
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polycyclic aromatic hydrocarbons in traditional Chinese medicines: an analytical method based on different medicinal parts, levels, distribution, and sources†
Bao Yu‡
a,
Dan Zhang‡a,
Li-Hong Tanb,
Sheng-Ping Zhaoa,
Jian-Wei Wanga,
Ling Yaoa and
Wei-Guo Cao*ac
aCollege of Traditional Chinese Medicine, Chongqing Medical University, Chongqing, China
bDepartment of Pharmaceutics, Chongqing Medical and Pharmaceutical College, Chongqing, China
cThe Laboratory of Traditional Chinese Medicine, Chongqing Medical University, Chongqing, China. E-mail: cwgzd2001@hotmail.com
First published on 16th January 2017
Abstract
Herein, we describe a rapid, easy, and cost-effective high-performance liquid chromatography method using UV and fluorescence detectors for the simultaneous analysis of 16 polycyclic aromatic hydrocarbons (PAHs) in traditional Chinese medicines (TCMs). Pretreatment involved different extraction methods depending on the different medicinal parts, and was followed by silica gel purification. The method was validated and used to assess PAHs contamination in 32 TCMs. In the samples analyzed, all 16 PAHs were present. Their total contents ranged from 19.5 to 1614.1 μg kg−1. Among all PAHs studied, phenanthrene was the most common and serious contaminant, followed by fluorene and fluoranthene. Leaves had the highest levels of the 16 PAHs, followed by roots and stems, seeds, flowers, and fruits. The diagnostic ratios and principle component analysis showed that the main sources of PAHs in TCMs were both pyrogenic and petrogenic. Furthermore, PAHs in roots and stems primarily originated from wood or coal combustion, as reported for the first time. Our results suggest that PAHs contamination in TCMs is widespread, and that the proposed method may be a useful tool for quality control of PAHs in TCMs, and for determining their potential health risks.
1. Introduction
Polycyclic aromatic hydrocarbons (PAHs), a class of over 200 different compounds containing two or more fused aromatic rings, are primarily formed by incomplete combustion or heat-induced decomposition of organic matter.1 They may interfere with the normal function of DNA2 and some have been proven highly carcinogenic, mutagenic, and genotoxic in experiments on animals.3 PAHs are widely present in the environment due to their hydrophobic properties, allowing adsorption onto atmospheric particles and direct deposition in sediments, soils, and plants.4 Traditional Chinese Medicines (TCMs), are largely herbal in nature and are susceptible to contamination with PAHs. Besides environmental pollution, different forms of processing undergone by TCMs, such as drying, can also lead to the PAH contamination.5 Due to their specific theory and long historical clinical practice, TCMs are gaining increasing attention; an estimated 1.5 billion people now use them worldwide.6 Therefore, it is important to monitor PAHs levels to evaluate the potential risks associated with human consumption of TCMs. The following 16 PAHs have been listed as priority organic pollutants by the United States Environmental Protection Agency (USEPA): acenaphthylene (ACL), anthracene (AN), acenaphthene (AC), benzo[a]pyrene (BaP), benzo[a]anthracene (BaA), benzo[b]fluoranthene (BbFA), benzo[k]fluoranthene (BkFA), benzo[g,h,i]perylene (BghiP), chrysene (CHR), dibenzo[a,h]anthracene (DBahA), fluoranthene (FA), fluorene (FL), indeno[1,2,3-c,d]pyrene (IP), naphthalene (NA), phenanthrene (PHE), and pyrene (PY).Various analytical methods can be used to quantify PAHs, of which high-performance liquid chromatography, coupled with fluorescence (HPLC-FLD) or ultraviolet (UV)-visible detection,5,7,8 as well as gas chromatography-tandem mass spectrometry (GC-MS/MS)9,10 are the two most common analytical techniques applied in recent years. The advantage of using UV and fluorescence detections in series with HPLC is that UV detection is required for acenaphthylene because it is inactive to fluorescence. Owing to the complex matrix of samples to be analyzed, optimization of the extraction and cleanup procedures is indispensable. Until now, pretreatment for PAHs in a matrix has commonly relied on a two-stage methodology, involving liquid–liquid extraction with various solvents (acetone, acetonitrile, hexane, cyclohexane, and methylene chloride) and a solid-phase extraction (SPE) cleanup using alumina, florisil, silica, and C18 cartridges, or gel permeation chromatography (GPC).10–14 A modified QuEChERS method has also been reported by Magdalena Surma et al.,15 which provided a significant reference method and guidance for the analysis of PAHs in TCMs.
However, TCMs have many different medicinal parts, including roots, stems, flowers, fruits, seeds, and leaves. Different medicinal parts have different types of matrices; therefore, different pretreatments should be established for different sample types. To the best of our knowledge, there had only been one study on PAHs determination in different parts of TCMs,9 wherein purification procedures were conducted using different SPE columns, which was complicated and costly. Therefore, this study aimed to develop and validate a simple and easy method for determining the levels of 16 EPA PAHs in TCMs based on medicinal parts. The pretreatment consisted of three different extraction methods—ultrasonic extraction, homogenization extraction, and oscillation extraction—followed by a silica gel cleanup. Subsequently, analysis was carried out using HPLC coupled to UV and fluorescence detectors in series, which ensured the detection of all 16 PAHs. In addition, we further discussed the distribution and source apportionment of PAHs in different types of TCMs, which has not been reported previously. This work could provide more information to reduce PAHs in TCMs from the source.
2. Experimental
2.1. Material and reagents
Thirty-two TCM samples, including six roots and stems, eight fruits, eight seeds, five flowers, and five leaves, were purchased from local market. They were manufactured at the place of origin and kept in cool and dry conditions. The TCMs were powdered, passed through a 0.5 mm metal sieve, and stored at 4 °C until analysis.All glassware was cleaned with detergent, followed by ultrapure water, and finally rinsed with solvents and dried in a hot air oven. A standard 16 PAHs mixture in benzene/methylene chloride solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) containing 2000 μg mL−1 of each component was purchased from Aladdin Co. (CA, USA). Hexane, acetone, acetonitrile, and methylene chloride were all HPLC grade and obtained from Sigma-Aldrich (St. Louis, MO, USA). Anhydrous sodium sulfate (Merck, India) was cleaned with solvents in a Soxhlet apparatus, dried at 110 °C for 3 h and stored in a sealed desiccator before use. Silica gel (100–200 mesh) was purchased from Supelco (Sigma-Aldrich, USA) and activated at 105 °C for 2 h before use. SPE columns (ProElut C18, 1 g/6 mL) used for purification were obtained from Dikma Technologies (Beijing, China). Ultra-high-quality water was produced by a Milli-Q water purification system (Millipore, Madrid, Spain).
:1, v/v) containing 2000 μg mL−1 of each component was purchased from Aladdin Co. (CA, USA). Hexane, acetone, acetonitrile, and methylene chloride were all HPLC grade and obtained from Sigma-Aldrich (St. Louis, MO, USA). Anhydrous sodium sulfate (Merck, India) was cleaned with solvents in a Soxhlet apparatus, dried at 110 °C for 3 h and stored in a sealed desiccator before use. Silica gel (100–200 mesh) was purchased from Supelco (Sigma-Aldrich, USA) and activated at 105 °C for 2 h before use. SPE columns (ProElut C18, 1 g/6 mL) used for purification were obtained from Dikma Technologies (Beijing, China). Ultra-high-quality water was produced by a Milli-Q water purification system (Millipore, Madrid, Spain).
2.2. Sample pretreatment
:2, v/v) were mixed in a conical flask and sonicated for 20 min at 70 kHz. All the mixtures above were subsequently centrifuged at 3000 rpm for 5 min, and the supernatants decanted and collected. The residues were extracted again with another 20 mL of their corresponding extraction solvent. The combined supernatants were concentrated by a rotary evaporator at 35 °C to around 1 mL for further purification.:1, v/v). Both eluents were combined and evaporated to near dryness in a rotary evaporator at 35 °C under vacuum. Seed extracts were cleaned up using C18 columns before purification on silica gel. About 1 mL of the seeds extract was directly transferred onto a C18 column pre-activated with 4 mL methanol and acetonitrile and eluted twice with 8 mL acetonitrile/acetone (3:2, v/v). The eluent was collected and the solvent was changed into methylene chloride, before further cleaning up on a homemade silica gel column, as described above. After removing solvents, the residue obtained was dissolved in 1 mL of acetonitrile and filtered through 0.45 μm polytetrafluoroethylene (PTFE) filters, and injected for HPLC-UV-FLD analysis.2.3. PAHs analysis
PAHs analysis was carried out using a Shimadzu HPLC apparatus equipped, in series, with a LC-20AD pump, SPD-20A UV-Vis detector, and RF-20A fluorescence detector. Chromatographic separation was performed on a LC-PAH Supelcosil™, an analytical column (25 cm × 4.6 mm, 5 μm film) using a 20 μL sample injection volume and a gradient flow using acetonitrile (A) and water (B) at 1.0 mL min−1. The gradient elution program was as follows: 0 → 6 min, isocratic 56% A; 6 → 17 min, linear gradient 56% A → 88% A; 17 → 38 min, linear gradient 88% A → 86% A; 38 → 39 min, linear gradient 86% A → 56% A; 39 → 45 min, isocratic 56% A. The temperature in the column oven was set to 35 °C. All 16 PAHs were determined using a UV-Vis detector set at 229 nm and the fluorescence detector with variations in the wavelength excitement (Ex) and emission (Em) as follows: (1) NA – Ex/Em 270/324, (2) AC – Ex/Em 270/324, (3) FL – Ex/Em 270/324, (4) PHE – Ex/Em 248/375, (5) AN – Ex/Em 248/375, (6) FA – Ex/Em 280/462, (7) PY – Ex/Em 270/385, (8) BaA – Ex/Em 270/385, (9) CHR – Ex/Em 270/385, (10) BbFA – Ex/Em 256/446, (11) BkFA – Ex/Em 292/410, (12) BaP – Ex/Em 292/410, (13) DBahA – Ex/Em 292/410, (14) BghiP – Ex/Em 292/410, and (15) IP – Ex/Em 302/507. Before analyzing a sample set, blank solvent was injected to insure that the system was free of contaminants or interfering peaks.2.4. Qualitation and quantification
The qualitation of PAHs in TCMs was achieved by comparing their retention times with those of standard PAHs. The external standard plot method was used for quantification. The stock solution containing 2000 μg mL−1 of all 16 PAHs was diluted successively in acetonitrile to construct an analytical curve, and each standard solution was prepared in duplicate.The limits of detection (LOD) and quantification (LOQ) of the method were calculated from the signal-to-noise (S/N) ratio of standard solutions. The LOD corresponded to the amount of analyte for which the S/N ratio of the peak area was equal to 3, while the LOQ corresponded to an S/N ratio of 10.
2.5. Recovery study
Recovery experiments were performed by spiking four representative samples (Rhizoma dioscoreae, Mulberry leaves, Fructus mume, and Fructus cannabis) from four TCM categories with three different concentrations of the standard PAHs solution (10, 50, and 100 μg kg−1). To ensure the proposed method was accurate for every TCM selected, the other selected TCMs were also spiked with 50 μg kg−1 of the standard solution to conduct the recovery study. The spiked samples and the unspiked controls were analyzed in triplicate. Recoveries were calculated from the differences in total amounts of each PAH between the spiked and unspiked samples.The repeatability was estimated for all PAHs during the recovery studies and expressed as the relative standard deviation (n = 3). The intra-day precision was determined by analysing the same standard mixture (100 μg kg−1 for each PAH) six times on the same day with the same instrument and the same operator, while the inter-day precision was calculated on the basis of the results from two different days and from the different operators. The result was also expressed as the relative standard deviation.
2.6. Source apportionment
Ratios of individual PAHs, such as the diagnostic ratios of AN/(AN + PHE), FA/(FA + PY), and BaA/(BaA + CHR), are frequently utilized as diagnostic tools to distinguish the PAH sources.16–18Moreover, principal component analysis (PCA) can be used to analyze the sources of PAHs. PCA is known as a dimensional reduction because the method is able to decrease the dimensionality of the primary set of data (measured PAH contents in TCM samples) and compress data into a lower dimensional matrix (principal components).19 By utilizing the orthogonal transformation method, principle components (PCs) were extracted with different factor loadings indicating correlations between each pollutant species and each PC.16,20 Each PC was further evaluated and recognized by source markers or profiles as reasonable pollution sources.
3. Results and discussion
3.1. Optimization of sample pretreatment
Four representative TCMs (Rhizoma dioscoreae, Mulberry leaves, Fructus mume, and Fructus cannabis) were chosen from four categories of TCM medicinal parts and used to conduct the optimization of extraction methods, including ultrasonic extraction, homogenization extraction, and oscillation extraction. The total yields of 16 PAHs (Σ16PAHs) and their average recoveries (by spiking 50 μg kg−1 for each PAH) were adopted to evaluate the methods. The optimization results are shown in Table S1.† For seeds, roots, and stems, it was observed that ultrasonic extraction had the highest total yield and average recovery, while oscillation and homogenization extraction were more suitable for fruits and leaves/flowers, respectively.
The solvents used to extract PAHs from plant samples were usually hexane, acetone, methylene chloride, acetonitrile, or a mixture thereof. Weak polar solvents such as hexane and methylene chloride were used in the experiment owing to the low polarity of PAHs and less polar interferences. However, for seeds, polar solvents acetonitrile and acetone were used, in order to reduce the extraction of fats in the sample and coordinate with subsequent C18 purification without exchanging solvents. Different parts of medicinal plants have different matrix categories, so different solvent systems should be used and optimized to ensure full extraction and high recoveries. For seeds, three solvents, including acetone, acetonitrile, and acetonitrile/acetone (3:2, v/v), were compared using the representative sample, Fructus cannabis. For the other groups, hexane, methylene chloride, and hexane/methylene chloride (1:1, v/v) were investigated to seek proper solvents (see Table S2†). In addition, different extraction times were also studied for every extraction method (see Table S3†). The final established extraction conditions were as described above.
Standard solution (1 mL, 200 ng mL−1) was transferred to pre-activated silica gel and C18 columns to conduct optimization experiments on choosing the proper eluent and eluent volume by comparing the total yield of 16 PAHs. As shown in Fig. S1 and S2,† hexane/methylene chloride (20 mL; 1:1, v/v) was a suitable eluent for silica gel and was enough for 8 mL acetonitrile/acetone (3:2, v/v) to wash away all PAHs attracted to the C18 column.
3.2. Method validation
HPLC elution conditions were optimized to ensure the good separation of all 16 PAHs; the chromatogram is shown in Fig. 1. In order to detect all 16 PAHs and obtain the best detection limit, fluorescence and UV detectors were connected in series. ACL was inactive in fluorescence and detected by UV at 229 nm, whereas the other 15 PAHs were detected by fluorescence detector. | ||
| Fig. 1 Chromatogram of the standard solution monitored by both (a) UV and (b) fluorescence detection (peaks identification: 1, NA; 2, ACL; 3, AC; 4, FL; 5, PHE; 6, AN; 7, FA; 8, PY; 9, BaA; 10, CHR; 11, BbFA; 12, BkFA; 13, BaP; 14, IP; 15, DBahA; and 16, BghiP). | ||
The calibration curve was obtained by regression of the peak area with standard solution concentration. As shown in Table 1, all calibration curves were highly linear (with correlation coefficient R2 ≥ 0.9994) in the range of concentration examined.
| PAH | Regression equation | Correlation coefficient (R2) | Linearity range (μg kg−1) | LOD (μg kg−1) | LOQ (μg kg−1) |
|---|---|---|---|---|---|
| NA | Y = 5.489 × 104X + 3.522 × 104 | 0.9994 | 0.5–1000 | 0.030 | 0.100 |
| ACL | Y = 2.201 × 10X − 3.647 × 10 | 0.9998 | 2.0–1000 | 0.300 | 1.000 |
| AC | Y = 1.031 × 105X − 5.876 × 104 | 0.9998 | 0.1–1000 | 0.015 | 0.050 |
| FL | Y = 3.673 × 105X − 2.364 × 105 | 0.9999 | 0.1–1000 | 0.015 | 0.050 |
| PHE | Y = 1.300 × 105X − 2.979 × 103 | 1.0000 | 0.1–1000 | 0.015 | 0.050 |
| AN | Y = 3.104 × 105X + 1.450 × 105 | 0.9999 | 0.1–1000 | 0.015 | 0.050 |
| FA | Y = 4.804 × 104X − 2.276 × 104 | 0.9996 | 0.5–1000 | 0.030 | 0.100 |
| PY | Y = 2.447 × 105X + 3.085 × 104 | 1.0000 | 0.5–1000 | 0.030 | 0.100 |
| BaA | Y = 2.683 × 105X + 1.515 × 105 | 0.9998 | 0.5–1000 | 0.030 | 0.100 |
| CHR | Y = 3.435 × 105X + 6.308 × 103 | 0.9999 | 0.5–1000 | 0.030 | 0.100 |
| BbFA | Y = 1.090 × 105X + 9.449 × 103 | 1.0000 | 0.5–1000 | 0.030 | 0.100 |
| BkFA | Y = 5.586 × 105X − 4.118 × 103 | 0.9999 | 0.5–1000 | 0.030 | 0.100 |
| BaP | Y = 5.431 × 105X + 2.681 × 105 | 1.0000 | 0.5–1000 | 0.030 | 0.100 |
| IP | Y = 1.884 × 105X − 4.345 × 104 | 0.9999 | 0.5–1000 | 0.030 | 0.100 |
| DBahA | Y = 1.989 × 105X − 9.106 × 104 | 0.9999 | 0.5–1000 | 0.030 | 0.100 |
| BghiP | Y = 1.644 × 104X + 3.417 × 103 | 0.9999 | 0.5–1000 | 0.030 | 0.100 |
The interval for the limit of detection (LOD) for all 16 PAHs was from 0.015 to 0.300 μg kg−1, while the limit of quantification (LOQ) ranged from 0.050 to 1.000 μg kg−1. All LODs were below 0.030 μg kg−1, except for ACL (0.300 μg kg−1), which was 10 times higher than the other PAHs, explained by the UV sensitivity being much lower than FLD. Compared with LOD values from previous reports,9,10,25 it was clear that the proposed method had sufficient sensitivity for the determination of PAHs in TCMs.
Recovery experiments were carried out by adding 10, 50, and 100 μg kg−1 of each analyte standard to four representative samples. The results are shown in Table 2. Overall, the spiked recoveries ranged from 66.7 to 97.5% for all PAHs, indicating that the method accuracy was satisfactory. Moreover, to ensure the method was accurate for every sample tested in this study, the other TCMs selected were also spiked with 50 μg kg−1 standard solution to conduct recovery studies. The results, presented in Table S4,† ranged from 69.5 to 108.2%, confirming good method accuracy for all TCM samples involved in the study. The relative standard deviations (RSDs) did not exceed 10% in any instance. The repeatability, and intra and inter-day precision results are also shown in Table 2; all were below 8% (RSD), demonstrating the high repeatability and precision of the method.
| PAHs | Recoveries (RSD) (%) | Repeatability/inter-day precision (2 d)/intra-day precision (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rhizoma dioscoreae | Mulberry leaves | Fructus mume | Fructus cannabis | ||||||||||
| Lowa | Mediumb | Highc | Low | Medium | High | Low | Medium | High | Low | Medium | High | ||
| a Low level was prepared by spiking the sample with 10 μg kg−1 of each PAH.b Medium level was prepared by spiking the sample with 50 μg kg−1 of each PAH.c High level was prepared by spiking the sample with 100 μg kg−1 of each PAH. | |||||||||||||
| NA | 72.3 (7.5) | 68.8 (8.9) | 74.1 (1.2) | 75.6 (2.8) | 79.2 (4.8) | 85.4 (2.3) | 72.6 (4.3) | 92.5 (4.7) | 94.5 (6.4) | 78.5 (3.5) | 79.5 (9.0) | 84.3 (7.4) | 3.5/2.9/4.1 |
| ACL | 66.7 (7.4) | 72.4 (7.6) | 81.6 (4.4) | 69.5 (2.6) | 83.3 (6.8) | 87.4 (1.0) | 87.5 (2.5) | 89.5 (4.0) | 96.2 (3.8) | 80.4 (6.0) | 85.5 (5.4) | 87.4 (2.9) | 2.9/3.8/3.2 |
| AC | 84.2 (5.2) | 72.6 (7.2) | 69.5 (4.0) | 80.5 (4.1) | 85.4 (3.7) | 82.4 (1.8) | 79.5 (4.9) | 82.7 (5.0) | 90.4 (4.1) | 79.5 (9.8) | 81.6 (7.8) | 80.5 (2.5) | 2.3/1.1/1.7 |
| FL | 76.5 (7.0) | 79.9 (4.0) | 80.4 (3.8) | 84.8 (7.5) | 88.9 (8.2) | 91.1 (2.6) | 76.5 (5.2) | 84.9 (5.2) | 94.2 (3.6) | 84.6 (2.8) | 84.5 (5.1) | 90.3 (4.3) | 1.9/4.3/2.8 |
| PHE | 85.5 (2.9) | 83.7 (5.6) | 90.2 (8.7) | 90.6 (6.0) | 87.5 (8.0) | 97.4 (3.0) | 92.4 (4.5) | 93.7 (3.4) | 93.3 (5.2) | 87.5 (3.6) | 85.7 (6.3) | 94.0 (7.4) | 5.4/6.9/5.2 |
| AN | 74.8 (5.5) | 83.6 (8.0) | 88.4 (6.6) | 92.3 (6.2) | 85.2 (6.5) | 95.2 (8.8) | 90.5 (5.8) | 94.5 (7.7) | 95.7 (5.1) | 89.5 (1.0) | 85.6 (5.1) | 95.5 (6.5) | 7.2/6.4/3.1 |
| FA | 81.4 (3.0) | 85.6 (2.7) | 85.3 (7.3) | 79.3 (5.9) | 81.4 (6.0) | 88.3 (8.0) | 80.8 (6.2) | 97.5 (3.2) | 94.1 (6.6) | 87.2 (2.6) | 89.7 (8.4) | 89.0 (3.7) | 3.8/3.6/5.2 |
| PY | 78.8 (3.5) | 85.4 (1.9) | 86.2 (7.0) | 92.4 (2.6) | 90.6 (6.2) | 89.5 (6.7) | 85.5 (4.4) | 87.6 (3.0) | 90.5 (8.0) | 86.5 (3.1) | 90.9 (9.9) | 89.8 (3.5) | 1.4/5.5/4.8 |
| BaA | 79.4 (3.8) | 83.4 (1.3) | 87.9 (2.9) | 84.8 (4.9) | 86.4 (5.9) | 90.4 (4.1) | 91.5 (1.1) | 89.5 (6.5) | 89.4 (4.5) | 89.6 (1.4) | 95.7 (7.1) | 85.3 (6.4) | 3.7/2.5/4.3 |
| CHR | 81.5 (8.8) | 83.8 (7.6) | 93.4 (5.7) | 81.4 (7.8) | 80.9 (2.7) | 96.3 (2.9) | 87.8 (1.1) | 93.5 (6.8) | 91.5 (4.3) | 90.4 (1.9) | 93.3 (2.9) | 83.8 (5.4) | 3.4/2.8/2.8 |
| BbFA | 89.5 (4.2) | 86.4 (5.9) | 84.6 (7.0) | 78.9 (5.4) | 88.7 (8.9) | 87.9 (6.5) | 83.2 (4.9) | 84.9 (8.3) | 92.7 (4.7) | 92.2 (7.4) | 92.7 (3.0) | 93.5 (5.0) | 2.6/3.5/2.9 |
| BkFA | 81.9 (6.0) | 87.3 (5.3) | 88.6 (6.4) | 79.4 (1.8) | 85.6 (7.4) | 82.5 (6.2) | 84.3 (4.8) | 89.2 (7.5) | 89.5 (6.2) | 87.4 (6.5) | 88.5 (5.0) | 90.3 (5.1) | 4.3/6.7/5.6 |
| BaP | 82.1 (5.1) | 77.5 (2.8) | 89.6 (8.5) | 82.4 (4.6) | 87.4 (8.0) | 94.4 (7.0) | 84.9 (7.5) | 85.4 (8.7) | 92.7 (5.9) | 85.9 (5.3) | 84.6 (5.1) | 81.4 (3.9) | 3.0/2.7/1.0 |
| IP | 89.7 (9.2) | 89.4 (9.2) | 92.4 (6.6) | 85.5 (4.0) | 83.3 (2.9) | 90.0 (5.4) | 87.2 (4.2) | 97.4 (3.4) | 84.6 (7.1) | 78.8 (6.8) | 87.3 (4.2) | 87.6 (4.4) | 4.5/2.0/1.8 |
| DBahA | 78.9 (9.0) | 81.3 (4.3) | 81.9 (2.9) | 86.3 (5.0) | 89.7 (4.7) | 88.9 (6.4) | 82.5 (2.7) | 85.7 (6.0) | 90.5 (9.0) | 80.5 (4.4) | 85.4 (3.5) | 85.5 (6.4) | 5.2/3.0/2.6 |
| BghiP | 74.5 (7.1) | 79.7 (3.7) | 81.8 (3.3) | 82.9 (5.9) | 92.3 (5.2) | 82.9 (4.5) | 81.7 (3.1) | 86.2 (8.8) | 83.3 (8.3) | 76.9 (3.9) | 87.8 (4.8) | 93.7 (4.8) | 6.6/5.2/5.9 |
3.3. Application to real samples
After validation, the proposed method was applied to evaluate the 16 PAHs contamination in 32 TCMs, including six roots and stems, eight fruits, eight seeds, five flowers, and five leaves. Table S5† shows the results obtained, expressed as the mean of three replications ± standard deviation (μg kg−1). Concentrations below the limit of detection were considered as not detected (nd).All 32 TCMs were contaminated with some PAHs, but with a large variability in each PAH level in different samples, which might be attributed to different PAH sources for each sample. The total levels of the 16 PAHs varied from 19.5 μg kg−1 (Fructus rubi) to 1614.1 μg kg−1 (Radix liquiritiae), with an average of 376.8 μg kg−1. Among all PAHs studied, PHE was the most common and serious contaminant (found in 32 of 32 samples; 100%), followed by FL (31 samples; 97%) and FA (31 samples; 97%). Similar results were found for PHE and FA in some fruit and herbal teas,26 for PHE, FA, ACL, FL and PY in 24 Chinese herbal medicines,9 and for PY, FA and NA in tea products and crude drugs.5 Furthermore, BghiP was detected only in Radix liquiritiae (4.8 μg kg−1), which had the lowest detection rates among the 16 PAHs.
The highest concentration of any PAH in the studied medicinal plants was that of PHE in Radix liquiritiae (586.4 μg kg−1). Radix liquiritiae was also the only TCM in which all 16 PAHs were detected, the total content of PAHs reaching 1614.1 μg kg−1. Cui et al.9 reported a similar result of 1842.8 μg kg−1, indicating that Radix liquiritiae may be vulnerable to PAH contaminants and that special attention should be paid to improve its quality. In particular, BaP, one of the most potent carcinogenic PAHs, was detected in 8 of 32 samples. Its contents in 8 samples ranged from 10.3 to 32.8 μg kg−1, all of which exceeded levels set for BaP by EU regulations for foods (2 μg kg−1).
3.4. Distribution analysis
Fig. 2 shows the distribution of mean values for the sums of 16 PAHs (Σ16PAHs), low molecular weight PAHs (ΣL-PAHs), and high molecular weight PAHs (ΣH-PAHs), as well as PAHs with the highest levels in the five medicinal parts of TCMs. Among the different medicinal parts, the highest level of Σ16PAHs was found in leaves (747.0 μg kg−1), followed by roots and stems (649.6 μg kg−1), seeds (255.7 μg kg−1), flowers (233.3 μg kg−1), and, finally, fruits (151.7 μg kg−1), which was consistent with the order for levels of ΣL-PAHs. However, for ΣH-PAHs, the highest levels were found in roots and stems, followed sequentially by leaves, seeds, flowers, and fruits. Moreover, the highest levels found were for PHE in leaves (236.8 μg kg−1), seeds (50.3 μg kg−1), roots and stems (180.0 μg kg−1), for NA in fruits (45.1 μg kg−1), and for ACL in flowers (45.7 μg kg−1). | ||
| Fig. 2 Distribution of total levels of 16 PAHs, light PAH levels, and heavy PAH levels, as well as PAHs with the highest contents in the different medicinal parts of TCMs. | ||
Leaves had the highest levels of Σ16PAHs and ΣL-PAHs among medicinal parts, which might be related to leaves taking longer time to grow and having a greater surface area than other parts, thus resulting in a longer exposure to PAHs and higher accumulation of PAHs.25,27 Flowers also have a high surface area, but the contamination was not as serious as in leaves due to their shorter growth cycle. Compared with fruits, the seeds, most of which were rich in fat, had higher levels of Σ16PAHs, ΣL-PAHs, and ΣH-PAHs. This could be explained by the lipophilic compounds contributing to the accumulation of hydrophobic PAHs in seeds and, consequently, causing a higher PAH contamination level.26 Roots and stems had the highest level of ΣH-PAHs and the second highest level of Σ16PAHs; possible reasons for this are soil-to-root transfer and atmosphere-to-plant pathway,27 or the different forms of processing to which the samples were submitted, such as the drying process. This will be discussed further in the next section (“Source analysis”).
From Table S5† and Fig. 2, it was obvious to see that low molecular weight PAHs, accounting for 45.8–94.9% of total PAHs with a mean value of 77.0%, predominated over high molecular weight PAHs in all TCM groups. The percentage of heavy PAHs, for which genotoxic, mutagenic, and carcinogenic properties have been stated, among all PAHs was generally low. Similar results have also been observed in the literature for tea samples,13 fruit and herbal teas samples,26 and in tea and coffee samples.28
3.5. Source analysis
PAHs originate from both the natural as well as anthropogenic sources, and anthropogenic sources were the main ones. Anthropogenic sources of PAHs can be categorized into pyrogenic and petrogenic groups.29 With pyrogenic sources, PAHs are generated by combustion of fossil fuel (coal and petroleum) and biomass. The petrogenic origin was from discharge of crude oil and petroleum products.30 This research focuses on the ratio method to conduct qualitative analysis of the sources of PAHs.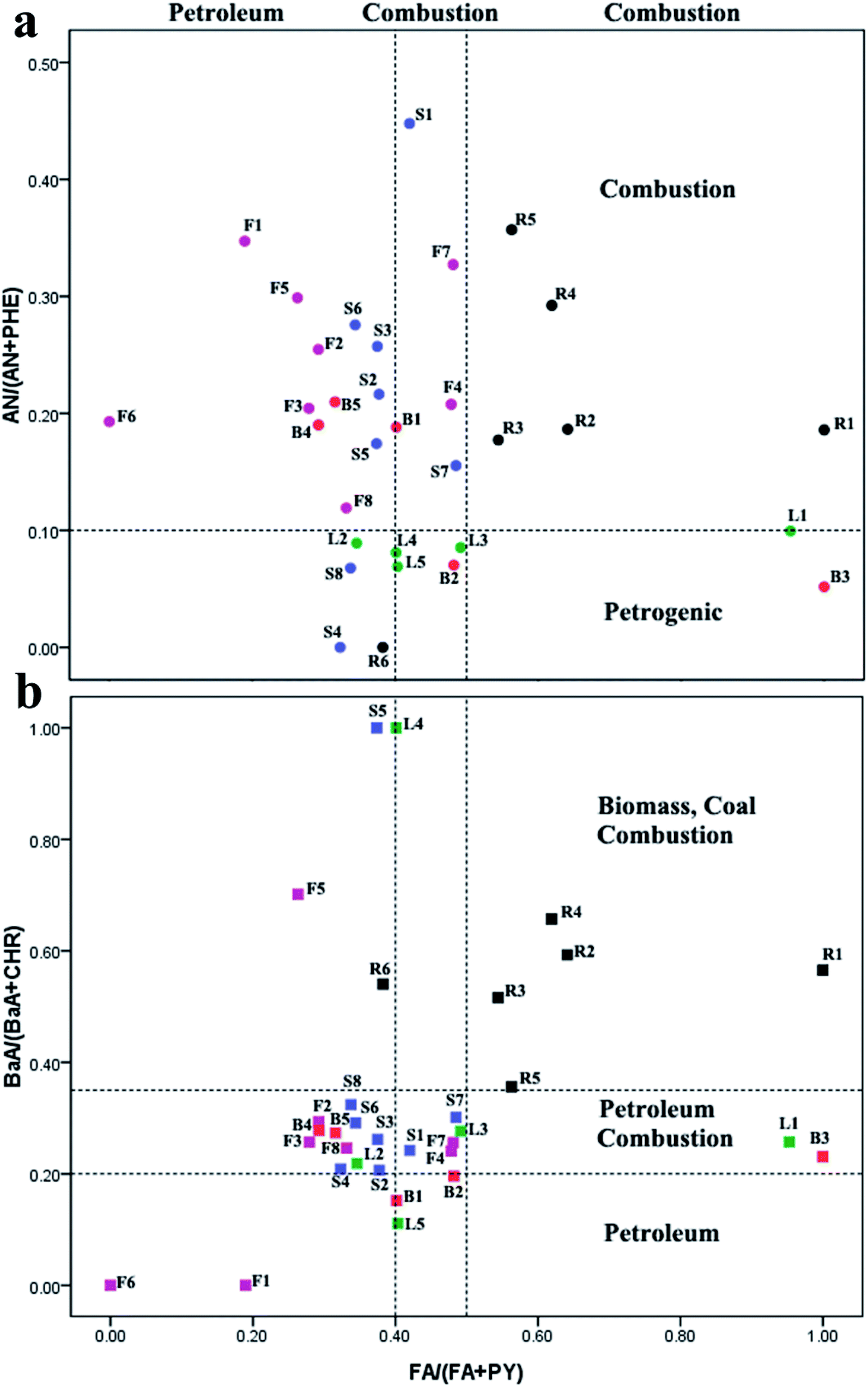 | ||
| Fig. 3 Cross plots for PAH diagnostic ratios in selected TCMs (a) AN/(AN + PHE) vs. FA/(FA + PY), (b) BaA/(BaA + CHR) vs. FA/(FA + PY). | ||
It is noteworthy that, in most root and stem samples (R1–R5), the ratios of FA/(FA + PY), AN/(AN + PHE), and BaA/(BaA + CHR) were all more than 0.5, 0.1, and 0.35, respectively. These ratios strongly implied sources from grass, wood, or coal combustion, which was consistent with the fact that most roots and stems are dried using combustion gases from burning wood or coal, and that the type of wood has a different influence on the PAH levels produced.32
| Species | PCA factor loadings | ||||
|---|---|---|---|---|---|
| Factor 1 | Factor 2 | Factor 3 | Factor 4 | Factor 5 | |
| a Bold loadings >0.53. | |||||
| NA | 0.176 | −0.122 | 0.473 | 0.752a | 0.192 |
| ACL | 0.724 | −0.144 | 0.322 | −0.328 | 0.199 |
| AC | 0.525 | −0.122 | 0.600 | 0.303 | −0.165 |
| FL | 0.618 | −0.433 | 0.421 | −0.338 | −0.158 |
| PHE | 0.897 | −0.240 | 0.065 | 0.101 | 0.243 |
| AN | 0.857 | 0.049 | −0.269 | 0.048 | 0.108 |
| FA | 0.943 | −0.158 | −0.159 | −0.070 | 0.044 |
| PY | 0.707 | −0.389 | 0.110 | −0.236 | −0.391 |
| BaA | 0.396 | 0.658 | 0.410 | −0.262 | 0.024 |
| CHR | 0.167 | 0.682 | 0.278 | −0.321 | 0.391 |
| BbFA | 0.655 | 0.520 | −0.089 | 0.094 | −0.302 |
| BkFA | 0.625 | 0.161 | −0.182 | −0.008 | −0.227 |
| BaP | 0.418 | 0.553 | 0.012 | 0.459 | −0.306 |
| IP | 0.885 | 0.195 | −0.242 | −0.097 | 0.036 |
| DBahA | 0.695 | −0.120 | −0.249 | 0.211 | 0.533 |
| BghiP | 0.866 | −0.095 | −0.347 | 0.120 | −0.091 |
| Contribution (%) | 45.848 | 12.736 | 9.419 | 8.830 | 6.480 |
| Possible source | Combustion (coal/wood) traffic emission | Vehicle exhaust | Petrogenic source | Petrogenic source | Coal combustion |
Factor 1 explained the total variance of 45.848% in the data, and was strongly related to ACL, AC, FL, PHE, AN, and FA. PHE and FA have generally been attributed to coal combustion,33,34 and ACL, AC and AN have been identified as tracers for PAH compounds emitted by grass or wood combustion.35,36 Additionally, this factor was also composed of high molecular weight PAHs with 4–6 rings, such as PY, BbFA, BkFA, IP, DBahA, and BghiP, and are basically known to be derived from the traffic emissions.36,37 BghiP has been identified as a tracer of gasoline emissions. IP and BkFA have been found in gasoline vehicle soot, and both gas and diesel engine emissions.38–40 Therefore, factor 1 was selected to represent mixed coal, wood combustion, and traffic emissions.
Factor 2 was responsible for 12.736% of the total variance, and was predominately composed of BaA, CHR, and BaP, which were indicative of diesel-powered vehicles sources.41 This factor could be the vehicle exhaust source category.
Factors 3 and 4 accounted for 9.419% and 8.830% of the total variance, respectively, with high loading values of AC and NA respectively, both suggesting petrogenic source.29,42 Factor 5 contributed 6.480% to the total variance, containing only one highly loaded component, DBahA, which might be related to coal combustion.43
Generally, the results of PCA analyses were in concordance with the evidence from the diagnostic ratios of PAHs, which revealed a mixture of pyrogenic and petrogenic-derived PAHs. Vehicular emissions, petrogenic sources, wood and coal combustion may be responsible for the PAHs found in TCMs in the present study.
4. Conclusion
Different extraction methods (ultrasonic extraction, homogenization extraction, and oscillation extraction) were adopted according to the different matrices contained in the different medicinal parts of TCMS, followed by silica gel cleanup and HPLC-UV-FLD detection for the analysis of PAHs in TCMs. The proposed method was proved to be easy, cheap, effective, and accurate, and was successfully applied to determine PAHs in 32 TCMs of different types, including roots and stems, leaves, flowers, seeds, and fruits. The contamination of PAHs in TCMs seemed especially serious in leaves, roots, and stems. All 32 TCMs were contaminated with some PAHs, but with a large variability concerning PAHs distribution in different samples or medicinal parts. PAHs diagnostic ratios and PCA analysis indicated that PAHs in TCMs were from both pyrogenic and petrogenic sources. Wood or coal combustion could be responsible for the majority of PAHs found in roots and stems. The consumption of TCMs, especially by patients suffering chronic diseases, may be a long-term process and, thus, needs strict quality control while no legislation to regulate the contamination of TCMs with PAHs. The method presented in this study could be promoted as a useful tool for the quantification of PAHs contents in TCMs. The data obtained could also provide valuable information for further studies of the maximum residue limits and potential health risk assessment.Acknowledgements
We thank Lianju Ma (College of Pharmacy, Chongqing Medical University) for the assistance in HPLC-FLD analysis.References
- Q. Yang, H. Chen and B. Li, PLoS One, 2015, 10, e0118141 Search PubMed.
- T. H. Kao, S. Chen, C. J. Chen, C. W. Huang and B. H. Chen, J. Agric. Food Chem., 2012, 60, 1380–1389 CrossRef CAS PubMed.
- T. Wenzl, R. Simon, E. Anklam and J. Kleiner, TrAC, Trends Anal. Chem., 2006, 25, 716–725 CrossRef CAS.
- R. Liu, Y. Dai and L. Sun, PLoS One, 2015, 10, e0120369 Search PubMed.
- A. Ishizaki, K. Sito and H. Kataoka, Anal. Methods, 2011, 3, 299–305 RSC.
- I. Hosbach, G. Neeb, S. Hager, S. Kirchhoff and B. Kirschbaum, Anaesthesia, 2003, 58, 282–283 CrossRef CAS PubMed.
- L. Veiga, H. Amorim, J. Moraes, M. Silva, R. Raices and S. Quiterio, Food Chem., 2014, 152, 612–618 CrossRef CAS PubMed.
- M. Fernández, S. Clavijo, R. Forteza and V. Cerdà, Talanta, 2015, 138, 190–195 CrossRef PubMed.
- Z. Cui, N. Ge, A. Zhang, Y. Liu, J. Zhang and Y. Cao, Anal. Bioanal. Chem., 2015, 407, 1989–1997 CrossRef CAS PubMed.
- L. Yu, Y. Cao, J. Zhang, Z. Cui and H. Sun, Food Addit. Contam., Part A, 2012, 29, 1800–1809 CrossRef CAS PubMed.
- K. Dost and C. İdeli, Food Chem., 2012, 133, 193–199 CrossRef CAS.
- H. Yin, Q. Tan, Y. Chen, G. Lv and X. Hou, Microchem. J., 2011, 97, 138–143 CrossRef CAS.
- D. Lin, Y. Tu and L. Zhu, Food Chem. Toxicol., 2005, 43, 41–48 CrossRef CAS PubMed.
- M. V. Russo, P. Avino, L. Perugini and I. Notardonato, RSC Adv., 2016, 6, 13920–13927 RSC.
- M. Surma, A. Sadowska-Rociek and E. Cieślik, Eur. Food Res. Technol., 2014, 238, 1029–1036 CrossRef CAS.
- R. M. Harrison, D. Smith and L. Luhana, Environ. Sci. Technol., 1996, 30, 825–832 CrossRef CAS.
- C. Bourotte, M.-C. Forti, S. Taniguchi, M. C. Bícego and P. A. Lotufo, Atmos. Environ., 2005, 39, 3799–3811 CrossRef CAS.
- M. B. Yunker, R. W. Macdonald, R. Vingarzan, R. H. Mitchell, D. Goyette and S. Sylvestre, Org. Geochem., 2002, 33, 489–515 CrossRef CAS.
- I. Primpas and M. Karydis, Environ. Monit. Assess., 2011, 178, 257–269 CrossRef CAS PubMed.
- R. K. Larsen and J. E. Baker, Environ. Sci. Technol., 2003, 37, 1873–1881 CrossRef CAS PubMed.
- B. Kumar, V. Verma, R. Gaur, S. Kumar, C. Sharma and A. Akolkar, Adv. Appl. Sci. Res., 2014, 5, 201–209 Search PubMed.
- M. W. Ashraf and A. Salam, Bull. Environ. Contam. Toxicol., 2012, 88, 543–547 CrossRef CAS PubMed.
- S. A. Tfouni, R. M. Machado, M. C. Camargo, S. H. Vitorino, E. Vicente and M. C. F. Toledo, Food Chem., 2007, 101, 334–338 CrossRef CAS.
- S. Singh and A. Vashishth, Environ. Monit. Assess., 2011, 177, 35–38 CrossRef CAS PubMed.
- H. Krajian and A. Odeh, Toxicol. Environ. Chem., 2013, 95, 942–953 CrossRef CAS.
- M. Ciecierska and M. Obiedzinski, Herba Pol., 2009, 55, 18–24 CAS.
- K. Srogi, Environ. Chem. Lett., 2007, 5, 169–195 CrossRef CAS.
- N. R. Bishnoi, U. Mehta, U. Sain and G. Pandit, Environ. Monit. Assess., 2005, 107, 399–406 CrossRef CAS PubMed.
- Z. Wang, M. Fingas, Y. Shu, L. Sigouin, M. Landriault, P. Lambert, R. Turpin, P. Campagna and J. Mullin, Environ. Sci. Technol., 1999, 33, 3100–3109 CrossRef CAS.
- Z. Ouyang, L. Gao, X. Chen, S. Yao and S. Deng, RSC Adv., 2016, 6, 71441–71449 RSC.
- A. Dvorská, G. Lammel and J. Klánová, Atmos. Environ., 2011, 45, 420–427 CrossRef.
- B. K. Larsson, Z. Lebensm.-Unters. Forsch., 1982, 174, 101–107 CrossRef CAS.
- U. M. Sofowote, B. E. McCarry and C. H. Marvin, Environ. Sci. Technol., 2008, 42, 6007–6014 CrossRef CAS PubMed.
- G.-L. Shi, Y.-Z. Tian, C.-S. Guo, Y.-C. Feng, J. Xu and Y. Zhang, J. Soils Sediments, 2012, 12, 1154–1163 CrossRef CAS.
- N. R. Khalili, P. A. Scheff and T. M. Holsen, Atmos. Environ., 1995, 29, 533–542 CrossRef CAS.
- Y. Zhang, C.-S. Guo, J. Xu, Y.-Z. Tian, G.-L. Shi and Y.-C. Feng, Water Res., 2012, 46, 3065–3073 CrossRef CAS PubMed.
- H.-Y. Chen, Y.-G. Teng and J.-S. Wang, Sci. Total Environ., 2012, 414, 293–300 CrossRef CAS PubMed.
- Y. Liu, L. Chen, Q.-H. Huang, W.-Y. Li, Y.-J. Tang and J.-F. Zhao, Sci. Total Environ., 2009, 407, 2931–2938 CrossRef CAS PubMed.
- M. P. Zakaria, H. Takada, S. Tsutsumi, K. Ohno, J. Yamada, E. Kouno and H. Kumata, Environ. Sci. Technol., 2002, 36, 1907–1918 CrossRef CAS PubMed.
- R. Boonyatumanond, M. Murakami, G. Wattayakorn, A. Togo and H. Takada, Sci. Total Environ., 2007, 384, 420–432 CrossRef CAS PubMed.
- P. Masclet, G. Mouvier and K. Nikolaou, Atmos. Environ., 1967, 1986(20), 439–446 Search PubMed.
- H. Soclo, P. Garrigues and M. Ewald, Mar. Pollut. Bull., 2000, 40, 387–396 CrossRef CAS.
- T. T. Dong and B.-K. Lee, Chemosphere, 2009, 74, 1245–1253 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24682f |
| ‡ Bao Yu and Dan Zhang contributed equally to this work; they are co-first authors. |
| This journal is © The Royal Society of Chemistry 2017 |