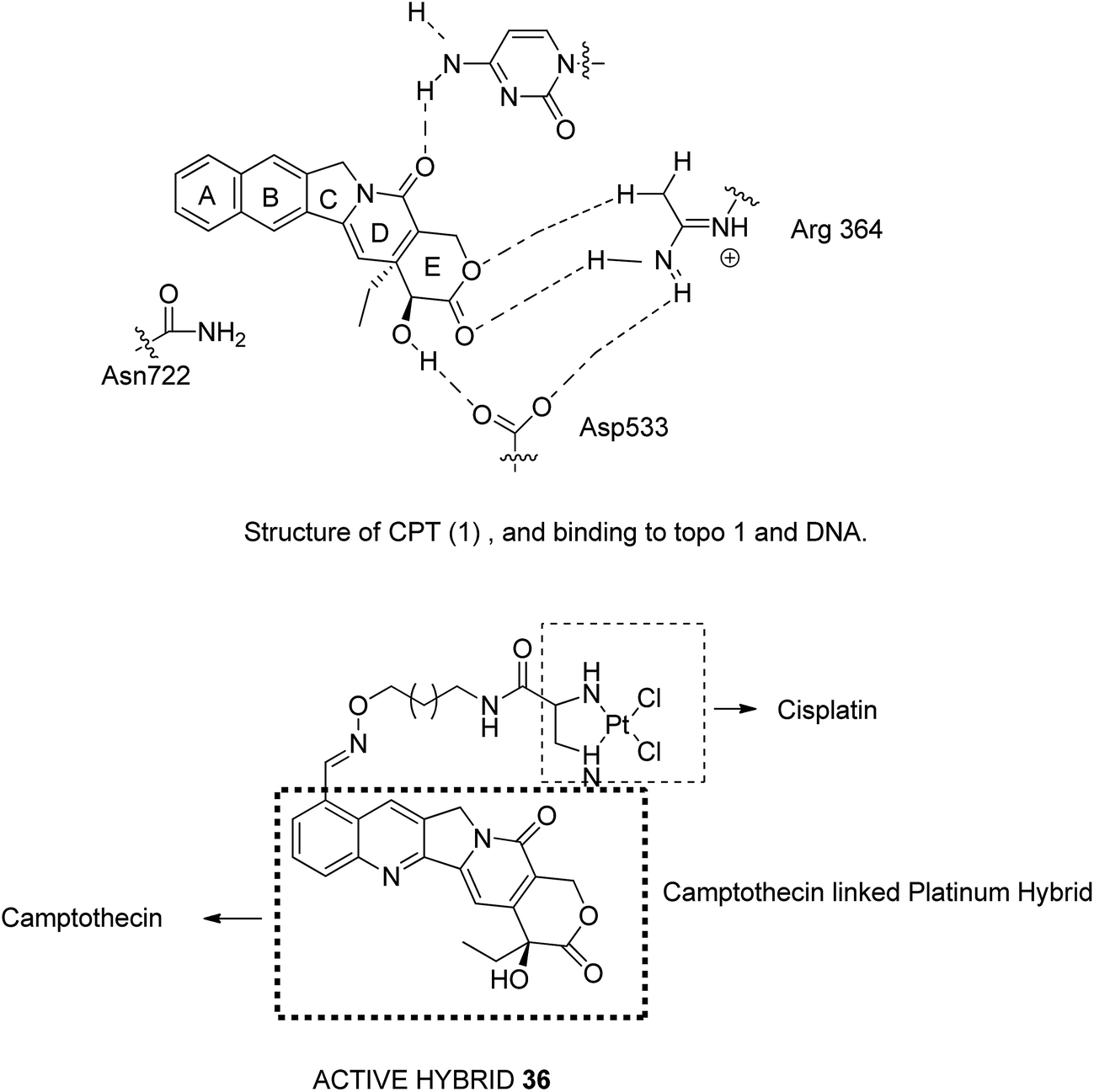
Camptothecin (CPT), a quinoline alkaloid with potent antitumor activities, was isolated from the Chinese plant Camptotheca acuminate by Wall et al. in 1996.32 It inhibits the DNA enzyme topoisomerase I by blocking the rejoining step of the cleavage/relegation of Topo-I. Topo I is an enzyme essential for cellular processes, participating in replication, transcription, and DNA repair. The major limitations related to CPT are low water solubility, inactivation of carboxylate salt, and short pharmacokinetic half-time. Thus, to overcome these limitations, Huang et al.33 synthesized E-ring-modified camptothecin. The substitution of methylene in the E-ring resulted in a seven-membered β-hydroxy lactone E-ring, which resulted in stereoselective inhibition of TOPO-I. Similarly, Cincinelli et al.34 synthesized camptothecin-linked platinum hybrids and assessed them against human tumor cell lines. Among these hybrids, compound 36 showed remarkable activity against the H460 tumor xenograft. This hybrid overcame the drawbacks such as off-target effects, poor pharmacokinetic profile of the platinum drug, and DNA cleavage reversibility of camptothecin (Fig. 22).
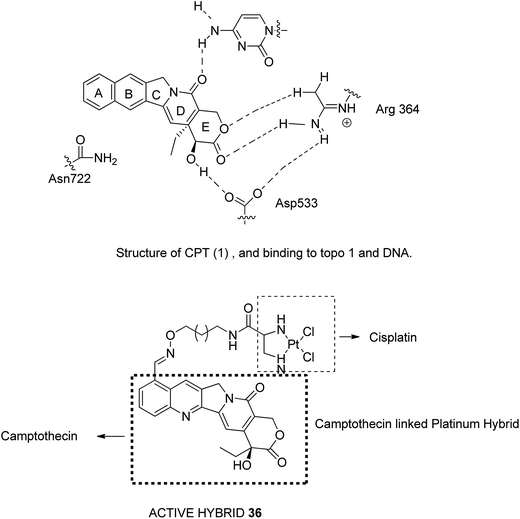 |
| | Fig. 22 Camptothecin-based hybrids. | |
4. Chalcone-based hybrids
Chalcone is an aromatic ketone and an enone that forms the central core for a variety of important biological compounds, which are known as chalcones or chalconoids. Chalcones have shown significant anticancer properties with their mode of action being similar to that of combretastatin. Chalcones bind to tubulin and prevent it from polymerizing microtubules, which is an important target for several anticancer agents. Chalcones can easily be prepared from substituted benzaldehydes that make it a privileged scaffold.35 The major advancements in the field of hybrid compounds derived from chalcones have been summarized below.
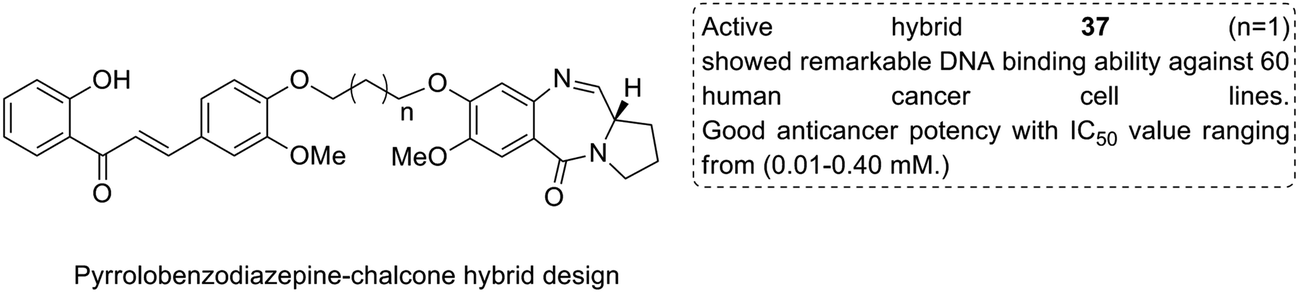
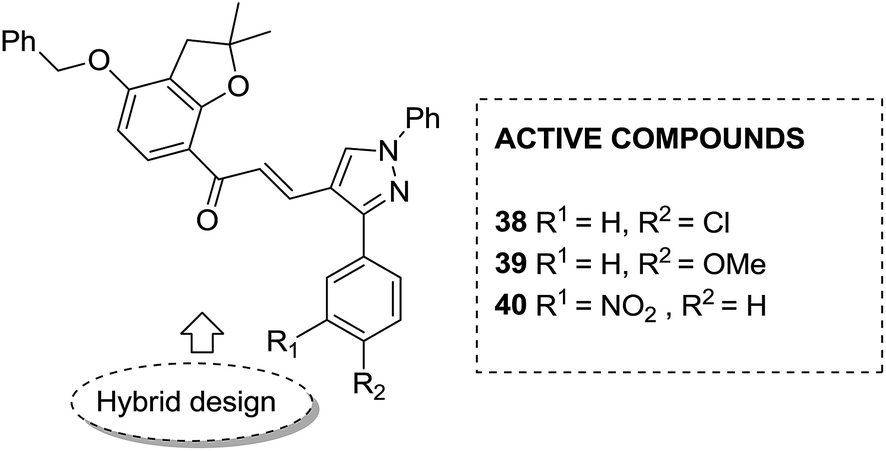
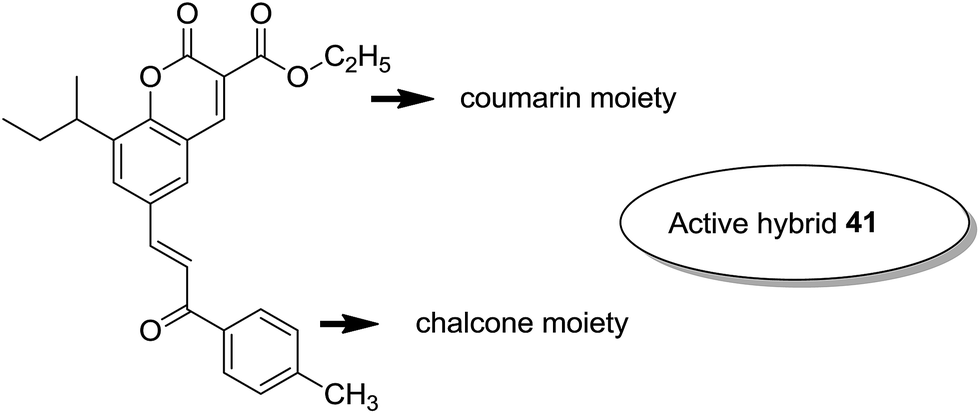
Considering the importance of chalcones, Kamal et al.36 employed a solid-phase synthetic protocol to prepare a new class of C8-linked pyrrolo[2,1-c][1,4] benzodiazepine–chalcone hybrids via utilizing an intramolecular aza-Wittig reductive cyclization approach. Among these hybrids, compound 37 showed promising anticancer activity. Solid-supported synthetic method and aldol condensation were also utilized during the synthesis of these conjugates (Fig. 23). Nagaraju et al. applied the Claisen-Schmidt condensation to synthesize a new class of hybrid chalcones.37 The synthesized compounds were tested for their anticancer activity against PC-3 (prostate cancer), HT-29 (colon cancer), B-16 (mouse macrophages), and NCI-H460 (lung cancer) cell lines. Compounds 38, 39, and 40 (IC50 = 8.4, 7.9, and 5.9 μM) showed significant anticancer activity, especially towards the PC-3 cell line. Since selectivity of a drug is an important parameter in cancer chemotherapy, further structural optimization of these compounds was carried out to improve the selectivity and it led to the preparation of more potential anticancer drug candidates (Fig. 24). Sashidhara et al. synthesized hybrids of coumarin and chalcones and carried out in vitro cytotoxicity studies against normal fibroblasts (NIH3T3) and a panel of four human cancer cell lines. Compound 41 was found to be the most potent, which showed about 30-fold more selectivity towards cervical carcinoma (C33A) cells over normal fibroblast NIH3T3 cells (Fig. 25).38
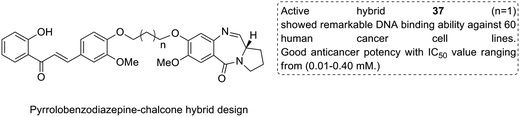 |
| | Fig. 23 C8-linked pyrrolo[2,1-c][1,4] benzodiazepine–chalcone hybrids. | |
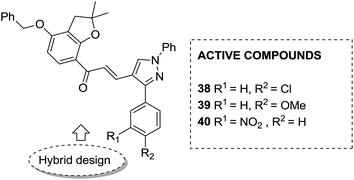 |
| | Fig. 24 Novel chalcone hybrids. | |
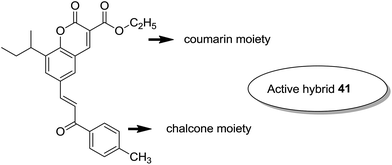 |
| | Fig. 25 Novel coumarin–chalcone hybrids. | |
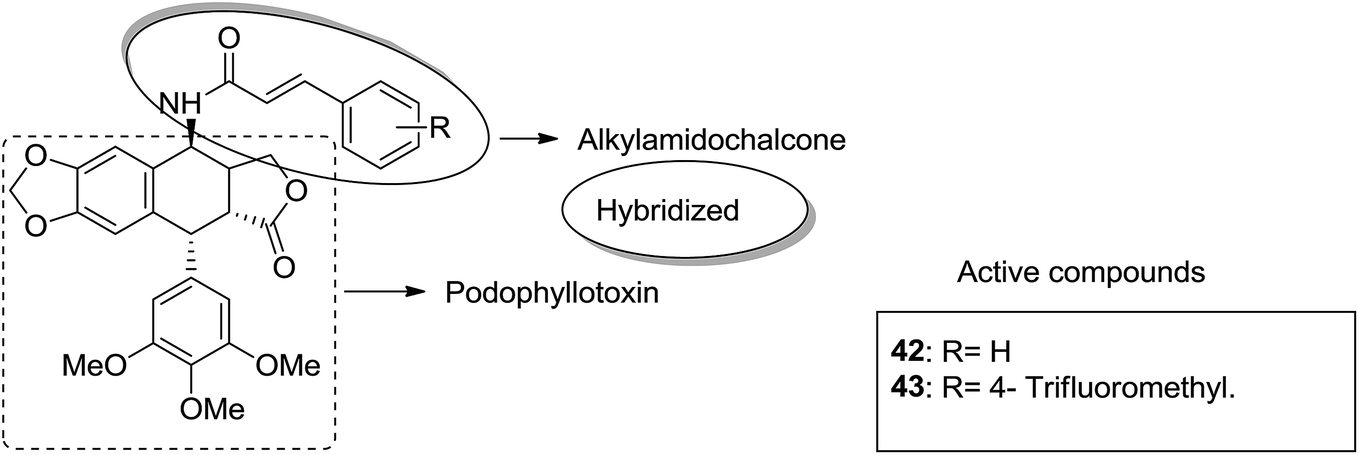
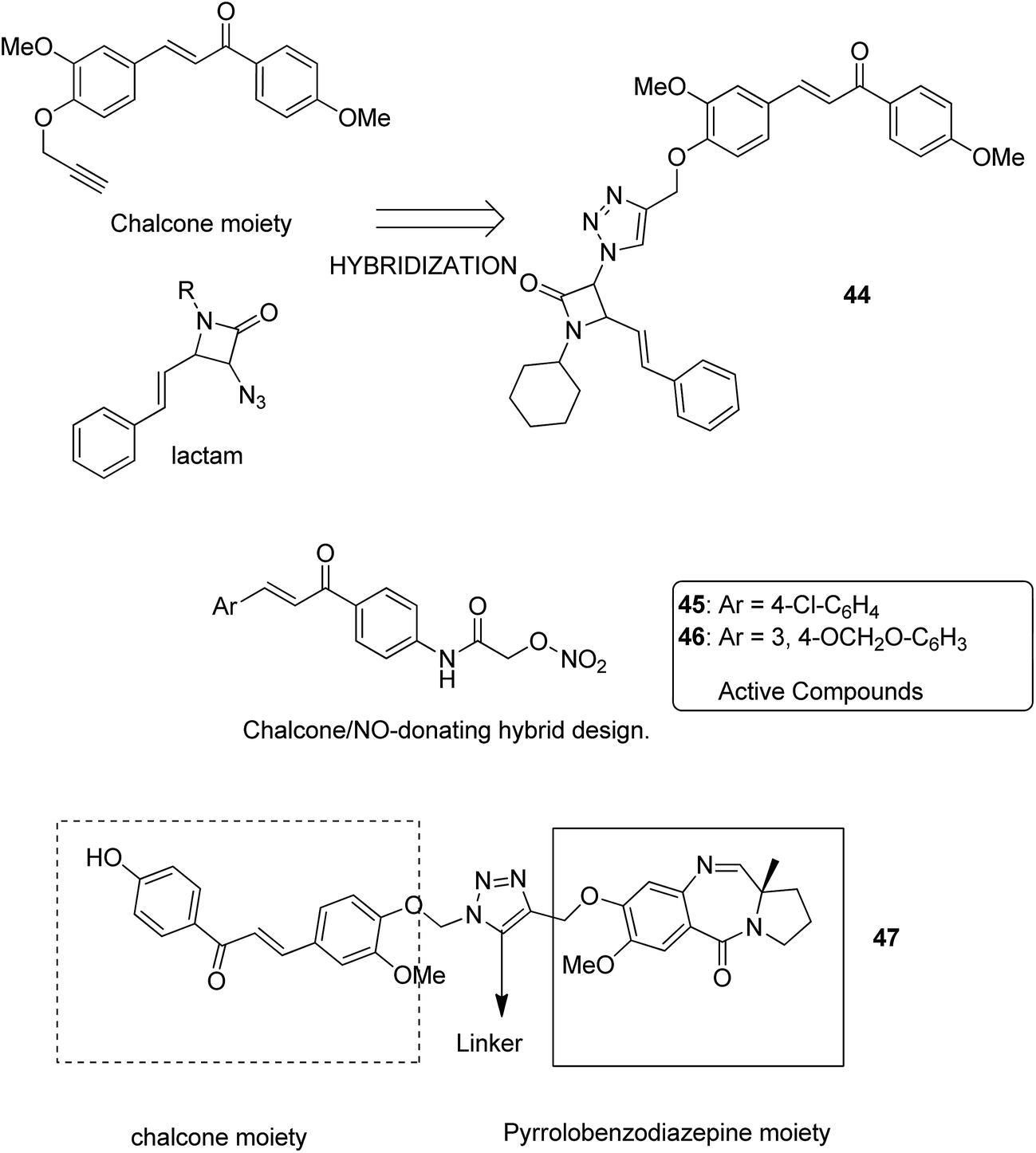
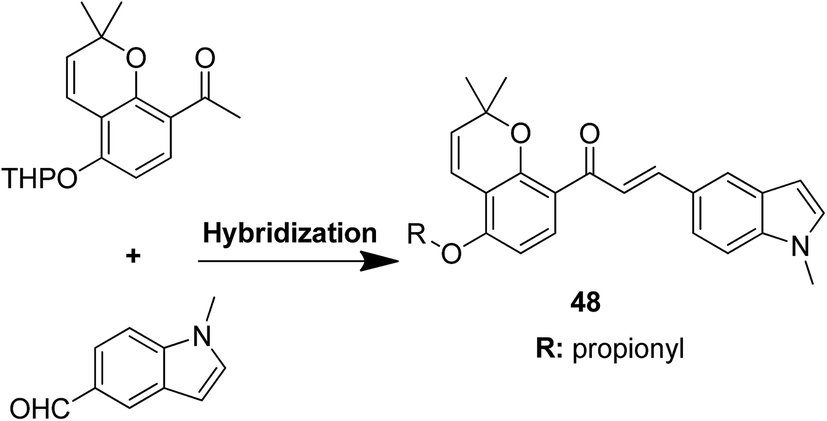
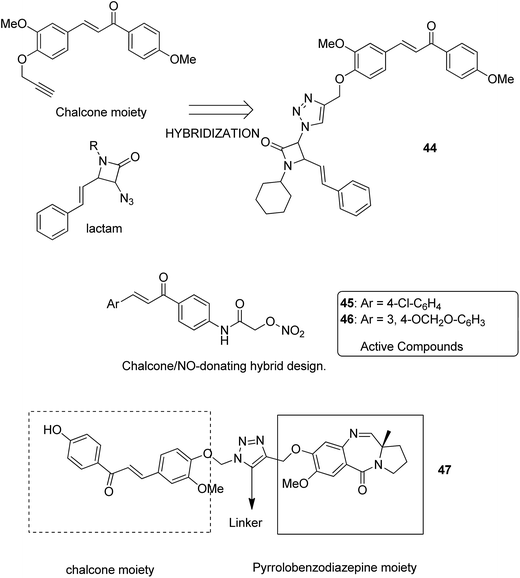
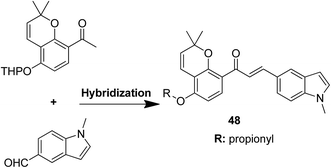
Podophyllotoxin is a naturally occurring antitumor lignan lactone isolated from the genus Podophyllum. It acts by inhibiting tubulin polymerization and is used for the treatment of various types of cancers. Considering the importance of podophyllotoxin, Kamal et al. synthesized podophyllotoxin tethered to 4-β-alkylamidochalcone and 4-β-cinnamido hybrids and assessed them for their anticancer activity in 5 human cancer cell lines.39 When compared to etoposide, hybrids 42 and 43 showed potent activity against the A-549 cancer cell line with the IC50 values of 2.7 and 2.1 μM, respectively. It induced caspase-3-dependent apoptotic cell death by arresting the cell cycle at the G2/M phase. The results were evaluated through flow cytometric analysis (Fig. 26). As is well-known, chalcones possess antitumor properties, triazoles have various biological activities (including anticancer profile), and β-lactam compounds (evergreen bioactive molecules) can widely and safely be used without significant toxicity. This information motivated Singh et al.4 to fuse these three moieties using azide–alkyne cycloaddition reactions and click chemistry approach. It resulted in the synthesis of novel 1,2,3-triazole-tethered β-lactam chalcone bifunctional hybrids, which were assessed against four human cancer cell lines. SAR studies showed that methoxy substitution at the ortho-position in ring A and para-position in ring B on chalcone enhanced the anticancer activity of these hybrids. Among the hybrids, compound 44 showed remarkable activity against A-549 (lung), PC-3 (prostate), THP-1 (leukemia), and Caco-2 (colon) cell lines with the IC50 value < 1, 67.1, <1, and 6.37 μM, respectively. In another study, nitrogen oxide (NO), which is an important mediator in biological systems, was found to prevent tumor cells from metastasizing and facilitated macrophages to kill the tumor cells. The nitric oxide-donating moieties such as nitrate ester, oximes, and furoxans were linked to amino chalcones to synthesize NO-donating chalcone hybrids as anticancer agents.40 Among the hybrids, compound 45 and 46 showed significant activity against colon and melanoma cancer cell lines and also showed anti-inflammatory activity with less ulcerogenic liability. The significant anticancer potential of chalcones and PBD encouraged Kamal et al.41 to design anticancer hybrids using the click chemistry approach. A novel series of A-C8/C-C-2 chalcone-linked pyrrolo [2,1-c][1,4] benzodiazepine hybrids were synthesized and connected by 1,2,3-triazole side-armed alkane spacers. These hybrids were then extensively evaluated towards the MCF-7 cancer cell lines. The results of the MTT proliferation assay revealed hybrid 47 as the most potent anti-proliferative agent. It showed its action through a GI cell cycle arrest with an effect on the protein cyclin D1 and CdK4 (Fig. 27). Novel pyrano chalcone derivatives containing indole moiety were synthesized and evaluated for their anticancer activities. Compound 48 emerged as the most potent cytotoxic agent against all cancer cell lines, with the IC50 values ranging from 0.22 to 1.80 μM. This compound also induced cell cycle arrest at the G2/M phase and inhibited the polymerization of tubulin (Fig. 28).42
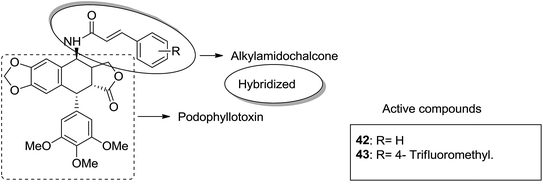 |
| | Fig. 26 4β-Alkylamidochalcone and 4β-cinnamido-linked podophyllotoxins hybrids. | |
 |
| | Fig. 27 1,2,3-Triazole-tethered β-lactam-chalcone bifunctional hybrids. | |
 |
| | Fig. 28 Pyrano chalcone derivatives. | |
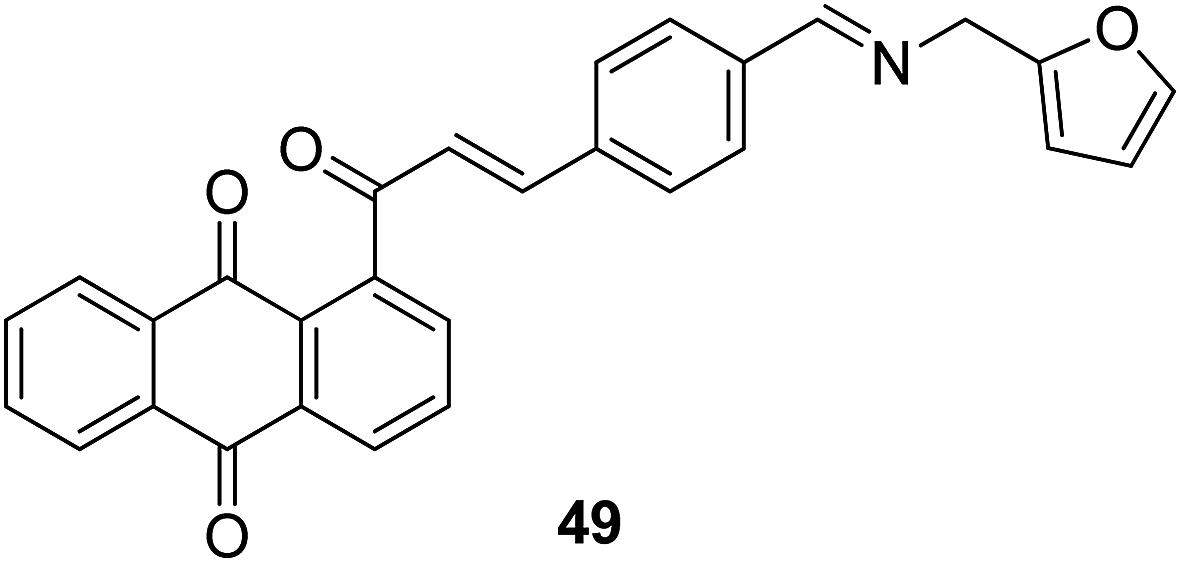
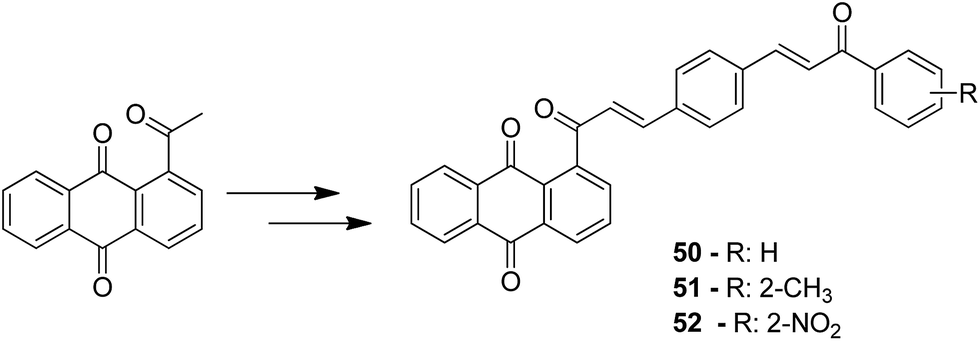
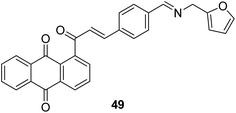
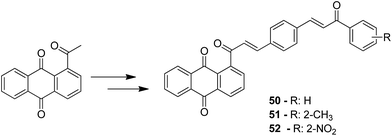
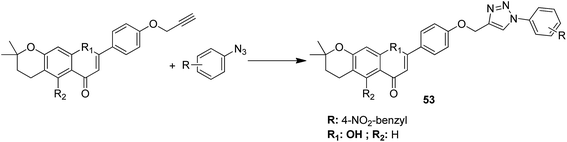
A novel series of chalcone hybrid analogues containing an anthraquinone scaffold and an imine fragment was synthesized and evaluated for anticancer activity against HeLa, LS174, and A549 cancer cells.38 The most potent compound 49 had an imino group linked to a furan ring. It was revealed from the mode of action studies that this compound induced the changes that were essential for apoptosis in HeLa cells (Fig. 29). Furthermore, a novel series of anthraquinone-based chalcone compounds was synthesized by the Claisen-Schmidt reaction using 1-acetylanthraquinone as the starting material. Compounds 50, 51, and 52 showed low cytotoxicity against healthy MRC-5 cell lines and inhibited HeLa cells with the IC50 values ranging from 2.36 to 2.73 μM. The flow cytometry studies confirmed that these compounds caused cell arrest at the S and G2/M phases and induced caspase-dependent apoptosis. These compounds also exhibited calf thymus DNA-binding activity (Fig. 30).39 Chinthala et al. synthesized a novel series of chalcone-triazole derivatives that were evaluated for in vitro anticancer activity against IMR32 (neuroblastoma), HepG2 (human hepatoma), MCF-7 (human breast adenocarcinoma), DU-145 (human prostate carcinoma), and A549 (human lung adenocarcinoma) cell lines. Compound 53 was found to be most potent as it showed significant anticancer activity against all the cell lines (Fig. 31).43
 |
| | Fig. 29 Anthraquinone-based chalcone analogue. | |
 |
| | Fig. 30 Anthraquinone-chalcone hybrids. | |
 |
| | Fig. 31 Novel chalcone triazoles. | |
5. Pyrrolo [2,1-c][1,4] benzodiazepine (PBD)-based hybrids
Pyrrolo [2,1-c][1,4] benzodiazepines (PBDs) are naturally occurring DNA interactive antitumor antibiotics isolated from various Streptomyces species. DC-81 and anthramycin are some of the typical examples of PBDs.44 The PBD cytotoxins exert powerful antitumor activity by covalently binding at the C-11 position of PBD and the C-2 amino group of the guanine residues within the minor groove of double-stranded DNA. Hybrids with antitumor agents linked to PBD have been discussed below.
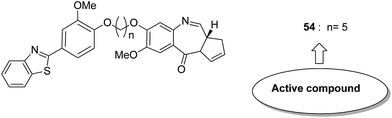
Considering the structural modification of PBD and the important anticancer activities of benzothiazoles as well as isosteric benzoxazoles, Kamal et al.45 synthesized a series of benzothiazole/benzoxazole–PBD hybrids. Alkane and alkylamide spacers attached to the conjugates of benzothiazole and benzoxazole-linked pyrrolobenzodiazepine were prepared and evaluated for their anticancer activity. The synthesized compounds were also investigated for DNA thermal denaturation studies and restriction endonuclease digestion. Among the hybrids, compound 54 showed potent anticancer activity via arresting or stopping the apoptosis. SAR studies showed that the presence of a sulphur atom in benzothiazole and the 5-carbon chain length resulted in high DNA-binding ability of minor groove binders with the SEQ-A and A375 cell line at a low concentration of 0.5 μM (Fig. 32).
 |
| | Fig. 32 Benzothiazole/benzoxazole-pyrrolo[2,1-c][1,4] benzodiazepine (PBD) hybrids. | |
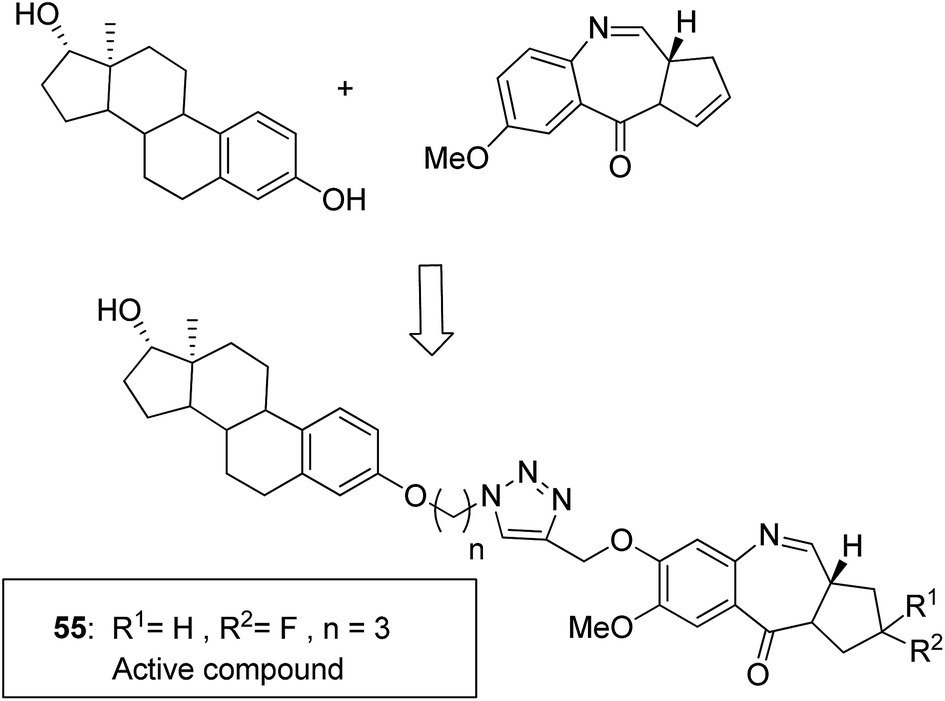
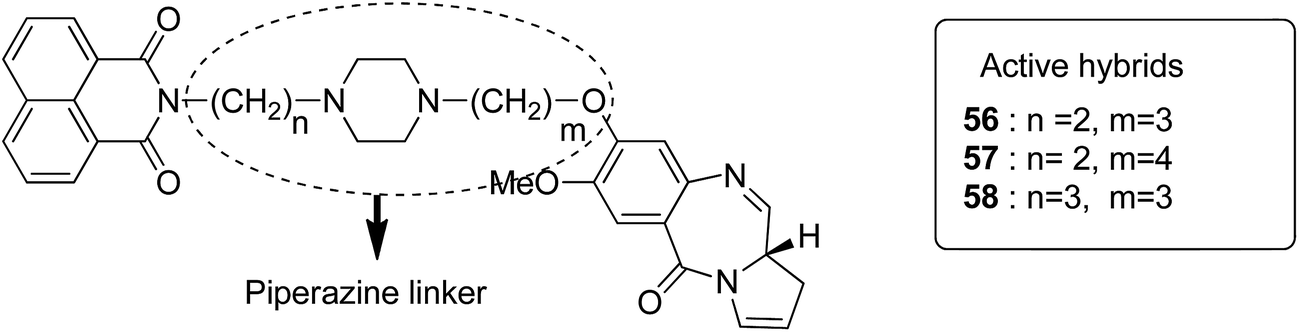
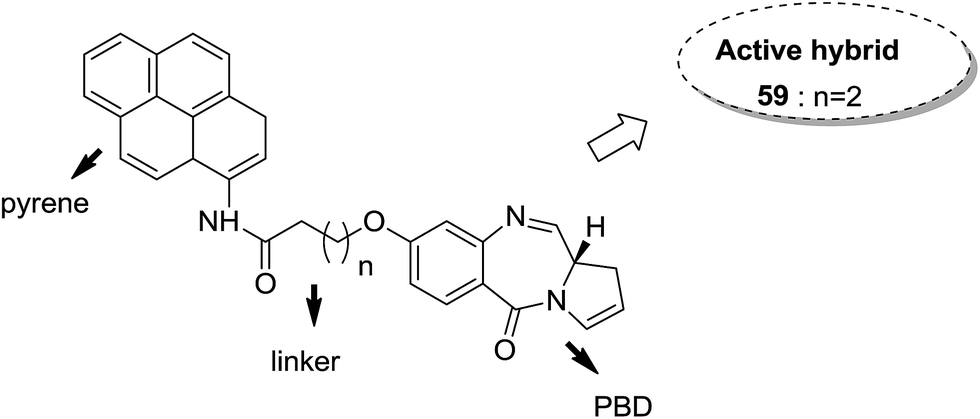
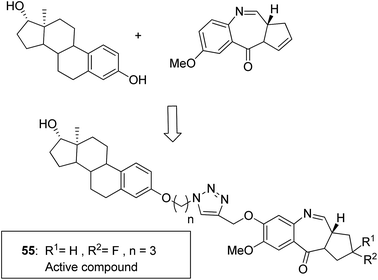
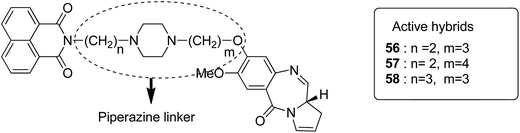
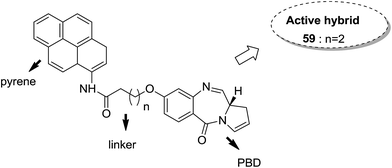
Moreover, estrogen receptor, a key biological target over the years, has attracted significant interest since it is expressed in many cancers. Therefore, estradiol and its related receptor can be used to direct a cytotoxic agent to the target cells. This information was utilized by Kamal et al. for the synthesis of estradiol-linked pyrrolobenzodiazepine (PBD) conjugates as anticancer agents.46 Among the 18 synthesized conjugates, compound 55 was most potent against the breast cancer cell line MCF-7. Active conjugate 55 was synthesized by the reduction of nitro groups using hydrated stannous chloride in the presence of methanol. Cyclization and deprotection were carried out using mercuric chloride and calcium carbonate. Fragmentation of nuclei followed by disruption of microtubules resulted in a CDK1 inhibitor property of the active compound (Fig. 33). Naphthalimides with charge transfer and stacking between base pairs act as DNA-intercalating agents with high antitumor activity. Using this information, Kamal et al. synthesized novel pyrrolobenzodiazepine–naphthalimide conjugates 56, 57, and 58 with high DNA binding affinity. These unique bi-functional compounds showed in vitro activity against some cancer cell lines (Fig. 34).47 In another study, the novel hybrids of pyrene-linked pyrrolo[2,1-c][1,4]benzodiazepines were synthesized48 and found to exhibit effective anticancer activity against various human tumor cell lines. Among all the synthesized conjugates, hybrid 59 showed maximum anticancer activity against 60 cancer cell lines (Fig. 35).
 |
| | Fig. 33 Estradiol-linked hybrid molecules. | |
 |
| | Fig. 34 PBD–napthalimide hybrid molecules. | |
 |
| | Fig. 35 Pyrene-linked hybrid molecule. | |
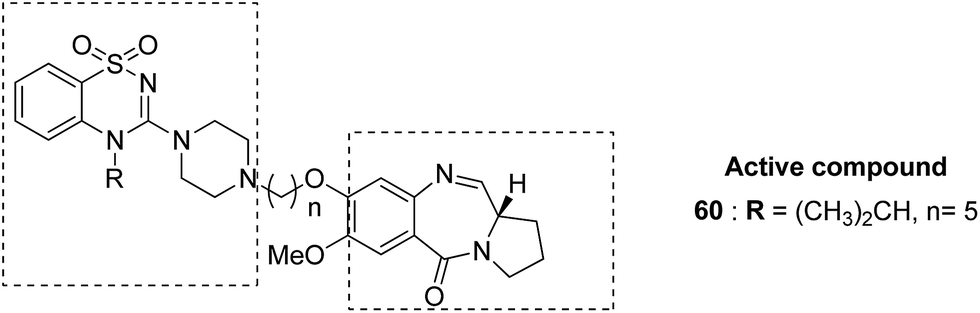
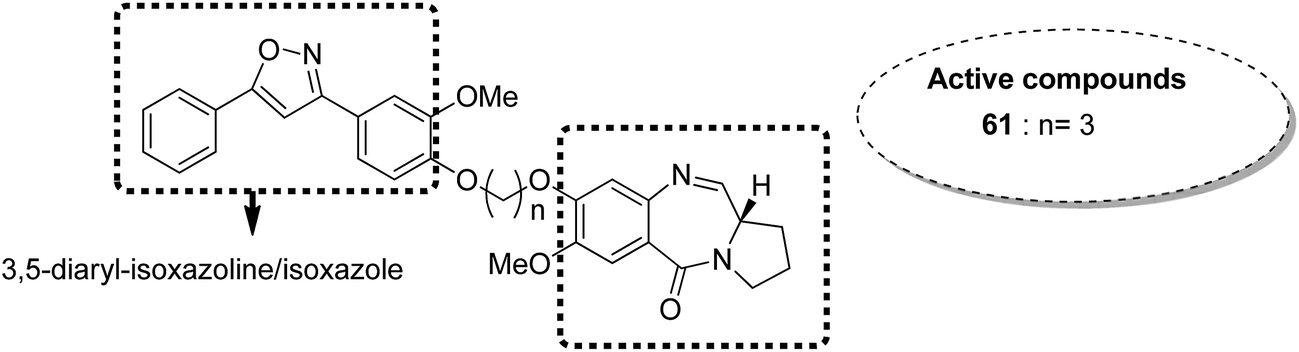
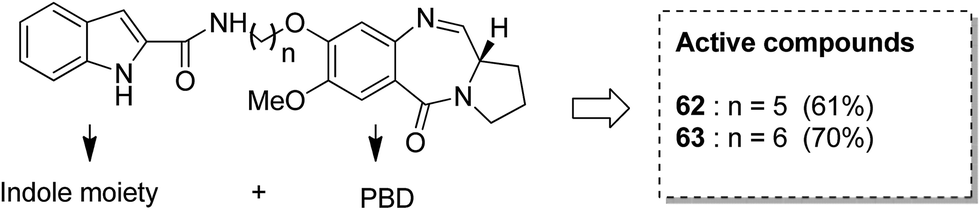
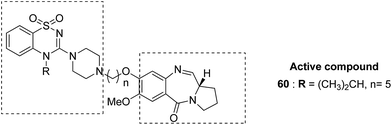
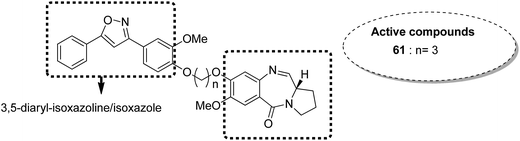
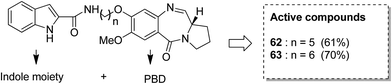
Benzothiadiazine–pyrrolobenzodiazepine-linked hybrids were synthesized and evaluated for cytotoxicity against many cancer cell lines. Among the synthesized PBD hybrids,49 compound 60 was found to be most effective as it increased the temperature of CT DNA by 6.7 °C after incubation for 36 hours. SAR studies showed that 1,2,4-benzothiadiazine was linked to the C-8 position of the PBD ring through a piperazinyl alkane spacer and analyzed as a new possible anticancer agent. This hybrid showed significant in vitro anticancer activity against breast, oral, and prostrate human cancer cell lines along with impressive DNA-binding strength (Fig. 36). Another study involved the synthesis of a series of 3,5-diaryl-isoxazoline/isoxazole-linked pyrrole [2,1-c][1,4]benzodiazepine (PBD) conjugates, which were evaluated for anticancer activity.50 Further investigations on these conjugates were performed for cell cycle disruption, which led to subsequent apoptotic cell death. The PBD conjugate 61 was selected for the preclinical studies since it was the most potent among all the synthesized compounds. The GI50 value for this compound was in the range of 0.1–2.15 μM. The improved anticancer activity was due to the linking of 3,5-diaryl-isoxazoline/isoxazole moiety to the PBD ring system (Fig. 37). Wang et al. synthesized novel hybrids of [2,1-c][1,4]benzodiazepine (PBD) and indole carboxylates.51 The carbon chain linkers were used to link the C-8 position of DC-81 with the indole 2-carbonyl moiety to form the high yield compounds 62 and 63. The preliminary tests confirmed that these compounds possessed potent anticancer activity. These compounds showed higher cytotoxicity and better DNA binding ability towards human melanoma A2058 cells. The NCI screening results suggested that these compounds can be used as potent broad spectrum anticancer agents that inhibit the growth of a variety of cancer cell lines (Fig. 38).
 |
| | Fig. 36 1,2,4-Benzothiadiazine-linked pyrrolo[2,1-c][1,4]benzodiazepine hybrids. | |
 |
| | Fig. 37 3,5-Diaryl-isoxazoline/isoxazole–pyrrolobenzodiazepine conjugates. | |
 |
| | Fig. 38 Pyrrolo[2,1-c][1,4]benzodiazepine and indole hybrids. | |
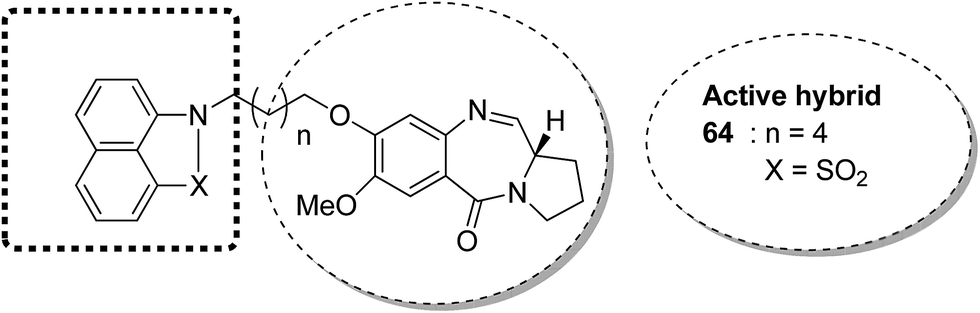
The novel conjugates of a series of benzo[c,d]indol and 2(1H)one-PBD were designed and synthesized as potential anticancer agents. The C-8 position of DC-81 was linked with a benzo[c,d]indol-2(1H)one moiety through different alkane spacers. The MTT assay method was used to evaluate these compounds for their anticancer activity in selected human cancer cell lines of lung, skin, colon, and prostate. Among these compounds, 64 was the most potent and showed promising anticancer activity as confirmed by Annexin V-FITC, Hoechst staining, caspase-3 activity, and DNA fragmentation analysis. It was confirmed via FACS analysis that compound 64 had cell cycle regulatory properties since cell death occurred due to cell cycle arrest at the SubG1 phase. The most important property of this compound was that it did not affect the normal cell line (HEK-293) (Fig. 39).52
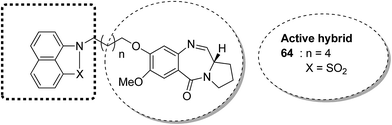 |
| | Fig. 39 Benzo[c,d]indolone–pyrrolobenzodiazepine hybrids. | |
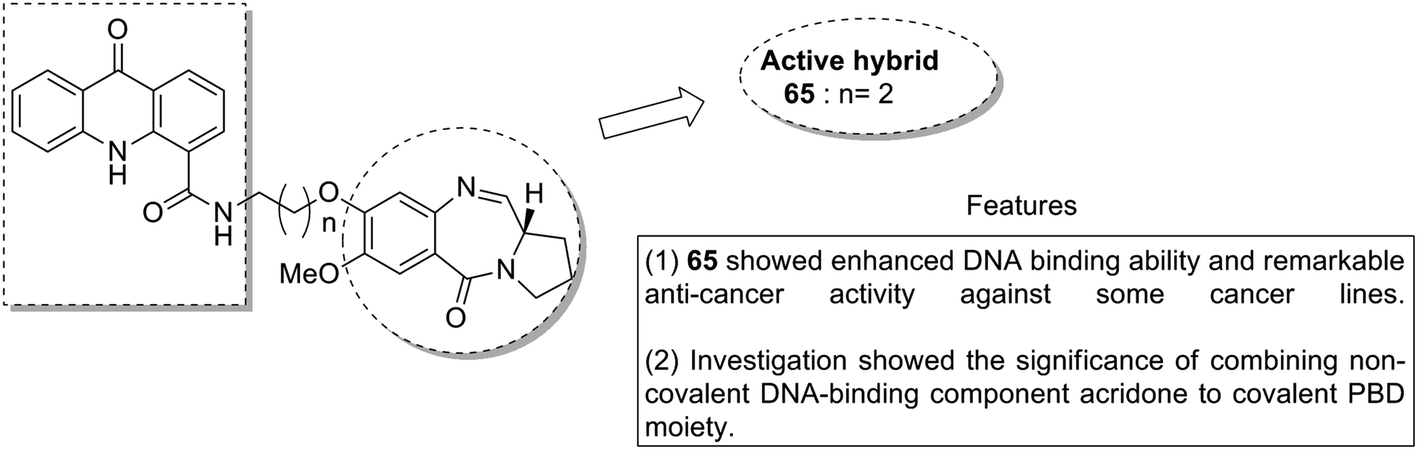
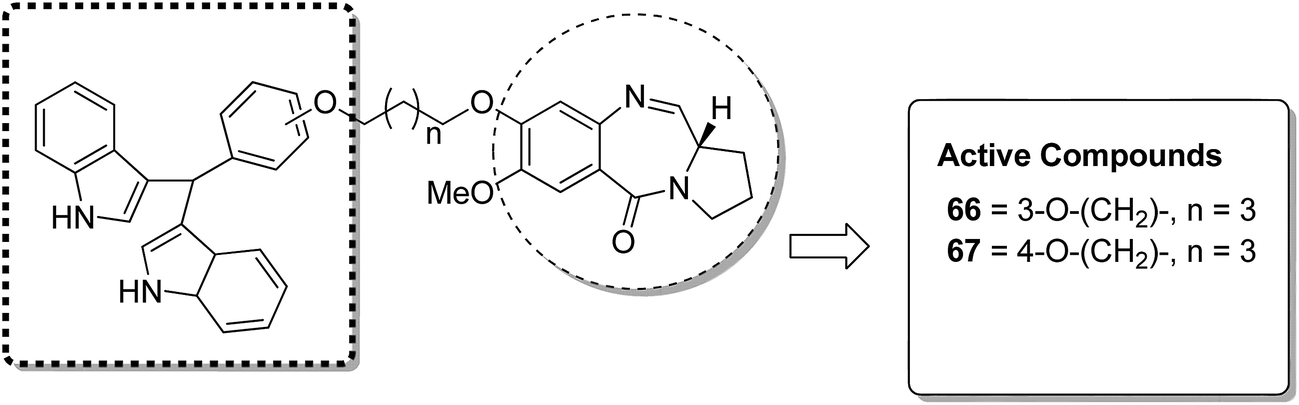
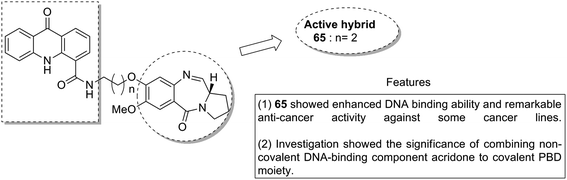
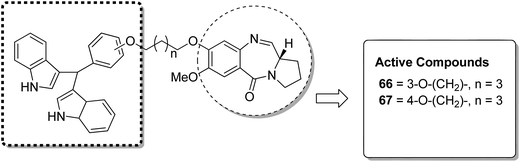
Acridine and acridone-based compounds consist of an important class of DNA-intercalating anticancer drugs. The anticancer hybrid compounds were prepared by linking the pyrrolobenzodiazepine hybrids with the acridone/acridine ring systems at the C8-position.53 Compound 65 was found to be the most potent among the synthesized compounds (Fig. 40). Moreover, another study was based on the synthesis of a novel series of bisindole–pyrrolobenzodiazepine conjugates, which were linked through different alkane spacers.54 The most potent compounds were 66 and 67, which showed significant anticancer activity and subjected to a detailed study towards the MCF-7 cell line. It was found via the flow cytometric (FACS) analysis that there was an increase in the sub-G1 phase cells and a decrease in the G2/M phase cells, which caused apoptosis in the MCF-7 cells. The apoptotic cell death was due to the down regulation of the levels of histone deacetylase protein, such as HDAC-1,2,3,8, and an increase in the levels of p21. The increased expression of cleaved-PARP and active caspase-7 in MCF-7 cells further confirmed the apoptotic nature of cell death (Fig. 41).
 |
| | Fig. 40 Pyrrolo[2,1-c][1,4]benzodiazepine–acridone/acridine hybrids. | |
 |
| | Fig. 41 Bisindole-linked PBD hybrid design. | |
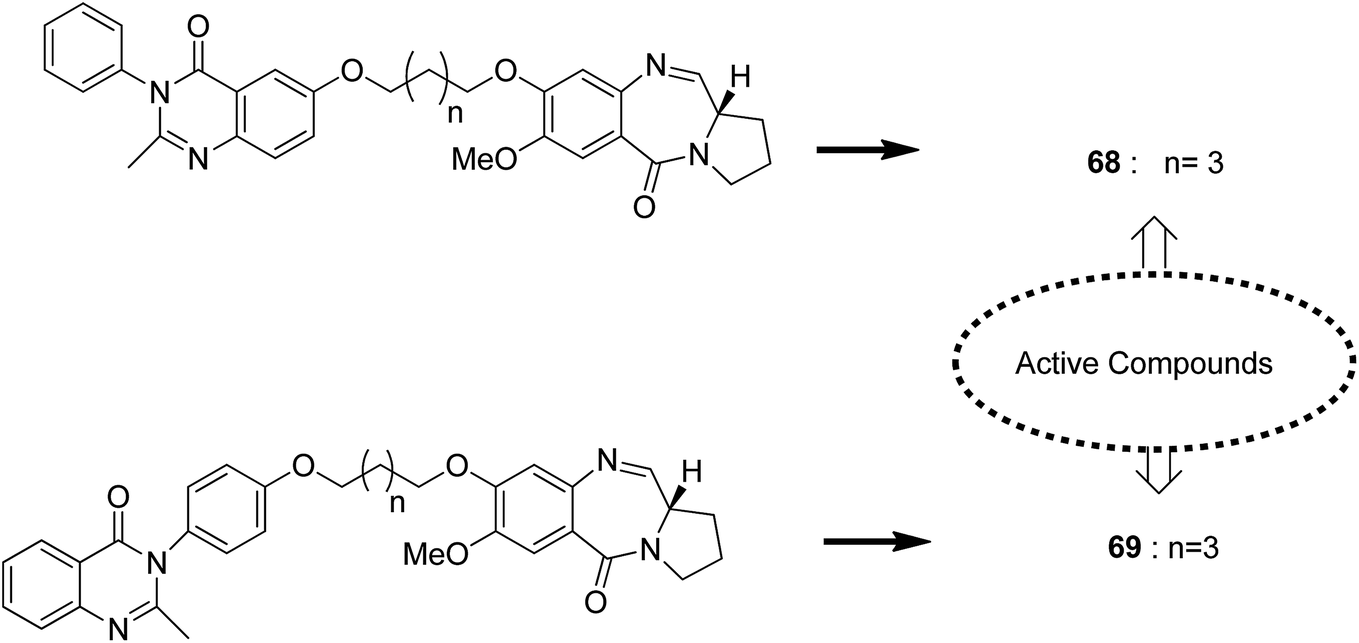
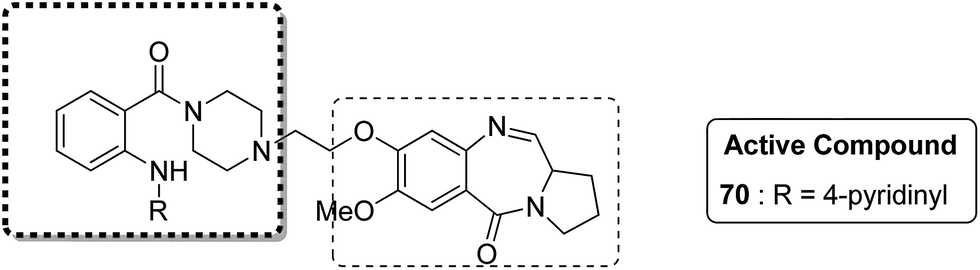
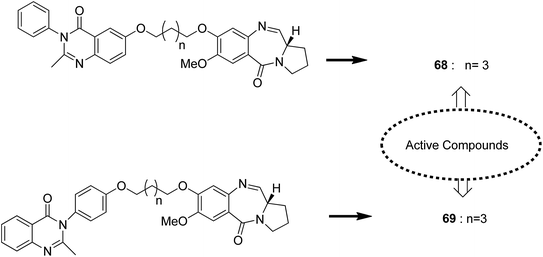
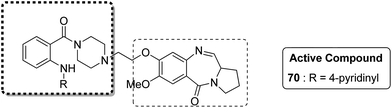
Novel hybrids of quinazoline linked to pyrrolobenzodiazepines were synthesized. Thermal denaturation studies showed that compounds 68 and 69 possessed better DNA binding ability as compared to DC-81. These compounds also exhibited crucial cytotoxicity towards A375 cells at 4 μM. The results were validated through an MTT assay (Fig. 42).55 In another study, a novel series of anthranilamide-fused pyrrolo [2,1-c] [1,4] benzodiazepine hybrids was synthesized and evaluated against the A374 cancer cell line. PBD hybrid 70 showed remarkable activity by inducing apoptosis through caspase-3 activation and increasing the level of p-53 (master regulator of apoptosis). SAR studies showed that in the active hybrid compound, the anthranilamide moiety was fused at the C-8 position of the A ring in the PBD ring system through a 4-piperazinyl alkane spacer, which resulted in effective anticancer activity (Fig. 43).56
 |
| | Fig. 42 Quinazoline-linked (PBD) hybrid design. | |
 |
| | Fig. 43 Anthranilamide–PBD hybrid design. | |
6. Coumarin-based hybrids
Coumarin, belonging to the benzopyrone chemical class, is a fragrant organic chemical compound. It is a colorless, crystalline substance in its standard state. Coumarin exhibits antitumour activity in vivo, with the effect believed to be due to its metabolites such as 7-hydroxycoumarin.57 Some coumarin-based hybrids have been discussed below.
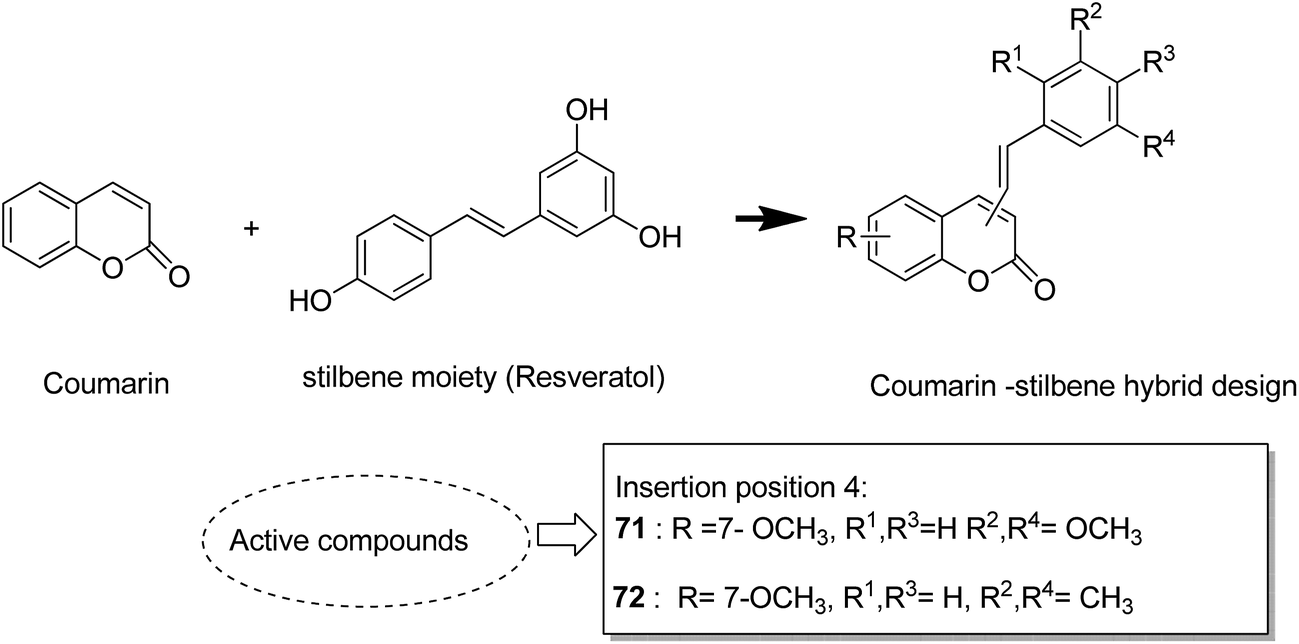
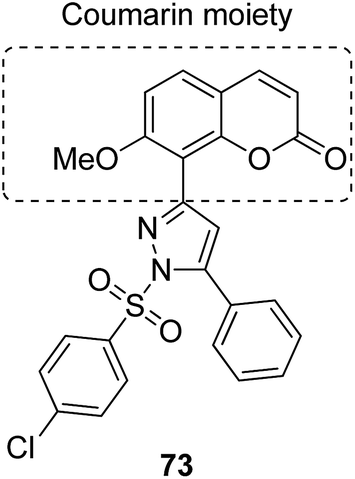
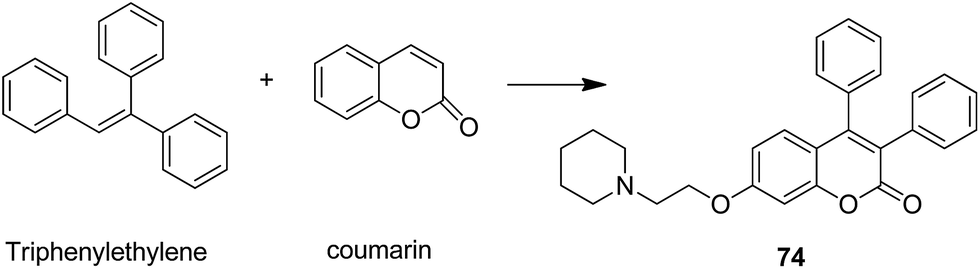
The naturally occurring coumarins and resveratrol possess significant anticancer activity. Belluti et al. synthesised and evaluated a new class of hybrid compounds by attaching a substituted trans-vinylbenzene moiety on a coumarin backbone that exhibited high antitumor activity.58 Compounds 71 and 72 were highly active compounds among this series, having excellent antiproliferative and proapoptotic activities. The excellent antitumor activity was due to the presence of the 7-methoxycoumarin nucleus, together with the 3,5-disubstitution in the trans-vinyl benzene moiety. Compounds 71 and 72 were tested against H460 lung carcinoma cells and they exhibited pharmacologically relevant antiproliferative activity. Comprehensive biological investigations revealed that these compounds had the ability to induce an appreciable level of apoptosis (Fig. 44). Amin et al.59 synthesized a novel series of coumarin–pyrazole hybrids by cyclizing coumarin chalcones with various substituted hydrazines through 1,4-addition on an α,β-unsaturated carbonyl system to produce the corresponding pyrazolines. The synthesized compounds were investigated against 60 cancer cell lines for their anticancer activity. Most of the compounds were effective against the breast cancer cells MCF7 and colon cancer cells HCT-116. Among these, compound 73 possessed the highest cytotoxicity, but it showed weak enzyme inhibitory activity against P13K (Fig. 45). Chen et al. designed and synthesized novel triphenylethylene–coumarin hybrid derivatives, having different amounts of amino side chains, under microwave radiations.60 The compounds with two amino side chains (except for morpholinyl) exhibited broad-spectrum and good antiproliferative activity against five tumor cell lines. These compounds also possessed low cytotoxicity in osteoblast. SAR analysis revealed that the amino alkyl chain had an important role against tumor cell proliferation as well as DNA interactions. Compound 74 showed good anticancer activity and significant binding with DNA. The study suggested that DNA might be one of the potential targets for triphenylethylene–coumarin hybrids as anticancer agents (Fig. 46).
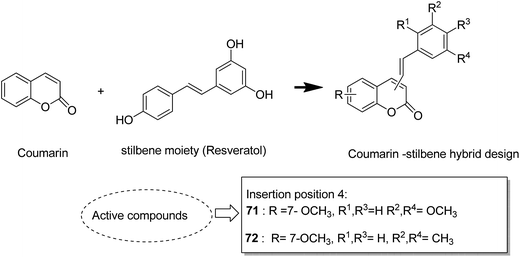 |
| | Fig. 44 Coumarin–stilbene hybrid molecules. | |
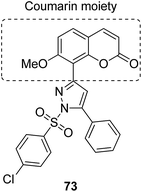 |
| | Fig. 45 Coumarin–pyrazoline hybrid design. | |
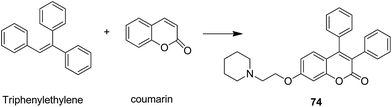 |
| | Fig. 46 Coumarin–triphenylethylene hybrid molecule. | |
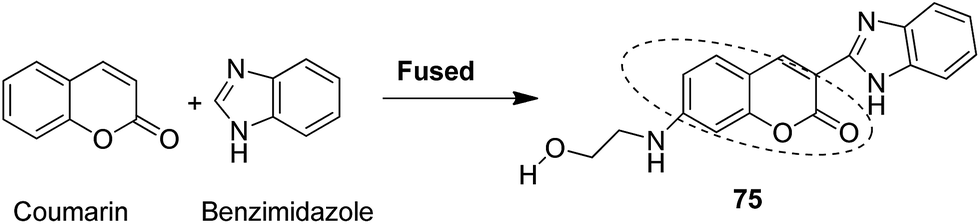
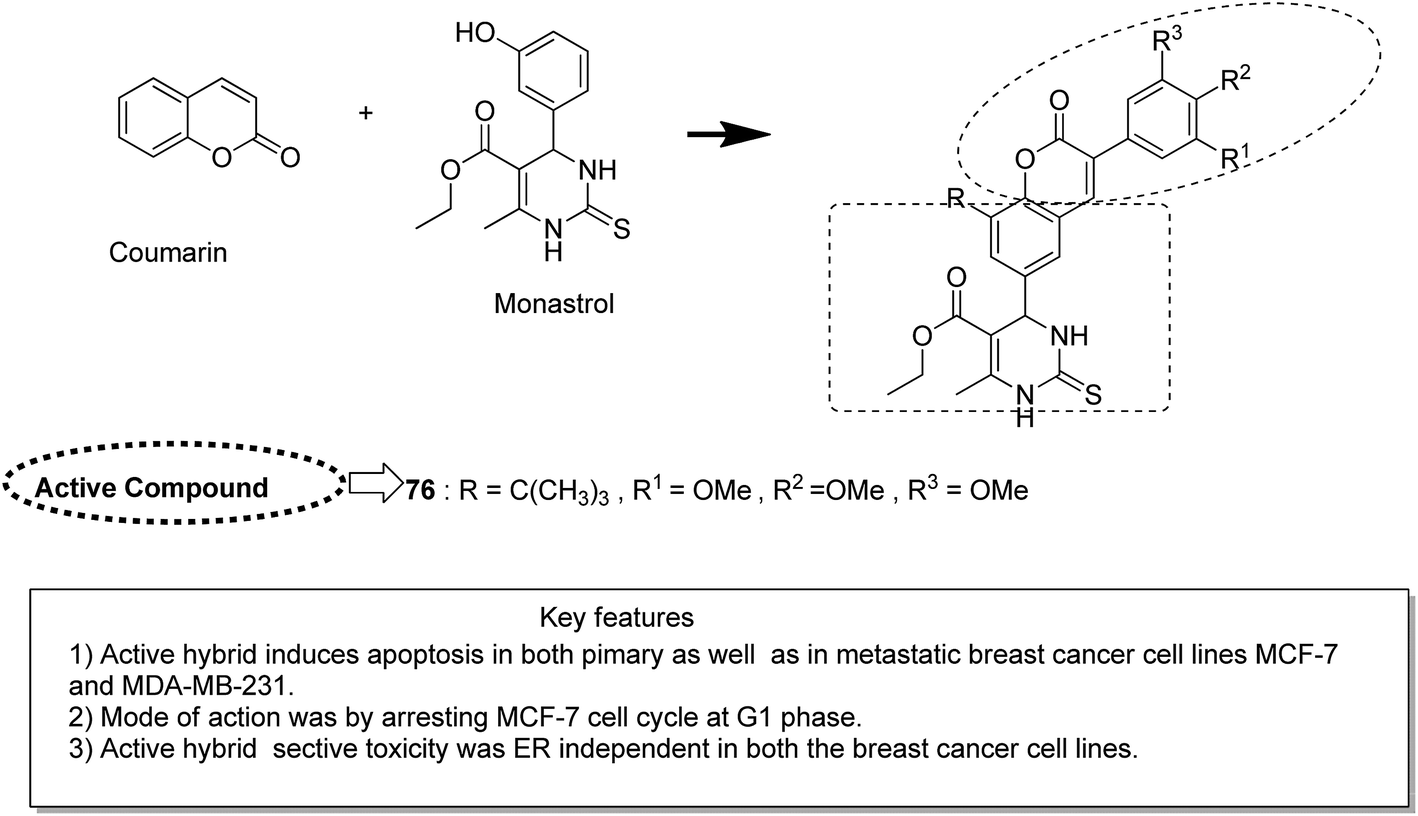
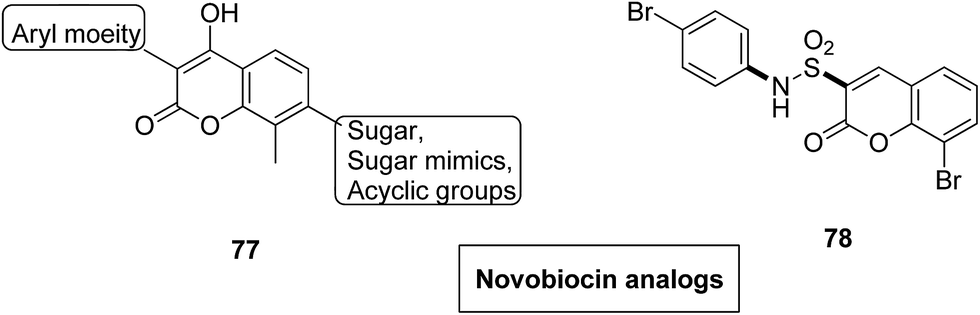
Novel hybrids of coumarin and benzimidazole were synthesized and evaluated as antitumor agents.61 Among the hybrids, compound 75 showed remarkable suppression for cancer cell lines of leukemia, colon, and breast cancer. The SAR studies revealed that the substitution at the 7-position of coumarin–benzimidazole hybrid and the H-bond interaction with TOPO 2, RNA, and DHFR resulted in its great activity and selectivity against cancer cell lines (Fig. 47). Derivatives of coumarin–monastrol (compound 76) were synthesized and assessed as anti-breast tumor specific agents.62 The increase in the number of methoxy groups on the benzene ring that substituted the coumarin ring resulted in a significant increase in the anticancer activity against breast cancer (Fig. 48). A series of selective Hsp90 inhibitors 77 was developed by the structural modification of novobiocin, which is an antibiotic that acts through inhibition of DNA gyrase.63 The SAR studies revealed that the anticancer activity was known to be in the nM range and was due to the replacement of novoise sugar and phenyl ring of the benzamido moiety with appropriate groups. The excellent anticancer activity was achieved via certain sugar replacements, such as pyranose, a sugar mimic, and piperidine, an aza sugar mimic. The coupling of arylsulfonamides with multifaceted coumarin led to the formation of a series of coumarin-3-sulfonamides that possessed potent anticancer activity against different cell lines. Compound 78 was synthesized by exploring various substituents at different positions of the phenyl and coumarin nuclei (Fig. 49).
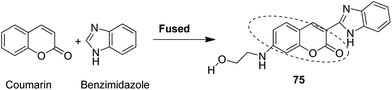 |
| | Fig. 47 Coumarin–benzimidazole hybrid molecule. | |
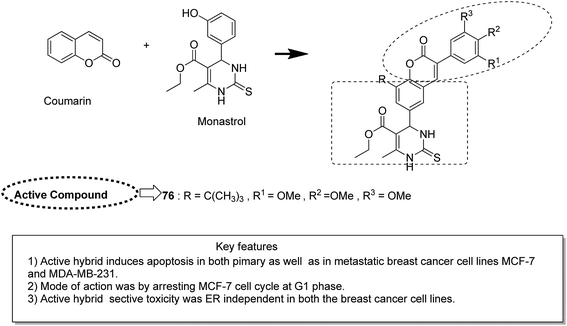 |
| | Fig. 48 Coumarin–monastrol hybrid molecule. | |
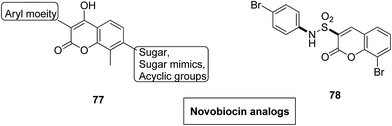 |
| | Fig. 49 Novel novobiocin analogs. | |
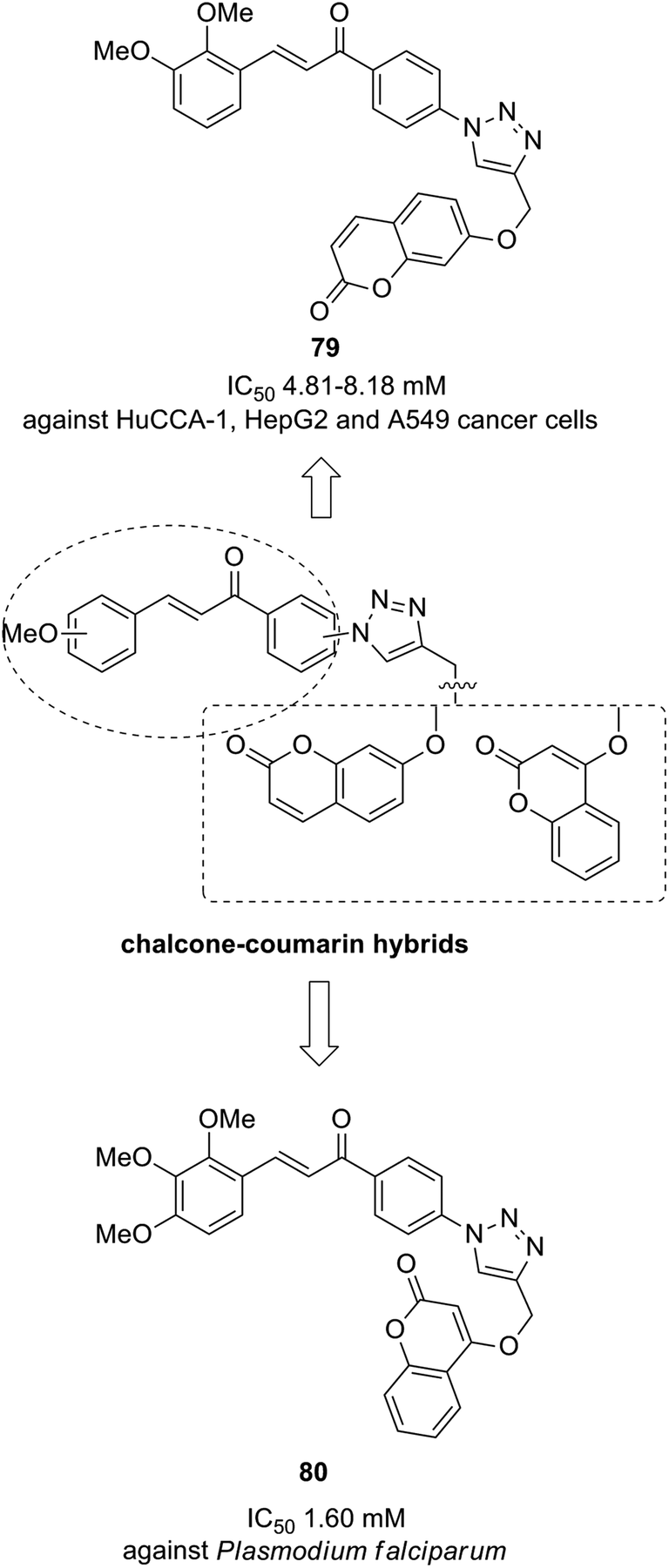
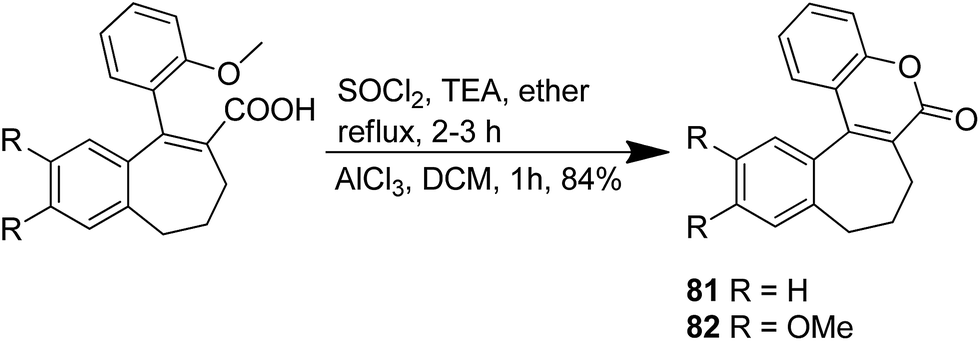
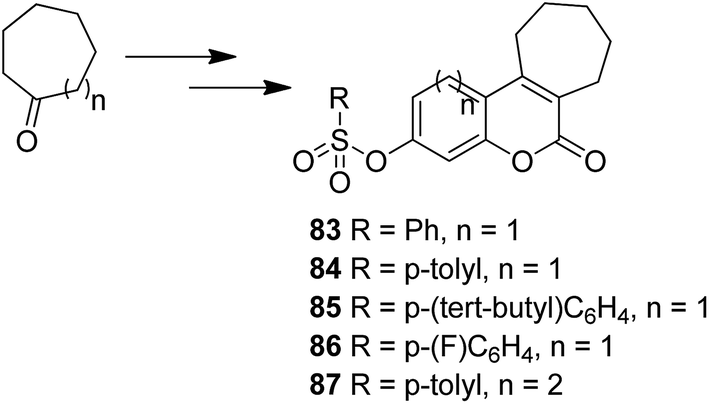
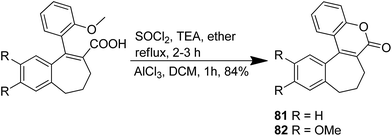
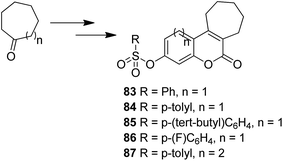
The novel chalcone–coumarin hybrids were prepared through azide/alkyne dipolar cycloaddition reaction by the linkage of 1,2,3-triazole ring.64 When compared to the standard drug etoposide, compounds 79 and 80 exhibited higher inhibitory efficacy in HepG2 cells. The compound 79 was found to be non-toxic to normal cells (Fig. 50). Yadagiri et al.65 synthesized a novel series of benzosuberone-bearing coumarin moieties. The synthesized structures were determined via analytical and spectral studies. Among the synthesized compounds, 81 exhibited significant anticancer activity against all the cancer cell lines such as A549, HeLa, MCF-7, and MDA-MB-231, whereas compound 82 was cytotoxic against HeLa and MDA-MB-231 cell lines (Fig. 51). A novel group of compounds was synthesized containing fused tricyclic coumarin sulfonate as the key molecule.66 On testing the compounds against 57 human cancer cell lines, 83, 84, and 87 were found to be more selective against leukemia and colon cancer subpanels, whereas compounds 85 and 86 showed broad-spectrum anticancer activities (Fig. 52).
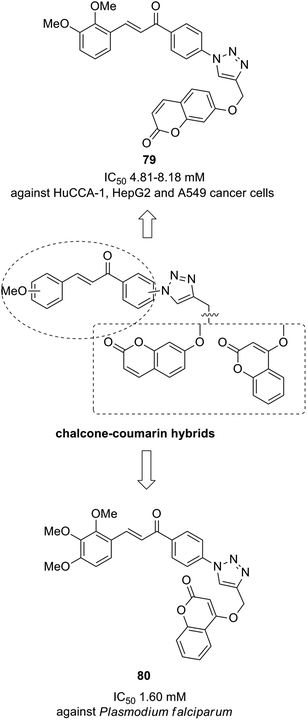 |
| | Fig. 50 Novel chalcone–coumarin hybrids. | |
 |
| | Fig. 51 Synthesis of novel benzosuberone-bearing coumarin moieties. | |
 |
| | Fig. 52 Fused tricyclic coumarin sulfonate derivatives. | |
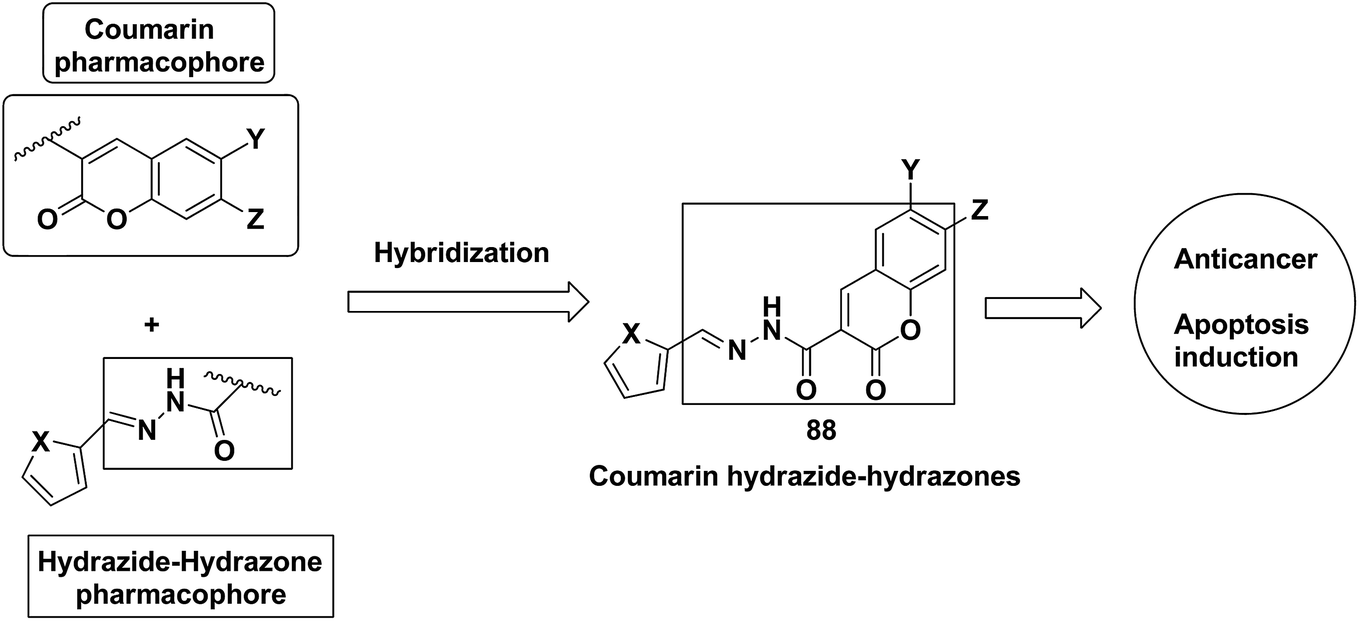
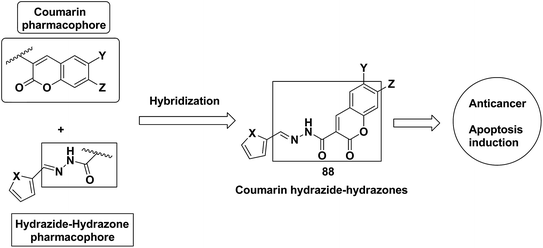
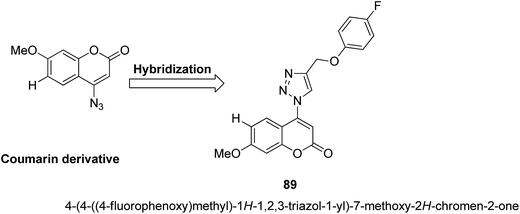
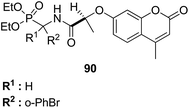
A series of novel coumarin-substituted hydrazide-hydrazone derivatives 88 was synthesized and evaluated for its anticancer activity against Panc-1, Hep-G2, and CCRF cell lines.67 The results revealed that 6-brominated coumarin hydrazide-hydrazone derivatives showed higher cytotoxicity than doxorubicin against resistant Panc-1 cells. Among these compounds, a few showed significant anticancer activity against all the tested cells, whereas others were only cytotoxic against the resistant Panc-1 cells (Fig. 53). Zhang et al.68 synthesized a series of 4-(1,2,3-triazol-1-yl) coumarin derivatives and evaluated it for anticancer activity against three human cancer cell lines: human breast carcinoma MCF-7 cell, colon carcinoma SW480 cell, and lung carcinoma A549 cell. The biological potency was increased by conducting structural optimization at the C-4 position of 1,2,3-triazole and the C-6 and C-7 positions of coumarin. Compound 89 exhibited excellent anticancer activity by arresting the G2/M cell cycle and inducing apoptosis (Fig. 54). A new method was developed for the synthesis of coumarin-containing α-aminophosphonates having two chiral centers.69 The single-crystal structure compounds were evaluated for their anticancer activity against human pulmonary carcinoma cell line (A549), human nasopharyngeal carcinoma (human KB), and human lung adenocarcinoma (MGC-803) cell lines. Compound 90 exhibited the best anticancer activity due to the influence of chirality in the DNA-binding assay since this compound exhibited a higher binding constant (Fig. 55).
 |
| | Fig. 53 Coumarin-substituted hydrazide-hydrazone derivatives. | |
 |
| | Fig. 54 4-(1,2,3-Triazol-1-yl) coumarin derivatives. | |
 |
| | Fig. 55 Coumarin containing aminophosphonates. | |
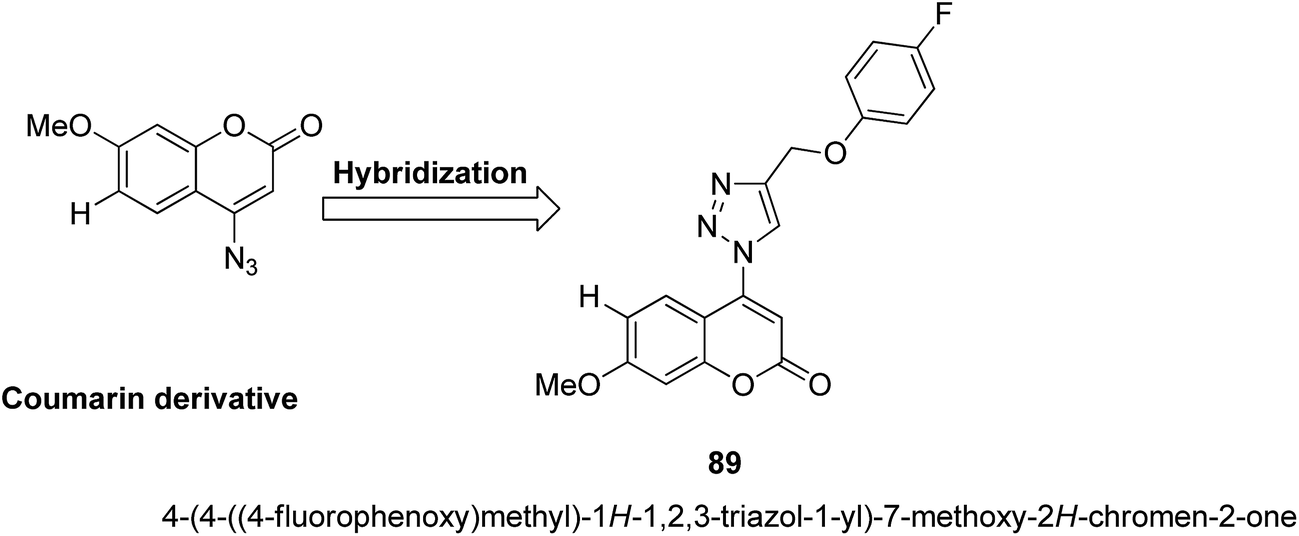
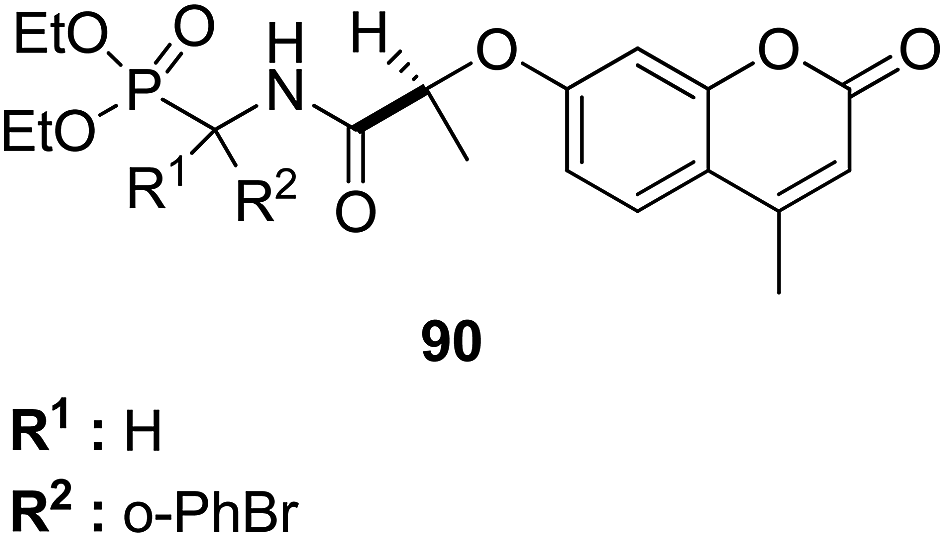
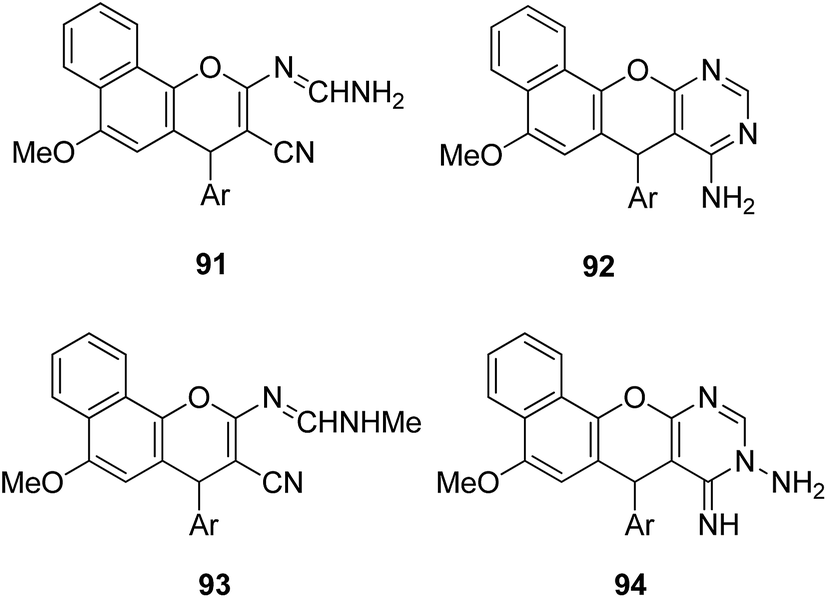
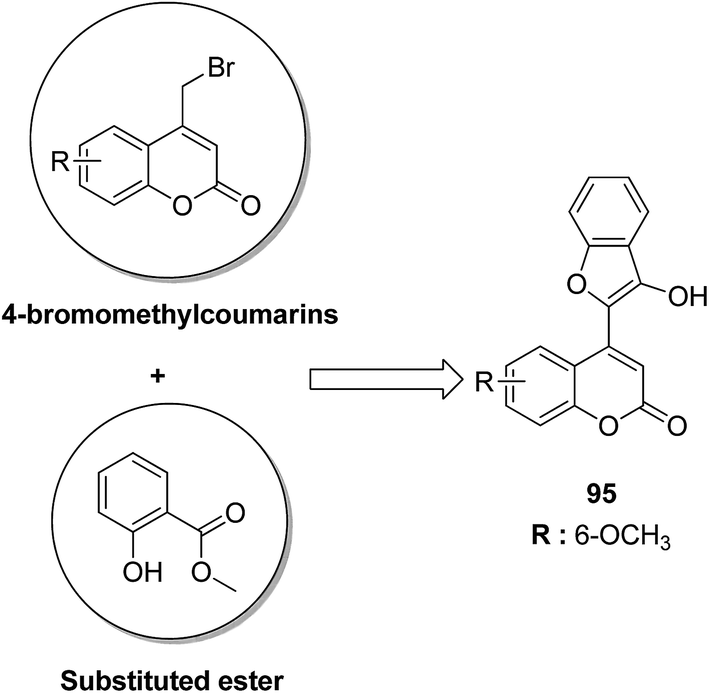
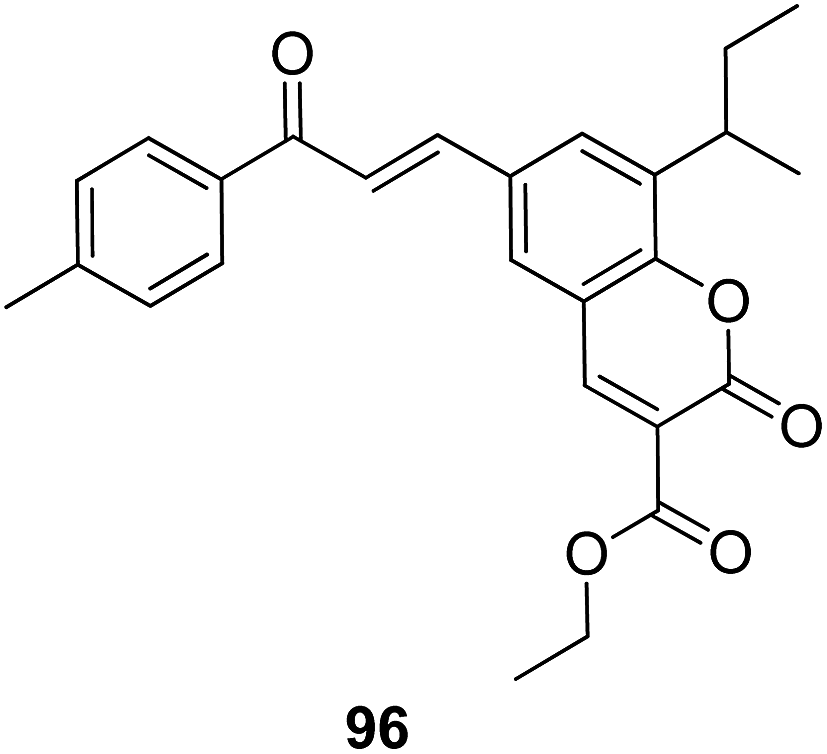
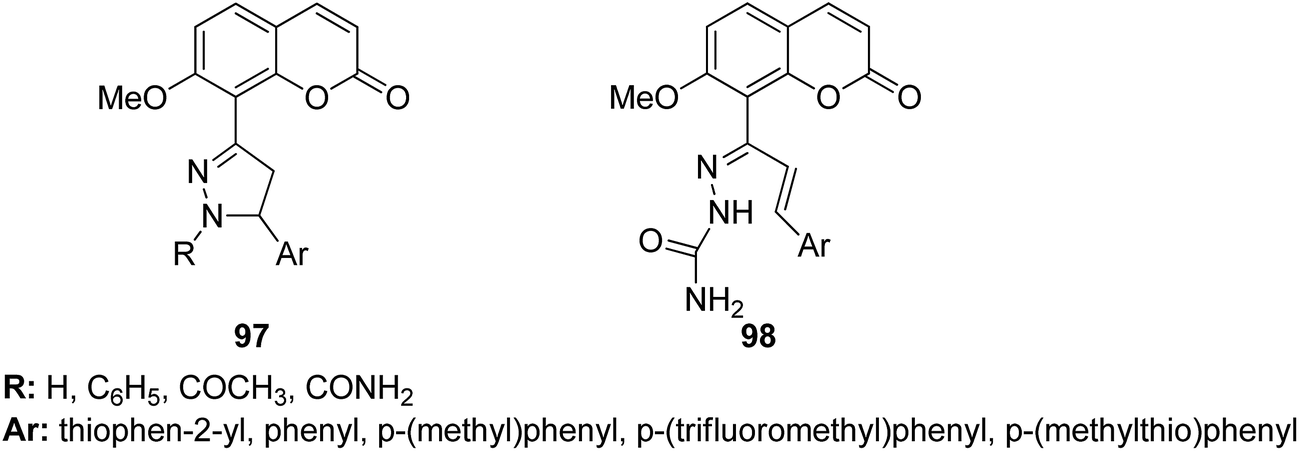
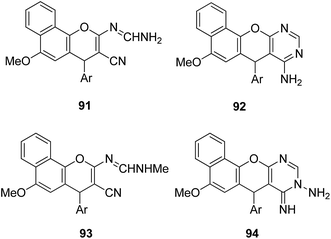
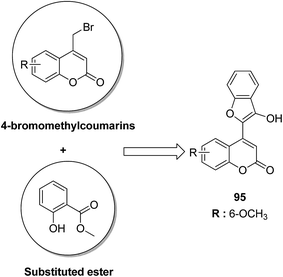
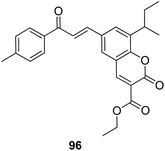
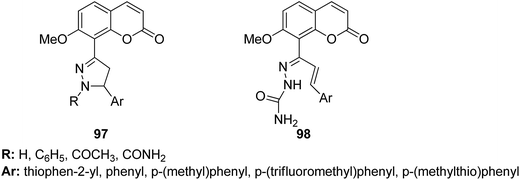
4-H-Benzo[h]chromene and 7H-benzo[h]chromeno[2,3-d]pyrimidine derivatives were synthesized and evaluated for anticancer activity via comparison with the standard drugs vinblastine, colchicine, and doxorubicin using MTT calorimetric assay.70 Compounds 91, 92, 93, and 94 were most potent against the three tumor cell lines MCF-7, HCT, and HepG-2. The SAR studies of 4H-benzo[h]chromenes with modifications at the 2- and 3-positions and 7H-benzo[h]chromeno[2,3-d]pyrimidine at the 2 and 3-positions revealed that the anticancer activity was significantly affected by the lipophilicity of the substituents at the 2- and 3- and 2 and 3-positions, respectively (Fig. 56). Various substituted benzoic acid methyl esters were prepared by reacting 4-bromo-methylcoumarins with methyl salicylate.71 The cytotoxicity studies revealed that compound 95 was more potent than the standard 5-fluorouracil (Fig. 57). Another study reported the mechanism of action and in vivo efficacy of the potent compound 96.72 Upon oral administration of this compound, a significant decrease in tumor level induced by HeLa cell xenografts was observed in nod SCID mice. This compound also inhibited the growth of cervical cancer cells (HeLa and C33A) by arresting the cell cycle at the G2/M phase and inducing apoptosis (Fig. 58). A novel series of coumarin–pyrazoline hybrids was synthesized along with a series of coumarins bearing non-cyclic isosteres of pyrazolines.73 The synthesized compounds were evaluated for cytotoxicity against the HepG2 cell line. The most active compounds 97 and 98 were examined for telomerase inhibition and apoptosis induction. The results revealed that the compounds inhibited telomerase up to 78.6% and induced apoptosis in a dose-dependent manner (Fig. 59).
 |
| | Fig. 56 Highly active chromene derivatives. | |
 |
| | Fig. 57 4-(3-Hydroxy-benzofuran-2-yl) coumarins. | |
 |
| | Fig. 58 Novel coumarin–chalcone hybrid. | |
 |
| | Fig. 59 Substituted coumarin–pyrazoline hybrids. | |
7. Colchicine-based hybrids
Colchicine is a classical tubulin-binding agent, which is isolated from Colchicum autumnale (meadow saffron). It binds to a second lower affinity site on tubulin and changes its secondary structure, which further hinders the formation of microtubules.74 Some chemical modifications in colchicine and its derivatives have been discussed below.
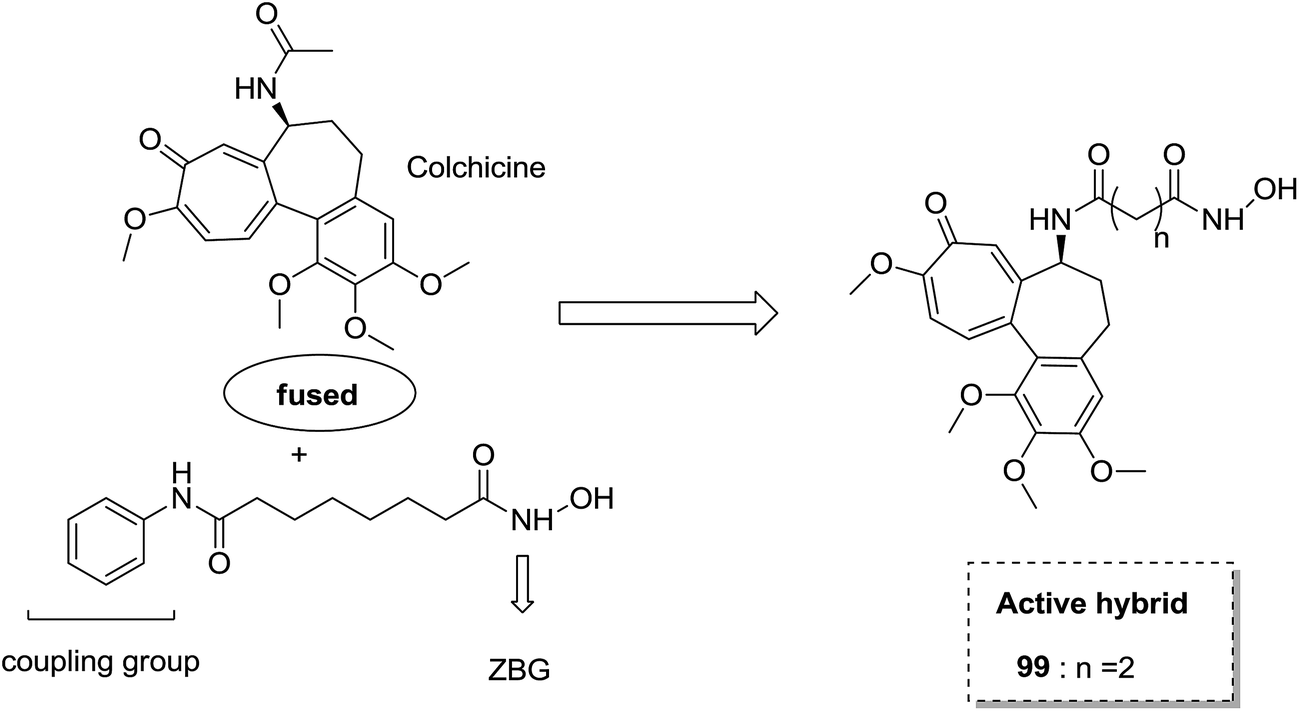
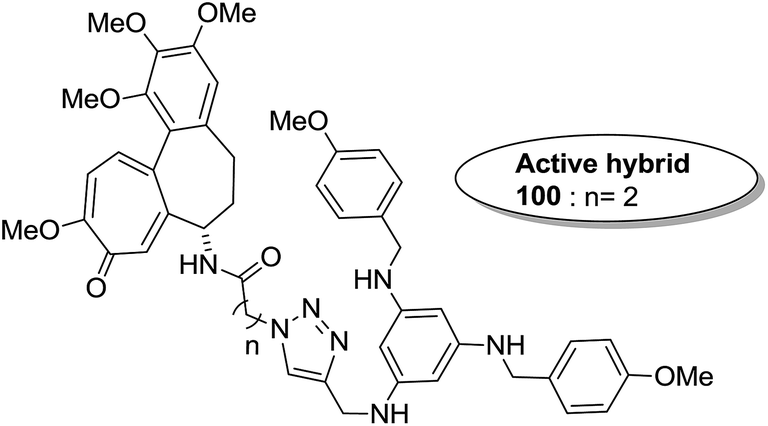
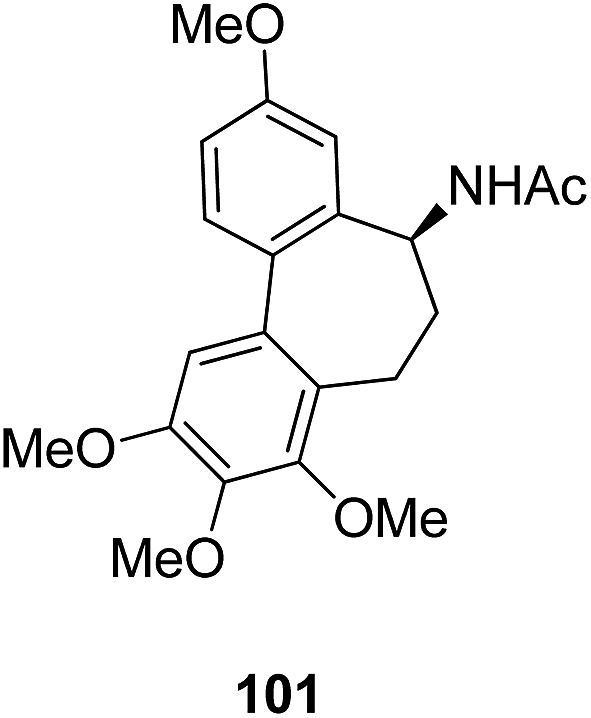
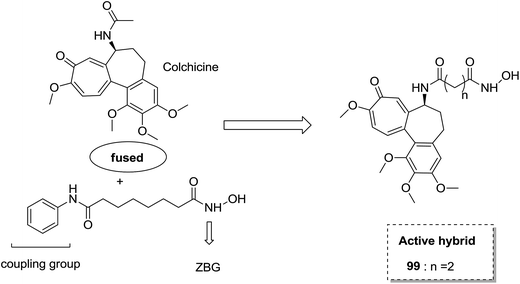
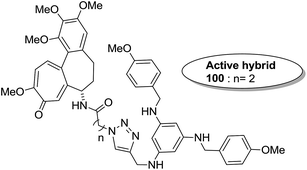
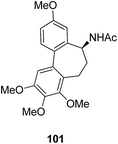
The synthesis and design of a new class of colchicine–SAHA hybrids were carried out by Zhang et al.75 These were the first designed molecules that were dual inhibitors of tubulin and HDAC. Compound 99 exhibited the strongest HDAC inhibitory activity and powerful antiproliferative activity against five cancer cell lines. The inhibitory activity of this compound was evaluated under both cell-free conditions and in in vitro cell cultures. The tubulin inhibition activity was not hampered by the linkage of SAHA to colchicines (Fig. 60). Malysheva et al.76 synthesized novel bivalent hybrids of colchicine and tubulizine using a 1,3 dipolar cycloaddition reaction. These hybrids were then assessed against HBL100 epithelial cells. The active compound 100 exhibited the IC50 value ranging from 0.599 to 2.93 μM, possessing effective cytotoxicity. SAR studies showed that this hybrid was prepared via copper-mediated 1,3-dipolar cycloaddition reaction, and a flexible spacer was used to connect the azide-containing diacetyl colchicine and acetylene-substituted tubulizine entities. The colchicine moiety was linked to 239Cyβ and the tubulizine moiety was linked to 12Cy β of the protein molecule (Fig. 61). In another study, a novel derivative of allocolchicine 101 was synthesized and tested for anticancer properties.77 The results suggested that compound 101 induced pro-death autophagy in pancreatic cancer cells and E6-1 leukemia cells without affecting normal human cells. It did not affect tubulin polymerization and increased the production of reactive oxygen species (ROS) in mitochondria that were isolated from pancreatic cancer cells. This compound is one of the best examples to demonstrate how a small change in the structure of colchicines apparently changes the mechanism of action and leads to improved selectivity (Fig. 62).
 |
| | Fig. 60 Colchicine–SAHA based hybrids. | |
 |
| | Fig. 61 Colchicine–tubulizine hybrids. | |
 |
| | Fig. 62 Novel allocolchicine analogue. | |
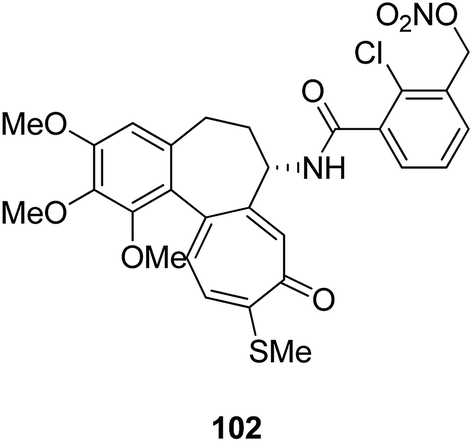
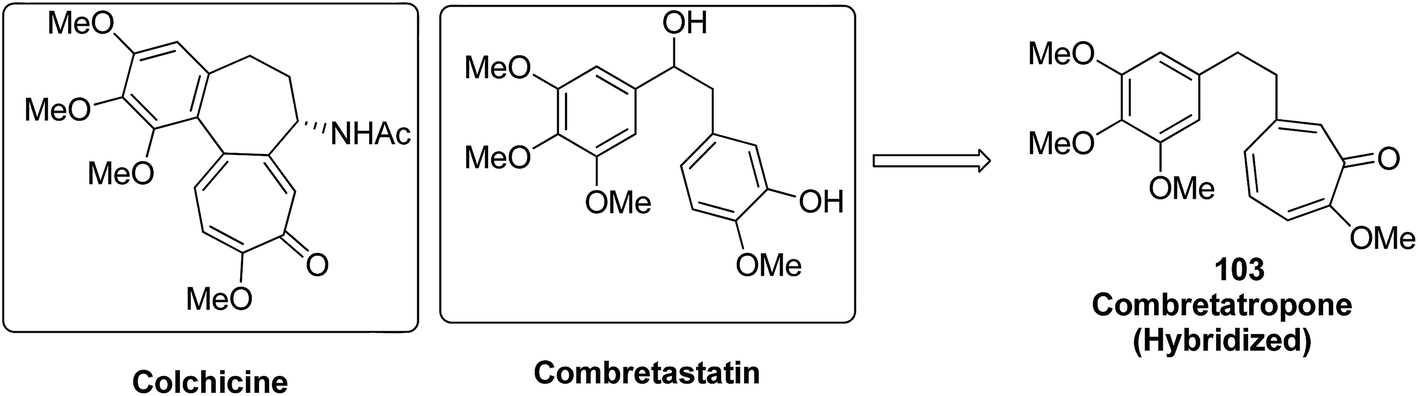
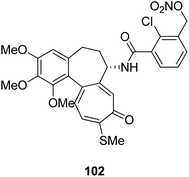
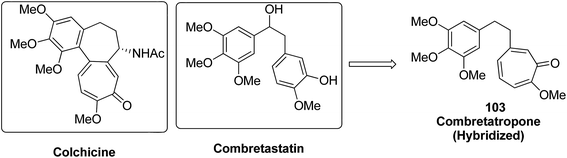
Moreover, a new series of colchicine derivatives was synthesized and evaluated for its anticancer activity.78 Compound 102 exhibited potent anticancer activity by modulating the microtubule integrity and arresting the cell cycle at the G2/M phase before apoptosis. It was observed from the optical density analysis study using purified tubulin that this compound had a higher repolymerizing activity than taxol. The results suggested that this compound had different microtubule integrity compared to colchicine (Fig. 63). Researchers have been developing various innovative strategies for designing better and effective cancer drugs that can act upon two or more targets at the same time. One of these strategies involves the incorporation of the colchicine pharmacophore into combretastatin to form a hybrid compound combretatropone 103 (Fig. 64). This compound was evaluated for its cytotoxic activity via examining its drug interaction studies with tubulin. Promising results were achieved in the preliminary phase of the study, and further results are being awaited as the drug is still under clinical trials.79
 |
| | Fig. 63 Novel colchicine derivative. | |
 |
| | Fig. 64 Colchicine–combretastatin hybrid compound. | |
8. Platinum-based hybrids
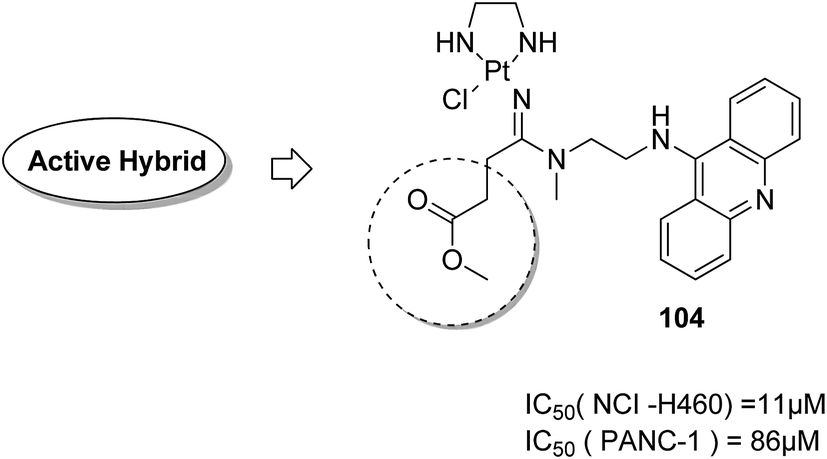
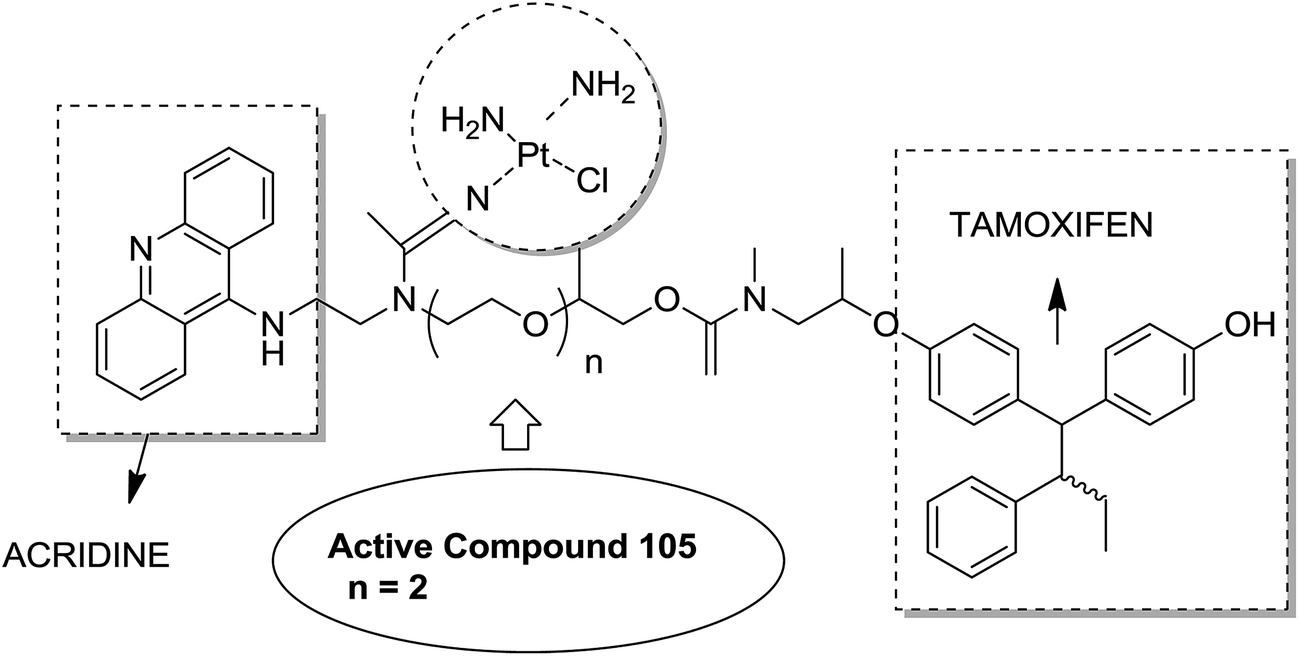
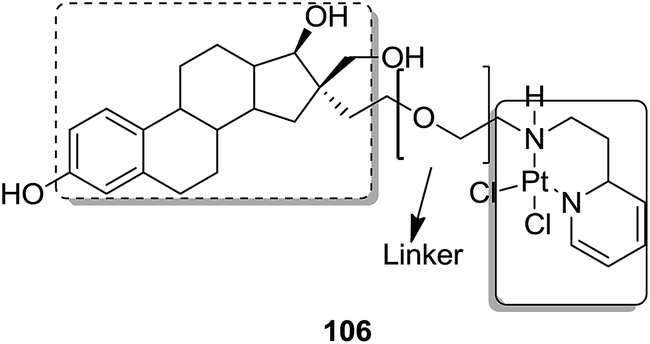
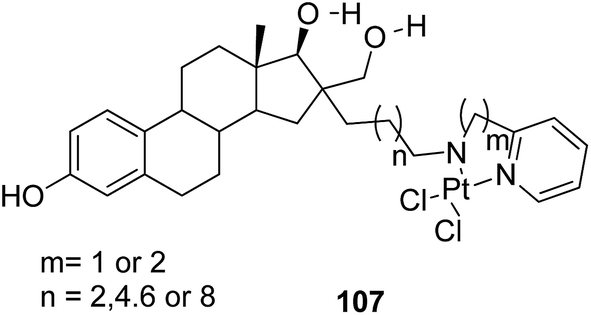
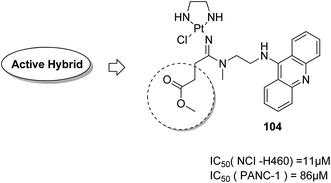
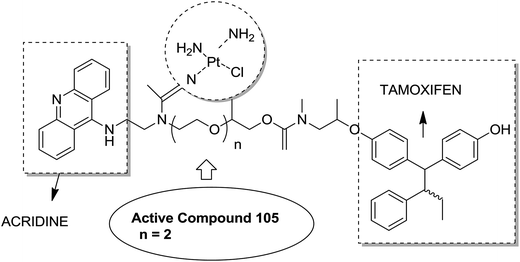
Graham et al. synthesized a novel series of carboxylic ester group-attached platinum–acridine hybrids.80 The synthesized compounds formed monofunctional-intercalative DNA molecules without cross-linking the DNA and were found to have significant antitumor activity. Among the conjugates, active hybrid 104 showed 6-fold higher activity against ovarian cancer (OVCAR-3) and breast cancer (MCF-7, MDA-MB-231) as compared to cisplatin (Fig. 65). Ding et al.81 synthesized a novel pharmacophore for the treatment of breast cancer using carbamate coupling chemistry by including a DNA-targeted platinum–acridine hybrid agent and estrogen receptor-targeted 4-hydroxy-N-desmethyltamoxifen (endoxifen). Upon evaluation in breast cancer cell lines, compound 105 was found to be most potent (Fig. 66). Moreover, the novel hybrids of 17β-estradiol platinum(II) 106 were synthesized from estrone through five-step chemical methods.82 SAR studies showed that the hybrid molecules were formed by linking different lengths of polyethylene glycol (PEG) chains and 2-(2-aminoethyl) pyridine ligand. MTT assay further revealed that hybrids with the longest PEG chain resulted in effective activity against MCF-7 and MDA-MB-231 breast cell cancer lines (Fig. 67). In another study, novel 17β-estradiol-linked platinum(II) hybrids 107 were synthesized and their biological activities were assessed against uterine and ovarian cancer cell lines.83 These novel conjugates showed better activity towards the estrogen receptor α (ERα). The newly synthesized estrogen-Pt(II) hybrids had the IC50 values ranging from 0.3 to 2.26 μM, which were far better than the natural ligand 17β-estradiol. The presence of the 16-β hydroxymethyl group provided more hydrogen bonds to the estrogen receptor, making the hybrids cytotoxic towards platinum-resistant endometrial and ovarian cell cancer cell lines (Fig. 68).
 |
| | Fig. 65 Platinum–acridine hybrid. | |
 |
| | Fig. 66 Platinum–acridine–endoxifen hybrid. | |
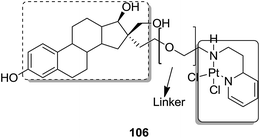 |
| | Fig. 67 17β-Estradiol–platinum(II) hybrid. | |
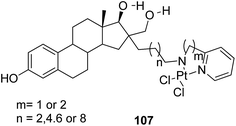 |
| | Fig. 68 Estrogen–platinum(II) hybrids. | |
9. Miscellaneous hybrids
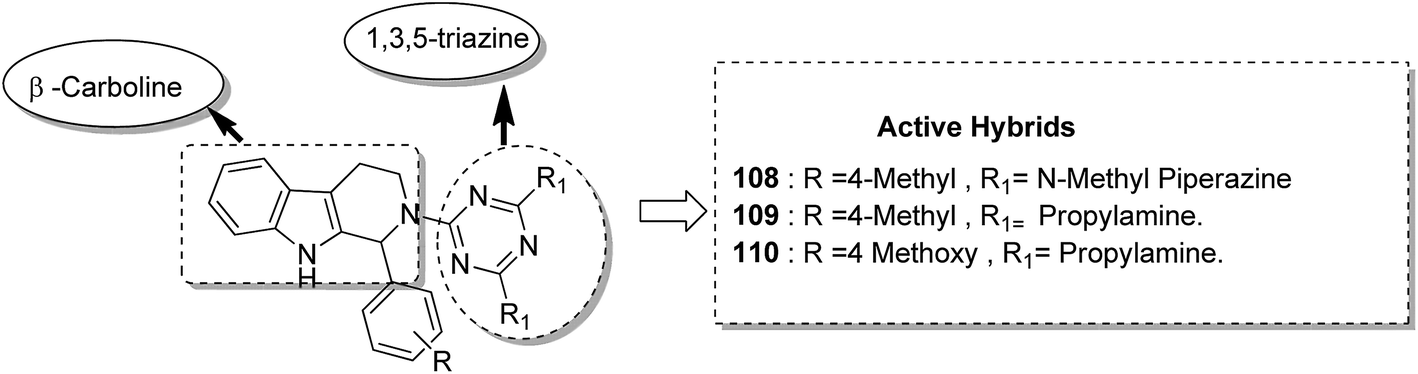
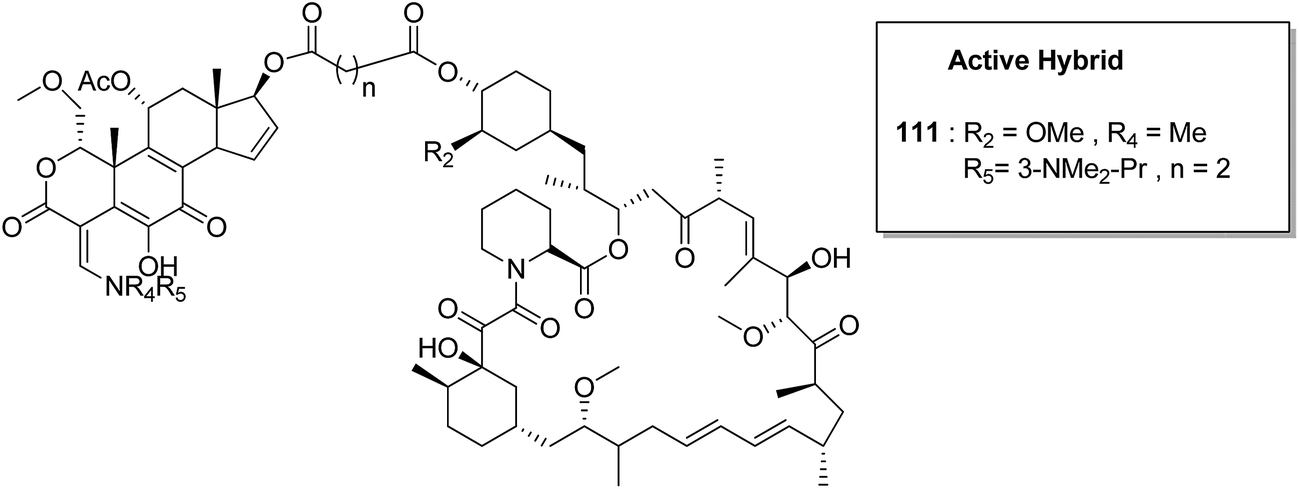
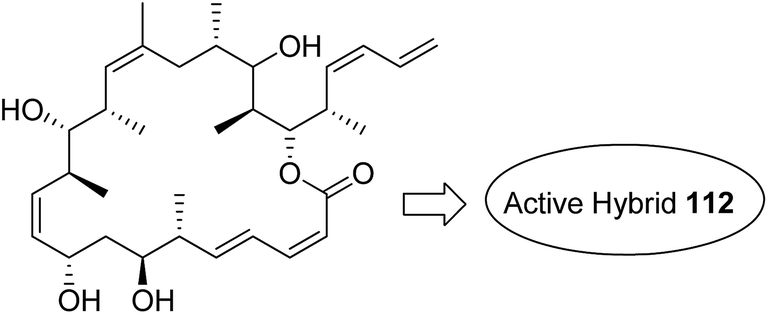
Kumar et al.84 synthesised a series of tetrahydro-β-carbolines and 1,3,5-triazine hybrids that led to the discovery of new racemic compounds 108, 109, and 110, which were selectively cytotoxic towards KB (oral cancer) cell lines. These compounds were evaluated for anticancer activity against a panel of eight human cancer cell lines and normal human fibroblasts (NIH3T3). Their IC50 values were reported to be 105.8, 664.7, and 122.2 μM. Although these compounds were active towards KB cell lines, their enantiopure forms were less active and non-selective. The enantiopure compounds were 2.5 times more selective towards MCF7 cells than towards normal fibroblast NIH3T3 cells and induced apoptosis in MCF7 and MDA MB231 cell lines (Fig. 69). Cancer is caused by the hyperactivation of the PI3K/AKT/mTOR signaling pathways. Promotion of tumor growth is due to the synergistic effect of P13K and mTOR. Thus, strong and synergistic anticancer activity can be achieved via combined targeting of PI3K and mTOR. The novel wortmannin–rapamycin conjugates linked with a prodrug linker were synthesized using this approach.85 This led to the synthesis of compound 111, which completely inhibited the growth of HT29 tumors at a dose of 15 mg kg−1. When administered alone or in combination with bevacizumab, this compound exhibited superior efficacy in the A498 renal tumor model (Fig. 70). Discodermolide and dictyostatin exhibit significant anticancer activity against cancer cells, and the mode of action is via accumulation of cells at the G2/M phase and cell death by apoptosis. Encouraged by this, Paterson et al.86 designed and synthesized the novel hybrids of dictyostatin and discodermolide 112, which showed improved cell growth inhibition in four human cancer cell lines including taxol-resistant NCI/ADR-Res cell line (Fig. 71).
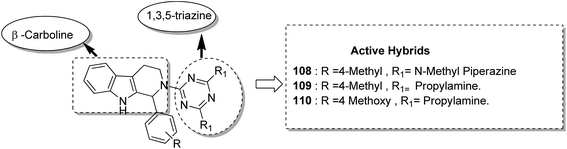 |
| | Fig. 69 (Tetrahydro-β-carboline)–1,3,5-triazine hybrids design. | |
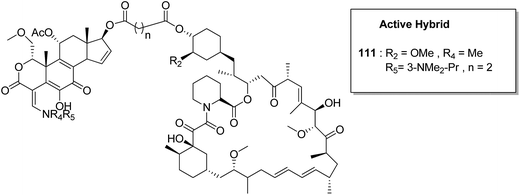 |
| | Fig. 70 Wortmannin–rapamycin hybrid. | |
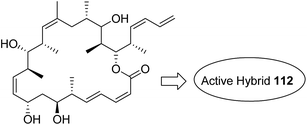 |
| | Fig. 71 Dictyostatin–discodermolide hybrid. | |
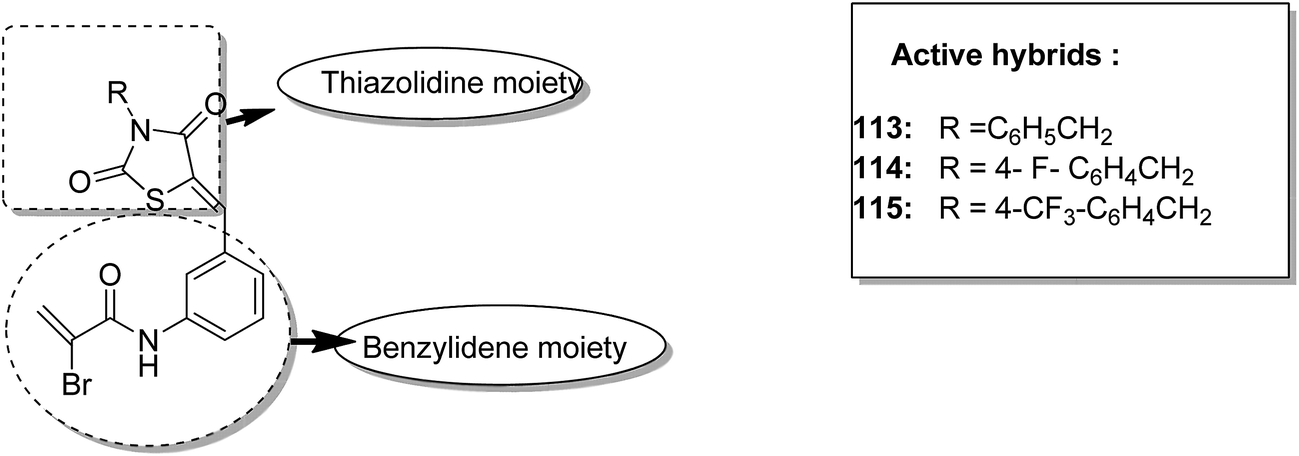
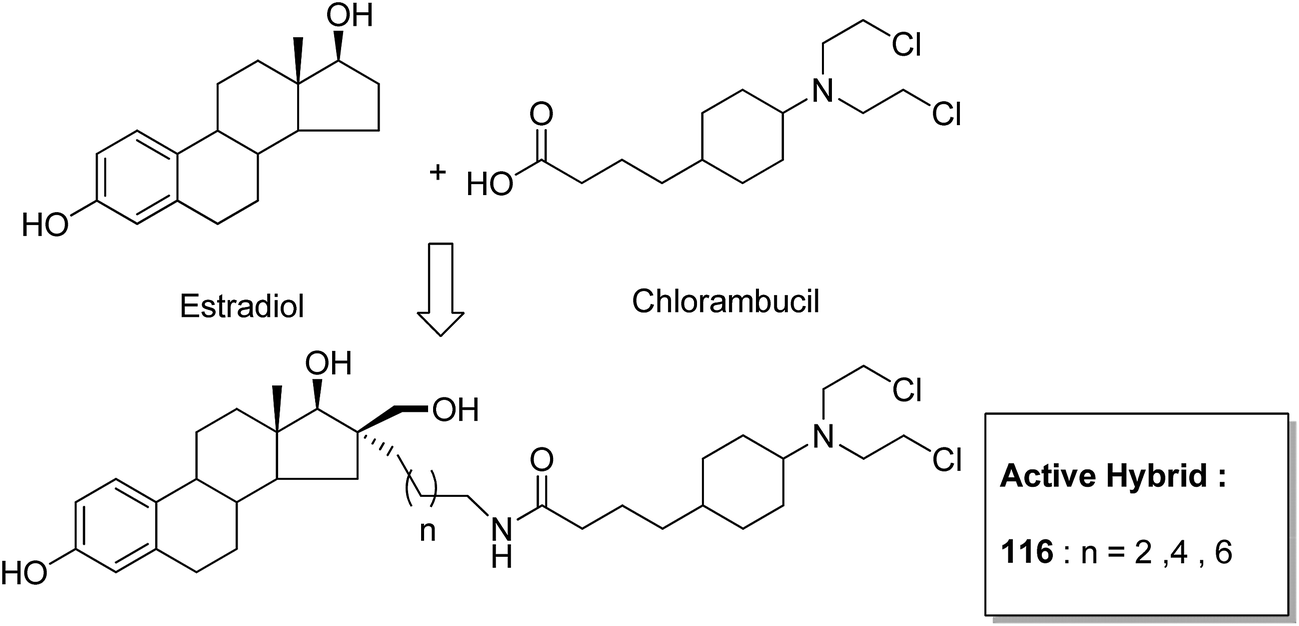
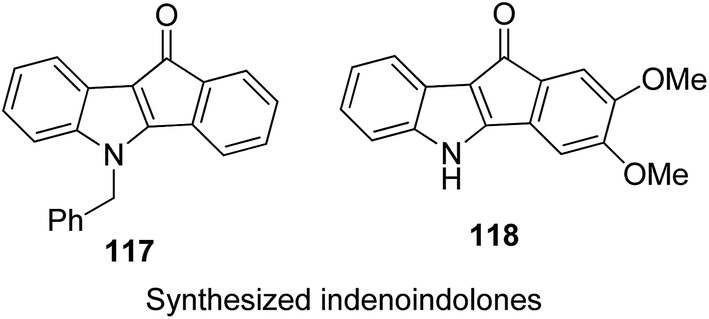
One of the methods to treat cancer is hybridization of two different bioactive molecules with different mechanisms of action. This approach led to the synthesis of novel anticancer agents by linking two series of N-3-substituted-5-arylidene thiazolidine-2,4-diones with α-bromoacryloylamido moiety at the para- or meta-position in the phenyl ring of the arylidene portion.87 Compounds 113, 114, and 115 were found to induce apoptotic cell death by activating multiple caspases and releasing cytochrome-c from mitochondria. These compounds contain a pair of Michael acceptors in their structures. When evaluated against HL-60 and U937 cells, these compounds showed certain apoptotic features such as cell shrinkage, chromatin condensation, and fragmented nuclei by activating the extrinsic and intrinsic pathways of cell death (Fig. 72). Gupta et al.88 synthesized a series of estradiol–chlorambucil hybrids and evaluated it for site-directed chemotherapy of breast cancer. Different hormone-dependent and hormone-independent breast cancer cell lines were used for the evaluation of anticancer efficacy of these newly synthesized compounds. When compared to chlorambucil, the novel hybrids showed significant in vitro anticancer activity. The length of the spacer chain between the carrier and drug molecule was revealed via structure–activity relationship (SAR) studies. Compound 116 showed significant cytotoxic activity against hormone-dependent (MCF-7) and hormone-independent (MDA-MB-436 and MDA-MB-486) breast cancer cell lines (Fig. 73). The novel indenoindolones were prepared as potential anticancer agents via scaffold hybridization of various natural and synthetic anticancer leads.89 When compared to etoposide and 5-fluorouracil, these compounds showed highly potent anticancer activity in kidney cancer cells (HEK 293) and low toxicity in corresponding normal cells. The apoptotic effect was attributed to the cell cycle arrest at the G2/M phase. Compounds 117 and 118 were found to exhibit higher cytotoxicity among all the synthesized indenoindolones. These compounds caused 50% cell death (LC50) of HEK 293 cells. On treatment of the cells with these compounds, increase in the punctate, bubble shape, and shrinkage nuclei of HEK 293 cells were observed. The exact biological target for anticancer activity of these indenoindolones is not yet known; however, the possible target may be proteins, such as cyclins and cyclin-dependent kinase (CDKs) families, p21 and p53, that are responsible for the G2/M phase of the cell cycle. Western blot analysis further revealed the increased levels of BAX/BCL-XL ratio and cleaved product of caspase-3 and PARP, indicating apoptotic effects of these compounds (Fig. 74).
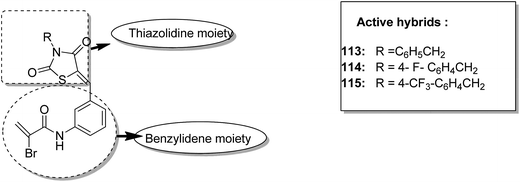 |
| | Fig. 72 Thiazolidine–benzylidene hybrid design. | |
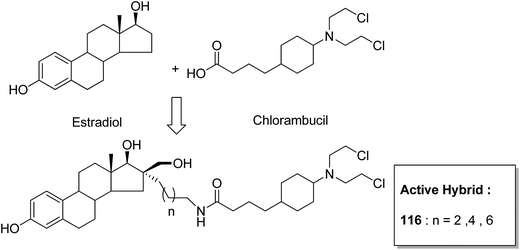 |
| | Fig. 73 Estradiol–chlorambucil hybrid. | |
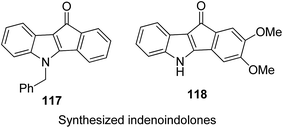 |
| | Fig. 74 Scaffold hybridization of indenoindolones. | |
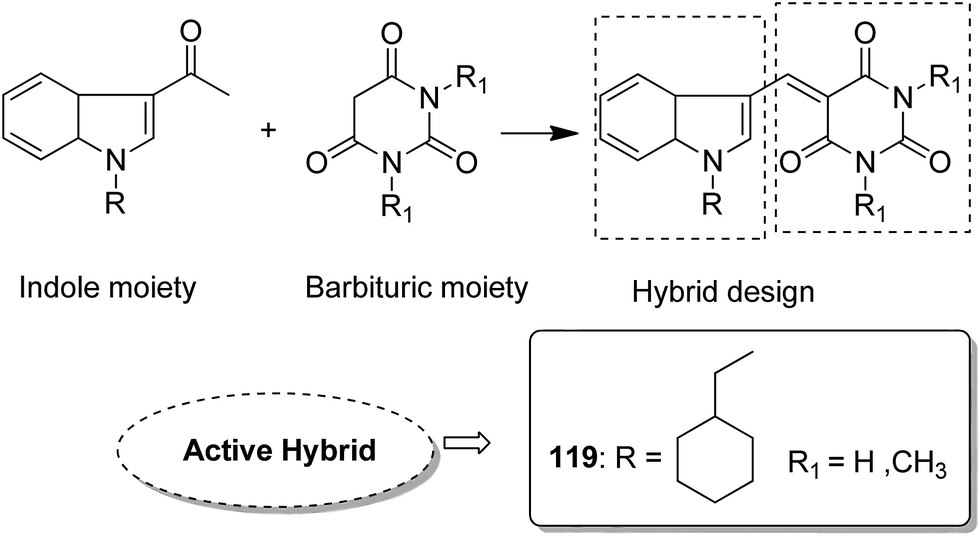
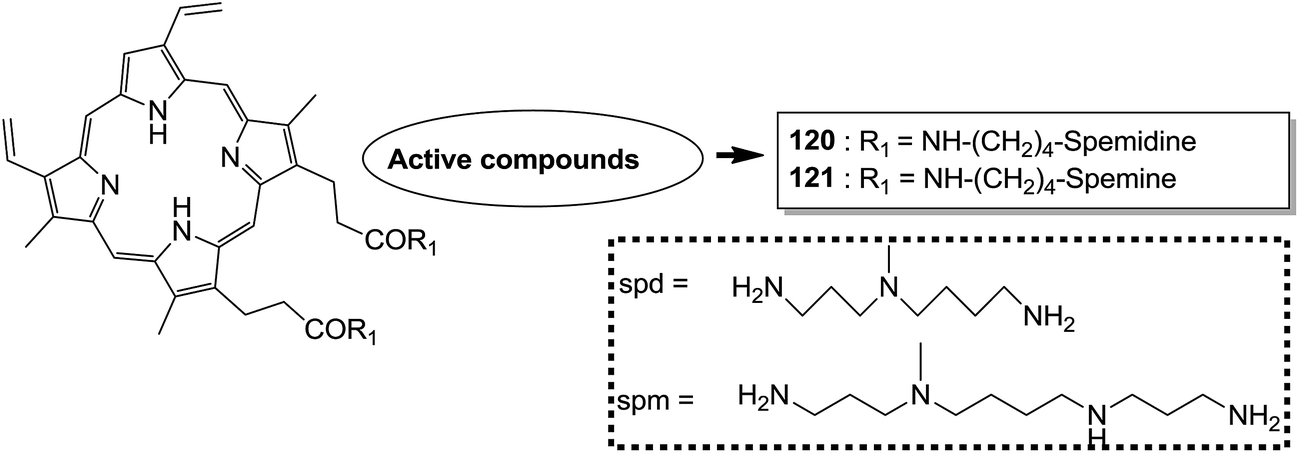
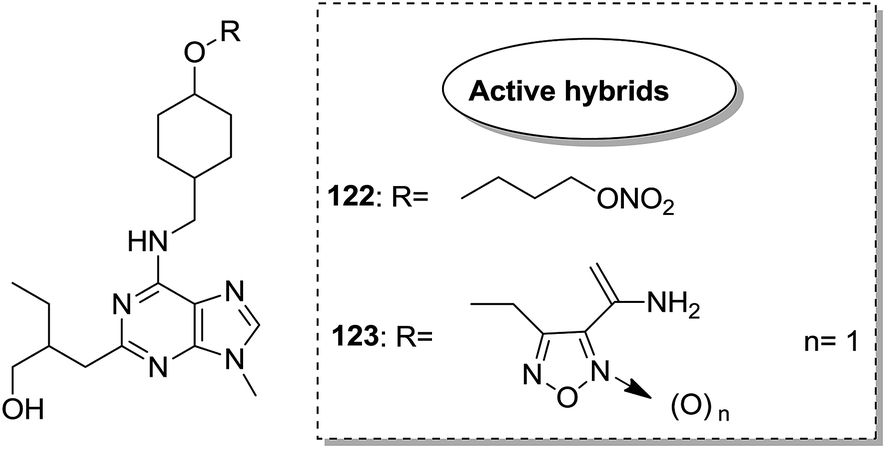
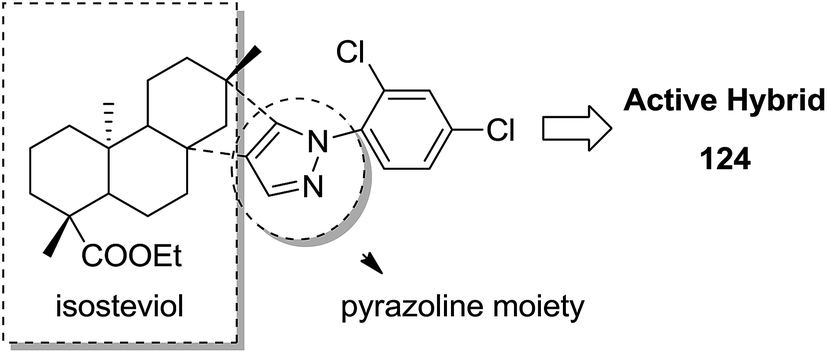
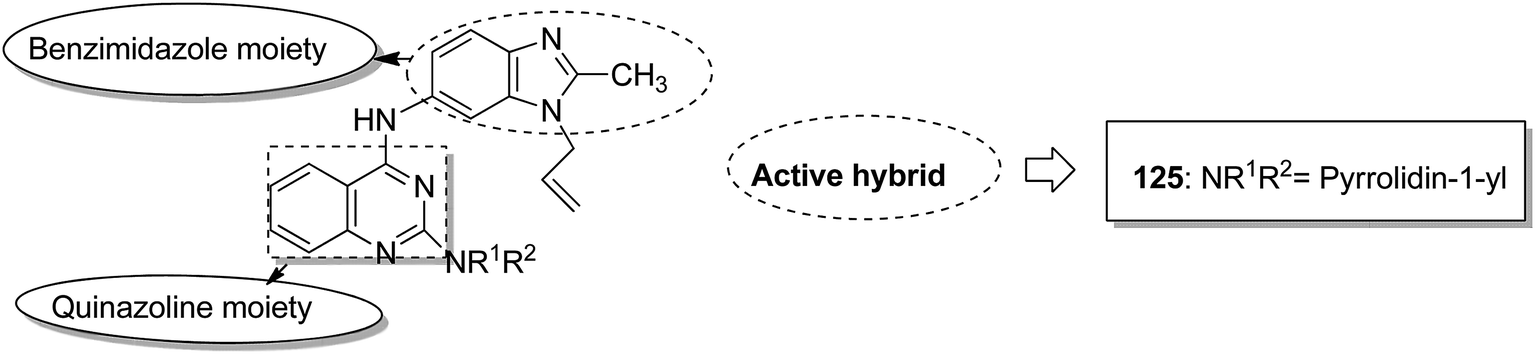
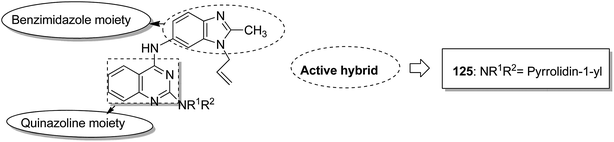
Indole and barbituric acids are well known for their different biological activities such as anticonvulsant, anti-hypnotic, anti-cancer, and anti-inflammatory. This information motivated Singh et al.90 to design and synthesize new hybrid molecules by combining the structural features of indole and barbituric acid. Compound 119 showed significant anticancer activity when evaluated over a 60 cell line panel of human cancer cells of non-small cell lung cancer (NSCLC), colon cancer, CNS cancer, ovarian cancer, renal cancer, and breast cancer (Fig. 75). Protoporphyrin is an efficient first-generation photosensitizer approved for photodynamic therapy (PDT) and treatment of neoplastic diseases. Sol et al. synthesized conjugates of tritolyporphyrin and protoporphyrin-IX, 120 and 121, by the five-step method.91 Photocytoxicity of protoporphyrin conjugate was tested against K562 human chronic myelogenous leukemia cells, and the conjugate exhibited better activity. Structure–activity relationship studies showed that these conjugates were composed of polyamine units of either spermidine or spermine, which were also attached to two molecules of polyamine-bearing protoporphyrin-IX. Presence of spermidine and spermine in the active conjugate protoporphyrin-IX increased the amphiphilic character and phototoxicity of the conjugate. Protoporphyrin-IX polyamine derivative with log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P value near 0 was soluble in water (Fig. 76). R-roscovitine, a 2,3,9-trisubstituted purine, is an inhibitor of cyclin-dependent kinase (CDK). CDKs regulate many biological functions such as cell division, transcription, neural cell physiology, and apoptosis, whereas nitric oxide (NO) also plays an important role in apoptosis, exerts pro-apoptotic or anti-apoptotic effects depending on the NO-donors. On this basis, Montanaro et al.92 designed and synthesized novel hybrids by linking R-roscovitine to different NO-donor molecules. These hybrids were assessed as pro-resolution agents. Among these hybrids, compounds 122 and 123 showed remarkable pro-apoptotic activity (Fig. 77). Zhu et al. synthesized novel isosteviol-fused pyrazoline and pyrazole hybrids via intramolecular 1,3-dipolar cycloaddition and condensation reactions.93 Novel hybrids were then assessed for their anticancer activity against malignant cancer cell lines such as SGC, 7901, A549, Raji, and HeLa. The hybrid compound 124 showed remarkable inhibition against malignant cancer cell lines with the IC50 values of 2.17, 3.18, 10.9, and 13.52 μM, respectively (Fig. 78). Sharma et al.94 synthesized benzimidazole–quinazoline hybrids and assessed them in vitro against 60 tumor cell lines. Among the hybrids, compound 125 showed remarkable activity against cancer cell lines. SAR studies showed that the substitution of alkyl or allyl groups on benzimidazole and aryl groups of quinazoline ring increased the lipid solubility of the hybrid compounds. The substitution of pyrroline moiety and other secondary amines at the 2-position of quinazoline also increased the activity of the compound (Fig. 79).
P value near 0 was soluble in water (Fig. 76). R-roscovitine, a 2,3,9-trisubstituted purine, is an inhibitor of cyclin-dependent kinase (CDK). CDKs regulate many biological functions such as cell division, transcription, neural cell physiology, and apoptosis, whereas nitric oxide (NO) also plays an important role in apoptosis, exerts pro-apoptotic or anti-apoptotic effects depending on the NO-donors. On this basis, Montanaro et al.92 designed and synthesized novel hybrids by linking R-roscovitine to different NO-donor molecules. These hybrids were assessed as pro-resolution agents. Among these hybrids, compounds 122 and 123 showed remarkable pro-apoptotic activity (Fig. 77). Zhu et al. synthesized novel isosteviol-fused pyrazoline and pyrazole hybrids via intramolecular 1,3-dipolar cycloaddition and condensation reactions.93 Novel hybrids were then assessed for their anticancer activity against malignant cancer cell lines such as SGC, 7901, A549, Raji, and HeLa. The hybrid compound 124 showed remarkable inhibition against malignant cancer cell lines with the IC50 values of 2.17, 3.18, 10.9, and 13.52 μM, respectively (Fig. 78). Sharma et al.94 synthesized benzimidazole–quinazoline hybrids and assessed them in vitro against 60 tumor cell lines. Among the hybrids, compound 125 showed remarkable activity against cancer cell lines. SAR studies showed that the substitution of alkyl or allyl groups on benzimidazole and aryl groups of quinazoline ring increased the lipid solubility of the hybrid compounds. The substitution of pyrroline moiety and other secondary amines at the 2-position of quinazoline also increased the activity of the compound (Fig. 79).
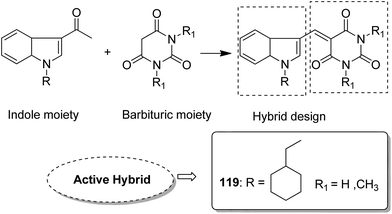 |
| | Fig. 75 Hybrids of indole and barbituric acids. | |
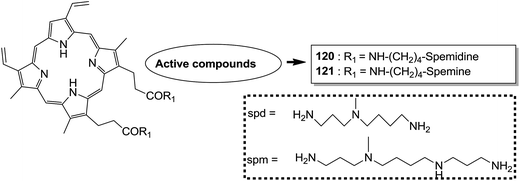 |
| | Fig. 76 Polyamine conjugates design. | |
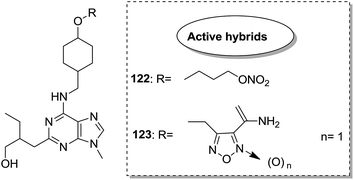 |
| | Fig. 77 Roscovitine–NO hybrid molecule. | |
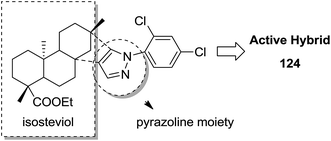 |
| | Fig. 78 Isosteviol-linked pyrazoline and pyrazole hybrids. | |
 |
| | Fig. 79 Benzimidazole–quinazoline hybrids. | |
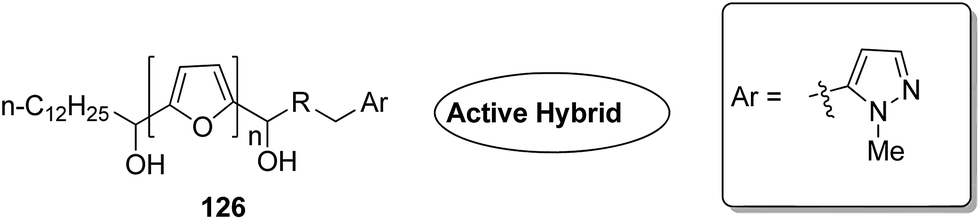
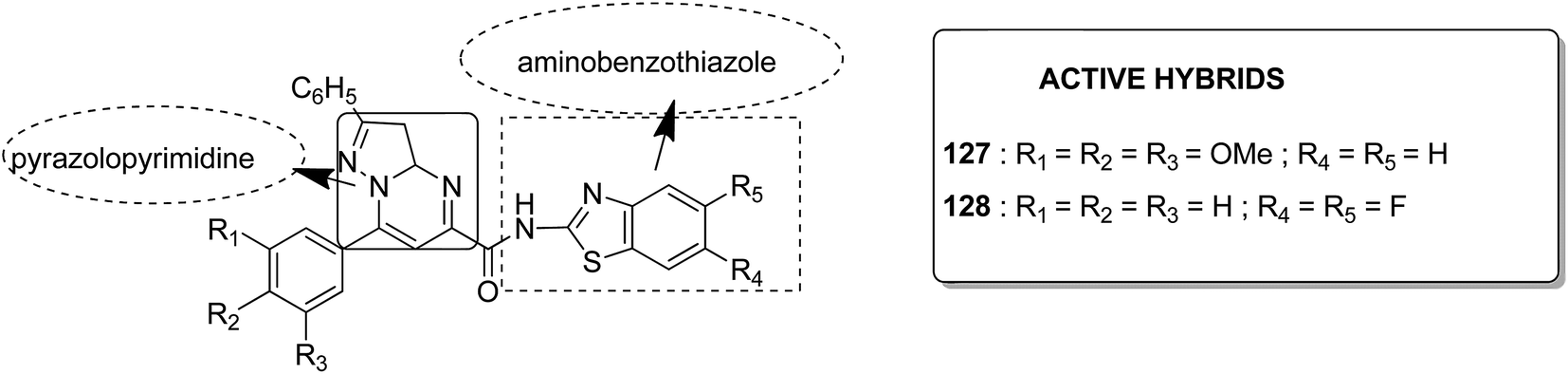
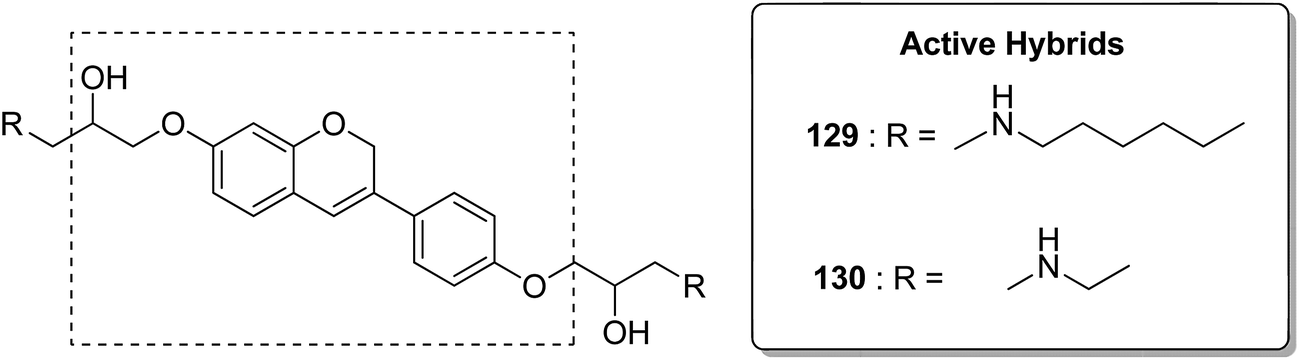
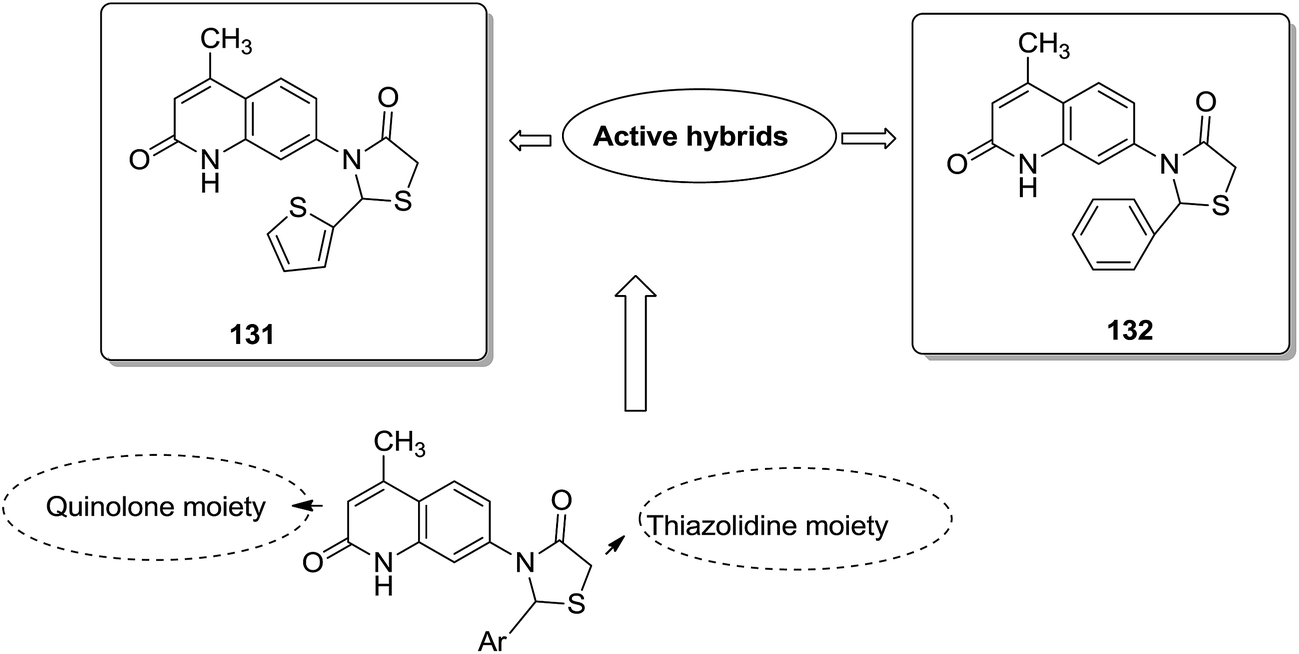
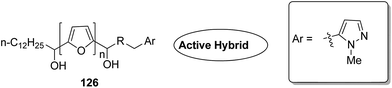
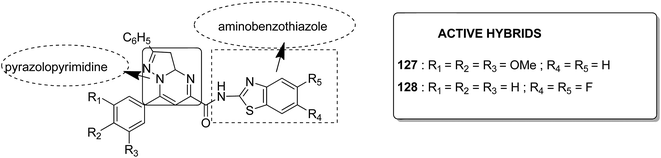
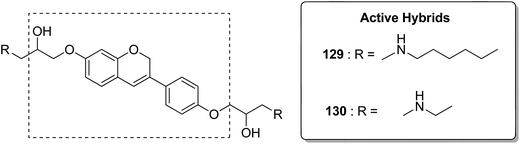
Acetogenins are polyketides having one to three tetrahydrofuran (THF) rings and are connected to the α,β-unsaturated-γ-lactone ring via a long hydrocarbon chain. They exhibit a broad range of biological activities including cytotoxic, immunosuppressive, and antitumor activities. They act by inhibiting mitochondrial NADH ubiquinone oxidoreductase, which leads to the suppression of ATP production and results in apoptosis of cancer cells. Encouraged by this information, α–β unsaturated γ-lactone-free nitrogen-containing hybrids 126 were synthesized by Kojima et al. and evaluated for cytotoxicity against 39 tumor cell lines. Among these, the L-methyl-pyrazol-5-yl derivative showed potential activity towards the NCI-H23 cancer cell line (Fig. 80).95 A novel series of pyrazolo [1,5-α] pyrimidine-linked aminobenzothiazole hybrids was synthesized and its anticancer activity was assessed against 5 human tumor cell lines.96 Among the synthesized compounds, hybrids 127 and 128 exhibited good anticancer activity, having the IC50 value between 2.01 and 7.01 μM and 1.94 and 3.46 μM, respectively. SAR studies further showed that in compounds 127 and 128, 7-phenyl ring of the pyrazolo [1,5-α] pyrimidine was substituted with 3,4,5-trrimethoxy, 4-fluoro, and 4-methoxy groups, which resulted in significant anticancer activity against 5 human cancer cell lines (A549, HeLa, MCF-7, HF2a, and ACHN) (Fig. 81). Novel isoflavene–propranolol hybrids were synthesized and evaluated as antitumor agents.3 Among the hybrids, compounds 129 and 130 exhibited remarkable activity against the SHEP and MDA-MB-231 cell lines. SAR showed that substitution of an isoflavene scaffold with a 1-isopropyl amine-2-propanol side chain resulted in enhancement of the antiproliferative and anti angiogenic properties (Fig. 82). Suthar et al.97 synthesized novel quinolone-linked thiazoline-4-one hybrids and evaluated them as antiinflammatory and anticancer agents. These hybrids were found to bind the active site NF-KB (which regulates genes involved in cancer and inflammation) for its suppression. Among the synthesized hybrid compounds, 131 and 132 showed remarkable activity against B7-549, HeLa, colon-205, and ACHN human cancer cell lines, which was attributed to the augmentation of apoptosis and NF-KB inhibition (Fig. 83).
 |
| | Fig. 80 Nitrogen containing analogs of acetogenins. | |
 |
| | Fig. 81 Pyrazolopyrimidine linked aminobenzothiazole hybrids. | |
 |
| | Fig. 82 Isoflavene–propranolol hybrids. | |
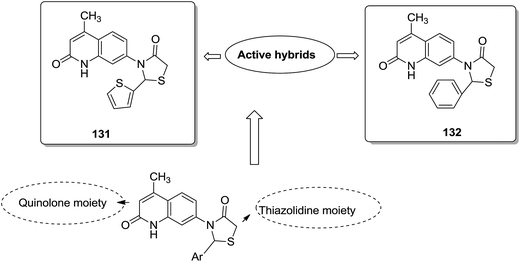 |
| | Fig. 83 Quinolone linked thiazolidine hybrid design. | |
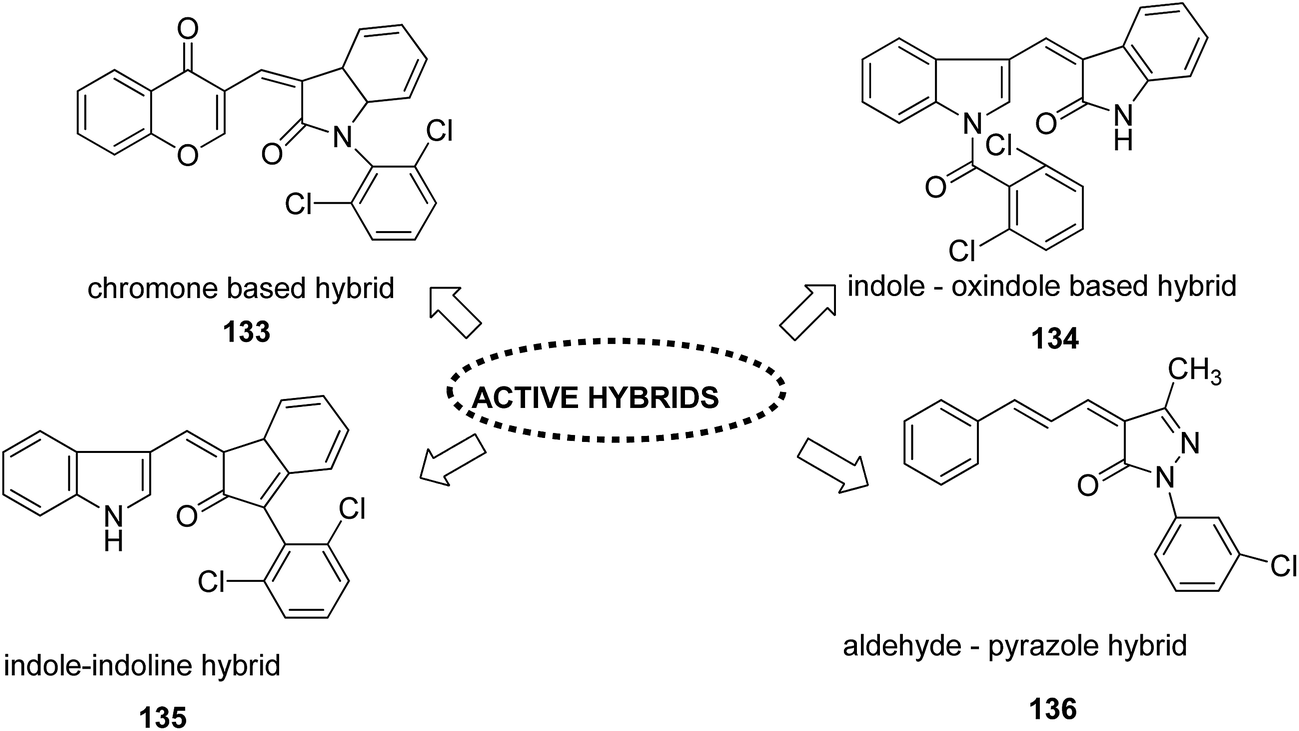
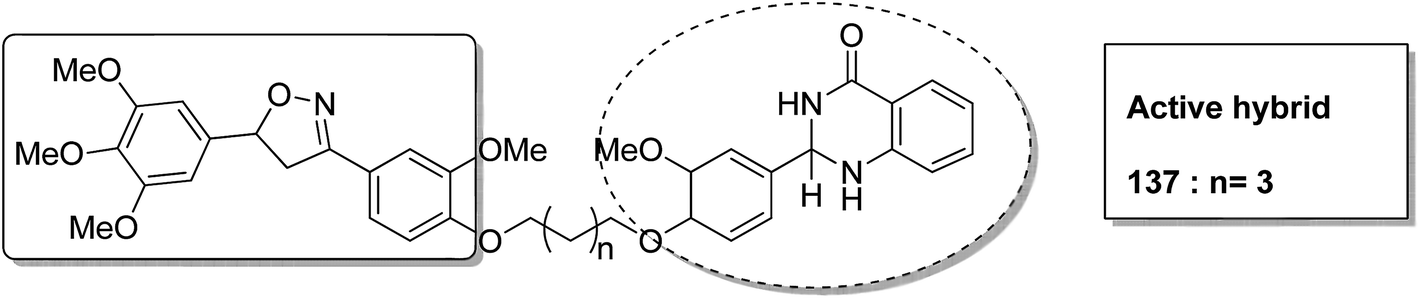
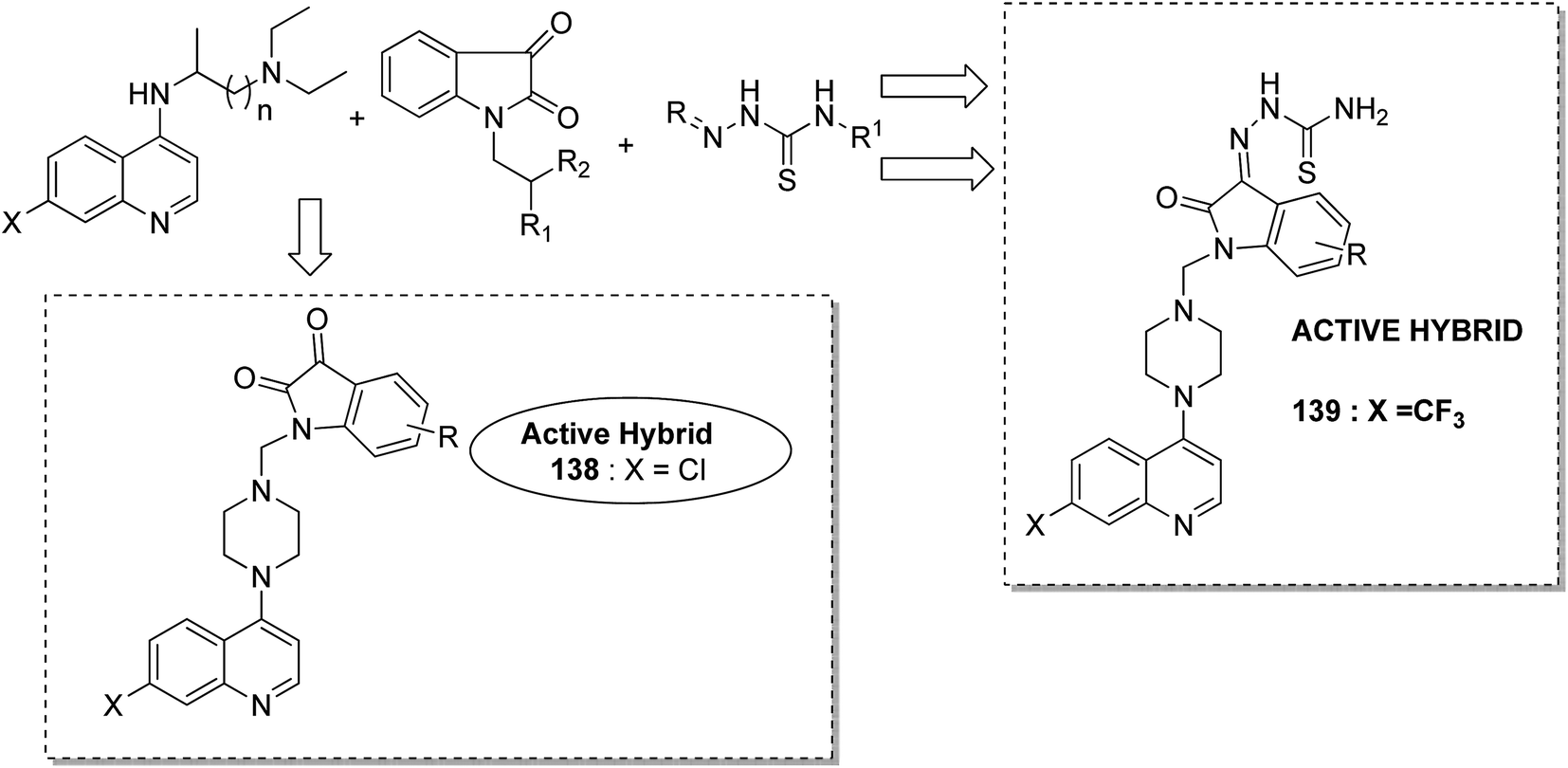
Different groups of hybrids were synthesized by linking chromone–pyrimidine, chromone–indolinone, chromone–pyrazole, indole–pyrimidine, indole–indolinone, and indole–pyrazole moieties.98 Among the synthesized hybrids, compounds 133, 134, 135, and 136 showed activity against tumor cell lines. SAR studies showed that the substitution of benzyl/2,6-dichlorophenyl/2,6-dichlorobenzyl groups with indole/indolinone moiety is responsible for an increase in the anticancer activity (Fig. 84). Quinazolinone, a natural product with a wide range of biological activities, is known to have inhibitory effects on tubulin polymerization and possesses anticancer activity. The cis-restricted analogues of CA-4 (isoxazoline/isoxazoles) also possess anti-tubulin activity. Thus, a series of 2,3-dihydroquinazoline-linked 3,5-diarylisoxazolineisoxazole hybrids were synthesized.99 Among the hybrids, compound 137 exhibited remarkable activity against 18 human cancer cell lines. The mode of action of this compound was G2/M cell cycle arrest and inhibition of B1 and CDK1 (Fig. 85). Solomon et al.100 synthesized a series of isatin scaffold-based hybrids of 4-piperazinyl quinolone and evaluated it against two breast cancer cell lines: MDA-MB468 and MCF-7. The compounds were also evaluated against the non-cancer cell lines 184B5 and MCF10A. Among the synthesized 4-piperazinoyl quinolone-linked isatin hybrid compounds, 138 and 139 showed remarkable activity against MCF cancer cells by inducing apoptosis (Fig. 86).
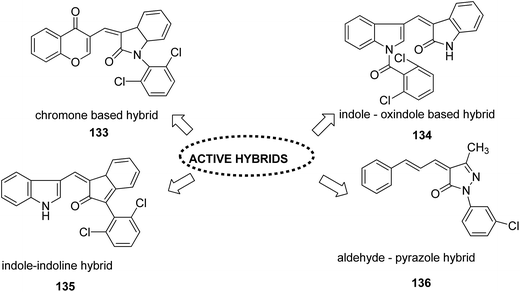 |
| | Fig. 84 Indole, pyazole, chromone and pyrimidine based hybrids. | |
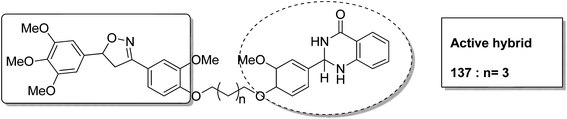 |
| | Fig. 85 3,5-Diaryl isoxazoline linked 2,3-dihydroquinazolinone hybrids. | |
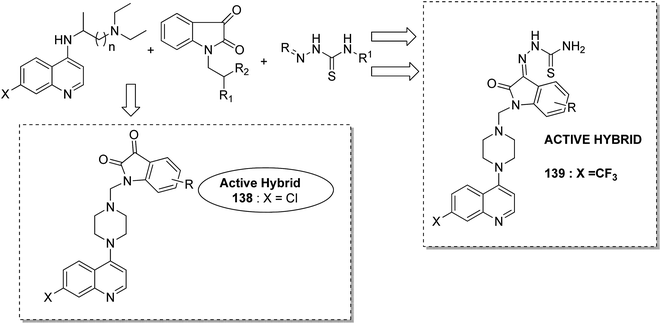 |
| | Fig. 86 Isatin scaffold based hybrids of 4-piperazinyl quinolone. | |
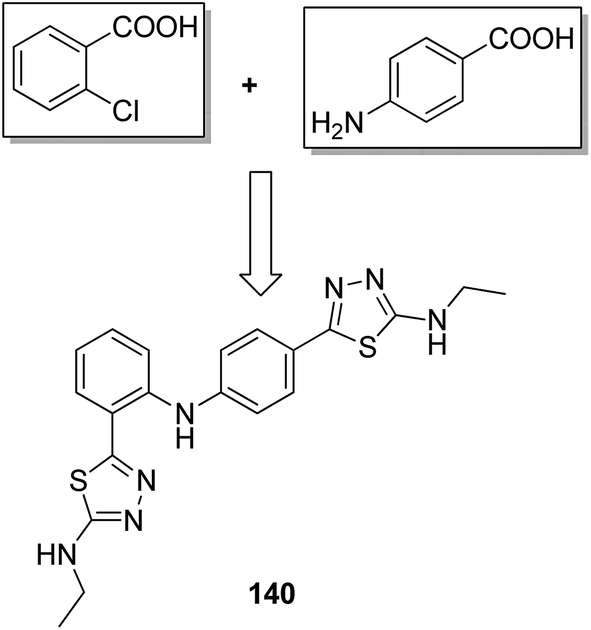
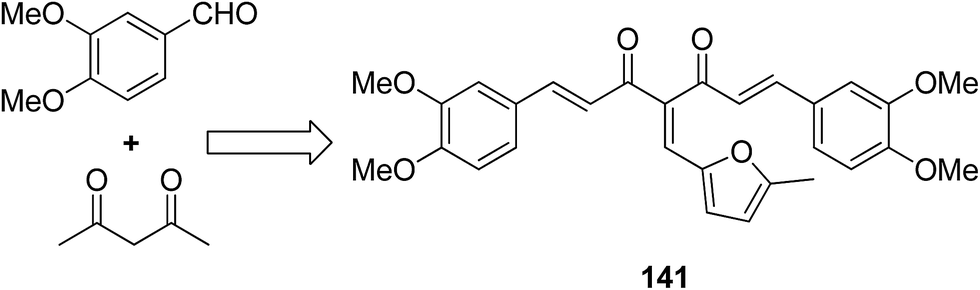
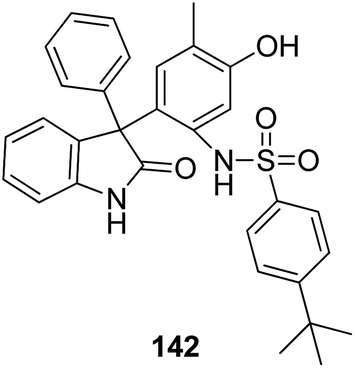
A novel series of 2,4′-bis substituted diphenylamines was synthesized and evaluated for EGFR tyrosine kinase inhibitor activity. Compound 140 was found to be the most potent inhibitor with 98% inhibition at 10 μM (Fig. 87).101 A series of curcumin analogs was synthesized and evaluated for anticancer activity.102 The results of cell growth inhibition assays showed that compound 141 effectively decreased the growth of a panel of lung cancer cells at sub-micromolar concentrations. This compound was found to significantly inhibit NF-KB activity as compared to the parent compound curcumin. This compound also reduced the tumorigenic potential of cancer cells in a clonogenic assay (Fig. 88). Various substitution patterns for arylsulfoanilides were identified through SAR studies, which were mediated by the partial depletion of intracellular Ca2+ stores.103 The substitution patterns resulted in development of a potent arylsulfoanilide–oxindole hybrid 142 that showed micromolar activity against lung cancer cells in a growth inhibition assay (Fig. 89).
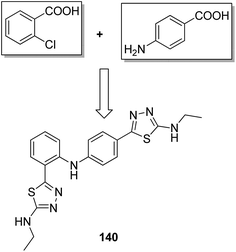 |
| | Fig. 87 2,4′-Bis substituted diphenylamines. | |
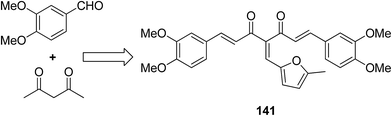 |
| | Fig. 88 4-Arylidene curcumin analogs. | |
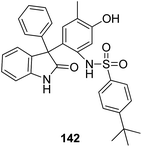 |
| | Fig. 89 Arylsulfoanilide–oxindole hybrids. | |
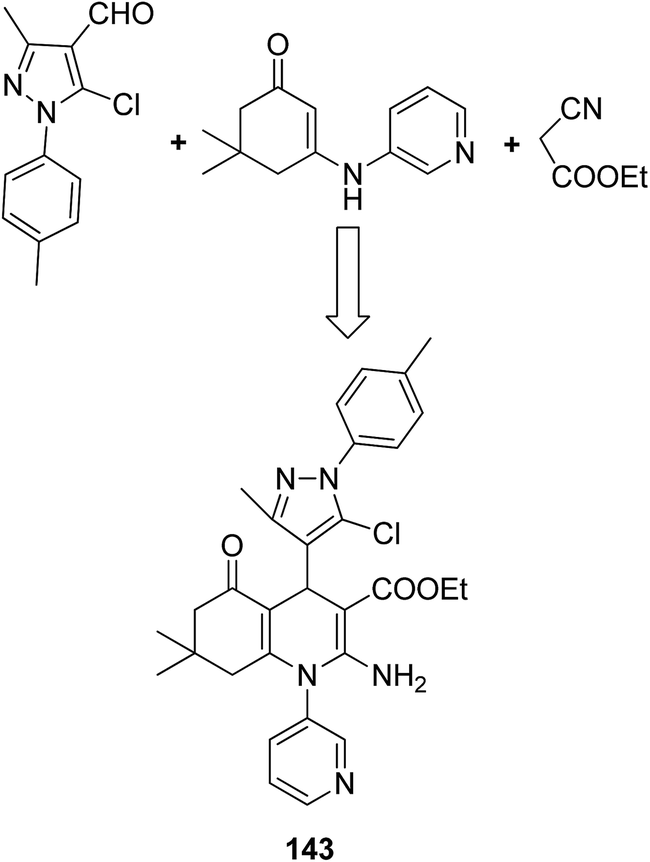
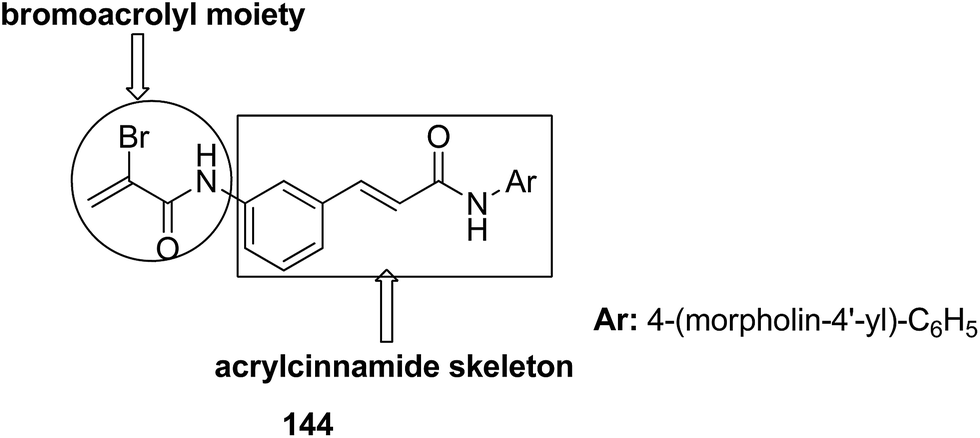
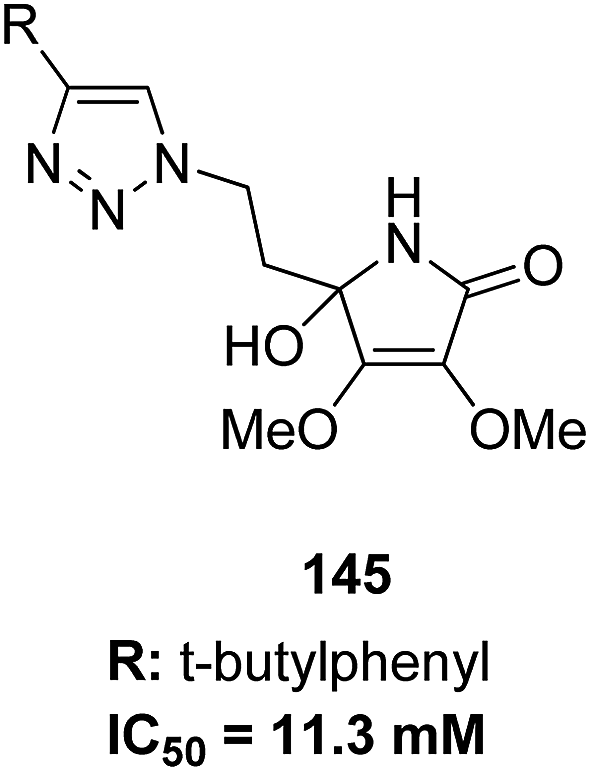
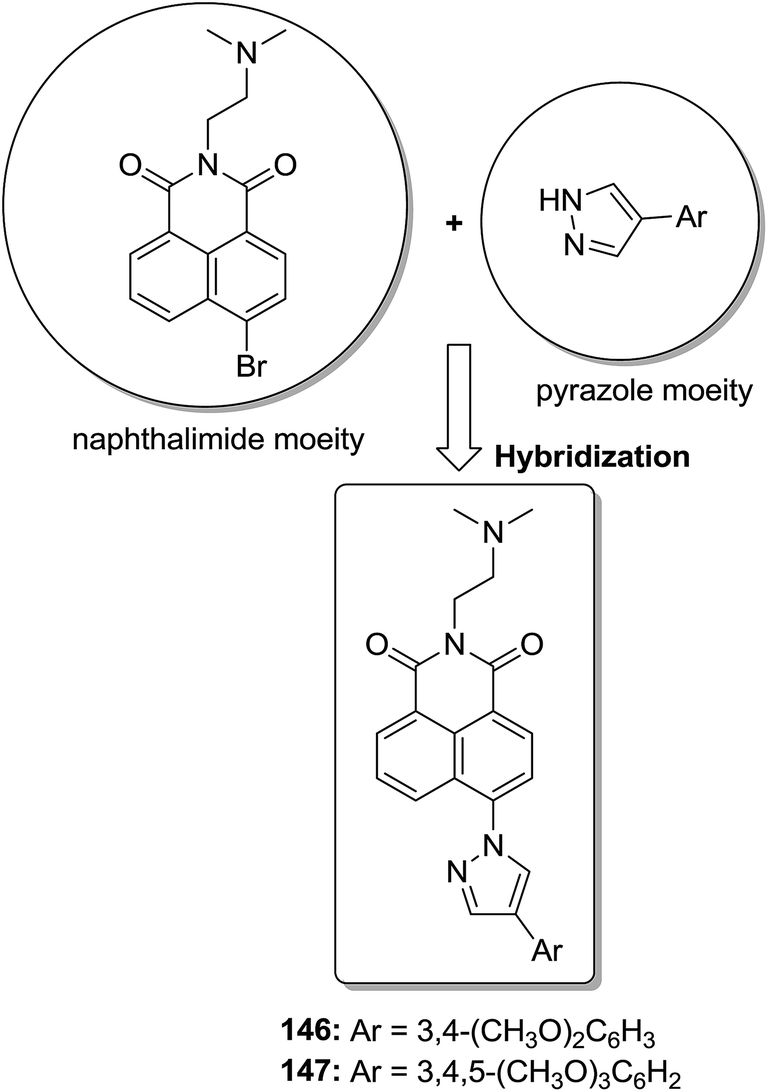
The molecular hybridization technique was used to design a new series of pyrazole–quinoline–pyridine hybrids.104 The compounds were synthesized using a base-catalyzed cyclocondensation reaction and tested for EGFR enzyme inhibitory activity. Compound 143 emerged as the most potent anticancer agent since it was bound to the active pocket of EGFR via three hydrogen bonds and one Π-cation (Fig. 90). Moreover, two pharmacophores, the acrylcinnamide skeleton and α-bromoacrolyl moiety, were combined together to synthesize a series of novel antiproliferative agents.105 The new derivatives were tested against a panel of seven human cancer cell lines. The SAR studies confirmed that compound 144 slightly increased the ROS production in HeLa cells and decreased the glutathione content in the treated cells. It was also observed that only thiol-containing antioxidants were able to protect the cells from induced cell death (Fig. 91). The hybridized 5-hydroxypyrrolidones were synthesized by reacting butenolides with ethanolic amines.106 On testing these compounds for anticancer activity in vitro, compound 145 exhibited the most potent anticancer activity against breast cancer cells. It was revealed from preliminary studies that the potency of this compound was due to ROS generation and activation of p38, which results in apoptosis of cancer cells (Fig. 92). Li et al.107 synthesized a novel series of 4-pyrazolyl-1,8-naphthalimide derivatives and evaluated them for in vitro anticancer activity. When compared to the standard drug amonafide, compounds 146 and 147 showed better cytotoxic activity against MCF-7 cells with the IC50 values of 0.51 μM and 0.79 μM, respectively. UV-vis, fluorescence, and circular dichroism (CD) spectroscopies were used to study the DNA-binding properties of these compounds. The results suggested that compound 147 exhibited a middle binding affinity with CT-DNA (Fig. 93).
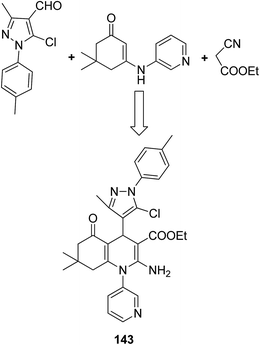 |
| | Fig. 90 Pyrazole–quinoline–pyridine hybrid. | |
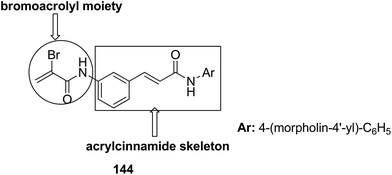 |
| | Fig. 91 Arylcinnamide hybrid derivatives. | |
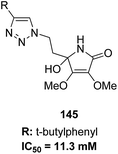 |
| | Fig. 92 Butenolides and polyfunctional pyrrolinones. | |
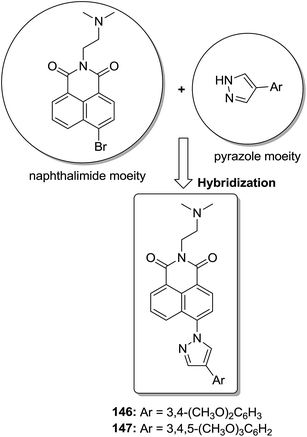 |
| | Fig. 93 Pyrazol–naphthalimide derivatives. | |
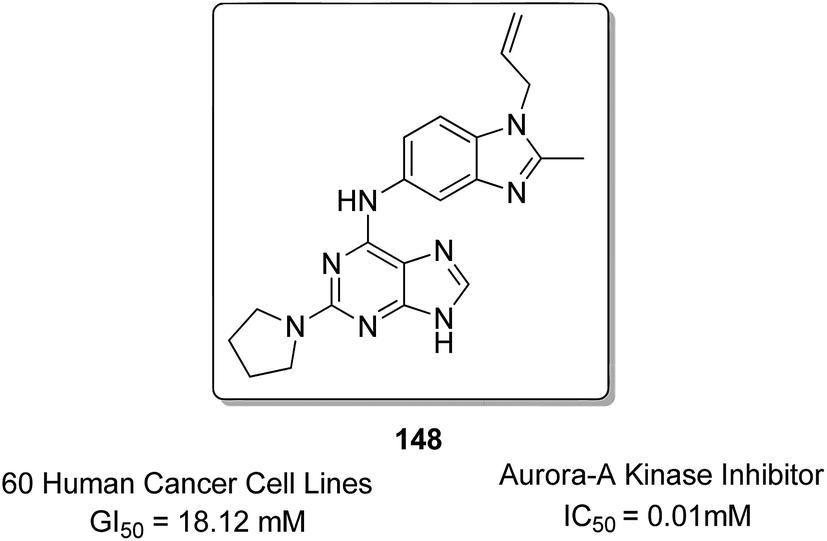
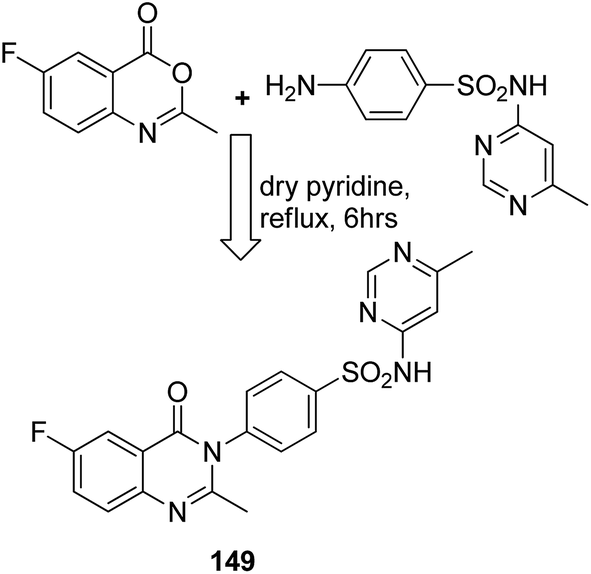
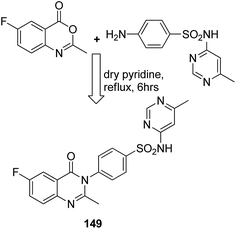
Sharma et al.108 synthesized a novel series of purine–benzimidazole hybrids through nucleophilic substitution at the C-6 position of the purine ring with versatile cyclic amines at the C-2 position. On evaluating the novel compounds against 60 human tumor cell lines at one dose concentration level, compound 148 was found to be 1.25 times more active than the standard control 5-FU, having a GI50 value of 18.12 μM. This compound was also a selective inhibitor of Aurora-A kinase inhibitors with an IC50 value of 0.01 μM (Fig. 94). Zayed et al. synthesized several novel fluorinated quinazoline–sulphonamide derivatives and evaluated them for cytotoxic activity. The anticancer activities were determined by in vitro MTT assay using a three-cell-line panel consisting of lung cancer cells, breast cancer cells, and normal kidney cells. Compound 149 exhibited significant anticancer activity, and the toxicity levels were also low as compared to those of the standard drug methotrexate, with an IC50 value of 2.51 μM on the NCI cell line (Fig. 95).109
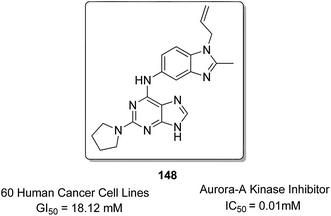 |
| | Fig. 94 Purine–benzimidazole hybrid compound. | |
 |
| | Fig. 95 Synthesis of fluorinated quinazolinone–sulphonamide hybrid compound. | |
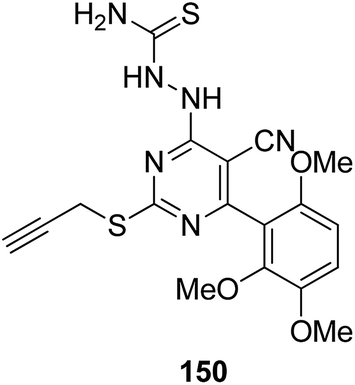
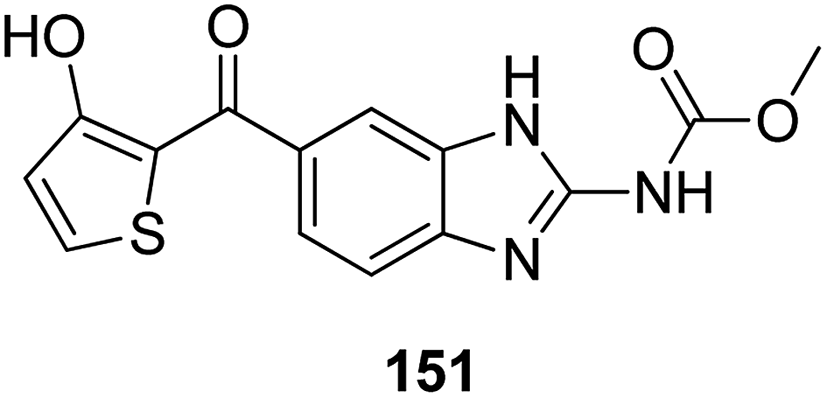
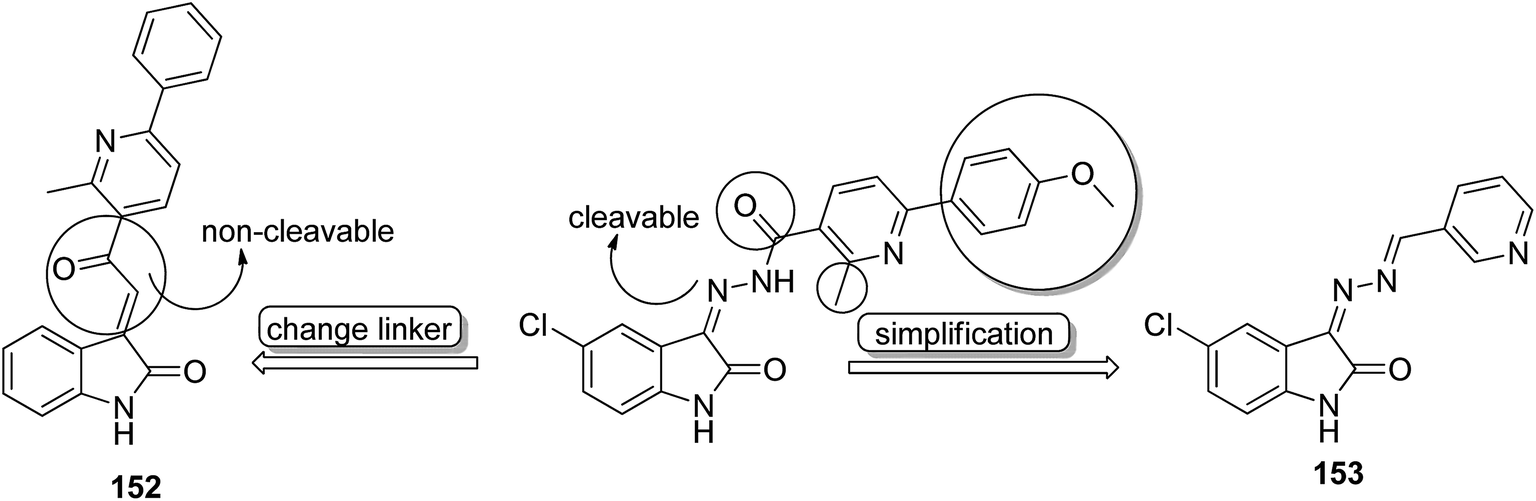
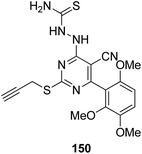
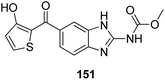
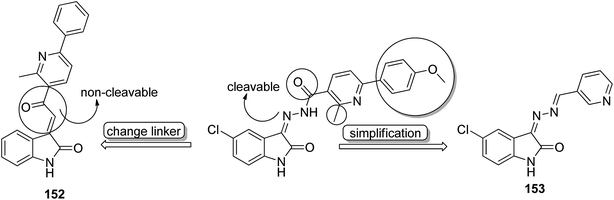
Histone lysine specific demethylase 1 (LSD1) was used as an anticancer drug target for the synthesis of a novel series of pyrimidine–thiourea hybrids.110 Compound 150 containing a terminal alkyne appendage was the most potent and selective LSD1 inhibitor in vitro since it exhibited strong cytotoxicity against LSD1 over-expressed gastric cancer cells. When the drug was orally administered, it showed a marked inhibition of cell migration without significant side effects (Fig. 96). A novel series of combretastatin–nocodazole hybrids were synthesized and evaluated for anticancer activity.111 The conjugates were prepared by replacing the thiophene ring in nocodazole moiety with a substituted phenyl ring from the combretastatin moiety. The synthesized compounds were tested as tubulin polymerization inhibitors and antiproliferative agents against the human lung cancer cell line A-549. Compound 151 was found to be most potent in both assays (Fig. 97). Eldehna et al.112 synthesized three different sets of isatin–pyridine hybrids and evaluated them for anti-tumor activity against HepG2, A549, and MCF-7 cancer cell lines. Discovery Studio 2.5 software was used for the analysis of quantitative–structure activity relationship (QSAR) studies. The results revealed that compound 152 was the most active hybrid against HepG2, whereas compound 153 exhibited equal potency as compared to the standard drug doxorubicin (Fig. 98).
 |
| | Fig. 96 Pyrimidine–thiourea hybrid. | |
 |
| | Fig. 97 Novel hybrid nocodazole analogue. | |
 |
| | Fig. 98 Isatin–pyridine hybrid compounds. | |
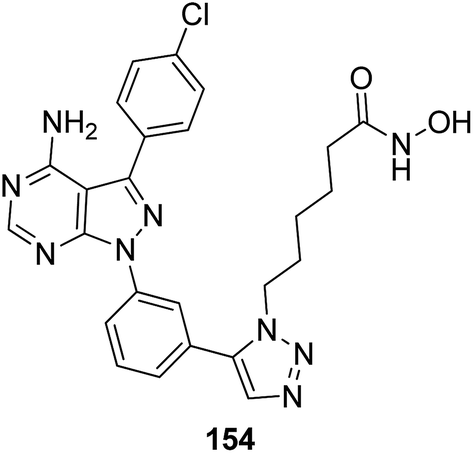
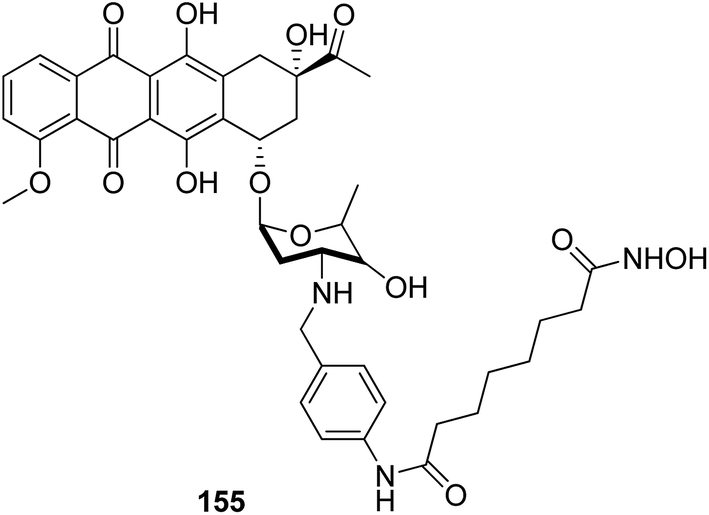
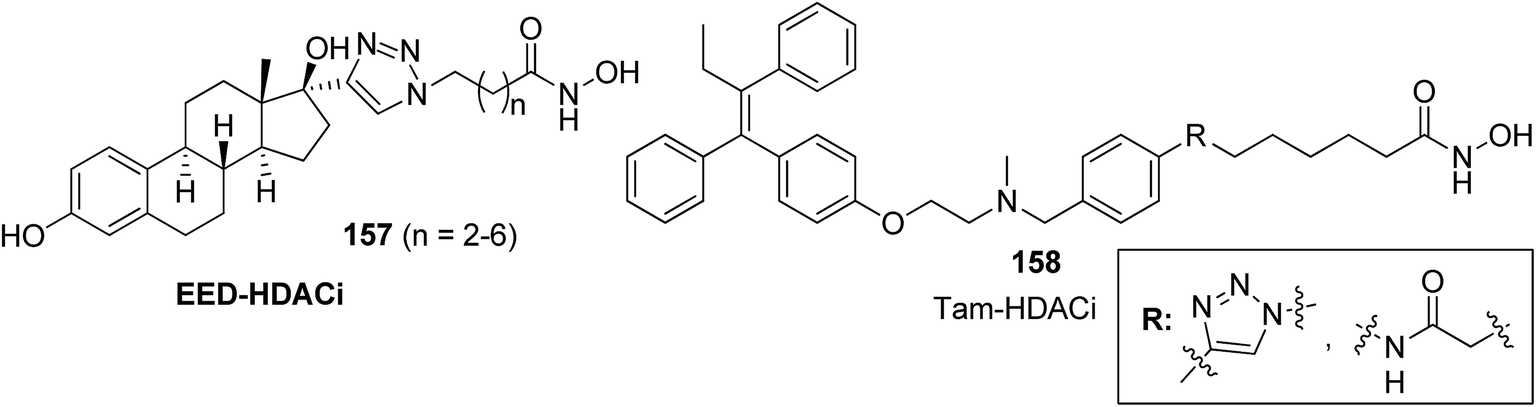
The development of the first chimeric c-Src kinase and HDAC inhibitor was described on the basis of synergism observed between a selective c-Src kinase inhibitor with an HDAC inhibitor. Further studies explored the high potency of the chimeric inhibitor 154 against cancer cell lines. This technique proved to be more efficacious than a dual-targeting strategy using discrete c-Src and HDAC inhibitors. The SAR studies described that the activity was due to the presence of a 1,5-triazole moiety synthesized using a ruthenium-mediated cycloaddition reaction (Fig. 99).113 As is well-known, the drugs that act upon multiple targets are far more superior to the single-target drugs provided the side effects are fewer. By exploring this basic idea, Guerrant et al.114 synthesized novel dual-acting histone deacetylase and topoisomerase II inhibitors. The source for the derivatization of these dual-acting agents originated from suberoylanilide hydroxamic acid (SAHA) and anthracycline daunorubicin, prototypical histone deacetylase (HDAC), and topoisomerase II (Topo II) inhibitors. Compound 155 was found to inhibit HDAC and Topo II in both cell-free as well as whole-cell assays. The compound also inhibited the proliferation of representative cancer cell lines (Fig. 100). A novel series of dual acting compounds was synthesized to inhibit the effect of histone deacetylase (HDAC) and 3-hydroxy-3-methyl-glutaryl coenzyme A reductase (HMGR) enzymes.115 The inhibition was induced by introducing a chelating hydroxamate group with zinc ion at the active site of HDAC. The thus formed statin hydroxamic acid 156 showed most promising anticancer activity against HDACs and HMGR with the IC50 values in the nanomolar range, without causing toxicity to the normal cells (Fig. 101).
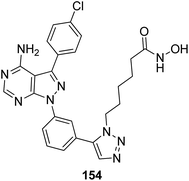 |
| | Fig. 99 Chimeric c-Src kinase and HDAC inhibitor. | |
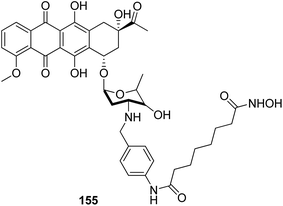 |
| | Fig. 100 Histone deacetylase and topoisomerase II dual-acting inhibitor. | |
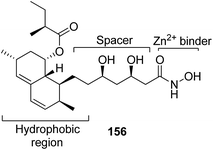 |
| | Fig. 101 Histone deacetylase and HMG-CoA reductase inhibitor. | |
A novel series of histone deacetylase inhibitors (HDACi) was synthesized by merging with either an antagonist or an agonist of the estrogen receptor (ER).116 The compounds were selectively active against breast cancer as they significantly inhibited HDAC at nanomolar concentrations by either agonizing or antagonizing ERα and ERβ. Basically, two types of hybrid conjugates were synthesized: ethynylestradiol–HDACi conjugates (EED-HDACi) 157 and tamoxifen–HDACi conjugates (Tam-HDACi) 158. The ER antagonist activities of Tam-HDACi were identical to those of tamoxifen, whereas EED–HDACi conjugates exhibited ER agonist activities as compared to the parent ethynylestradiol. The in silico docking analysis revealed that Tam–HDACi conjugates were more potent and selective against MCF-7 cancer cell lines than MDA-MB-231, DU145, and Vero cancer cell lines (Fig. 102). A novel series of curcumin–thalidomide hybrids was synthesized for the treatment of human multiple myeloma (MM) and biologically characterized against MMIS, RPMI8226, U266 (human multiple myeloma), and A549 (human lung cancer) cells.117 The results revealed that compounds 159 and 160 were highly effective against all the human MM cell lines. The mechanistic studies explained that these compounds induced the production of ROS and caused G1/S arrest, leading to apoptosis and cell death. These compounds also exhibited inhibitory effects on NFkB activation in A549 cells (Fig. 103). Manohar et al. synthesized novel molecular hybrids of 4-aminoquinoline and triazine as potential anticancer agents. Among the synthesized compounds, 161 and 162 were significantly active against NCI 60 human tumor cell lines. The results suggested that compound 161 activated caspase 8 and 9 but did not cause migration of phosphatidylserine, DNA fragmentation, and effector caspase activation. Compound 162 exhibited significant migration of phosphatidylserine that indicated cell apoptosis. This compound also activated caspase 9, but was unable to activate caspase 8. Furthermore, neither DNA fragmentation nor activation of effector caspases was observed (Fig. 104).118
 |
| | Fig. 102 Estrogen receptor modulated histone deacetylase inhibitors. | |
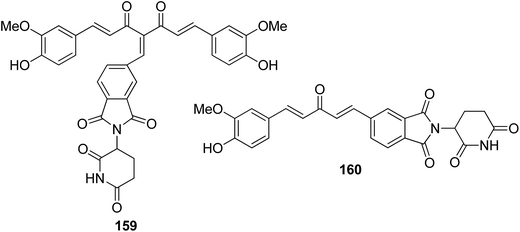 |
| | Fig. 103 Curcumin–thalidomide hybrid compounds. | |
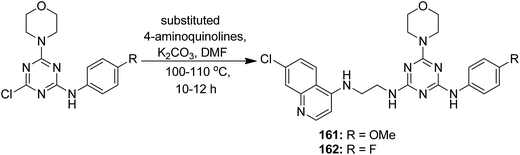 |
| | Fig. 104 Synthesis of novel 4-aminoquinoline–triazine hybrids. | |
Thiourea derivatives have been known to exhibit excellent anticancer activity. The studies also suggest that benzenesulfonamide derivatives also possess apoptotic properties. To further explore the tumor inhibiting properties of these compounds, both moieties were incorporated together to develop a novel series of hybrid compounds. Structural analysis was carried out on the basis of elemental analysis, IR, 1H-NMR, 13C-NMR, and mass spectral data. Compounds 163, 164, 165, and 166 were found to be most active against all cell lines (except for breast cancer cell line), when compared with the standard drugs DCF and doxorubicin (Fig. 105). Molecular docking studies were used to explain the mechanism of action of these compounds. These studies suggested that the cytotoxic activity was due to their action as MK-2 enzyme inhibitors.119 A simple and convenient method was used to synthesize the novel hybrid compounds of quinazoline and sulfonamides. The in vitro anticancer evaluation of the synthesized compounds was performed against four cancer cell lines, and 167 and 168 were found to be most active (Fig. 106). The results suggested that these hybrid compounds have a potential to be potent anticancer agents.120 In a latest study, a newly developed ligand-free simple and inexpensive one-pot method was employed to design rosuvastatin-based novel indole derivatives. The reaction involved copper-catalyzed coupling-cyclization of rosuvastatin-based alkyne with o-iodoanilides. PEG-400, copper(I) iodide, and potassium chromate were also used to complete the reaction. The synthesized compounds were highly active against cancer cell lines. Furthermore, increase in p21 mRNA expression and apoptotic effects in the embryos of zebrafish were also observed. The design of rosuvastatin–indole hybrid compounds 169 is shown in Fig. 107.121
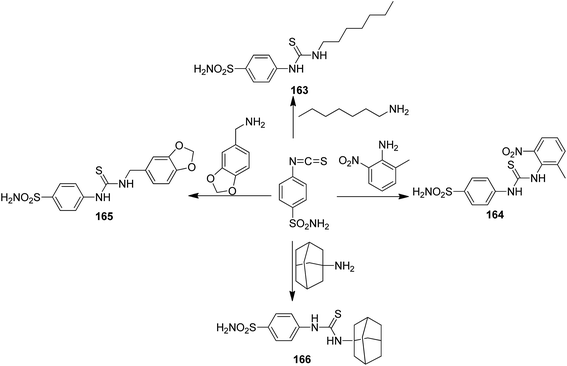 |
| | Fig. 105 Highly active hybrid compounds of thioureido–benzenesulfonamides. | |
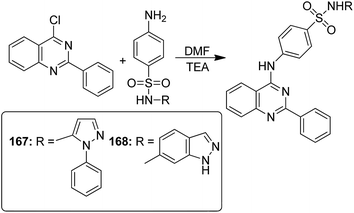 |
| | Fig. 106 Synthesis of novel quinazoline–sulfonamide hybrids. | |
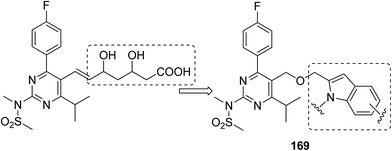 |
| | Fig. 107 Cu-catalyzed rosuvastatin–indole derivatives. | |
10. Future perspective
The hybrid anticancer agents have been proven to be a boon for pharmaceutical industries. The design of these drugs is not only efficacious but also cost-effective. Imagination and creativity along with skills are necessary to construct a new hybrid drug. The research in this field is still ongoing throughout the world. The future hybrid drugs should be designed in a way that they can tackle more than one type of cancer by showing multi-target effect with less toxicity. Moreover, the techniques such as use of blended or fully integrated chemical structures and formation of vectorized anticancer hybrids may prove to be useful. It will be better to synthesize the drugs that release its active ingredients at the site of action for more specific results. The design of drug formulations can also be changed in a way that the carrier molecules show high affinity for its cognate receptors. A number of studies are being carried out in this field, and in coming days, we will surely see advancement in the design and discovery of novel hybrid anticancer molecules.
11. Conclusion
This review article summarized the intellectual advances in designing anticancer hybrids via utilizing molecular hybridization technique. Literature survey assigns the merits of molecular hybridization to novel hybrids that have been designed and discovered from the fusion of two or more active moieties or pharmacophore units. Molecular hybridization results in compounds with changed selectivity profile, less toxicity, dual actions, and new molecular scaffolds. In most of the studies, the main reason for using molecular hybridization is for targeting tubulin as microtubules play a critical role in cellular functions such as cell division, intracellular transport, cell death, and cell shape. Thus, in different hybrid designs, one of the active moieties is microtubule inhibitor such as chalcones, combretastatin, and colchicines, and promising results have been obtained. Isatin and coumarin, the heteroaryl-based drugs, were also reported with inhibitory activity for the pyrrolo[2,1-c][1,4]benzodiazepine (PBD). The DNA interactive antitumor antibiotics obtained from streptomyces species hybrids were also synthesized that resulted in significant activity. Overall, the scope of molecular hybridization is quite broad for medicinal chemists around the globe. This technique has been utilized to synthesize multi-target drugs and tackle complex diseases such as cancer.
References
- K. Patel, C. Karthikeyan, V. R. Solomon, N. S. H. N. Moorthy, H. Lee, K. Sahu, G. S. Deora and P. Trivedi, Lett. Drug Des. Discovery, 2011, 8, 308–311 CrossRef CAS.
- S. A. Rostom, Bioorg. Med. Chem., 2006, 14, 6475–6485 CrossRef CAS PubMed.
- E. M. Yee, E. Pasquier, G. Iskander, K. Wood, D. S. Black and N. Kumar, Bioorg. Med. Chem., 2013, 21, 1652–1660 CrossRef CAS PubMed.
- P. Singh, R. Raj, V. Kumar, M. P. Mahajan, P. Bedi, T. Kaur and A. Saxena, Eur. J. Med. Chem., 2012, 47, 594–600 CrossRef CAS PubMed.
- Z. Chen, X. Liang, H. Zhang, H. Xie, J. Liu, Y. Xu, W. Zhu, Y. Wang, X. Wang and S. Tan, J. Med. Chem., 2010, 53, 2589–2600 CrossRef CAS PubMed.
- C. Viegas-Júnior, A. Danuello, V. d. S. Bolzani, E. J. Barreiro and C. A. M. Fraga, Curr. Med. Chem., 2007, 14, 1829–1852 CrossRef.
- S. Fortin and G. Bérubé, Expert Opin. Drug Discovery, 2013, 8, 1029–1047 CrossRef CAS PubMed.
- R. Morphy, C. Kay and Z. Rankovic, Drug Discovery Today, 2004, 9, 641–651 CrossRef CAS PubMed.
- J. Walsh and A. Bell, Curr. Pharm. Des., 2009, 15, 2970–2985 CrossRef CAS PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. Bedi and K. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS PubMed.
- D. A. Erlanson, R. S. McDowell and T. O'Brien, J. Med. Chem., 2004, 47, 3463–3482 CrossRef CAS PubMed.
- Y.-C. Duan, Y.-C. Ma, E. Zhang, X.-J. Shi, M.-M. Wang, X.-W. Ye and H.-M. Liu, Eur. J. Med. Chem., 2013, 62, 11–19 CrossRef CAS PubMed.
- A. Kamal, A. Mallareddy, M. J. Ramaiah, S. Pushpavalli, P. Suresh, C. Kishor, J. Murty, N. S. Rao, S. Ghosh and A. Addlagatta, Eur. J. Med. Chem., 2012, 56, 166–178 CrossRef CAS PubMed.
- L.-X. Liu, X.-Q. Wang, J.-M. Yan, Y. Li, C.-J. Sun, W. Chen, B. Zhou, H.-B. Zhang and X.-D. Yang, Eur. J. Med. Chem., 2013, 66, 423–437 CrossRef CAS PubMed.
- X.-Q. Wang, L.-X. Liu, Y. Li, C.-J. Sun, W. Chen, L. Li, H.-B. Zhang and X.-D. Yang, Eur. J. Med. Chem., 2013, 62, 111–121 CrossRef CAS PubMed.
- A. Husain, M. Rashid, M. Shaharyar, A. A. Siddiqui and R. Mishra, Eur. J. Med. Chem., 2013, 62, 785–798 CrossRef CAS PubMed.
- M. Rashid, A. Husain and R. Mishra, Eur. J. Med. Chem., 2012, 54, 855–866 CrossRef CAS PubMed.
- V. R. Solomon, C. Hu and H. Lee, Bioorg. Med. Chem., 2009, 17, 7585–7592 CrossRef CAS PubMed.
- Y.-C. Duan, Y.-C. Zheng, X.-C. Li, M.-M. Wang, X.-W. Ye, Y.-Y. Guan, G.-Z. Liu, J.-X. Zheng and H.-M. Liu, Eur. J. Med. Chem., 2013, 64, 99–110 CrossRef CAS PubMed.
- S. Grosse, V. Mathieu, C. Pillard, S. Massip, M. Marchivie, C. Jarry, P. Bernard, R. Kiss and G. Guillaumet, Eur. J. Med. Chem., 2014, 84, 718–730 CrossRef CAS PubMed.
- K.-P. Shao, X.-Y. Zhang, P.-J. Chen, D.-Q. Xue, P. He, L.-Y. Ma, J.-X. Zheng, Q.-R. Zhang and H.-M. Liu, Bioorg. Med. Chem. Lett., 2014, 24, 3877–3881 CrossRef CAS PubMed.
- N. Abbassi, E. M. Rakib, H. Chicha, L. Bouissane, A. Hannioui, C. Aiello, R. Gangemi, P. Castagnola, C. Rosano and M. Viale, Arch. Pharm., 2014, 347, 423–431 CrossRef CAS PubMed.
- K. Kaur, V. Kumar, A. K. Sharma and G. K. Gupta, Eur. J. Med. Chem., 2014, 77, 121–133 CrossRef CAS PubMed.
- D.-Q. Xue, X.-Y. Zhang, C.-J. Wang, L.-Y. Ma, N. Zhu, P. He, K.-P. Shao, P.-J. Chen, Y.-F. Gu and X.-S. Zhang, Eur. J. Med. Chem., 2014, 85, 235–244 CrossRef CAS PubMed.
- M. F. Cardoso, P. C. Rodrigues, M. E. I. Oliveira, I. L. Gama, I. M. da Silva, I. O. Santos, D. R. Rocha, R. T. Pinho, V. F. Ferreira and M. C. B. de Souza, Eur. J. Med. Chem., 2014, 84, 708–717 CrossRef CAS PubMed.
- R. Pingaew, P. Mandi, C. Nantasenamat, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Eur. J. Med. Chem., 2014, 81, 192–203 CrossRef CAS PubMed.
- G. S. Hassan, Med. Chem. Res., 2014, 23, 388–401 CrossRef CAS.
- R. Bollu, J. D. Palem, R. Bantu, V. Guguloth, L. Nagarapu, S. Polepalli and N. Jain, Eur. J. Med. Chem., 2015, 89, 138–146 CrossRef CAS PubMed.
- R. Sunke, P. V. Babu, S. Yellanki, R. Medishetti, P. Kulkarni and M. Pal, Org. Biomol. Chem., 2014, 12, 6800–6805 CAS.
- K. S. S. Praveena, E. V. V. S. Ramarao, N. Y. S. Murthy, S. Akkenapally, C. G. Kumar, R. Kapavarapu and S. Pal, Bioorg. Med. Chem. Lett., 2015, 25, 1057–1063 CrossRef CAS PubMed.
- N. Kuntala, J. R. Telu, V. Banothu, S. B. Nallapati, J. S. Anireddy and S. Pal, MedChemComm, 2015, 6, 1612–1619 RSC.
- M. E. Wall, M. Wani, C. Cook, K. H. Palmer, A. a. McPhail and G. Sim, J. Am. Chem. Soc., 1966, 88, 3888–3890 CrossRef CAS.
- Q. Huang, L. Wang and W. Lu, Eur. J. Med. Chem., 2013, 63, 746–757 CrossRef CAS PubMed.
- R. Cincinelli, L. Musso, S. Dallavalle, R. Artali, S. Tinelli, D. Colangelo, F. Zunino, M. De Cesare, G. L. Beretta and N. Zaffaroni, Eur. J. Med. Chem., 2013, 63, 387–400 CrossRef CAS PubMed.
- A. Boumendjel, A. McLeer-Florin, P. Champelovier, D. Allegro, D. Muhammad, F. Souard, M. Derouazi, V. Peyrot, B. Toussaint and J. Boutonnat, BMC Cancer, 2009, 9, 242 CrossRef PubMed.
- A. Kamal, N. Shankaraiah, S. Prabhakar, C. R. Reddy, N. Markandeya, K. L. Reddy and V. Devaiah, Bioorg. Med. Chem. Lett., 2008, 18, 2434–2439 CrossRef CAS PubMed.
- M. Nagaraju, E. Gnana Deepthi, C. Ashwini, M. Vishnuvardhan, V. L. Nayak, R. Chandra, S. Ramakrishna and B. Gawali, Bioorg. Med. Chem. Lett., 2012, 22, 4314–4317 CrossRef CAS PubMed.
- K. V. Sashidhara, A. Kumar, M. Kumar, J. Sarkar and S. Sinha, Bioorg. Med. Chem. Lett., 2010, 20, 7205–7211 CrossRef CAS PubMed.
- A. Kamal, A. Mallareddy, P. Suresh, V. L. Nayak, R. V. Shetti, N. S. Rao, J. R. Tamboli, T. B. Shaik, M. Vishnuvardhan and S. Ramakrishna, Eur. J. Med. Chem., 2012, 47, 530–545 CrossRef CAS PubMed.
- M. A. Mourad, M. A. Aziz, G. E.-D. A. Abuo-Rahma and H. H. Farag, Eur. J. Med. Chem., 2012, 54, 907–913 CrossRef CAS PubMed.
- A. Kamal, S. Prabhakar, M. J. Ramaiah, P. V. Reddy, C. R. Reddy, A. Mallareddy, N. Shankaraiah, T. L. N. Reddy, S. Pushpavalli and M. Pal-Bhadra, Eur. J. Med. Chem., 2011, 46, 3820–3831 CrossRef CAS PubMed.
- G. Wang, C. Li, L. He, K. Lei, F. Wang, Y. Pu, Z. Yang, D. Cao, L. Ma and J. Chen, Bioorg. Med. Chem., 2014, 22, 2060–2079 CrossRef CAS PubMed.
- Y. Chinthala, S. Thakur, S. Tirunagari, S. Chinde, A. K. Domatti, N. K. Arigari, K. Srinivas, S. Alam, K. K. Jonnala and F. Khan, Eur. J. Med. Chem., 2015, 93, 564–573 CrossRef CAS PubMed.
- A. Correa, I. Tellitu, E. Domínguez, I. Moreno and R. SanMartin, J. Org. Chem., 2005, 70, 2256–2264 CrossRef CAS PubMed.
- A. Kamal, K. S. Reddy, M. N. A. Khan, R. V. Shetti, M. J. Ramaiah, S. Pushpavalli, C. Srinivas, M. Pal-Bhadra, M. Chourasia and G. N. Sastry, Bioorg. Med. Chem., 2010, 18, 4747–4761 CrossRef CAS PubMed.
- A. Kamal, M. K. Reddy, M. J. Ramaiah, J. S. Reddy, Y. Srikanth, D. Dastagiri, E. V. Bharathi, S. Pushpavalli, P. Sarma and M. Pal-Bhadra, Bioorg. Med. Chem., 2011, 19, 2565–2581 CrossRef CAS PubMed.
- A. Kamal, R. Ramu, V. Tekumalla, G. R. Khanna, M. S. Barkume, A. S. Juvekar and S. M. Zingde, Bioorg. Med. Chem., 2008, 16, 7218–7224 CrossRef CAS PubMed.
- A. Kamal, G. Ramesh, O. Srinivas and P. Ramulu, Bioorg. Med. Chem. Lett., 2004, 14, 471–474 CrossRef CAS PubMed.
- A. Kamal, M. N. A. Khan, K. S. Reddy, S. K. Ahmed, M. S. Kumar, A. Juvekar, S. Sen and S. Zingde, Bioorg. Med. Chem. Lett., 2007, 17, 5345–5348 CrossRef CAS PubMed.
- A. Kamal, J. S. Reddy, M. J. Ramaiah, D. Dastagiri, E. V. Bharathi, M. A. Azhar, F. Sultana, S. Pushpavalli, M. Pal-Bhadra and A. Juvekar, Eur. J. Med. Chem., 2010, 45, 3924–3937 CrossRef CAS PubMed.
- J.-J. Wang, Y.-K. Shen, W.-P. Hu, M.-C. Hsieh, F.-L. Lin, M.-K. Hsu and M.-H. Hsu, J. Med. Chem., 2006, 49, 1442–1449 CrossRef CAS PubMed.
- A. Kamal, G. Ramakrishna, V. L. Nayak, P. Raju, A. S. Rao, A. Viswanath, M. Vishnuvardhan, S. Ramakrishna and G. Srinivas, Bioorg. Med. Chem., 2012, 20, 789–800 CrossRef CAS PubMed.
- A. Kamal, O. Srinivas, P. Ramulu, G. Ramesh and P. P. Kumar, Bioorg. Med. Chem. Lett., 2004, 14, 4107–4111 CrossRef CAS PubMed.
- A. Kamal, Y. Srikanth, M. J. Ramaiah, M. Khan, M. K. Reddy, M. Ashraf, A. Lavanya, S. Pushpavalli and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2012, 22, 571–578 CrossRef CAS PubMed.
- A. Kamal, E. V. Bharathi, M. J. Ramaiah, D. Dastagiri, J. S. Reddy, A. Viswanath, F. Sultana, S. Pushpavalli, M. Pal-Bhadra and H. K. Srivastava, Bioorg. Med. Chem., 2010, 18, 526–542 CrossRef CAS PubMed.
- A. Kamal, E. V. Bharathi, M. J. Ramaiah, J. S. Reddy, D. Dastagiri, A. Viswanath, F. Sultana, S. Pushpavalli, M. Pal-Bhadra and A. Juvekar, Bioorg. Med. Chem. Lett., 2010, 20, 3310–3313 CrossRef CAS PubMed.
- Z. Nofal, M. El-Zahar and S. Abd El-Karim, Molecules, 2000, 5, 99–113 CrossRef CAS.
- F. Belluti, G. Fontana, L. D. Bo, N. Carenini, C. Giommarelli and F. Zunino, Bioorg. Med. Chem., 2010, 18, 3543–3550 CrossRef CAS PubMed.
- K. M. Amin, A. A. Eissa, S. M. Abou-Seri, F. M. Awadallah and G. S. Hassan, Eur. J. Med. Chem., 2013, 60, 187–198 CrossRef CAS PubMed.
- H. Chen, S. Li, Y. Yao, L. Zhou, J. Zhao, Y. Gu, K. Wang and X. Li, Bioorg. Med. Chem. Lett., 2013, 23, 4785–4789 CrossRef CAS PubMed.
- K. Paul, S. Bindal and V. Luxami, Bioorg. Med. Chem. Lett., 2013, 23, 3667–3672 CrossRef CAS PubMed.
- K. V. Sashidhara, S. R. Avula, K. Sharma, G. R. Palnati and S. R. Bathula, Eur. J. Med. Chem., 2013, 60, 120–127 CrossRef CAS PubMed.
- S. Sandhu, Y. Bansal, O. Silakari and G. Bansal, Bioorg. Med. Chem., 2014, 22, 3806–3814 CrossRef CAS PubMed.
- R. Pingaew, A. Saekee, P. Mandi, C. Nantasenamat, S. Prachayasittikul, S. Ruchirawat and V. Prachayasittikul, Eur. J. Med. Chem., 2014, 85, 65–76 CrossRef CAS PubMed.
- B. Yadagiri, U. D. Holagunda, R. Bantu, L. Nagarapu, C. G. Kumar, S. Pombala and B. Sridhar, Eur. J. Med. Chem., 2014, 79, 260–265 CrossRef CAS PubMed.
- M. I. El-Gamal and C.-H. Oh, Eur. J. Med. Chem., 2014, 84, 68–76 CrossRef CAS PubMed.
- T. Nasr, S. Bondock and M. Youns, Eur. J. Med. Chem., 2014, 76, 539–548 CrossRef CAS PubMed.
- W. Zhang, Z. Li, M. Zhou, F. Wu, X. Hou, H. Luo, H. Liu, X. Han, G. Yan and Z. Ding, Bioorg. Med. Chem. Lett., 2014, 24, 799–807 CrossRef CAS PubMed.
- Y.-J. Li, M.-Y. Ye, R.-Z. Huang, G.-Y. Yao, Y.-M. Pan, Z.-X. Liao and H.-S. Wang, Med. Chem. Res., 2014, 23, 3144–3156 CrossRef CAS.
- A. M. El-Agrody, A. M. Fouda and A.-A. M. Al-Dies, Med. Chem. Res., 2014, 23, 3187–3199 CrossRef CAS.
- P. Boregowda, S. Kalegowda, V. P. Rasal, J. Eluru and E. Koyye, Org. Chem. Int., 2014, 2014, 297586 Search PubMed.
- N. Singh, J. Sarkar, K. V. Sashidhara, S. Ali and S. Sinha, Apoptosis, 2014, 19, 1017–1028 CrossRef CAS PubMed.
- K. M. Amin, S. M. Abou-Seri, F. M. Awadallah, A. A. Eissa, G. S. Hassan and M. M. Abdulla, Eur. J. Med. Chem., 2015, 90, 221–231 CrossRef CAS PubMed.
- C. Dumontet and B. I. Sikic, J. Clin. Oncol., 1999, 17, 1061 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, L. Tong, Y. Luo, M. Su, Y. Zang, J. Li, W. Lu and Y. Chen, Bioorg. Med. Chem., 2013, 21, 3240–3244 CrossRef CAS PubMed.
- Y. B. Malysheva, S. Combes, D. Allegro, V. Peyrot, P. Knochel, A. E. Gavryushin and A. Y. Fedorov, Bioorg. Med. Chem., 2012, 20, 4271–4278 CrossRef CAS PubMed.
- K. Larocque, P. Ovadje, S. Djurdjevic, M. Mehdi, J. Green and S. Pandey, PLoS One, 2014, 9, e87064 Search PubMed.
- S.-K. Kim, S.-M. Cho, H. Kim, H. Seok, S.-O. Kim, T. K. Kwon and J.-S. Chang, Exp. Mol. Med., 2013, 45, e19 CrossRef PubMed.
- S. R. Punganuru, H. R. Madala and K. S. Srivenugopal, Med. Chem., 2016, 2016, 165–173 Search PubMed.
- L. A. Graham, J. Suryadi, T. K. West, G. L. Kucera and U. Bierbach, J. Med. Chem., 2012, 55, 7817–7827 CrossRef CAS PubMed.
- S. Ding, X. Qiao, G. L. Kucera and U. Bierbach, Chem. Commun., 2013, 49, 2415–2417 RSC.
- J. Provencher-Mandeville, C. Descoôteaux, S. K. Mandal, V. Leblanc, É. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2008, 18, 2282–2287 CrossRef CAS PubMed.
- V. Gagnon, M.-È. St-Germain, C. Descôteaux, J. Provencher-Mandeville, S. Parent, S. K. Mandal, E. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2004, 14, 5919–5924 CrossRef CAS PubMed.
- R. Kumar, L. Gupta, P. Pal, S. Khan, N. Singh, S. B. Katiyar, S. Meena, J. Sarkar, S. Sinha and J. K. Kanaujiya, Eur. J. Med. Chem., 2010, 45, 2265–2276 CrossRef CAS PubMed.
- S. Ayral-Kaloustian, J. Gu, J. Lucas, M. Cinque, C. Gaydos, A. Zask, I. Chaudhary, J. Wang, L. Di and M. Young, J. Med. Chem., 2009, 53, 452–459 CrossRef PubMed.
- I. Paterson, G. J. Naylor and A. E. Wright, Chem. Commun., 2008, 4628–4630 RSC.
- R. Romagnoli, P. G. Baraldi, M. K. Salvador, M. E. Camacho, J. Balzarini, J. Bermejo and F. Estévez, Eur. J. Med. Chem., 2013, 63, 544–557 CrossRef CAS PubMed.
- A. Gupta, P. Saha, C. Descôteaux, V. Leblanc, É. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2010, 20, 1614–1618 CrossRef CAS PubMed.
- M. Kashyap, D. Das, R. Preet, P. Mohapatra, S. R. Satapathy, S. Siddharth, C. N. Kundu and S. K. Guchhait, Bioorg. Med. Chem. Lett., 2012, 22, 2474–2479 CrossRef CAS PubMed.
- P. Singh, M. Kaur and P. Verma, Bioorg. Med. Chem. Lett., 2009, 19, 3054–3058 CrossRef CAS PubMed.
- V. Sol, F. Lamarche, M. Enache, G. Garcia, R. Granet, M. Guilloton, J. Blais and P. Krausz, Bioorg. Med. Chem., 2006, 14, 1364–1377 CrossRef CAS PubMed.
- G. Montanaro, M. Bertinaria, B. Rolando, R. Fruttero, C. D. Lucas, D. A. Dorward, A. G. Rossi, I. L. Megson and A. Gasco, Bioorg. Med. Chem., 2013, 21, 2107–2116 CrossRef CAS PubMed.
- S.-L. Zhu, Y. Wu, C.-J. Liu, C.-Y. Wei, J.-C. Tao and H.-M. Liu, Eur. J. Med. Chem., 2013, 65, 70–82 CrossRef CAS PubMed.
- A. Sharma, V. Luxami and K. Paul, Bioorg. Med. Chem. Lett., 2013, 23, 3288–3294 CrossRef CAS PubMed.
- N. Kojima, T. Fushimi, N. Maezaki, T. Tanaka and T. Yamori, Bioorg. Med. Chem. Lett., 2008, 18, 1637–1641 CrossRef CAS PubMed.
- A. Kamal, J. R. Tamboli, V. L. Nayak, S. Adil, M. Vishnuvardhan and S. Ramakrishna, Bioorg. Med. Chem. Lett., 2013, 23, 3208–3215 CrossRef CAS PubMed.
- S. K. Suthar, V. Jaiswal, S. Lohan, S. Bansal, A. Chaudhary, A. Tiwari, A. T. Alex and A. Joesph, Eur. J. Med. Chem., 2013, 2013(63), 589–602 CrossRef PubMed.
- P. Singh, M. Kaur and W. Holzer, Eur. J. Med. Chem., 2013, 2010(45), 4968–4982 Search PubMed.
- A. Kamal, E. V. Bharathi, J. S. Reddy, M. J. Ramaiah, D. Dastagiri, M. K. Reddy, A. Viswanath, T. L. Reddy, T. B. Shaik and S. Pushpavalli, Eur. J. Med. Chem., 2013, 2011(46), 691–703 Search PubMed.
- V. R. Solomon, C. Hu and H. Lee, Bioorg. Med. Chem., 2010, 18, 1563–1572 CrossRef CAS PubMed.
- S. M. Abou-Seri, Eur. J. Med. Chem., 2010, 45, 4113–4121 CrossRef CAS PubMed.
- X. Qiu, Y. Du, B. Lou, Y. Zuo, W. Shao, Y. Huo, J. Huang, Y. Yu, B. Zhou and J. Du, J. Med. Chem., 2010, 53, 8260–8273 CrossRef CAS PubMed.
- A. Natarajan, Y. Guo, F. Harbinski, Y.-H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979–4982 CrossRef CAS PubMed.
- C. B. Sangani, J. A. Makawana, X. Zhang, S. B. Teraiya, L. Lin and H.-L. Zhu, Eur. J. Med. Chem., 2014, 76, 549–557 CrossRef CAS PubMed.
- R. Romagnoli, P. G. Baraldi, M. K. Salvador, M. Chayah, M. E. Camacho, F. Prencipe, E. Hamel, F. Consolaro, G. Basso and G. Viola, Eur. J. Med. Chem., 2014, 81, 394–407 CrossRef CAS PubMed.
- N. Devender, K. Ramakrishna, S. K. Shukla, R. Konwar and R. P. Tripathi, Eur. J. Med. Chem., 2014, 82, 106–119 CrossRef CAS PubMed.
- S. Li, S. Xu, Y. Tang, S. Ding, J. Zhang, S. Wang, G. Zhou, C. Zhou and X. Li, Bioorg. Med. Chem. Lett., 2014, 24, 586–590 CrossRef CAS PubMed.
- A. Sharma, V. Luxami and K. Paul, Eur. J. Med. Chem., 2015, 93, 414–422 CrossRef CAS PubMed.
- M. F. Zayed, H. E. Ahmed, S. Ihmaid, A.-S. M. Omar and A. S. Abdelrahim, J. Taibah Univ. Sci., 2015, 10, 333–339 Search PubMed.
- L.-Y. Ma, Y.-C. Zheng, S.-Q. Wang, B. Wang, Z.-R. Wang, L.-P. Pang, M. Zhang, J.-W. Wang, L. Ding and J. Li, J. Med. Chem., 2015, 58, 1705–1716 CrossRef CAS PubMed.
- S. S. Kale, G. S. Jedhe, S. N. Meshram, M. K. Santra, E. Hamel and G. J. Sanjayan, Bioorg. Med. Chem. Lett., 2015, 25, 1982–1985 CrossRef CAS PubMed.
- W. M. Eldehna, A. Altoukhy, H. Mahrous and H. A. Abdel-Aziz, Eur. J. Med. Chem., 2015, 90, 684–694 CrossRef CAS PubMed.
- K. S. Ko, M. E. Steffey, K. R. Brandvold and M. B. Soellner, ACS Med. Chem. Lett., 2013, 4, 779–783 CrossRef CAS PubMed.
- W. Guerrant, V. Patil, J. C. Canzoneri and A. K. Oyelere, J. Med. Chem., 2012, 55, 1465–1477 CrossRef CAS PubMed.
- J.-B. Chen, T.-R. Chern, T.-T. Wei, C.-C. Chen, J.-H. Lin and J.-M. Fang, J. Med. Chem., 2013, 56, 3645–3655 CrossRef CAS PubMed.
- B. E. Gryder, M. K. Rood, K. A. Johnson, V. Patil, E. D. Raftery, L.-P. D. Yao, M. Rice, B. Azizi, D. F. Doyle and A. K. Oyelere, J. Med. Chem., 2013, 56, 5782–5796 CrossRef CAS PubMed.
- K. Liu, D. Zhang, J. Chojnacki, Y. Du, H. Fu, S. Grant and S. Zhang, Org. Biomol. Chem., 2013, 11, 4757–4763 CAS.
- S. Manohar, A. Pepe, C. E. V. Gerena, B. Zayas, S. V. Malhotra and D. S. Rawat, RSC Adv., 2014, 4, 7062–7067 RSC.
- M. M. Ghorab, M. S. Alsaid, M. S. Al-Dosary, Y. M. Nissan and S. M. Attia, Chem. Cent. J., 2016, 10, 1 CrossRef PubMed.
- M. M. Ghorab, M. S. Alsaid, M. S. Al-Dosari, M. G. El-Gazzar and M. K. Parvez, Molecules, 2016, 21, 189 CrossRef PubMed.
- K. S. Kumar, B. Rajesham, M. S. Ramulu, B. Bhaskar, S. N. Dash, M. A. Ashfaq, R. Nagarapu, A. A. Khan, S. Lehtonen and M. Pal, RSC Adv., 2016, 6, 100487–100493 RSC.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Saurabh Dhiman
a,
Malleshappa N. Noolvi
b,
Harun M. Patel
c and
Varun Bhardwaj
*a
a,
Saurabh Dhiman
a,
Malleshappa N. Noolvi
b,
Harun M. Patel
c and
Varun Bhardwaj
*a