
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
TEMPO-catalyzed oxidative homocoupling route to 3,2′-biindolin-2-ones via an indolin-3-one intermediate†
Bo Yin,
Panpan Huang,
Yingbing Lu* and
Liangxian Liu*
Key Laboratory of Organo-Pharmaceutical Chemistry of Jiangxi Province, Gannan Normal University, Ganzhou 341000, PR China. E-mail: lxliu@xmu.edu.cn
First published on 3rd January 2017
Abstract
A combinative C2-selective arylation, and C3-selective carbonylation of free indole derivatives, by means of TEMPO catalysis and a silver oxidant under non-directing group conditions, was successful demonstrated. This new methodology is both atom and step efficient and is applicable to a broad scope of substrates, allowing the synthesis of a range of synthetically valuable 3,2′-biindolin-2-ones in moderate to excellent yields.
Introduction
Biindole scaffolds are important motifs in an array of natural products with diverse biological activities,1–3 exemplified by the TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) antagonist bisindigotin 1 (Fig. 1) from the Chinese medicinal herb Isatis indigotica.4 This herbal plant has long been used as a folk medicine in China for treatment of viral diseases and diseases with an inflammatory nature.4 In addition, indirubin, a bis-indole scaffold and its derivatives, are present in some traditional Chinese medicines and inhibit cyclin-dependent kinases (CDKs).5 These medicines have historically been used to treat chronic diseases including leukemia.6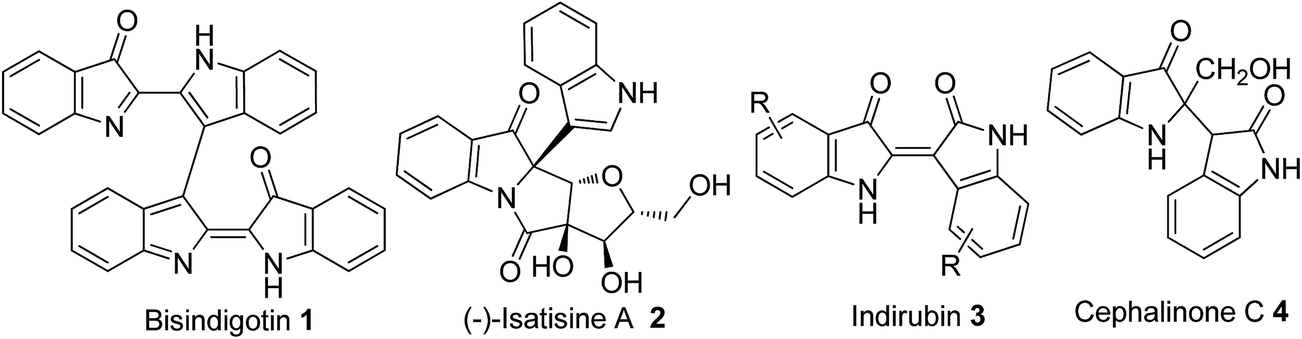 | ||
| Fig. 1 Representative natural products with a 2-substituent indolin-3-one structural unit. | ||
Accordingly, synthesis and functionalization of biindolyls have attracted much attention over decades.7–9 Recently, some progress on construction of the 2,3′-linked7 and 3,3′-linked8 biindolyl scaffolds was made, including the palladium- or copper-catalyzed intermolecular coupling reaction and iodine-induced dimerization of indoles. For example, Zhang and co-workers reported a mild and selective method for dimerization of indoles by palladium catalysis to give 2,3′-biindolyls in high yields at room temperature.9b However, most of these procedures require expensive metal catalysts and high loading of metal oxidants. In addition, the regioselectivity control of C2 arylation can be quite challenging under non-directing group conditions. From the viewpoints of atom economy, cost efficiency and green chemistry, atmospheric oxygen is obviously superior to other reagents, and thus represents the quintessential oxidant.10 In the past decades, most efforts have been directed to the development of transition metal-based catalysts. In contrast, much less attention has been paid to the development of non-metallic oxidation systems, largely ignoring their inherent advantages.10c The radical TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl radical) and its derivatives are well-established catalysts for oxidation processes, and now used extensively in organic synthesis and industrial applications as a mild, safe, and economical alternative to heavy metal reagents as highly selective oxidation catalysts for the production of pharmaceuticals, flavors, fragrances, agrochemicals, and a variety of other specialty chemicals.11 As part of our ongoing investigations on environmentally benign, selective, and controllable C–H bond functionalization, we studied the TEMPO-catalyzed oxidative homocoupling of indoles in air.12 Herein, we report the first successful example of TEMPO-catalyzed oxidative homocoupling of indoles affording substituted C2–C3′ bisindole-3-ones 9 (Scheme 1).
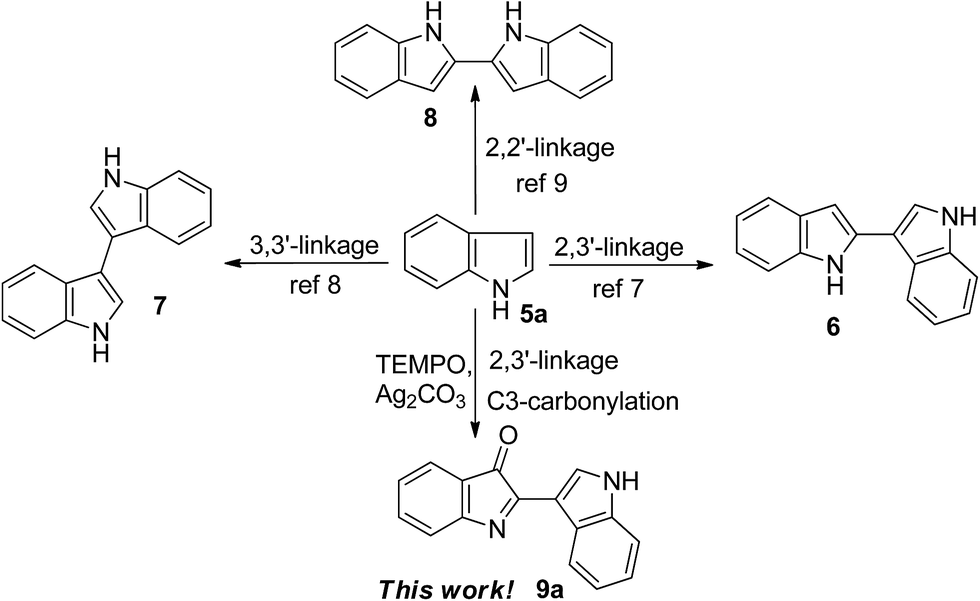 | ||
| Scheme 1 Homocoupling of indole. | ||
Results and discussion
Preliminary studies were carried out at 80 °C in an open tube, using commercially available unsubstituted indole (5a) in the presence of TEMPO and K3PO4. A variety of transition-metal catalysts, including FeCl3·6H2O, FeSO4, ZrCl4, ZnCl2, CoCl2, CuI, CuCl, and Ag2CO3, were screened. It was found that Ag2CO3 was the most efficient catalyst for this reaction, which gave the desired product 9a in 51% yield (Table 1, entry 8); while other transition-metal catalysts did not undergo the conversion under the reaction conditions. The structure of 9a was confirmed by X-ray crystallography (see ESI†). Among silver sources tested, including Ag2CO3, AgNO3, AgOTf, and AgBF4, were tested in DMF using TEMPO as the oxidant at 80 °C for 12 h, and Ag2CO3 was found to be the most effective catalyst (Table 1, entries 8–12). The amount of Ag2CO3 also has a large influence on the yield of 9a. In the absence of Ag2CO3 catalyst, the desired product was not obtained under these conditions, whereas, at 35 mol% of Ag2CO3, the desired product is obtained in 57% yield, and at lower or higher loading, the yield decreases (Table 1, entries 12–16). It was also found that the use of TEMPO as an oxidant is critical to the reaction. In the absence of TEMPO, the homocoupling reaction did not proceed. However, the slightly more loading amounts of TEMPO result in a dramatic influence on the yield (Table 1, entries 17–20). For example, 81% yield of 9a was obtained when using 0.15 mol% of TEMPO (Table 1, entry 19). Further assessment of the reaction solvents indicated that DMF was the optimal solvent, while other solvents gave lower yields or were ineffective (Table 1, entries 21–25).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | TEMPO (mol%) | Solvent | Base (mol%) | Yieldb (%) |
| a Condition: 5a (0.3 mmol), solvent (1 mL), 80 °C, 12 h, under open air.b Isolated yields. | |||||
| 1 | FeCl3·6H2O (15) | 10 | DMF | K3PO4 (10) | 0 |
| 2 | FeSO4 (15) | 10 | DMF | K3PO4 (10) | 0 |
| 3 | ZrCl4 (15) | 10 | DMF | K3PO4 (10) | 0 |
| 4 | ZnCl2 (15) | 10 | DMF | K3PO4 (10) | 0 |
| 5 | CoCl4 (15) | 10 | DMF | K3PO4 (10) | 0 |
| 6 | Cul (15) | 10 | DMF | K3PO4 (10) | Trace |
| 7 | CuCl2 (15) | 10 | DMF | K3PO4 (10) | Trace |
| 8 | Ag2CO3 (15) | 10 | DMF | K3PO4 (10) | 51 |
| 9 | AgNO3 (15) | 10 | DMF | K3PO4 (10) | 24 |
| 10 | AgOTf (15) | 10 | DMF | K3PO4 (10) | 35 |
| 11 | AgBF4 (15) | 10 | DMF | K3PO4 (10) | 22 |
| 12 | — | 10 | DMF | K3PO4 (10) | 0 |
| 13 | Ag2CO3 (5) | 10 | DMF | K3PO4 (10) | 10 |
| 14 | Ag2CO3 (25) | 10 | DMF | K3PO4 (10) | 53 |
| 15 | Ag2CO3 (35) | 10 | DMF | K3PO4 (10) | 57 |
| 16 | Ag2CO3 (45) | 10 | DMF | K3PO4 (10) | 55 |
| 17 | Ag2CO3 (35) | — | DMF | K3PO4 (10) | 0 |
| 18 | Ag2CO3 (35) | 5 | DMF | K3PO4 (10) | 27 |
| 19 | Ag2CO3 (35) | 15 | DMF | K3PO4 (10) | 81 |
| 20 | Ag2CO3 (35) | 20 | DMF | K3PO4 (10) | 81 |
| 21 | Ag2CO3 (35) | 15 | DMSO | K3PO4 (10) | 67 |
| 22 | Ag2CO3 (35) | 15 | Toluene | K3PO4 (10) | 0 |
| 23 | Ag2CO3 (35) | 15 | Pyridine | K3PO4 (10) | 0 |
| 24 | Ag2CO3 (35) | 15 | 1,4-Dioxane | K3PO4 (10) | Trace |
| 25 | Ag2CO3 (35) | 15 | ClCH2CH2Cl | K3PO4 (10) | 0 |
| 26 | Ag2CO3 (35) | 15 | DMF | NaOAc (10) | 87 |
| 27 | Ag2CO3 (35) | 15 | DMF | LiOH (10) | 0 |
| 28 | Ag2CO3 (35) | 15 | DMF | NaHCO3 (10) | 30 |
| 29 | Ag2CO3 (35) | 15 | DMF | K2CO3 (10) | 48 |
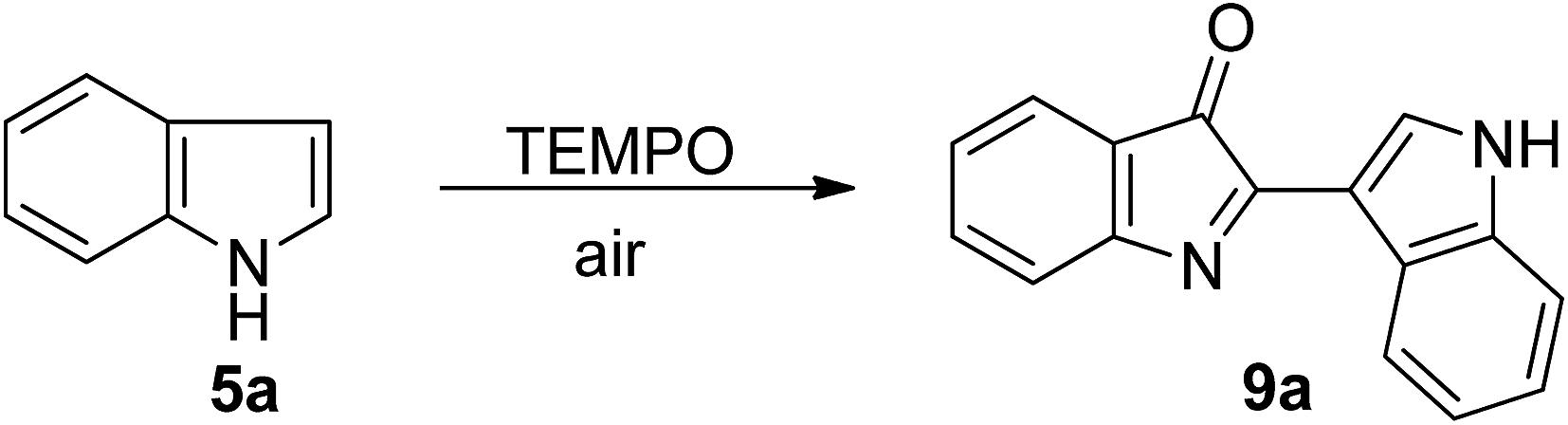
Finally, we examined a series of bases (Table 1, entries 26–29). The observation revealed that NaOAc is slightly better than K3PO4 and other bases, such as LiOH, NaHCO3, and K2CO3, are inferior to K3PO4. After a great deal of screening on different parameters we found that the combinative C2-selective arylation and C3-selective carbonylation of indole by using TEMPO (15 mol%) in air as catalyst, Ag2CO3 (35 mol%) as an oxidant, and NaOAc (10 mol%) as base in DMF at 80 °C led to the highest efficiency (87% yield, Table 1, entry 26).
With a set of optimized conditions in hand, we next examined the indole scope of this TEMPO-catalyzed oxidative homocoupling reaction. As shown in Table 2, the reaction can tolerate a variety of functional groups at the 4, 5, 6, and 7 positions of indoles, such as F, Cl, Br, CH3, CH3O, BnO, CO2CH3, and CN, and the corresponding reactions proceeded smoothly to afford the desired products in moderate to excellent yields with high regioselectivity. The substituent effect on the indole ring was then investigated. The results have shown that electronegativities of substituents played a major role in governing the reactivity of the substrates. Electron-donating substitutents showed better results than electron-withdrawing substitutents in this transformation. For example, 7-substituted indole derivatives with electron-donating substituents (CH3, OCH3, and OBn) afforded the desired 9l–n in yields ranging from 76% to 90%, while 7-substituted indole derivatives with electron-withdrawing substituents (Cl and Br) provided the desired products in 61 and 72% yields, respectively. It is worth noting that substrate with a strong electron-withdrawing substitutents at C4-position, such as CO2CH3 and CN, gave 9o and 9p in 63% and 38% yields, respectively. This is particularly important, since substrates with a strong electron-withdrawing group, such as a nitrile group, disfavored the homocoupling of indoles and there were few examples reported.7,8
|
|||
|---|---|---|---|
| Entry | R | Product | Yieldb [%] |
| a Reaction conditions: indole (0.3 mmol), TEMPO (15 mol%), Ag2CO3 (35 mol%), NaOAc (10 mol%), DMF (1 mL), 80 °C.b Isolated yields. | |||
| 1 | H | 9a | 87 |
| 2 | 5-F | 9b | 66 |
| 3 | 5-Br | 9c | 75 |
| 4 | 5-CH3 | 9d | 89 |
| 5 | 5-OCH3 | 9e | 71 |
| 6 | 5-OBn | 9f | 78 |
| 7 | 6-F | 9g | 73 |
| 8 | 6-Cl | 9h | 64 |
| 9 | 6-CH3 | 9i | 79 |
| 10 | 7-Cl | 9j | 61 |
| 11 | 7-Br | 9k | 72 |
| 12 | 7-CH3 | 9l | 90 |
| 13 | 7-OCH3 | 9m | 76 |
| 14 | 7-OBn | 9n | 82 |
| 15 | 4-CN | 9o | 38 |
| 16 | 4-CO2CH3 | 9p | 63 |
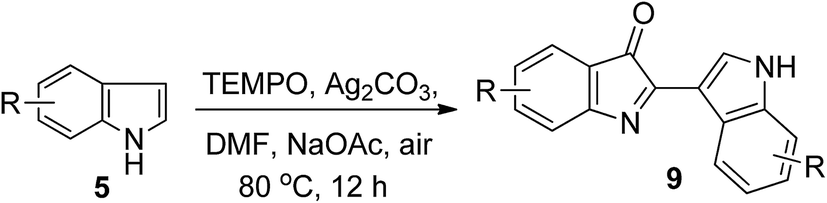
To gain some mechanistic insight into the process of this reaction, a series of control experiments were conducted (Scheme 2). Because the TEMPO-catalyzed oxidative homocoupling reaction was performed in air, the role of O2 in this reaction was explored by conducting several control experiments. Under an O2 atmosphere, the reaction yield was not increased, but a more rapid conversion of the starting material to the reaction product was observed by TLC detection compared to that performed under air conditions. However, only trace amount of the product was obtained under an argon atmosphere, even a long reaction time. These results indicated that O2 is essential for the TEMPO-catalyzed transformation. In addition, in the control experiment of 5a with TEMPO but without Ag2CO3, compound 9a was obtained only in 12% yield. The result indicated that Ag2CO3 act as terminal oxidant. Under the optimized conditions, N–CH3 indole was chosen as a substrate instead of indole. To our surprise, no conversion was observed, indicating that the substituents at the N1-position of the indole had a great influence on the reactivity.
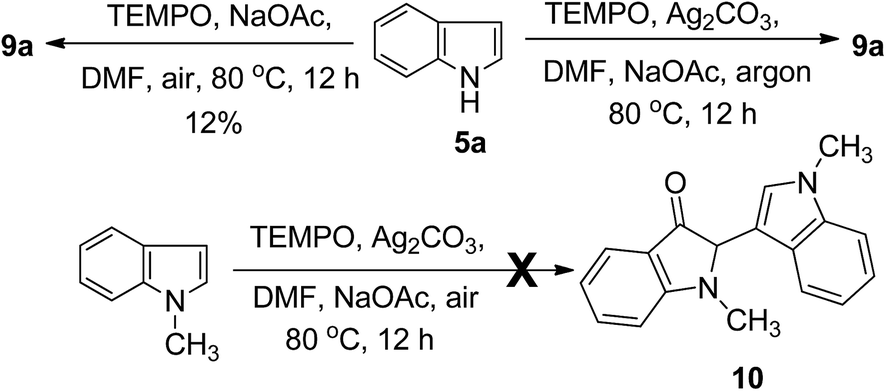 | ||
| Scheme 2 Mechanistic studies. | ||
Although the detailed mechanism remained unclear at the current stage, a plausible reaction pathway based on the basis of the results described above and relevant literature13 is outlined in Scheme 3. First, indole was oxidized slowly in the presence of TEMPO, Ag2CO3 and O2 into indenone 11 which should be unstable and was never isolated.13a,d Then, a rapid nucleophilic addition of another indole molecule on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of this intermediate gave the intermediate 12, which was oxidized rapidly to afford product 9a.
N bond of this intermediate gave the intermediate 12, which was oxidized rapidly to afford product 9a.
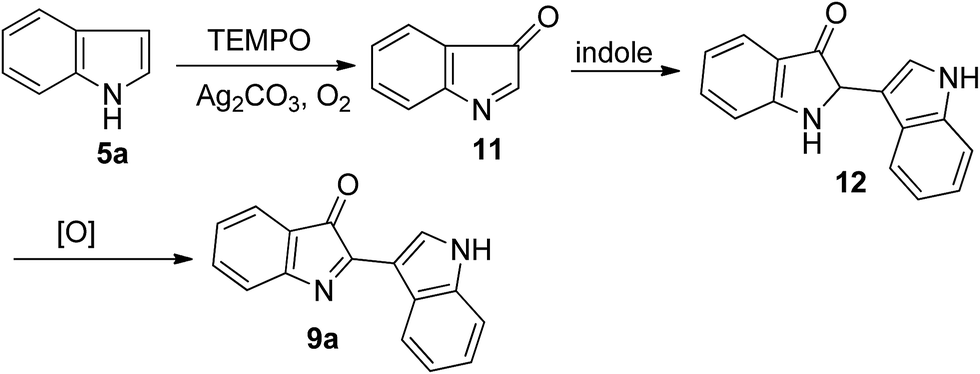 | ||
| Scheme 3 Plausible reaction pathway. | ||
To verify such a mechanistic scenario, we attempted to obtained the putative intermediate 12 or its derivatives. Fortunately, trace amounts of 13 can be determined in the reaction mixtures when 7-methyl-1H-indole was subjected to the standard reaction conditions, which showed us some clues on the reaction intermediate. In the next step, to obtain more information concerning the reaction pathway, we separated and collected the intermediate 13, and subjected it to react with TEMPO under standard reaction conditions, and this gave product 9l in 93% yield (Scheme 4). This result showed that 13 is the intermediate of the dimeric reaction.
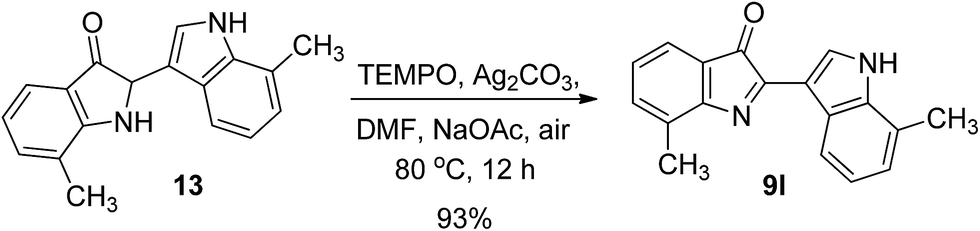 | ||
| Scheme 4 Investigation of the reaction mechanism. | ||
Conclusions
In conclusion, we have developed a general and efficient method for the synthesis of 3,2′-biindolin-2-ones via a TEMPO-catalyzed oxidative homo dimerization in moderate to excellent yields with high regioselectivity. The advantages of this new method are broad substrate scope, operational simplicity, and high atom-economy. Moreover, the high halogen compatibility of the process can provide a facile access to halo-substituted 3,2′-biindolin-2-ones.Acknowledgements
The authors are grateful to the NSF of China (No. 21462002), Natural Science Foundation of Jiangxi Province (No. 20161BAB203096) for financial support.Notes and references
- (a) R. J. Sundberg, Indoles, Academic Press, San Diego, 1996 Search PubMed; (b) S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873–2920 CrossRef CAS PubMed; (c) L. Joucla and L. Djakovitch, Adv. Synth. Catal., 2009, 351, 673–714 CrossRef CAS; (d) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608–9644 CrossRef CAS PubMed; (e) G. Bartoli, G. Bencivenni and R. Dalpozzo, Chem. Soc. Rev., 2010, 39, 4449–4465 RSC; (f) F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS.
- (a) L. S. Feng, M. L. Liu, S. Zhang, Y. Chai, B. Wang, Y. B. Zhang, K. Lv, Y. Guan, H. Y. Guo and C. L. Xiao, Eur. J. Med. Chem., 2011, 46, 341–348 CrossRef CAS PubMed; (b) S. Nam, A. Scuto, Y. Yang, W. Y. Chen, S. Park, H. S. Yoo, H. Konig, R. Bhatia, X. L. Cheng, K. H. Merz, G. Eisenbrand and R. Jove, Mol. Oncol., 2012, 6, 276–283 CrossRef CAS PubMed; (c) Y. K. Chan, H. H. Kwok, L. Y. Chan, K. S. Y. Leung, J. Shi, N. K. Mak, R. N. S. Wong and P. Y. K. Yue, Biochem. Pharmacol., 2012, 83, 598–607 CrossRef CAS PubMed; (d) S. Nam, W. Wen, A. Schroeder, A. Herrmann, H. Yu, X. L. Cheng, K. Z. Merz, G. Eisenbrand, H. Z. Li, Y. C. Yuan and R. Jove, Mol. Oncol., 2013, 7, 369–378 CrossRef CAS PubMed.
- (a) F. P. Guengerich, J. L. Sorrells, S. Schmitt, J. A. Krauser, P. Aryal and L. Meijer, J. Med. Chem., 2004, 47, 3236–3241 CrossRef CAS PubMed; (b) K. Vougogiannopoulou, Y. Ferandin, K. Bettayeb, V. Myrianthopoulos, O. Lozach, Y. Z. Fan, C. H. Johnson, P. Magiatis, A. L. Skaltsounis, E. Mikros and L. Meijer, J. Med. Chem., 2008, 51, 6421–6431 CrossRef CAS PubMed; (c) S. J. Choi, J. E. Lee, S. Y. Jeong, I. Im, S. D. Lee, E. J. Lee, S. K. Lee, S. M. Kwon, S. G. Ahn, J. H. Yoon, S. Y. Han, J. Kim and Y. C. Kim, J. Med. Chem., 2010, 53, 3696–3706 CrossRef CAS PubMed; (d) C. Pergola, N. Gaboriaud-Kolar, N. Jestädt, S. König, M. Kritsanida, A. M. Schaible, H. K. Li, U. Garscha, C. Weinigel, D. Barz, K. F. Albring, O. Huber, A. L. Skaltsounis and O. Werz, J. Med. Chem., 2014, 57, 3715–3723 CrossRef CAS PubMed.
- X. Y. Wei, C. Y. Leung, C. K. C. Wong, X. Y. Shen, R. N. C. Wong, Z. W. Cai and N. K. Mak, J. Nat. Prod., 2005, 68, 427–429 CrossRef CAS PubMed.
- (a) R. Hoessel, S. Leclerc, J. A. Endicott, M. E. M. Nobel, A. Lawrie, P. Tunnah, M. Leost, E. Damiens, D. Marie, D. Marko, E. Niederberger, W. C. Tang, G. Eisenbrand and L. Meijer, Nat. Cell Biol., 1999, 1, 60–67 CrossRef CAS PubMed; (b) V. Myrianthopoulos, P. Magiatis, Y. Ferandin, A. L. Skaltsounis, L. Meijer and E. Mikros, J. Med. Chem., 2007, 50, 4027–4037 CrossRef CAS PubMed; (c) A. Beauchard, H. Laborie, H. Rouillard, O. Lozach, Y. Ferandin, R. L. Guevel, C. Guguen-Guillouzo, L. Meijer, T. Besson and V. Thiery, Bioorg. Med. Chem., 2009, 17, 6257–6263 CrossRef CAS PubMed.
- Z. Xiao, Y. Hao, B. Liu and L. Qian, Leuk. Lymphoma, 2002, 43, 1763–1768 CAS.
- For the construction of C2–C3′ biindolyl, please see: (a) A. Pezzella, L. Panzella, A. Natangelo, M. Arzillo, A. Napolitano and M. d'Ischia, J. Org. Chem., 2007, 72, 9225–9230 CrossRef CAS PubMed; (b) Z.-J. Liang, J. L. Zhao and Y. H. Zhang, J. Org. Chem., 2010, 75, 170–177 CrossRef CAS PubMed; (c) Y. X. Li, K. G. Ji, H. X. Wang, S. Ali and Y. M. Liang, J. Org. Chem., 2011, 76, 744–747 CrossRef CAS PubMed.
- For the construction of C3–C3′ biindolyl, please see: (a) Y. Li, W. H. Wang, S. D. Yang, B. J. Li, C. Feng and Z. J. Shi, Chem. Commun., 2010, 46, 4553–4555 RSC; (b) T. M. Niu and Y. H. Zhang, Tetrahedron Lett., 2010, 51, 6847–6851 CrossRef CAS.
- For the construction of C2–C2′ biindolyl, please see: (a) A. Pezzella, L. Panzella, O. Crescenzi, A. Napolitano, S. Navaratman, R. Edge, E. J. Land, V. Barone and M. D'lschia, J. Am. Chem. Soc., 2006, 128, 15490–15498 CrossRef CAS PubMed; (b) P. A. Keller, N. R. Yepuri, M. J. Kelso, M. Mariani, B. W. Skelton and A. H. White, Tetrahedron, 2008, 64, 7787–7795 CrossRef CAS; (c) X. H. Xu, G. K. Liu, A. Azuma, E. Tokunaga and N. Shibata, Org. Lett., 2011, 13, 4854–4857 CrossRef CAS PubMed; (d) S. Liu and X. J. Hao, Tetrahedron Lett., 2011, 52, 5640–5642 CrossRef CAS; (e) L. Panzella, A. Pezzella, A. Napolitano and M. D'lschia, Org. Lett., 2007, 9, 1411–1414 CrossRef CAS PubMed; (f) C. A. Meric, Y. You, D. M. McInnes, A. L. Zechman, M. M. Miller and Q. L. Deng, Tetrahedron, 2001, 57, 5199–5212 CrossRef; (g) W. J. Zhang, Z. Liu, S. M. Li, T. T. Yang, Q. B. Zhang, L. Ma, X. P. Tian, H. B. Zhang, C. G. Huang, S. Zhang, J. H. Ju, Y. M. Shen and C. S. Zhang, Org. Lett., 2012, 14, 3364–3367 CrossRef CAS PubMed; (h) N. R. Yepuri, R. Haritakul, P. A. Keller, B. W. Skelton and A. H. White, Tetrahedron Lett., 2009, 50, 2501–2504 CrossRef CAS.
- (a) D. Lenoir, Angew. Chem., Int. Ed., 2006, 45, 3206–3210 CrossRef CAS PubMed; (b) S. S. Stahl, Science, 2005, 309, 1824–1826 CrossRef CAS PubMed; (c) Y. H. Kuang, H. Rokubuichi, Y. Nabae, T. Hayakawa and M. Kakimoto, Adv. Synth. Catal., 2010, 352, 2635–2642 CrossRef CAS.
- For reviews on the application of TEMPO and its derivatives in organic synthesis, see: (a) R. A. Sheldon, I. W. C. E. Arends, G. J. T. Brink and A. Dijksman, Acc. Chem. Res., 2002, 35, 774–781 CrossRef CAS PubMed; (b) R. A. Sheldon and I. W. C. E. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS; (c) J. M. Bobbitt, C. Bruckner and N. Merbouh, Org. React., 2009, 74, 103–424 CAS; (d) C. Bruckner, in Stable Radicals, ed. R. G. Hicks, John Wiley, United Kingdom, 2010, pp. 433–456 Search PubMed; (e) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed; (f) R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS; (g) O. Garcia-Mancheno and T. Stopka, Synthesis, 2013, 45, 1602–1611 CrossRef; (h) Z. G. Zhou and L. X. Liu, Curr. Org. Chem., 2014, 18, 459–474 CrossRef CAS.
- (a) F. Lin, Y. Chen, B. S. Wang, W. B. Qin and L. X. Liu, RSC Adv., 2015, 5, 37018–37022 RSC; (b) W. B. Qin, Q. Chang, Y. H. Bao, N. Wang, Z. W. Chen and L. X. Liu, Org. Biomol. Chem., 2012, 10, 8814–8821 RSC; (c) Y. H. Bao, J. Y. Zhu, W. B. Qin, Y. B. Kong, Z. W. Chen, S. B. Tang and L. X. Liu, Org. Biomol. Chem., 2013, 11, 7938–7945 RSC; (d) W. B. Qin, J. Y. Zhu, Y. B. Kong, Y. H. Bao, Z. W. Chen and L. X. Liu, Org. Biomol. Chem., 2014, 12, 4252–4259 RSC; (e) W. B. Qin, Q. Chang, H. Q. Luo, Y. H. Bao, Z. W. Chen and L. X. Liu, Curr. Org. Synth., 2013, 10, 492–499 CrossRef CAS.
- (a) C. Ganachaud, V. Garfagnoli, T. Tron and G. Iacazio, Tetrahedron Lett., 2008, 49, 2476–2478 CrossRef CAS; (b) J. Bergman, S. Bergman and J. O. Lindström, Tetrahedron Lett., 1998, 39, 4119–4122 CrossRef CAS; (c) Y. H. Liu and W. W. McWhorter Jr, J. Org. Chem., 2003, 68, 2618–2622 CrossRef CAS PubMed; (d) P. Astolfi, M. Panagiotaki, C. Rizzoli and L. Greci, Org. Biomol. Chem., 2006, 4, 3282–3290 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and NMR data of the prepared compounds. CCDC 1453369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra24834a |
| This journal is © The Royal Society of Chemistry 2017 |