
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
External oxidant-free cross-coupling of arylcopper and alkynylcopper reagents leading to arylalkyne†
Sheng Wanga,
Yaosen Mina,
Xiaowei Zhanga and
Chanjuan Xi *ab
*ab
aKey Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology, Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: cjxi@tsinghua.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 30th May 2017
Abstract
External oxidant-free oxidative cross-coupling between arylcopper and alkynylcopper has been performed, which provides a new way for the formation of arylalkyne with high selectivity.
Introduction
Arylalkynes are useful building blocks in organic synthesis as well as basic functional groups in various bioactive compounds and natural products.1 The most common approach to arylalkynes is transition-metal-catalyzed cross-coupling reaction such as Sonogashira reaction2 or Stille reaction3 between arylhalides and terminal alkynes or alkynyltin compounds. Recently, the oxidative cross-coupling reaction has been considered as a convenient method to synthesize arylalkynes under mild conditions utilizing various transition metals as catalysts in the presence of oxidants.4 Although this reaction provides a straightforward strategy for the synthesis of the unsymmetrical alkynes, realizing oxidative cross-coupling between arylmetal and alkylmetal in high efficiency with oxidants is still difficult since potential side reactions such as homo-coupling of the arylmetals or the alkylmetals as well as direct reaction between organometallic reagents and oxidants. During the course of our study on the carbon–carbon bond formation utilizing alkynylcoppers,5 we found a direct cross-coupling of alkynylcopper and arylcopper reagents to afford arylalkynes with excellent selectivity without external oxidant, which possibly underwent disproportionation mechanism and was worthy of investigation (Scheme 1). | ||
| Scheme 1 Direct coupling reaction of organocoppers. | ||
Results and discussion
Initially, we used phenylcopper 2a generated from phenyl magnesium 1a with CuCl·2LiCl salts6 and (phenylethynyl)copper 3a as starting materials. Reaction of 2a and 3a in the THF solvent at room temperature for 12 h afforded 4aa in 5% yield (Table 1, entry 1). During the reaction, we noticed the reaction mixture was a slurry. Therefore, we tried to add some additives such as hexamethyl-phosphoramide (HMPA), tetramethylethylenediamine (TMEDA), 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU), 4-dimethylaminopyridine (DMAP) and pyridine, respectively in the vessel to improve the solubility of organometallic reagents (entries 2–6). Only pyridine was found to enhance the yield to 18% (entry 6). The reaction was screened in different temperature (entries 6–9) and we found that when the reaction was treated at 50 °C, the product 4aa was formed in 75% isolated yield (entry 7). In the above reactions, the homo-coupling products have not been obtained and copper mirror on the wall of the reaction vessel was observed.
|
|||
|---|---|---|---|
| Entry | Temp (°C) | Additive | Yield [%] of 4aab |
| a Reaction conditions: phenylcopper 2a prepared in situ from the corresponding phenylmagnesium bromide 1a (0.50 mmol, 1.0 M in THF, 1.0 mL) with the 0.6 mL 1 M CuCl·2LiCl in THF-solution; (phenylethynyl)copper 3a (0.5 mmol, 82 mg), N2, 12 h.b GC yield, yield based on n-dodecane, isolated yield was given in parenthesis.c Diphenyl was obtained around in 60% GC yield. | |||
| 1 | rt | — | 5 |
| 2 | rt | HMPA | Trace |
| 3 | rt | TMEDA | Trace |
| 4 | rt | DBU | Trace |
| 5 | rt | DMAP | Trace |
| 6 | rt | Pyridine | 18 |
| 7 | 50 | Pyridine | 85 (75) |
| 8 | 67 | Pyridine | 72 |
| 9 | 80 | Pyridine | 70 |
| 10c | 50 | Pyridine/DDQ | Trace |
| 11c | 50 | Pyridine/DCE | 9 |
| 12c | 50 | Pyridine/DBE | 6 |
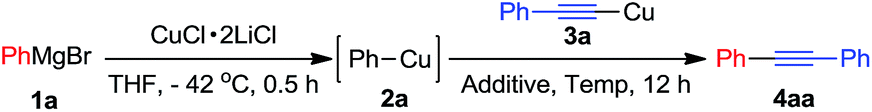
Theoretically, oxidative coupling of two organometallic reagents forming C–C bond, the oxidants are definitely necessary to take away two redundant electrons from organometallics. Therefore, dichloroethane (DCE), 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) and dibromoethane (DBE) were examined as the oxidants under the same reaction conditions. In each case, trace product 4aa and homo-coupling product diphenyl was detected in GC-MS (entries 10–12). Compared with oxidative cross-coupling reaction in the presence of external oxidant, our reaction may take place through an intermolecular disproportionation of arylcopper and alkynylcopper. The formation of copper mirror on the wall of the reaction vessel further confirmed the intermolecular disproportionation.
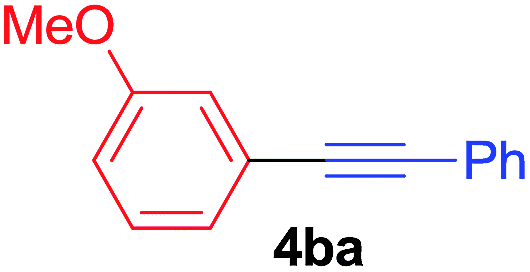
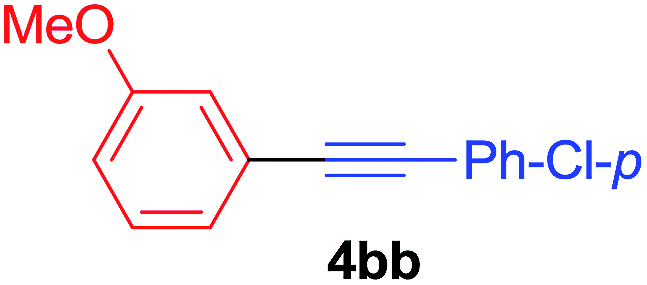
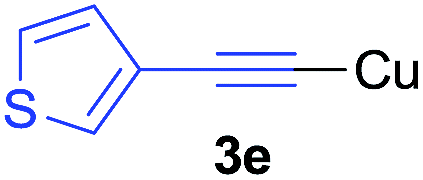
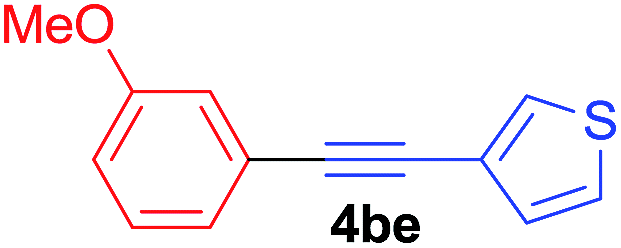
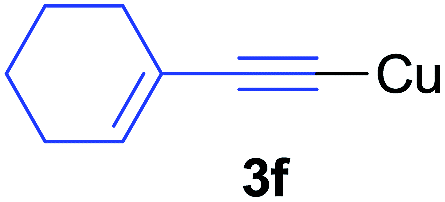
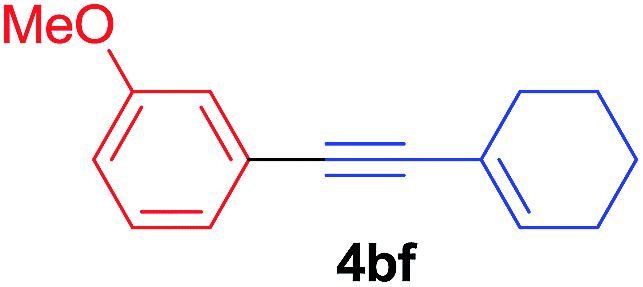
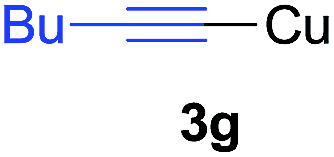
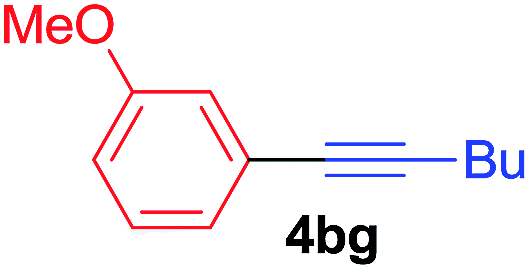
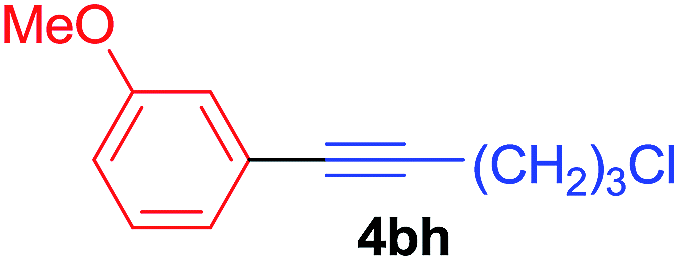
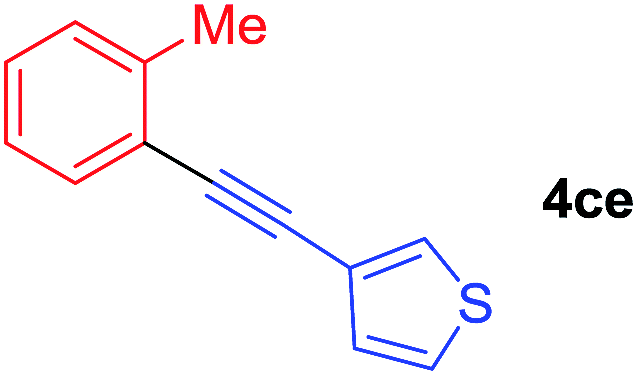
Under the optimized conditions, the experiments about functional-group compatibility were carried out. Firstly, aryl substituted alkynylcoppers 3 including phenyl, 4-chlorophenyl, 4-fluorophenyl, 4-methylphenyl, and thiophen-3-yl groups were used to react with (3-methoxyphenyl)copper 2b and a range of alkynes 4 were obtained in good yields (entries 1–5). Then, we tried (cyclohex-1-en-1-ylethynyl)copper 3f and desired product 4bf was obtained in 66% yield. Furthermore, employment of hex-1-yn-1-ylcopper 3g and (5-chloropent-1-yn-1-yl)copper 3h also afforded product 4bg and 4bh in 58% and 42% yields, respectively.
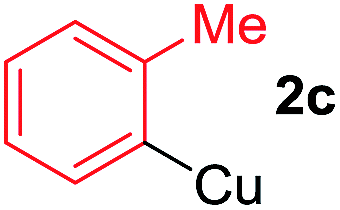
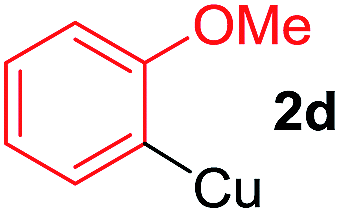
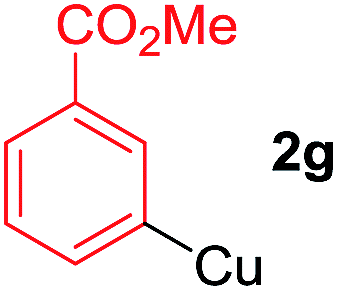
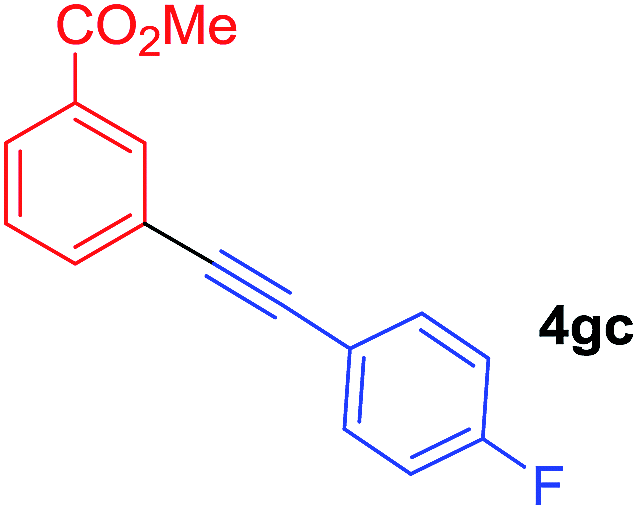
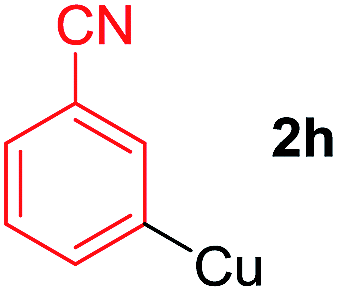
To establish the full scope of the reaction, compatibility of various arylcoppers in cross coupling reaction was studied. The representative results are summarized in Table 3. The reaction generally lead to satisfactory yields with electron-rich arylcoppers (entries 1–3). While arylcoppers bearing electron-poor groups such as Cl, CO2Me, and CN afforded the corresponding alkyne in diminished yields (entries 4–6) as well as formation of undesired self-coupling product from arylcoppers. Notably, the reaction seemed no sensitive to steric hindrance and the results as shown in Table 2 (entry 1) and Table 3 (entries 2–3).

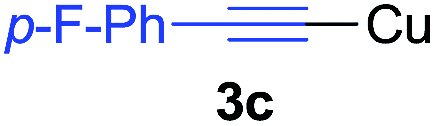
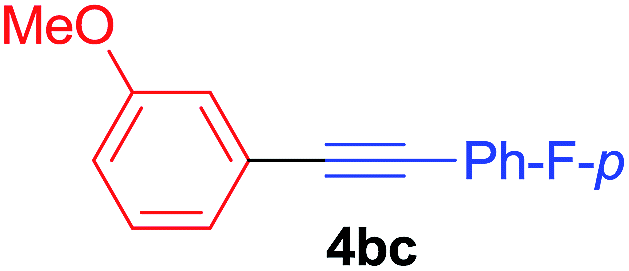
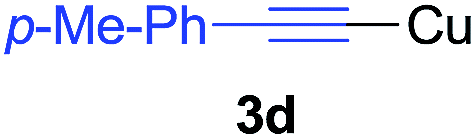
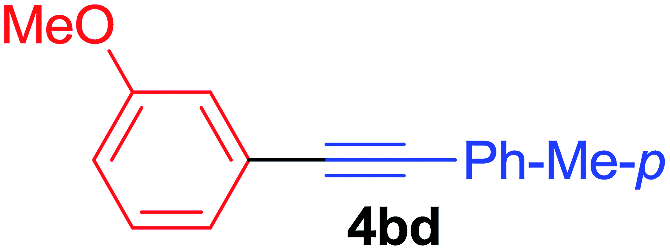
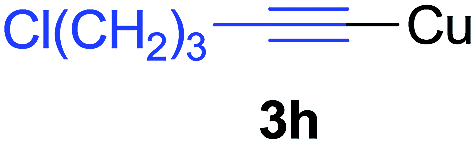
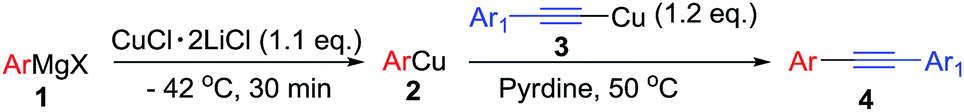
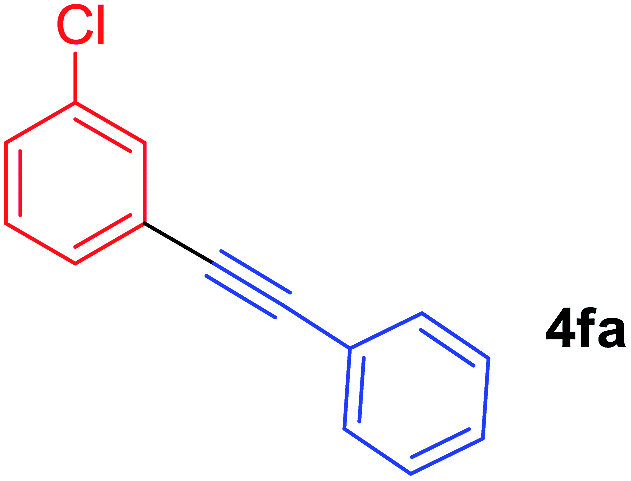
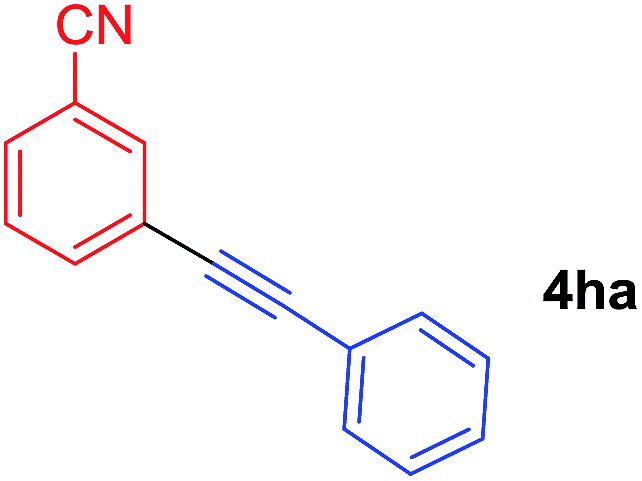
Arylmagnesium reagents are sensitive, which can be generated from aryl halides in situ. From practical point of view, aryl halides could be converted into the corresponding arylmagnesium reagents (1f, 1h, 1i, and 1j) by iodine/magnesium exchange from iodoarenes and iPrMgCl·LiCl.7 It was then transmetallated with CuCl·2LiCl (in THF solution) to give the corresponding arylcopper reagents, which was treated subsequently with the compound 3a to generate the alkyne (4fa, 4ha, 4ia, and 4ja) (Scheme 2), respectively.
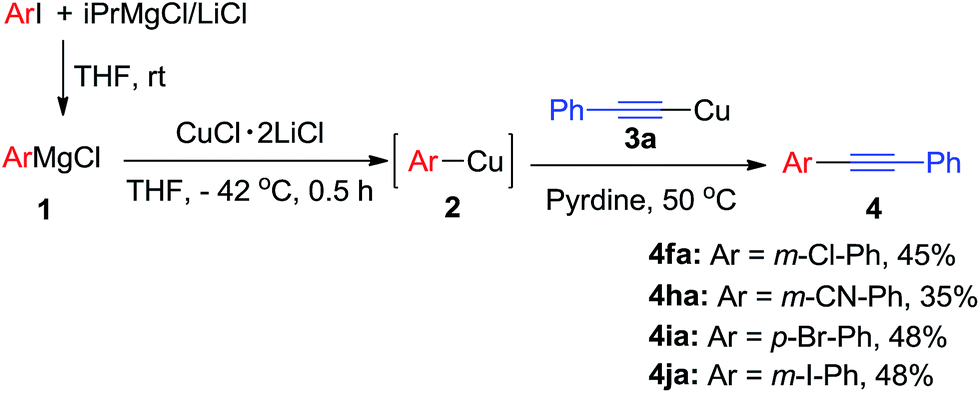 | ||
| Scheme 2 Use of PhMgCl prepared by I/Mg exchange with iPrMgCl. | ||
We also tried to use alkylcopper reagents in the reaction. As an example, reaction of cyclohexylcopper with alkynylcopper in the presence of pyridine at −20 °C for 6 h and product 4ka was isolated in 44% (Scheme 3) as well as bicyclohexane was detected in GC-MS.
 | ||
| Scheme 3 Cross-coupling between alkylcopper and alkynylcopper. | ||
Based on the above results, a possible mechanism for the oxidative cross-coupling of an arylmagnesium and an alkynylcopper is proposed in Scheme 4. First, transmetallation of arylmagnesium compound 1 with CuCl yields arylcopper 2. An alkynylcopper 3′ in the presence of pyridine coordinates to the copper of the arylcopper 2 to form intermediate 5, which undergoes disproportionation reaction leading to intermediate 6 and formation of copper powder. Finally, reductive elimination yields the desired cross-coupling product 4 with concomitant precipitation of another molecular copper powder from the reaction mixture.
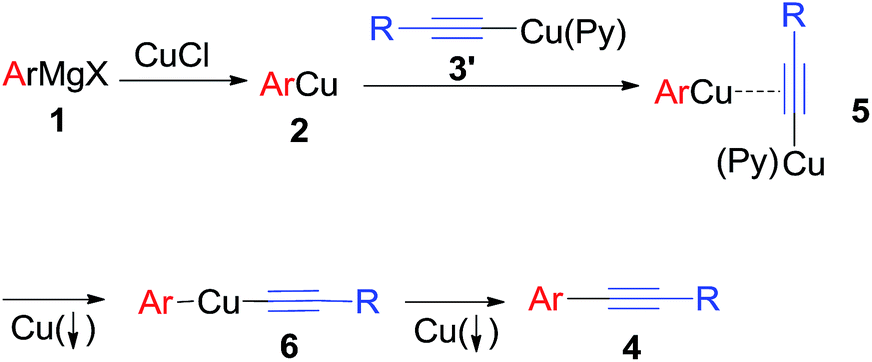 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusions
We have reported a new oxidative coupling of the arylcopper and alkynylcopper, which provides a complementary way for formation of arylalkyne. It is the first procedure for oxidative cross-coupling of two different organocopper reagents without external oxidant.Experimental section
General procedure for preparation of alkynylcopper reagents
Alkynylcopper reagents were prepared according to literature reported.8 To round-bottom flask equipped with a magnetic stirrer were added CuSO4·5H2O (2.5 g, 10 mmol), 100 mL H2O, 10 mL 28% aqueous ammonia and 1.4 g NH2OH·HCl (1.4 g, 20 mmol), and the solution was cooled to 0 °C. Then terminal alkyne (10 mmol) dissolved in 60 mL EtOH was added. Yellow precipitation appeared immediately and the mixture was stirred vigorously for 5 min, followed by filtration. The solid was washed with H2O (15 mL × 2), EtOH (15 mL × 2) and Et2O (15 mL × 3) and dried in vacuum.Preparation of CuCl·2LiCl (1 M in THF)
1 M CuCl·2LiCl solution was prepared according to literature reported.6 To nitrogen-flushed 50 mL Schlenk-tube containing a magnetic stirrer was added LiCl (0.95 g, 20 mmol) and was heated to 130 °C for 1 h in high vaccum. Then Schlenk-tube was cooled to room temperature and charged with CuCl (0.98 g, 10 mmol, 99.999% metal basis) under nitrogen flow. The Schlenk-tube was heated to 130 °C for 5 h in high vaccum. After cooling to room temperature, 10 mL fresh-distilled THF was added and the mixture was stirred vigorously overnight under nitrogen atmosphere protecting from light with aluminium foil. Colorless solution was obtained and stored away from light.Typical procedure to generate arylcopper reagents
General procedure of cross coupling between arylcopper reagents and alkynylcopper reagents
To a solution of arylcopper reagents (0.5 mmol) was added alkynylcopper reagents (0.6 mmol). 2 mL pyridine (extra dry) was added and the mixture was heated to 50 °C for 12 h. To quench the reaction, 5 mL HCl (1 M) was added, extracted with EtOAc (5 mL × 3). The combined organic layer was washed with saturated NaCl solution, dried by Na2SO4, evaporated and purified by flash chromatography.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21472106 and 91645120) and the National Key Basic Research Program of China (973 program) (2012CB933402).Notes and references
- (a) K. C. Nicolaou and W. M. Dai, Angew. Chem., Int. Ed., 1991, 30, 1387 CrossRef; (b) P. J. Stang and F. Diederich, Modern Acetylene Chemistry, Wiley-VCH, Weinheim, 1995 Search PubMed; (c) M. C. Joshi and D. S. Rawat, Chem. Biodiversity, 2012, 9, 459 CrossRef CAS PubMed; (d) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2008, 47, 3096 CrossRef CAS PubMed; (e) G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed; (f) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed.
- (a) H. Doucet and J. Hierso, Angew. Chem., Int. Ed., 2007, 46, 834 CrossRef CAS PubMed; (b) M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642 CrossRef CAS PubMed; (c) K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS; (d) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef; (e) K. Okuro, M. Furuune, M. Mura and M. Nomura, Tetrahedron Lett., 1992, 33, 5363 CrossRef CAS.
- (a) J. K. Stille, Angew. Chem., Int. Ed., 1986, 25, 508 CrossRef; (b) S.-K. Kang, J.-S. Kim and S.-C. Choi, J. Org. Chem., 1997, 62, 4208 CrossRef CAS PubMed; (c) J.-H. Li, B.-X. Tang, L.-M. Tao, Y.-X. Xie, Y. Liang and M.-B. Zhang, J. Org. Chem., 2006, 71, 7488 CrossRef CAS PubMed.
- (a) C. Liu, D. Liu and A. Lei, Acc. Chem. Res., 2014, 47, 3459 CrossRef CAS PubMed; (b) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (c) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761 RSC; (d) K. M. Liu, L. Y. Liao and X. F. Duan, Chem. Commun., 2015, 51, 1124 RSC; (e) K. M. Liu, J. Wei and X. F. Duan, Chem. Commun., 2015, 51, 4655 RSC; (f) J. Yu, J. Liu, G. Shi, C. Shao and Y. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4079 CrossRef CAS PubMed; (g) Y. Zhu, T. Xiong, W. Han and Y. Shi, Org. Lett., 2014, 16, 6144 CrossRef CAS PubMed; (h) C. Theunissen and G. Evano, Org. Lett., 2014, 16, 4488 CrossRef CAS PubMed; (i) T. Yasukawa, H. Miyamura and S. Kobayashi, Org. Biomol. Chem., 2011, 9, 6208 RSC.
- J. Jiao, S. Wang, C. Chen, Q. Liu and C. Xi, Organometallics, 2016, 35, 1415 CrossRef CAS.
- S. R. Dubbaka, M. Kienle, H. Mayr and P. Knochel, Angew. Chem., Int. Ed., 2007, 46, 9093 CrossRef CAS PubMed.
- (a) T. Klatt, J. T. Markiewicz, C. Sämann and P. Knochel, J. Org. Chem., 2014, 79, 4253 CrossRef CAS PubMed; (b) R. L. Bao, R. Zhao and L. Shi, Chem. Commun., 2015, 51, 9744 RSC.
- D. Gallego, A. Brück, E. Irran, F. Meier, M. Kaupp, M. Driess and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 15617 CrossRef CAS PubMed.
- G. C. Edwin Raja, F. M. Irudayanathan, H. Kim, J. Kim and S. Lee, J. Org. Chem., 2016, 81, 5244 CrossRef CAS PubMed.
- J. Mao, G. Xie, M. Wu, J. Guo and S. Ji, Adv. Synth. Catal., 2008, 350, 2477 CrossRef CAS.
- S. Urgaonkar and J. G. Verkade, J. Org. Chem., 2004, 69, 5752 CrossRef CAS PubMed.
- M. Chen, X. Zheng, W. Li, J. He and A. Lei, J. Am. Chem. Soc., 2010, 132, 4101 CrossRef CAS PubMed.
- K. Kiyokawa, N. Tachikake, M. Yasuda and A. Baba, Angew. Chem., Int. Ed., 2011, 50, 10393 CrossRef CAS PubMed.
- H. Huang, H. Jiang, K. Chen and H. Liu, J. Org. Chem., 2008, 73, 9061 CrossRef CAS PubMed.
- K. Park, G. Bae, J. Moon, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2010, 75, 6244 CrossRef CAS PubMed.
- C. Lin, Y. Wang and C. Lee, Eur. J. Org. Chem., 2010, 4368 CAS.
- A. Orita, H. Taniguchi and J. Otera, Chem.–Asian J., 2006, 1, 430 CrossRef CAS PubMed.
- G. Cahiez, O. Gager and J. Buendia, Angew. Chem., Int. Ed., 2010, 49, 1278 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra for all compounds. See DOI: 10.1039/c7ra03348f |
| This journal is © The Royal Society of Chemistry 2017 |