
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrophilic PAN based carbon nanofibres with improved graphitic structure and enhanced mechanical performance using ethylenediamine functionalized graphene†
Zhenyu Li,
Omid Zabihi,
Jinfeng Wang,
Quanxiang Li,
Jiemin Wang,
Weiwei Lei and
Minoo Naebe*
Institute for Frontier Materials, Deakin University, Victoria 3216, Australia. E-mail: minoo.naebe@deakin.edu.au; Tel: +61 403667708
First published on 12th January 2017
Abstract
Polyacrylonitrile (PAN) reinforced with nano-carbons such as graphene (Gr) and carbon nanotubes (CNTs) provides great opportunity for the development of low-cost and high-performance carbon materials. However, the poor dispersion and weak interaction between the carbon nanofillers and the surrounding PAN matrix prevent the final carbonized materials from reaching their full potential. Herein, we demonstrate a chemical approach using ethylenediamine (EDA) acting as a linker between graphene nanoplatelets and PAN for improved mechanical performance. The as-prepared CNFs exhibit a higher carbon yield and tensile modulus as well as improved graphitic structure compared to pristine PAN and PAN/Gr nanofibres. Furthermore, EDA can act as a N source for N-doping during the carbonization, enabling CNFs with hydrophilicity performance.
1. Introduction
Continuous carbon nanofibres (CNFs) derived from carbonized electrospun polymeric precursor nanofibres have received much attention in diverse fields such as catalysis,1 sensor,2 absorption/separation,3 and biomedical applications,4 driven by their high surface area, robust mechanical properties, excellent chemical resistance, and outstanding electrical/thermal conductivity.5 Among various polymeric precursors, polyacrylonitrile (PAN) is well-known to produce CNFs with fairly high carbon yield and tensile strength.6 However, most of the obtained CNFs still suffer a relatively low tensile modulus and modest electrical/thermal conductivity owing to the imperfection of the graphitic structure during the carbonization.The conventional method to improve the graphitic structures of carbon fibres is to increase the carbonization temperature,7,8 which requires specialized and expensive equipment, leading to high energy consumption and cost.9 Therefore, low-cost approaches for carbon fibres with improved graphitic structures and enhanced mechanical performance at relatively low carbonization temperature are of much interest. Nano-carbons such as carbon nanotubes (CNTs),10 graphene oxide (GO),11 and graphene (Gr),12 possess exceptional mechanical, electrical, thermal, and optical performances, making them appealing for the development of high performance composites and fibres.13,14 Taking the advantages of large surface area and long range graphitic structure, large interfacial area will be created between the carbon nanofillers and surrounding polymeric matrix. At this stage, the polymer chains around carbon nanofillers can be re-organized for several radii of gyration15 leading to improved graphitic structures during the carbonization.16 It has been shown that the graphitic structure of CNFs can be greatly improved at relative low carbonized temperature with the addition of low volume fractions of Gr or double wall carbon nanotubes.17 However, the dispersion of the nanofillers within the surrounding polymeric matrix is still quite poor due to the weak interactions between nanofiller and matrix, resulting in modest graphitic structure within the final CNFs. Although the dispersion of the carbon nanofillers can be greatly improved by surface oxidization, the intrinsic high oxygen contents of oxidized carbon nanofillers will lead to high weight loss during the carbonization, inhibiting their applications in fabricating CNFs with high carbon yield.
Ethylenediamine (EDA) has obtained particular attention in modifying carbon nanomaterials such as hydrogenated fullerenes,18 carbon nanotubes,19 and graphene20 via Benkeser hydrogenation. In the case of PAN matrix, the amine groups of EDA can also react with nitrile group of PAN to form the amine-terminated polymers.21 Thus the two amine groups on both sides of the ethylene can act as a linker between carbon nanofiller and PAN nitrile group. Prompted by such appealing performance of EDA, in this study, we demonstrated an effective approach to improve the dispersion and interaction between carbon nanofillers and PAN matrix using EDA. Our results show that the derived CNFs exhibited enhanced mechanical performance and graphitic order at relatively low carbonized temperature in contrast to those derived from PAN nanofibres and PAN/Gr composite nanofibres. What's more, EDA can also act as N source for N-doping during the carbonization, surprisingly changing the surface wettability.
2. Experimental section
2.1. Materials
Polyacrylonitrile (PAN: Mw ∼ 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Sigma-Aldrich) and N,N-dimethyl formamide (DMF, Sigma-Aldrich) were used as received. Ethylenediamine (EDA, Sigma-Aldrich) was dried by KOH (Sigma-Aldrich) for 24 h before use. Graphene nanoplatelet (grade C, surface area of ∼518 m2 g−1 as measured by BET) was supplied XG Sciences (Michigan, USA). Lithium chip (MTI) was stored in glove box under Ar atmosphere prior to use.
000, Sigma-Aldrich) and N,N-dimethyl formamide (DMF, Sigma-Aldrich) were used as received. Ethylenediamine (EDA, Sigma-Aldrich) was dried by KOH (Sigma-Aldrich) for 24 h before use. Graphene nanoplatelet (grade C, surface area of ∼518 m2 g−1 as measured by BET) was supplied XG Sciences (Michigan, USA). Lithium chip (MTI) was stored in glove box under Ar atmosphere prior to use.
2.2. Synthesis of Gr@EDA
Three lithium chips (∼99 mg) were dissolved in 80 mL of dry ethylenediamine (EDA), in a nitrogen-purged four-necked round-bottomed flask (250 mL) with vigorous stirring, at room temperature for ∼4 h. Then, 80 mg of Gr was added into the solution under vigorous stirring and nitrogen purge. After 24 h at 50 °C, the solution was quenched by bubbling air for 1 h. The suspension was filtered using a 0.2 μm polycarbonate membrane and washed with copious acetone, ethanol, distilled water, and acetone in turn. Gr@EDA samples were then kept in vacuum oven at 50 °C for 3 days before use.2.3. Grafting of Gr@EDA on PAN for FTIR-ATR measurement
In a typical process, 0.1 g of PAN was added into 20 mL of DMF in flask (100 mL). The solution was kept at 90 °C for 12 h under vigorous stirring and cooled down to room temperature. 0.1 g of Gr–EDA was added into 20 mL of DMF and sonicated for 3 h. Then the Gr–EDA/DMF solution was added into the PAN/DMF solution and stirred for 6 h, followed by heating to 70 °C for 24 h under vigorous stirring. At this stage, Gr@EDA will be grafted onto PAN. The PAN@Gr@EDA sample was obtained by dropping the PAN@Gr@EDA solution into an acetone coagulation bath through wet-spinning method. Acetone was used to rinse the samples for three times. Before the FTIR measurement, the Gr–EDA–PAN sample was kept in vacuum oven at 50 °C for 3 d.2.4. Preparation of the PAN@Gr@EDA and PAN/Gr solution for electrospinning
In a typical process, 1.1 g of PAN was added into 9 mL of DMF in vial (20 mL). The solution was then kept at 90 °C for 12 h under vigorous stirring and cooled down to room temperature. Gr@EDA (1 wt% respect to the PAN) was added into 1 mL of DMF and sonicated (Unisonics, Australia) for 3 h before adding it into the PAN/DMF solution. The resulting solution was stirred for 72 h in which the solution was sonicated for 6 h to disperse the Gr@EDA within PAN matrix. Then, the solution was heated up to 70 °C for 24 h under vigorous stirring to graft Gr@EDA on PAN. Gr/PAN sample was also prepared following the same protocol using 11% PAN/DMF solution containing 1% Gr.2.5. Electrospinning process
Electrospinning was conducted in air with the relative humidity (30–40%) with a set up described previously.22 The spinning solutions were loaded into a plastic syringe (23 gauge needle). The flow rate was fixed at 0.6 mL h−1 by a syringe pump (KDS 200, KD Scientific Inc.). A high-voltage (20 kV) and a grounded rotating drum was used (surface linear speed 12.8 m s−1). The distance between the tip and the collector was 21 cm.2.6. Thermal stabilisation and carbonization
The aligned nanofibre mats were heated to 250 °C from room temperature with the heating rate of 3 °C min−1 and kept for 2 h in air for thermal stabilisation. Consequently, the oxidised nanofibre mats were taken out of the oven and cooled down to the room temperature in air. The stabilised nanofiber mats were carbonised under N2 atmosphere using a tube furnace. Samples were heated to 650 °C or 850 °C (2 °C min−1) and dwell for 2 h.2.7. Characterization
TEM and SEM images were taken on a JEOL-2100 TEM with an acceleration voltage of 200 kV and a Supra 55VP SEM, respectively. X-ray diffraction was carried out with a Scintag XDS 2000 diffractometer with Cu Kα radiation. FTIR-ATR was performed using a Bruker Vertex 70 FTIR spectrometer in ATR mode with a resolution of 4 cm−1. Raman was used to characterize the graphitic structure of the carbonized samples via inVia Raman microscope from Renishaw with 514 nm laser as an excitation source. Tensile test was carried out using a Favimat instrument (Textechno, Monchengladbach, Germany). The gauge length for testing was fixed at 1 cm and the strain rate was 1.0 mm min−1. Thermogravimetric analyses (TGA) were carried out on a Pyris-1 thermogravimetric analyzer (Perkin Elmer, USA) with a heating rate of 10 °C min−1 under air or N2 atmosphere. Rheometer test was performed using a TA DHR 3 rheometer with cone–plate geometry at 25 °C. A cone with a diameter of 40 mm and a tilt angle of 2° was used, and gap width was set to be 50 μm. The solutions were placed between the cone and plate and were soaked for five minutes before testing. The contact angles were measured using a contact angle goniometer (KSV CAM 101). The water drop for the test was 5 mL in volume. X-ray photoelectron spectra (XPS) were recorded on an ESCLAB MKII using Al as the excitation source.3. Results and discussion
Benkeser hydrogenation process has been selected to graft EDA on graphene nanoplatelets,20,23 in which the lithium reacts with EDA firstly and generates the monolithium amid derivative (Li–EDA). Then Li–EDA will attack the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of Gr to form the EDA covalently bonded graphene (EDA@Gr). TEM, Raman and FTIR have been used to characterize the Gr and EDA@Gr (see ESI Fig. S1–S3†). From the TEM images (Fig. S1†), it can be seen that the overlap phenomenon of Gr diminished after EDA grafting, confirming that EDA grafting can improve the dispersion within ethanol solvent. From the Raman spectra (Fig. S2†), the reduced ID/IG ratio confirms destruction of the sp2 character and formation of the defects in commercial graphene nanoplatelets after EDA grafting. FTIR spectra (Fig. S3†) demonstrate two new and weak peaks at 2920 cm−1 and 2855 cm−1 corresponding to the typical C(sp3)–H stretching vibration, which is consistent with the previous studies.23 In addition, the presence of strong C–C stretching bands (1200 cm−1) and C–N stretching bands (1150 cm−1), confirm the presence of EDA on Gr. The chemical functionalization of Gr was further confirmed by TGA analysis (ESI Fig. S4†). A difference of ∼13% weight loss between pure Gr and EDA@Gr at 600 °C shows the thermal decomposition of EDA, confirming the presence of EDA on graphene nanoplatelets.
C of Gr to form the EDA covalently bonded graphene (EDA@Gr). TEM, Raman and FTIR have been used to characterize the Gr and EDA@Gr (see ESI Fig. S1–S3†). From the TEM images (Fig. S1†), it can be seen that the overlap phenomenon of Gr diminished after EDA grafting, confirming that EDA grafting can improve the dispersion within ethanol solvent. From the Raman spectra (Fig. S2†), the reduced ID/IG ratio confirms destruction of the sp2 character and formation of the defects in commercial graphene nanoplatelets after EDA grafting. FTIR spectra (Fig. S3†) demonstrate two new and weak peaks at 2920 cm−1 and 2855 cm−1 corresponding to the typical C(sp3)–H stretching vibration, which is consistent with the previous studies.23 In addition, the presence of strong C–C stretching bands (1200 cm−1) and C–N stretching bands (1150 cm−1), confirm the presence of EDA on Gr. The chemical functionalization of Gr was further confirmed by TGA analysis (ESI Fig. S4†). A difference of ∼13% weight loss between pure Gr and EDA@Gr at 600 °C shows the thermal decomposition of EDA, confirming the presence of EDA on graphene nanoplatelets.
The chemical bonds between EDA@Gr and PAN to form PAN@EDA@Gr has been confirmed by FTIR (ESI Fig. S4†). Before grafting, the characteristics bands of PAN at 2243 cm−1 and 1450 cm−1 corresponding to CN and CH vibrating bands can be seen. The peak observed at 2939 cm−1 can be assigned to CH stretching for PAN. Additionally, the characteristic peak of CO stretching of DMF at 1660 cm−1 (ref. 24) is observed. After grafting, a new peak at 1595 cm−1 corresponding to the CN vibrating band appeared confirming the chemical bonds between nitrile group of PAN and the amine group of EDA. At this stage, the two amine groups on both sides of the ethylene can be regarded as a linker between Gr and PAN nitrile group, simultaneously (Scheme 1).
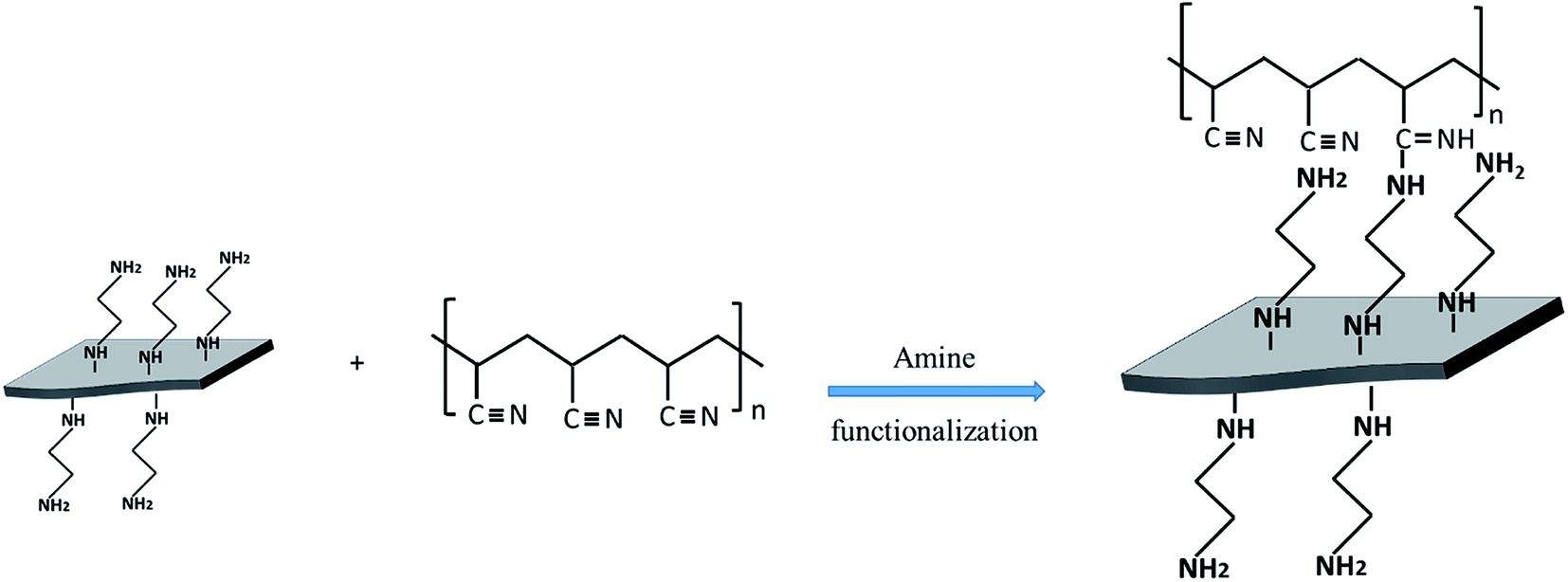 | ||
| Scheme 1 Mechanism of bonding between functionalised Gr and PAN. | ||
Fig. 1a–c present the typical SEM images of the electrospun PAN, PAN–Gr, and PAN@EDA@Gr nanofibres, respectively, indicating smooth PAN, PAN–Gr, and PAN–EDA–Gr fibres are produced via electrospinning route with the average diameter of 450 ± 20 nm, 420 ± 20 nm, and 380 ± 30 nm, respectively. The thinner diameters of the PAN–Gr is due to lower viscosity of the spun solution as a result of Gr inclusion. In contrast to the PAN–Gr, the diameter of PAN@EDA@Gr is thinner, implying the stronger interactions between PAN and Gr@EDA. To clearly see the distribution of Gr and Gr@EDA within the PAN fiber, TEM was used to characterize the samples (Fig. 1d–f). Different from PAN nanofibres, a darker core surrounded by a lighter skin structure can be detected from PAN–Gr and PAN@EDA@Gr nanofibres, confirming that Gr and EDA–Gr were incorporated inside the PAN fibres.
 | ||
| Fig. 1 SEM and TEM images of the (a and d) PAN, (b and e) PAN–Gr, and (c and f) PAN@EDA@Gr nanofibres, respectively. | ||
To better unveil the reason for smaller diameters of PAN–Gr and PAN@EDA@Gr, the viscosities of those three solutions have been measured as shown in Fig. 2, displaying that all solutions exhibit non-newtonian behaviours and that shear thinning phenomena is more conspicuous for pure PAN solution. Furthermore, the decrease in viscosity by adding Gr and EDA@Gr, means that more PAN chains have been absorbed on their surfaces,25 resulting in the less inter-entangled PAN chains within the solution and the weak shear thinning region at high shear force. The lowest viscosity of PAN@EDA@Gr in DMF solution, means the stronger interactions between PAN and EDA@Gr.
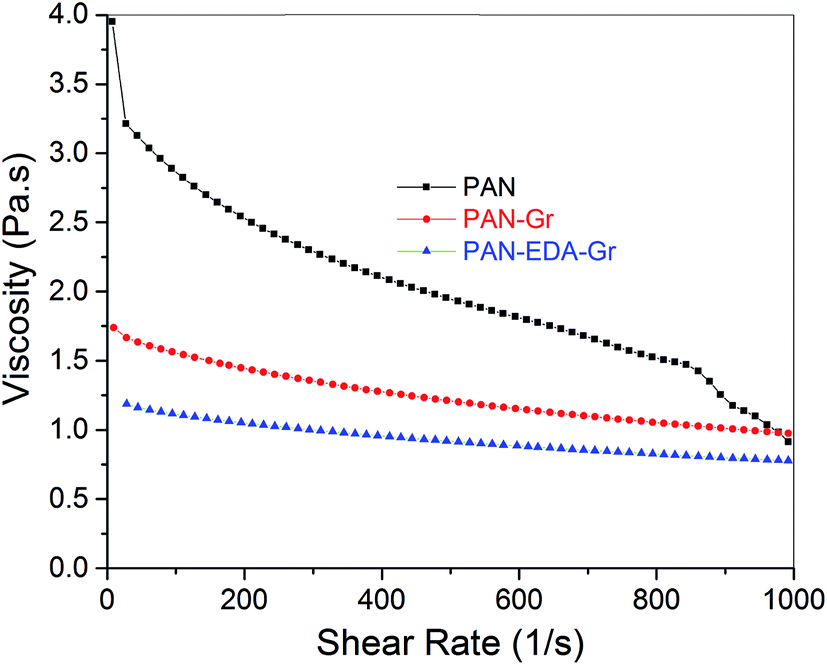 | ||
| Fig. 2 Viscosity versus shear rate for PAN, PAN–Gr, and PAN@EDA@Gr solutions in DMF solvent. Concentration of PAN/DMF in solutions was fixed at 1.1 g/10 mL. | ||
Fig. 3 shows the FTIR spectra in ATR mode of as-prepared nanofibres before and after stabilisation. Before the stabilisation, PAN distinct peaks at 2939, 2243, and 1454 cm−1 corresponding to CH stretching, CN and CH vibrating bands can be detected, respectively (Fig. 3a). After the stabilisation, a new peak at 1595 cm−1 assigned to conjugated CN stretching appeared due to the cyclization and dehydrogenation of the nanofiber, and the intensity of CN vibrating band (2243 cm−1) in stabilized nanofibres reduced.26 From Fig. 3b, the degree of stabilisation or extent of reaction [EOR = I1595/(I1595 + I2242)] can be quantified using the intensity of the peak at 1595 and 2243 cm−1, respectively. As expected, the PAN–Gr and PAN@EDA@Gr nanofibres exhibited higher EOR value of ∼0.94 and ∼0.95, respectively. But EOR of pristine PAN is only ∼0.92 at 250 °C in air for 3 h.
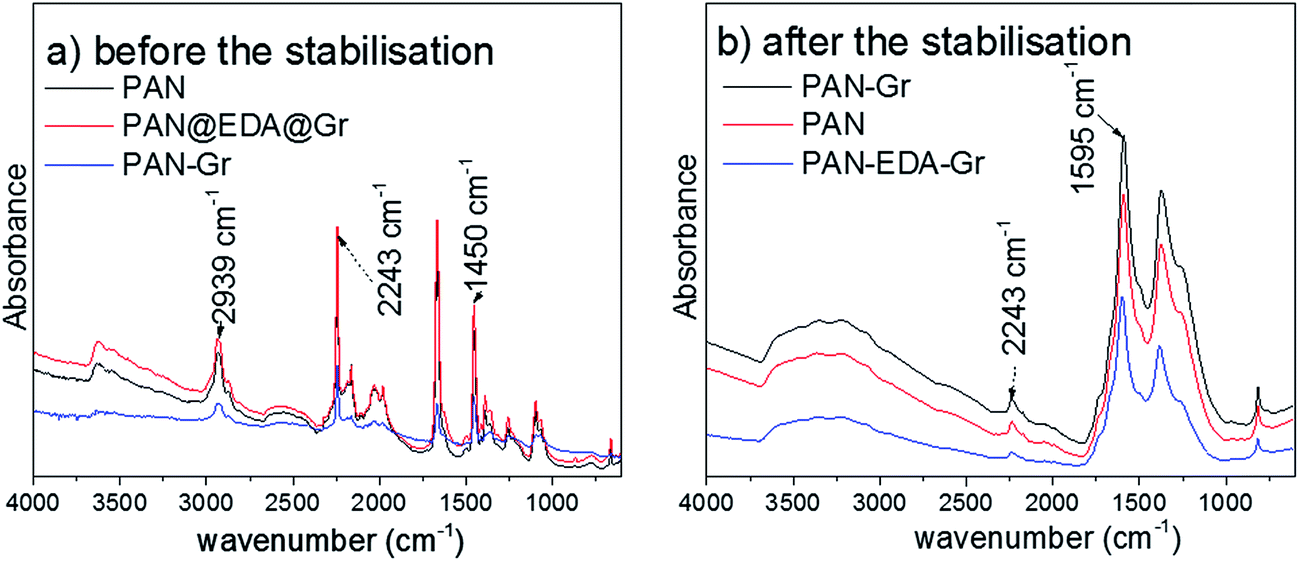 | ||
| Fig. 3 ATR infrared spectra of as-prepared fibres before (a) and after (b) thermal stabilization at 250 °C in air for 3 h. | ||
Morphologies of nanofiber samples under different carbonized temperature i.e. 650 and 850 °C have been observed using SEM as shown in Fig. 4. In contrast to their precursors, the diameters of CNFs reduced as the carbonized temperature increase. By increase in carbonization temperature from 650 to 850 °C, the diameters of fibres change from 380 ± 30 nm to 330 ± 30 nm, respectively. Same trend can be observed for CNFs derived from PAN–Gr and PAN@EDA@Gr as well.
 | ||
| Fig. 4 SEM images of the CNFs at different carbonized temperature derived from PAN (a and d), PAN–Gr (b and e), and PAN@EDA@Gr (c and f) precursors. | ||
The effect of adding Gr and Gr@EDA on carbon yield of PAN precursor has been evaluated via TGA, in which the carbon yield can be calculated as the weight loss value at 800 °C (Fig. 5a). From the TGA curves, the carbon yield for PAN, PAN–Gr, and PAN@EDA@Gr can be defined as 51%, 60%, and 67%, respectively. The high carbon yield of PAN–Gr and PAN@EDA@Gr derived CNFs lies in the barrier effect of Gr and Gr–EDA, which can effectively obstruct the diffusion of volatile products from the carbonized PAN nanofiber to the gas phase, therefore slowing down the decomposition process. The higher carbon yield of PAN@EDA@Gr compared to PAN–Gr confirms the stronger interaction between PAN and Gr@EDA than that of PAN–Gr composites. XRD analysis was conducted to better understand the influence of Gr and Gr@EDA on the evolution of the graphitic structure during carbonization. As the carbonized temperature was 650 °C, no peaks could be observed for PAN fibres derived CNFs, whereas PAN–Gr and PAN@EDA@Gr derived CNFs exhibited strong (002) peak, confirming the enhanced graphitic structure (Fig. 5b). As the carbonized temperature is 850 °C, one new (100) peak appeared, confirming further improvement in graphitic structure (Fig. 5c). Such enhanced graphitic structure lies in the nucleating effect of Gr and EDA@Gr during the carbonization.16 To further support our data, Raman spectroscopy was employed and results are shown in Fig. 5d and e. Two apparent peaks at 1330 and 1585 cm−1, corresponding to the D band and G band, respectively, can be observed. The D-band represents the defects in carbon structure, and the G-band is related to the sp2 carbon–carbon stretching vibrations (sp2 carbon atoms) in graphite layers.27 It can be clearly seen that the addition of Gr and EDA@Gr significantly improve the intensity of G band, which is consistent with the XRD data discussed above. The ratio R = ID/IG was calculated by fitting the D and G peaks using Gaussian curves (ESI Fig. S6†) superimposed on a linear and results have been illustrated in Fig. 4f. The ID/IG ratio for sample containing EDA@Gr is lower than those of pure PAN and PAN–Gr, indicating the higher graphitic structure at the same carbonization temperature.
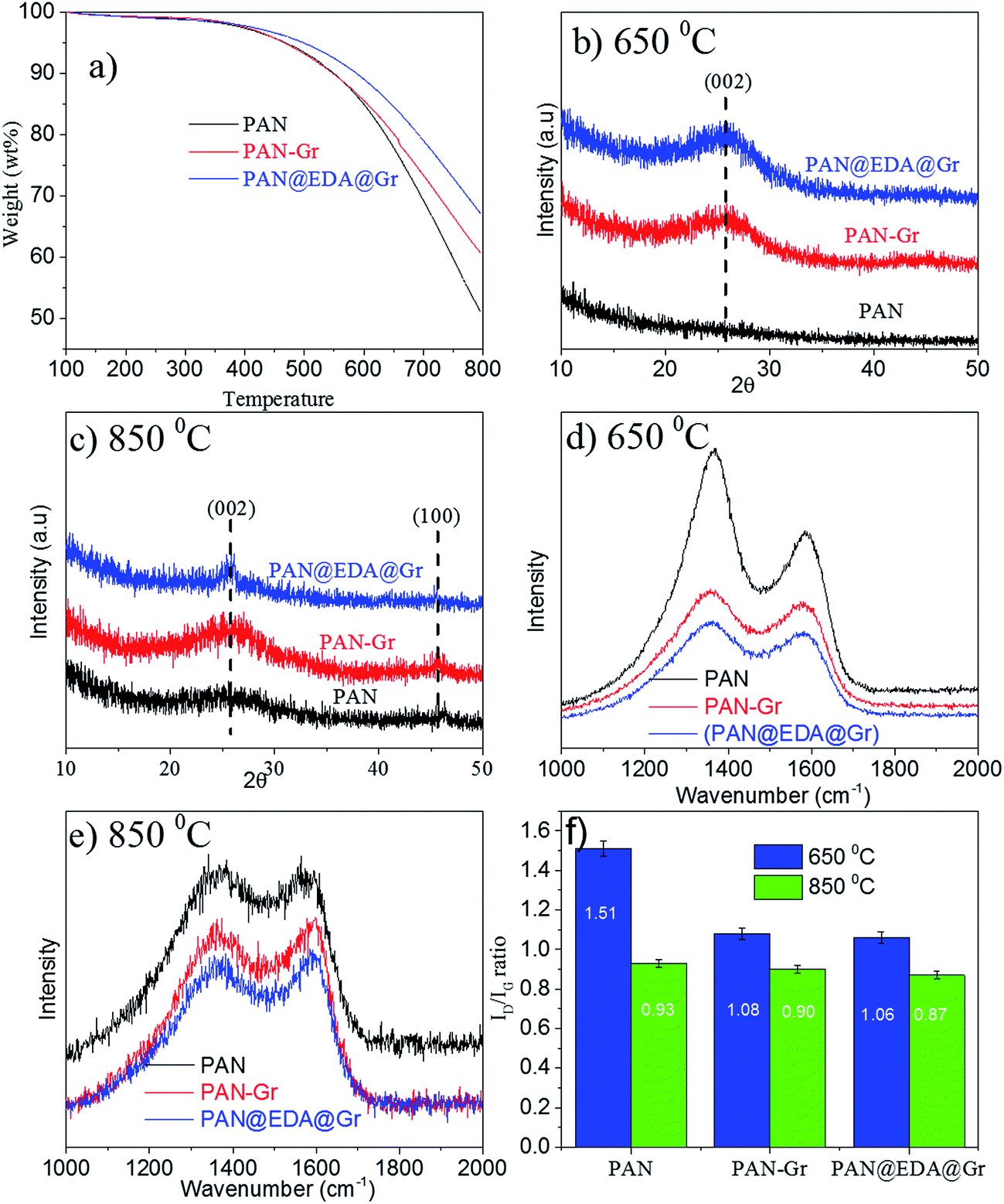 | ||
| Fig. 5 (a) Carbon yield of PAN, PAN–Gr, and PAN@EDA@Gr derived CNFs at 800 °C, (b) and (c) are XRD of the samples carbonized at 650 °C and 850 °C, respectively, (d) and (e) are Raman spectra of samples carbonized at 650 °C and 850 °C, respectively, (f) is the calculated ratio of ID/IG at different carbonized temperatures. | ||
The influence of Gr and Gr@EDA on the mechanical properties of the final CNFs were also investigated. The Young modulus of CNFs carbonised at 650 and 850 °C are presented in Table 1 and the typical stress–stain (S–S) curves are shown in Fig. 6. It is important to emphasize that our S–S curves are not the straight line like other carbon materials28 owing to the not well alignment of the fibres within the film. Thus the Young's modulus was calculated by stress/strain when the straight line emerge. As the carbonized temperature is 650 °C, the Young's modulus of CNFs derived from PAN, PAN–Gr and PAN@EDA@Gr were increased from 200 ± 20 MPa, to 250 ± 30 MPa, and to 400 ± 20 MPa. While the Young's modulus for PAN–Gr and PAN@EDA@Gr derived CNFs at 850 °C can reach 520 ± 40 MPa and 620 ± 40 MPa, respectively, which is ∼127% and 150% higher than that of PAN-derived CNFs. Such increase in the mechanical performances could be due to the reinforcing effect of Gr(EDA–Gr)29 as well as reduced diameter of nanofibres.30 Comparing with PAN–Gr derived CNFs, PAN@EDA@Gr derived CNFs exhibit higher Young's modulus, assigned to the stronger interactions between PAN and Gr@EDA.
| Samples | Young's modulus at 650 °C (MPa) | Young's modulus at 850 °C (MPa) |
|---|---|---|
| PAN derived CNFs | 200 ± 20 | 400 ± 50 |
| PAN–Gr derived CNFs | 250 ± 30 | 520 ± 40 |
| PAN–Gr–EDA derived CNFs | 400 ± 20 | 620 ± 40 |
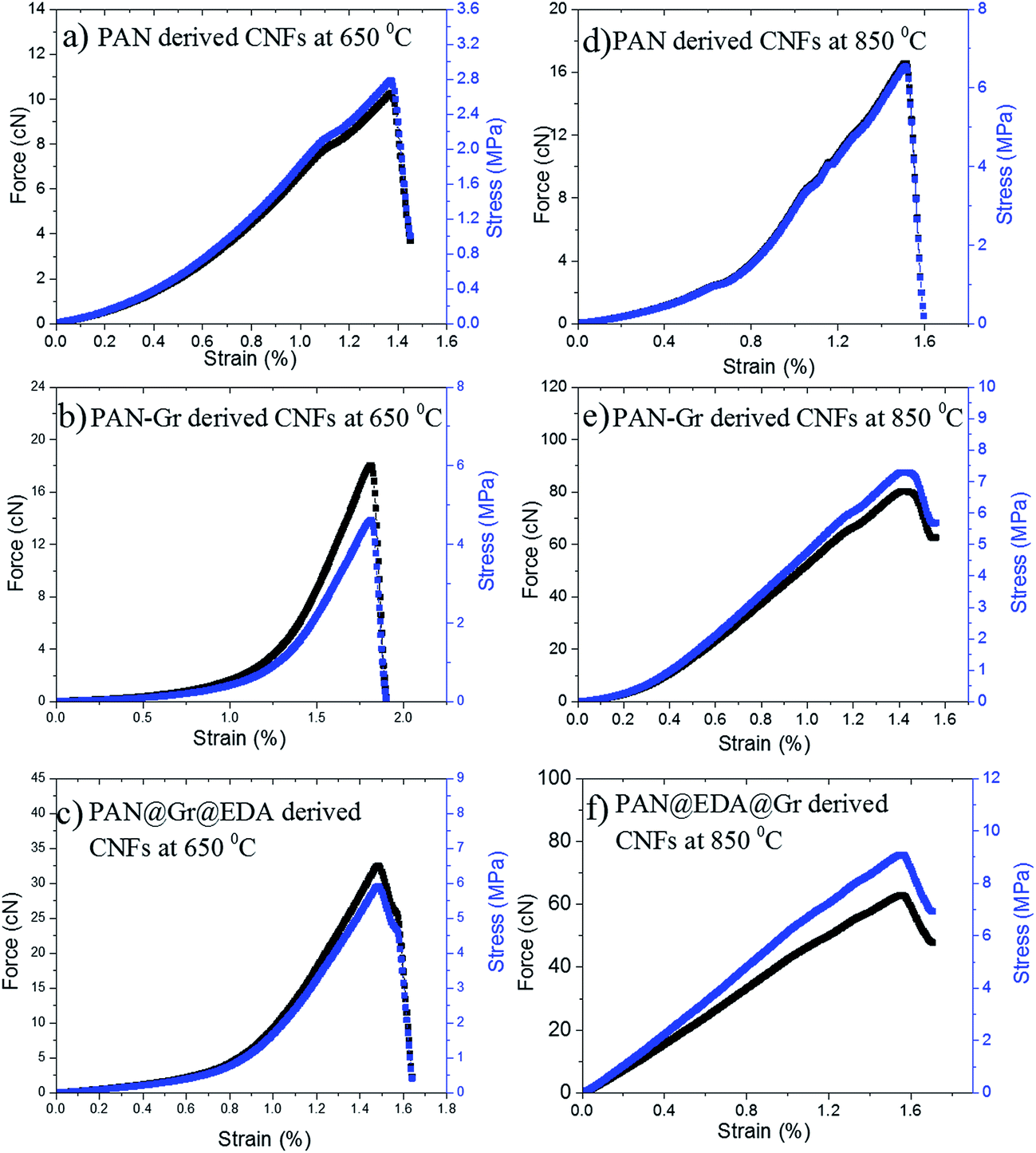 | ||
| Fig. 6 Stress–stain (S–S) curves of PAN, PAN–Gr, and PAN@EDA@Gr derived CNFs at different carbonized temperatures. | ||
The effects of adding Gr and Gr@EDA nanofillers on the wettability of CNFs carbonized at 850 °C were also investigated using water contact angle (WCA) as shown in Fig. 7. For pure PAN derived CNFs, the WCA is 142.8 ± 0.5° (Fig. 7a). As for the PAN–Gr derived CNFs, the WCA decreased to 131.5 ± 0.1° (Fig. 7b). For PAN–Gr–EDA derived CNFs (Fig. 7c), a significant decrease to 0° was observed, namely, hydrophilic CNFs have been obtained. Such phenomenon was ascribed to the N-doping during the carbonization with the EDA acting as N source, which change the wettability of the final product.31 To further prove our hypothesis, X-ray photoelectron spectroscopy (XPS) has been used to analyse the element composition within the final samples sintered at 850 °C as shown in Fig. 8. From the survey spectra, three typical peaks corresponding to the binding energies of C1s, N1s and O1s can be observed (Fig. 8a–c), with the N atomic rate of 3.14 at% (PAN: 850 °C), 3.22 at% (PAN–Gr: 850 °C), and 3.58 at% (PAN@EDA@Gr: 850 °C). Those data can confirm that the adding of Gr and Gr–EDA can improve the N content in the final samples. Such enhanced N content in the final samples change the surface wettability. Fig. 8A–C also demonstrate the high-resolution N1s spectra of the samples, in which the peaks can be deconvoluted into four individual peaks located at 398.28 eV, 399.75 eV and 400.94 eV, respectively, corresponding to the pyridinic N (398.2 eV), pyrrolic N (399.5 eV), quaternary N (401.1 eV) and commonly oxidized N (402.6 eV), respectively. The contents of each N form are illustrated in Table 2. From Table 2, we can found that quaternary N in PAN@EDA@Gr derived DNFs is higher than those in PAN and PAN–Gr derived the CNFs and the reason is in research.
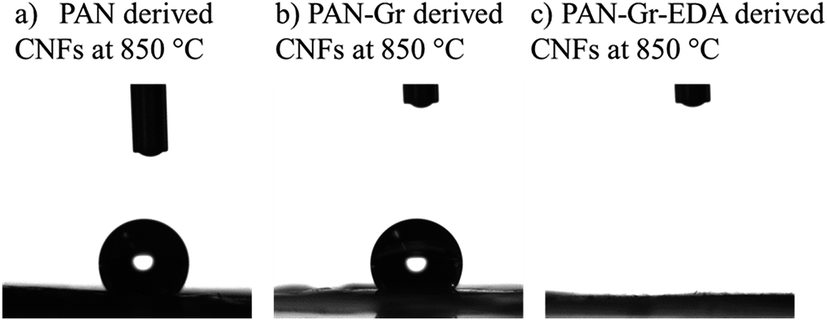 | ||
| Fig. 7 Profiles of water contact angle on derived CNFs carbonized at 850 °C derived from (a) pure PAN, (b) PAN–Gr, and (c) PAN@EDA@Gr. | ||
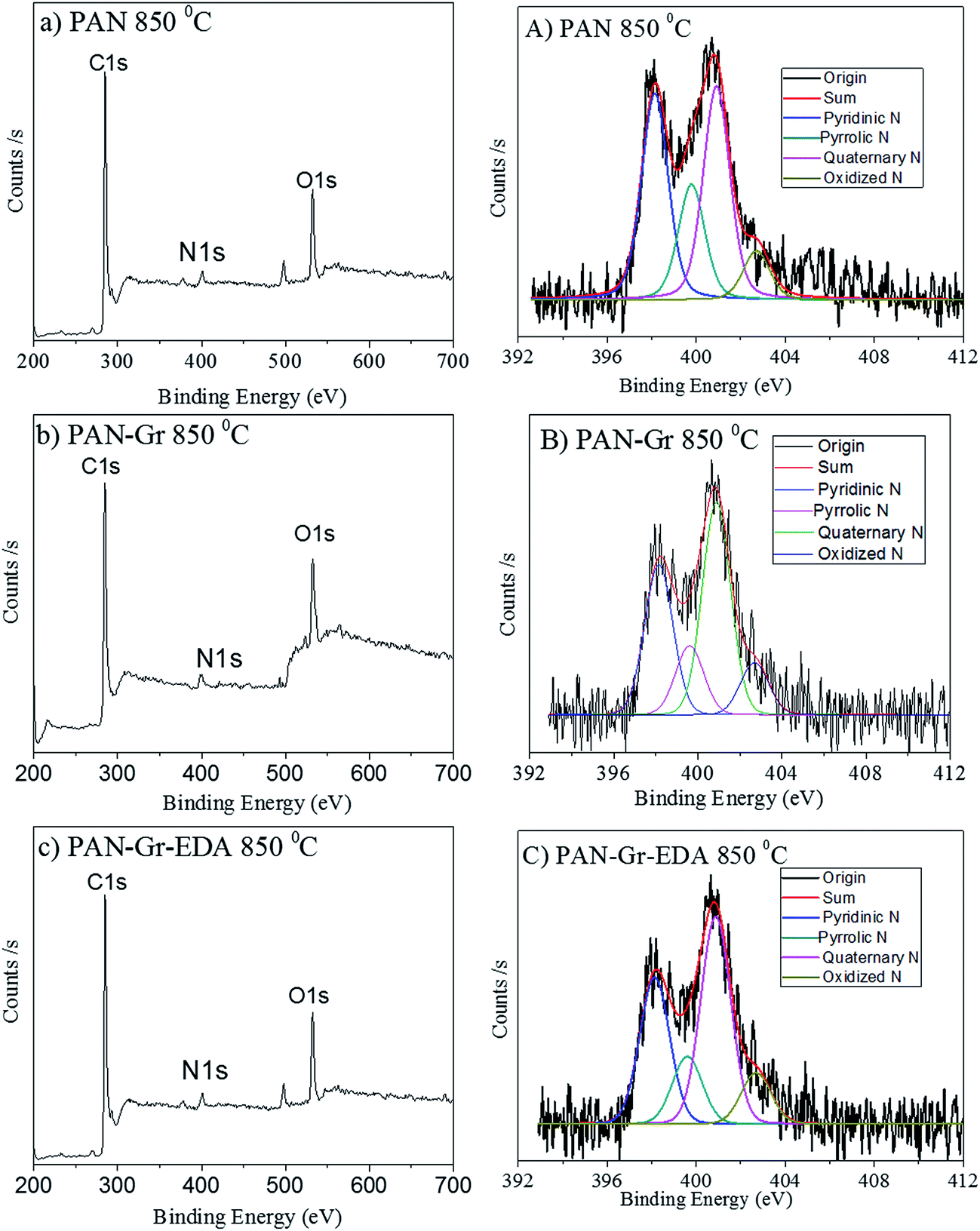 | ||
| Fig. 8 XPS survey spectra of the samples (a–c) and their corresponding N1s high resolution (A–C). | ||
| Samples (850 °C) | Pyridinic N (at%) | Pyrrolic N (at%) | Quaternary N (at%) | Oxidized N (at%) |
|---|---|---|---|---|
| PAN | 35.2 | 19.8 | 36.6 | 8.4 |
| PAN–Gr | 35.7 | 19.7 | 35.5 | 9.1 |
| PAN–Gr–EDA | 31.0 | 14.2 | 44.0 | 10.8 |
4. Conclusion
In summary, we have used EDA as a linker between Gr and PAN for high-performance CNFs. The improved dispersion and interaction between Gr and PAN lead to the higher carbon yield, improved Young's modulus and higher graphitic structure during the carbonization. Moreover, the EDA can act as the N source for N-doping during the carbonization, thus the PAN–Gr–EDA derived CNFs also exhibited hydrophilic performance. This study provides an effective approach for development of high-strength CNFs carbonized at low temperature with significant wettability.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by the Australian Research Council World Class Future Fibre Industry Transformation Research Hub (IH140100018).References
- M.-X. Wang, Z.-H. Huang, K. Shen, F. Kang and K. Liang, Catal. Today, 2013, 201, 109–114 CrossRef CAS.
- X. Mao, F. Simeon, G. C. Rutledge and T. A. Hatton, Adv. Mater., 2013, 25, 1309–1314 CrossRef CAS PubMed.
- G. Singh, D. Rana, T. Matsuura, S. Ramakrishna, R. M. Narbaitz and S. Tabe, Sep. Purif. Technol., 2010, 74, 202–212 CrossRef CAS.
- M. Wu, Q. Wang, X. Liu and H. Liu, Carbon, 2013, 51, 335–345 CrossRef CAS.
- L. F. Zhang, A. Aboagye, A. Kelkar, C. L. Lai and H. Fong, J. Mater. Sci., 2014, 49, 463–480 CrossRef CAS.
- E. Fitzer, Carbon, 1989, 27, 621–645 CrossRef.
- H. Khayyam, M. Naebe, A. Bab-Hadiashar, F. Jamshidi, Q. Li, S. Atkiss, D. Buckmaster and B. Fox, Appl. Energy, 2015, 158, 643–655 CrossRef CAS.
- R. Gupta and R. Goel, Microelectron. Reliab., 1991, 31, 1–5 CrossRef.
- H. Khayyam, M. Naebe, O. Zabihi, R. Zamani, S. Atkiss and B. Fox, IEEE Transactions on Industrial Informatics, 2015, 11, 887–896 CrossRef.
- M. Naebe, J. Wang, Y. Xue, X. Wang and T. Lin, J. Appl. Polym. Sci., 2010, 118, 359–365 CrossRef CAS.
- A.-T. Chien, H. C. Liu, B. A. Newcomb, C. Xiang, J. M. Tour and S. Kumar, ACS Appl. Mater. Interfaces, 2015, 7, 5281–5288 CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- D. W. Li, G. H. Li, P. F. Lv, N. Ullah, C. Wang, Q. Q. Wang, X. W. Zhang and Q. F. Wei, RSC Adv., 2015, 5, 30602–30609 RSC.
- O. Zabihi, M. Ahmadi, M. A. Bagherjeri and M. Naebe, RSC Adv., 2015, 5, 98692–98699 RSC.
- E. K. Lin, W.-l. Wu and S. K. Satija, Macromolecules, 1997, 30, 7224–7231 CrossRef CAS.
- S. Prilutsky, E. Zussman and Y. Cohen, Nanotechnology, 2008, 19, 165603–165612 CrossRef PubMed.
- D. Papkov, A. Goponenko, O. C. Compton, Z. An, A. Moravsky, X.-Z. Li, S. T. Nguyen and Y. A. Dzenis, Adv. Funct. Mater., 2013, 23, 5763–5770 CrossRef CAS.
- A. Peera, R. K. Saini, L. B. Alemany, W. E. Billups, M. Saunders, A. Khong, M. S. Syamala and R. J. Cross, Eur. J. Org. Chem., 2003, 2003, 4140–4145 CrossRef.
- X. Tang, Y. Zhao, Q. Jiao and Y. Cao, Fullerenes, Nanotubes, Carbon Nanostruct., 2010, 18, 14–23 CrossRef CAS.
- S. Wang, J. Wang, W. Zhang, J. Ji, Y. Li, G. Zhang, F. Zhang and X. Fan, Ind. Eng. Chem. Res., 2014, 53, 13205–13209 CrossRef CAS.
- H. Y. Zhang, C. Y. Xin, X. T. Wang and K. Wang, Int. J. Hydrogen Energy, 2016, 41, 12019–12028 CrossRef CAS.
- Z. Li, J. Cai, P. Cizek, H. Niu, Y. Du and T. Lin, J. Mater. Chem. A, 2015, 3, 16162–16167 CAS.
- X. Tang, Y. Zhao, Q. Jiao and Y. Cao, Fullerenes, Nanotubes, Carbon Nanostruct., 2010, 18, 14–23 CrossRef CAS.
- S. Cetiner, H. Karakas, R. Ciobanu, M. Olariu, N. U. Kaya, C. Unsal, F. Kalaoglu and A. S. Sarac, Synth. Met., 2010, 160, 1189–1196 CrossRef CAS.
- S. Jain, J. G. P. Goossens, G. W. M. Peters, M. van Duin and P. J. Lemstra, Soft Matter, 2008, 4, 1848–1854 RSC.
- S. Lee, J. Kim, B.-C. Ku, J. Kim and H.-I. Joh, Adv. Chem. Eng. Sci., 2012, 2, 8 Search PubMed.
- F. Cesano, D. Scarano, S. Bertarione, F. Bonino, A. Damin, S. Bordiga, C. Prestipino, C. Lamberti and A. Zecchina, J. Photochem. Photobiol., A, 2008, 196, 143–153 CrossRef CAS.
- Y. I. Jhon, C. Kim, M. Seo, W. J. Cho, S. Lee and Y. M. Jhon, Sci. Rep., 2016, 6, 2032–2039 Search PubMed.
- Z. Xu and C. Gao, Mater. Today, 2015, 18, 480–492 CrossRef CAS.
- D. Papkov, Y. Zou, M. N. Andalib, A. Goponenko, S. Z. D. Cheng and Y. A. Dzenis, ACS Nano, 2013, 7, 3324–3331 CrossRef CAS PubMed.
- G.-P. Hao, N. R. Sahraie, Q. Zhang, S. Krause, M. Oschatz, A. Bachmatiuk, P. Strasser and S. Kaskel, Chem. Commun., 2015, 51, 17285–17288 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TEM, Raman spectra, FTIR spectra and TGA curves of the Gr and Gr–EDA compound, FTIR spectra of the PAN and PAN@EDA@Gr samples, Raman spectra and computer decompositions of the derived CFs with different carbonized temperatures. See DOI: 10.1039/c6ra24719a |
| This journal is © The Royal Society of Chemistry 2017 |