
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of well-dispersive 2.0 nm Pd–Pt bimetallic nanoclusters supported on β-cyclodextrin functionalized graphene with excellent electrocatalytic activity†
Xin Ran‡
a,
Long Yang‡a,
Qing Qu*a,
Shunling Lia,
Ying Chenb,
Limei Zuoa and
Lei Li*b
aSchool of Chemical Science and Technology, Yunnan University, Kunming 650091, China. E-mail: quqing@ynu.edu.cn; Fax: +86 871 65036538; Tel: +86 871 65035798
bLaboratory for Conservation and Utilization of Bio-Resources, Yunnan University, Kunming, 650091, China. E-mail: leelei@ynu.edu.cn
First published on 12th January 2017
Abstract
In this work, ultrasmall Pd–Pt bimetallic nanoclusters with a uniform size of 2.0 nm monodispersed on β-cyclodextrin functionalized reduced graphene oxide (β-CD-RGO) were successfully prepared in aqueous solution at room temperature within 30 min without the need for any organic solvent or high temperature. The integration of β-CD and RGO was responsible for the formation of the monodispersed 2.0 nm Pd–Pt bimetallic nanoclusters. Inspired by the monodisperse, ultrasmall, and pristine properties of the Pd–Pt clusters, the Pd–Pt@β-CD-RGO nanohybrid displayed enhanced activity for methanol and ethanol oxidation in alkaline media in comparison with the commercial Pd/C catalysts. This facile and simple method is of significance for the preparation of bimetallic nanocatalysts with high catalytic activity on suitable supporting materials.
1. Introduction
Over the past decades, extensive research has been devoted to electrocatalysis studies to improve the performance of noble metal catalysts.1 One of the most promising strategies is to design and synthesize noble metals with ultrafine sizes because of their increased surface area and the number of edge and corner atoms, which could greatly improve their catalytic activities.2,3 In particular, ultrasmall noble metal nanoclusters (≤2 nm) with several to hundreds of atoms, have received significant attention.4 However, the surface energy increases with decreasing particle size, which usually leads to serious aggregation of the small particles in order to minimize the total surface energy.2,5 To overcome this aggregation, the metal particles must be anchored to suitable supports.6,7 For this reason, extensive interests have been drawn to the uses of sp2-hybridized carbon materials as the supporting materials.2,3,7 Among all the available supporting materials, graphene is considered as the most promising candidate for growing or anchoring metal nanoparticles because of its high surface area (2630 m2 g−1), high conductivity, and superior chemical inertness.7,8 Moreover, theoretical research based on first-principles calculations indicates that metal atoms can interact with and thereby bind with the sp2-hybridized carbons of graphene because more interaction states and transmission channels are generated between both.3,9–11 This implies that graphene may be an ideal substrate for growing and anchoring metal nanoparticles for high-performance electrocatalytic or electrochemical devices.The Pt-based bimetallic nanostructures have attracted considerable attention in recent years because these materials are highly promising catalysts.12–14 In comparison with monometallic Pt nanostructures, Pt-based bimetallic nanostructures show enhanced catalytic activities.1,13,15 The catalytic properties of the bimetallic nanostructures are dependent on their sizes, shapes, and compositions. Fine control of the structural features, sizes, and compositions is highly favorable for improving catalytic performance Pt-based catalysts.13,14 Among various Pt-based bimetallic nanostructures, Pt–Pd bimetallic nanostructures are considered as the most promising metallic nanostructures due to their superior catalytic activity.16 Up to now, there are numerous studies have been reported13,14,17–27 by controlling the morphology and composition to improve the catalytic activity of Pt–Pd bimetallic nanocatalyst. It is well known that the catalytic activities of noble metal particles are highly size dependent.4 For instance, ultrafine Pd nanoparticles with a size of 3.5 nm supported on graphene oxide (GO) reported by Xie's group have displayed enhanced electrocatalytic activity for formic acid and ethanol electrooxidation.2 Tang's group demonstrated that 1.8 nm Au clusters could be supported on reduced GO (RGO), which displayed high electrocatalytic performance towards oxygen reduction reactions.7 Mao's group reported electroless deposition of 4.0 nm Pd clusters on graphdiyne oxides with high catalytic activity for the reduction of 4-nitrophenol.3 Recently, a “soft” nitriding method to grow in situ 1.6 nm Au clusters on carbon reported by He's group exhibit superior activity for methanol oxidation.4 However, it is still highly desirable to develop a facile and general approach for the fabrication of ultrafine bimetallic nanocatalysts such as Pd–Pt clusters with ultrasmall sizes (≤2 nm) though some monometallic clusters supported on carbon materials have been synthesized by different approaches if considering the potential industrial applications.
Herein, we report a facile aqueous approach (Scheme 1) to prepare 2.0 nm Pd–Pt bimetallic nanoclusters supported on β-cyclodextrin functionalized reduced graphene oxide (β-CD-RGO) at room temperature within 30 min without the need for any organic solvent or high temperature. The introduction of β-CD is used to immobilize Pd–Pt clusters on RGO and to avoid aggregation. Most importantly, it was found that the as-prepared Pd–Pt@β-CD-RGO nanohybrid exhibited higher electrocatalytic activities towards methanol and ethanol oxidation.
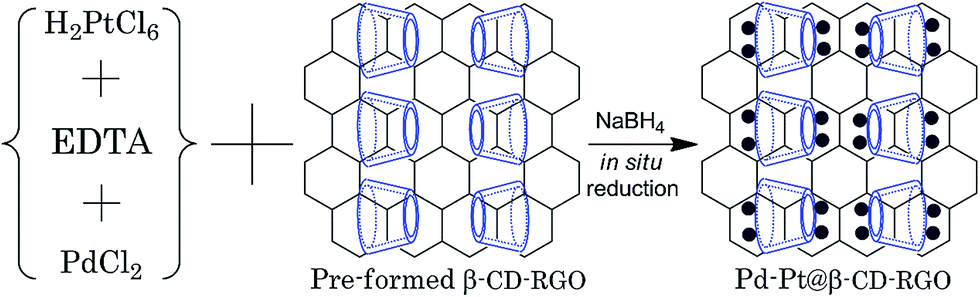 | ||
| Scheme 1 Illustration for the preparation of the Pd–Pt@β-CD-RGO nanohybrid using the in situ reduction method. | ||
2. Experimental
2.1. Chemicals and materials
Graphite oxide was purchased from Nanjing XFNANO Materials Tech Co., Ltd (Nanjing, China). The PdCl2 (99.99%), H2PtCl6 (99.99%), Nafion (5.0 wt%), and commercial Pd/C (10 wt%) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). The β-CD and ethylene diamine tetraacetic acid disodium salt (EDTA) were obtained from Aladdin Chemical Reagent Lo., Ltd (Shanghai, China). All other reagents were of analytical grade and used without further purification. Milli-Q ultrapure water (Millipore, ≥18.25 MΩ cm) was used throughout the experiments.2.2. Preparation of the β-CD-RGO nanocomposite
Graphene oxide (GO) sheets were exfoliated from graphite oxide by ultrasonication in ultrapure water at room temperature for 1 h. A water-based wet-chemical route was adopted to prepare the β-CD-RGO composite. In a typical experiment, 20 mL of 1.0 mg mL−1 β-CD aqueous solution was mixed with 20 mL of 0.5 mg mL−1 GO aqueous suspension and stirred at room temperature for 30 min. After that, a small amount of NaOH aqueous solution (1.0 M) was used to adjust the pH of the mixture to 12.0. Finally, the suspension was transferred to a flask and stirred at 90 °C in an oil bath for 4.0 h. After the reaction, the stable black suspension of the β-CD-RGO mixture was centrifuged at a relative centrifugal force (RCF) of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600g. After washing with ultrapure water for three times, the resulting β-CD-RGO composite was collected by freeze-drying. Additionally, the RGO was prepared with the similar procedure in the absence of β-CD.
600g. After washing with ultrapure water for three times, the resulting β-CD-RGO composite was collected by freeze-drying. Additionally, the RGO was prepared with the similar procedure in the absence of β-CD.
2.3. Synthesis of Pd–Pt@β-CD-RGO nanohybrid
Initially, a certain amount of β-CD-RGO freeze-dried powder was dispersed in ultrapure water with the aid of ultrasonication to obtain a concentration of 1.0 mg mL−1 β-CD-RGO homogeneous suspension. In a typical synthesis of Pd–Pt@β-CD-RGO, 3.0 mL of 10.0 mM PdCl2, 3.0 mL of 10.0 mM H2PtCl6, and 2.0 mL of 0.1 M EDTA were mixed together and stirred to form a homogeneous solution. After adjusting the solution pH to 9.0, twenty milliliter of 1.0 mg mL−1 β-CD-RGO aqueous suspension was added into the above mixture and stirred for 5 min. Then, three milliliter of 200 mM sodium borohydride solution was added to the mixture and stirred for 30 min. After centrifuging and washing with ultrapure water for three times, the obtained products were collected by freeze-drying. In addition, the Pd–Pt@RGO, Pd@β-CD-RGO, and Pt@β-CD-RGO were prepared with the similar procedure.2.4. Material characterization
The morphologies of the prepared samples were characterized by a JEM 2100 transmission electron microscopy (TEM, JEOL, Japan) with an accelerating voltage of 200 kV. High-resolution TEM (HRTEM) measurements, high-angle annular dark-field scanning TEM (HAADF-STEM), and elemental mapping were made on a JEM-2100F HRTEM (JEOL, Japan) operating at 200 kV. The samples for TEM and HRTEM characterization were prepared by depositing a drop of the diluted suspension on a copper grid coated by amorphous carbon. X-ray diffraction (XRD) patterns were recorded on a D8 ADVANCE (BRUKER, Germany) diffractometer using Cu-Kα radiation with a Ni filter (λ = 0.154059 nm at 30 kV and 15 mA). X-ray photoelectron spectroscopy (XPS) measurements were performed with Al Kα X-ray radiation as the X-ray source for excitation, which were carried out on an ESCALAB-MKII spectrometer (VG Co., United Kingdom). Fourier transform infrared (FTIR) study was performed over the wavenumber, range of 4000–400 cm−1 by a Thermo Fisher SCIENTIFIC Nicolet IS10 (Massachusetts, USA) FTIR impact 410 spectrophotometer using KBr pellets. Thermogravimetric analysis (TGA) was carried out on a Q50 TGA (TA Instruments, New Castle, USA), at a heating rate of 5 °C min−1 from 35 to 800 °C in nitrogen.2.5. Electrochemical measurements
All the electrochemical measurements were carried out with a CHI 660E Electrochemical Workstation from Chenhua Instrument (Shanghai, China) and conducted using a three-electrode system, with the modified glassy carbon electrode (GCE, 3 mm in diameter) as working electrode, a platinum wire as the counter electrode, a saturated calomel electrode as the reference electrode. Before the modifications, GCE was polished carefully with 0.3 and 0.05 μm Al2O3 powder to obtain a mirror-like surface, and then followed by sonication in ethanol and ultrapure water respectively. Then, the GCE was coated with the as-produced samples (5.0 μg) and dried at room temperature. Finally, 5.0 μl of Nafion (0.1 wt%) was coated on the surface of the modified GCE and dried before electrochemical experiments. Methanol and ethanol electro-oxidation measurements were performed in a solution of 1.0 M KOH containing 1.0 M methanol or 1.0 M ethanol at a scan rate of 50 mV s−1. The methanol or ethanol solution was deoxygenated with N2 bubbling for 30 min before electrochemical experiments. The electrochemically active surface area (ECSA) of each sample was estimated by cyclic voltammetry (CV) measurement carried out in 0.5 M H2SO4 solution under N2 protection with a sweep rate of 50 mV s−1. Mass current densities were normalized by the loaded Pt + Pd amount.3. Results and discussion
The β-CD-RGO composite was successfully prepared as proved by FTIR and TGA characterizations. As shown in Fig. S1,† the β-CD displayed the typical CD absorption features. It is found that the FTIR spectrum of RGO is featureless except the stretching vibrations of –OH (3400 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C conjugation (1630 cm−1), and C–C band (1190 cm−1), etc. Whereas the FTIR spectra of β-CD-RGO exhibit the typical CD absorption features of the coupled C–O/C–C stretching/O–H bending vibrations at 1032 cm−1, the coupled C–O–C stretching/O–H bending vibrations at 1152 cm−1, the C–H/O–H bending vibrations at 1410 cm−1, the CH2 stretching vibrations at 2925 cm−1, and O–H stretching vibrations at 3400 cm−1. The presence of these peaks confirmed that the β-CD molecules were successfully attached to the surface of the RGO.28 Additionally, the TGA was used to determine the amount of β-CD molecules on the surface of RGO. As shown in Fig. S2,† the β-CD showed an abrupt mass loss when the temperature was approximately 260 °C. For the pristine RGO, the loss in mass (17.0 wt%) at a temperature of approximately 600 °C was ascribed to the pyrolysis of the remaining oxygen-containing functional groups in RGO.28 The β-CD@RGO composite revealed an abrupt mass loss when the temperature was approximately 260 °C because of the decomposition of β-CD;29 the mass loss reached about 45.0 wt% when the temperature was 600 °C. The amount of β-CD grafted to RGO was estimated to be approximately 28.0 wt%.
C conjugation (1630 cm−1), and C–C band (1190 cm−1), etc. Whereas the FTIR spectra of β-CD-RGO exhibit the typical CD absorption features of the coupled C–O/C–C stretching/O–H bending vibrations at 1032 cm−1, the coupled C–O–C stretching/O–H bending vibrations at 1152 cm−1, the C–H/O–H bending vibrations at 1410 cm−1, the CH2 stretching vibrations at 2925 cm−1, and O–H stretching vibrations at 3400 cm−1. The presence of these peaks confirmed that the β-CD molecules were successfully attached to the surface of the RGO.28 Additionally, the TGA was used to determine the amount of β-CD molecules on the surface of RGO. As shown in Fig. S2,† the β-CD showed an abrupt mass loss when the temperature was approximately 260 °C. For the pristine RGO, the loss in mass (17.0 wt%) at a temperature of approximately 600 °C was ascribed to the pyrolysis of the remaining oxygen-containing functional groups in RGO.28 The β-CD@RGO composite revealed an abrupt mass loss when the temperature was approximately 260 °C because of the decomposition of β-CD;29 the mass loss reached about 45.0 wt% when the temperature was 600 °C. The amount of β-CD grafted to RGO was estimated to be approximately 28.0 wt%.
The Pd–Pt@β-CD-RGO and the related materials were characterized by TEM. As shown in Fig. 1A, the TEM image of Pd@β-CD-RGO indicates that the Pd clusters with uniform size could be obtained by using β-CD-RGO as the support. The high-resolution TEM (HRTEM) image (Fig. 1E) shows that the size of the Pd cluster is approximately 2.1 ± 0.3 nm with narrow size distribution, as demonstrated by the size distribution histogram (Fig. 1G). The well-dispersive Pd clusters might be attributed to abundant –OH groups on β-CD that are favorable for the coordination of cationic Pd2+ precursor via electrostatic interactions, followed by the in situ formation of the corresponding clusters on the surface of RGO. A similar function of –OH groups was also reported by Zhang et al. for the immobilization of Pd nanoparticles on hydroxypropyl-β-CD modified C60.30 The TEM image of Pt@β-CD-RGO was illustrated in Fig. 1B. However, the Pt clusters showed an increased size with serious aggregation when using anionic PtCl62− precursor. Thus, the nucleation of nanocatalysts on the β-CD-RGO is somewhat dependent on electrostatic interactions. These results are similar to that reported by Liu et al. using nitrided carbon as support to load metal clusters.4 In their work, the positive charged N-doped carbon could load and stabilize Au clusters to avoid overgrowth due to its strong affinity to anionic AuCl4−. Fig. 1C shows the representative TEM image of the Pd–Pt@β-CD-RGO nanohybrid. It can be seen that the Pd–Pt bimetallic nanoclusters with a uniform size are well monodispersed on the surface of β-CD-RGO. The HRTEM image (Fig. 1F) reveals that the size of the Pd cluster is approximately 2.0 ± 0.2 nm with narrow size distribution, as proved by the size distribution histogram (Fig. 1H). The size of Pd–Pt in the bimetallic Pd–Pt@β-CD-RGO material is also very small. It is well known that Pd and Pt are very easy to form an alloy through the co-reduction of Pd and Pt salts in the solution phase because they have a very small lattice mismatch of only 0.77% and similar redox potential Pd2+/Pd (0.62 V versus RHE); PtCl62−/Pt (0.74 V versus RHE).20 Thus, the very small size and uniform dispersibility of Pd–Pt clusters in the Pd–Pt@β-CD-RGO material may be ascribed to the fact that the Pd and Pt formed an alloy or heterostructure. The HRTEM image shows that the Pd–Pt clusters contain defects (several steps, corners, and edges), which can act as highly active sites in catalytic reactions.13,20 In order to further investigate the effect of surface properties of the support on the formation of Pd–Pt clusters, a control experiment was carried out by using RGO as support under the same conditions except for the absence of β-CD. As shown in Fig. 1D, the TEM image of Pd–Pt@RGO reveals that the Pd–Pt bimetallic clusters exhibit an obviously increased size and serious aggregations. This confirms that β-CD plays a key role in controlling the size and the monodispersion of the Pd–Pt clusters. Therefore, well-dispersive Pd–Pt clusters with a uniform size and a narrow distribution could be obtained using β-CD-RGO as the composite support.
 | ||
| Fig. 1 TEM images of Pd@β-CD-RGO (A), Pt@β-CD-RGO (B), Pd–Pt@β-CD-RGO (C), and Pd–Pt@RGO (D); the HRTEM images of Pd@β-CD-RGO (E) and Pd–Pt@β-CD-RGO (F); the corresponding size distribution histograms of Pd@β-CD-RGO (G) and Pd–Pt@β-CD-RGO (H). | ||
The crystal structures of the Pd@β-CD-RGO, Pt@β-CD-RGO, and Pd–Pt@β-CD-RGO composites were investigated by wide-angle X-ray diffraction XRD. The XRD patterns (Fig. 2A) of the synthesized materials show four peaks corresponding to {111}, {200}, {220}, and {311} of face-centered cubic (fcc) Pd/Pt. The ratio (3.3) between the intensities of the {111} and {200} peaks is higher than the value reported for some conventional samples (approximately 2.2),31,32 indicating that the Pd–Pt clusters are mainly covered with {111} facets. Moreover, a broad diffraction peak (002) centered at 2θ = 23.9° was observed, which was ascribed to the reduction of GO.33 The corresponding energy dispersive X-ray (EDX) spectra revealed the peaks of C, O, Pd, and Pt elements, indicating the existence of bimetallic Pd–Pt clusters on the surface of β-CD-RGO (Fig. 2B). The Pd–Pt@β-CD-RGO composite was also characterized by high-angle annular dark-field scanning TEM (HAADF-STEM) (Fig. 2C). It is difficult to see the Pd–Pt clusters in the Pd–Pt@β-CD-RGO composite due to small size. The positional distribution of Pd, Pt, C, and O in the Pd–Pt@β-CD-RGO was revealed by element mapping analysis. Fig. 3A–D show that the Pd, Pt, C, and O elements are uniformly distributed in the Pd–Pt@β-CD-RGO composite. The Pd–Pt nanostructures are usually alloy structure14 or heterostructure13 in published works. However, it is difficult to distinguish whether the present Pd–Pt cluster is alloy structure due to the too small size. This can be ascribed to the fact that resolution of the instrument is not enough to distinguish the too small Pd–Pt clusters.
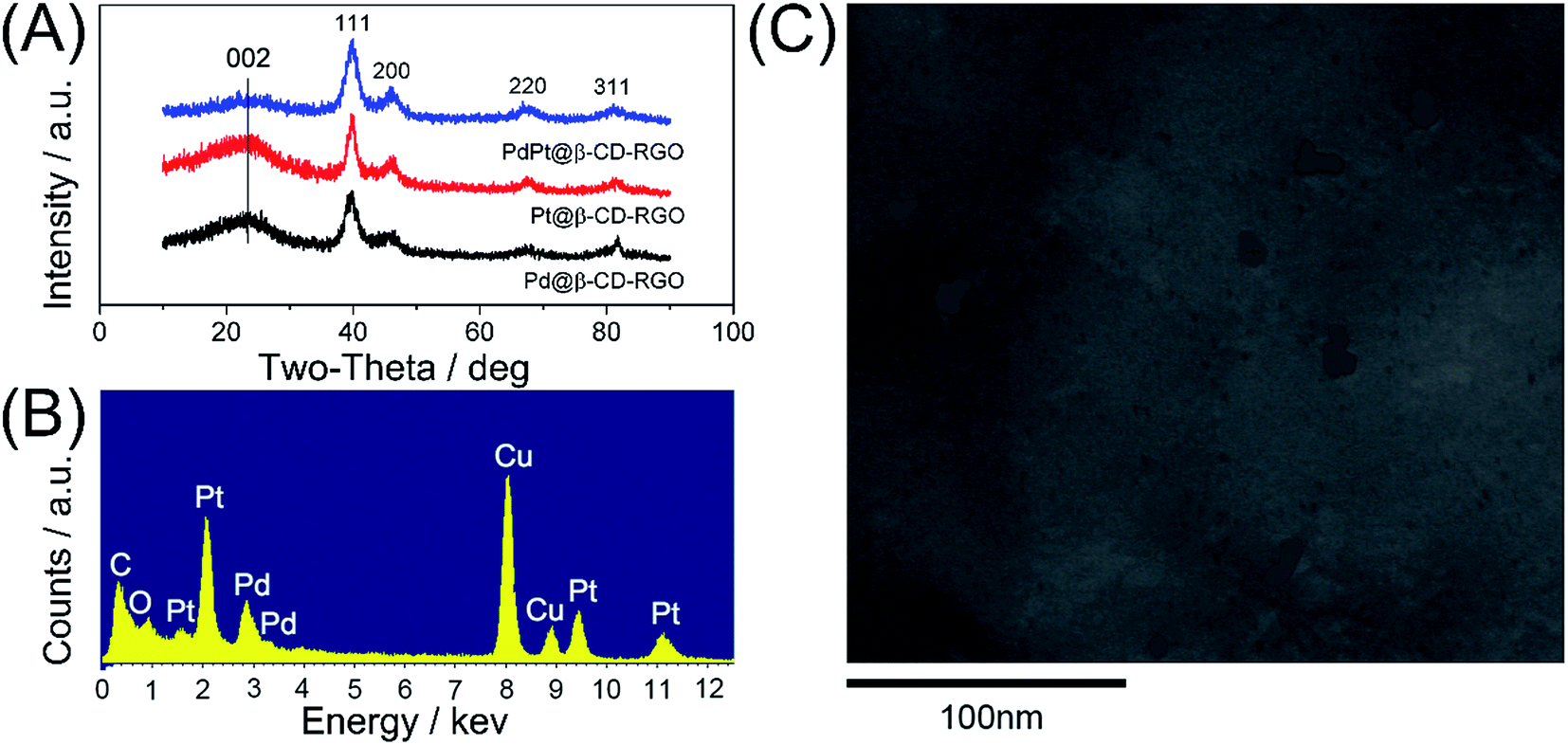 | ||
| Fig. 2 (A) XRD patterns of Pd@β-CD-RGO, Pt@β-CD-RGO, and Pd–Pt@β-CD-RGO. (B) EDX of spectrum of Pd–Pt@β-CD-RGO. (C) HAADF-STEM image of Pd–Pt@β-CD-RGO. | ||
 | ||
| Fig. 3 The elemental maps of C (A), O (B), Pd (C), and Pt (D) in Pd–Pt@β-CD-RGO. | ||
XPS analysis was used to study the electronic structure and compositions of the Pd–Pt@β-CD-RGO composite. As shown in Fig. 4A, the survey spectrum confirms the presence of Pd, Pt, C, and O in the Pd–Pt@β-CD-RGO composite. The Na signal is attributed to residual NaOH. Fig. 4B is the C 1s spectrum, which reveals that there are three components of carbon bond, namely sp2 C (CC, 284.6 eV), sp3 C (C–C, 285.3 eV), and C–O (286.8 eV) are presented in the composite.34–36 The high-resolution spectrum of Pd 3d is illustrated in Fig. 4C. The peaks centered at 335.2 and 340.5 eV can be assigned to 3d5/2 and 3d3/2 peaks, respectively, which are attributed to metallic Pd.2,20,37 In addition, the spectrum of Pt 4f is depicted in Fig. 4D. The 4f7/2 and 4f5/2 peaks at approximately 71.2 and 74.5 eV are ascribed to metallic Pt.20
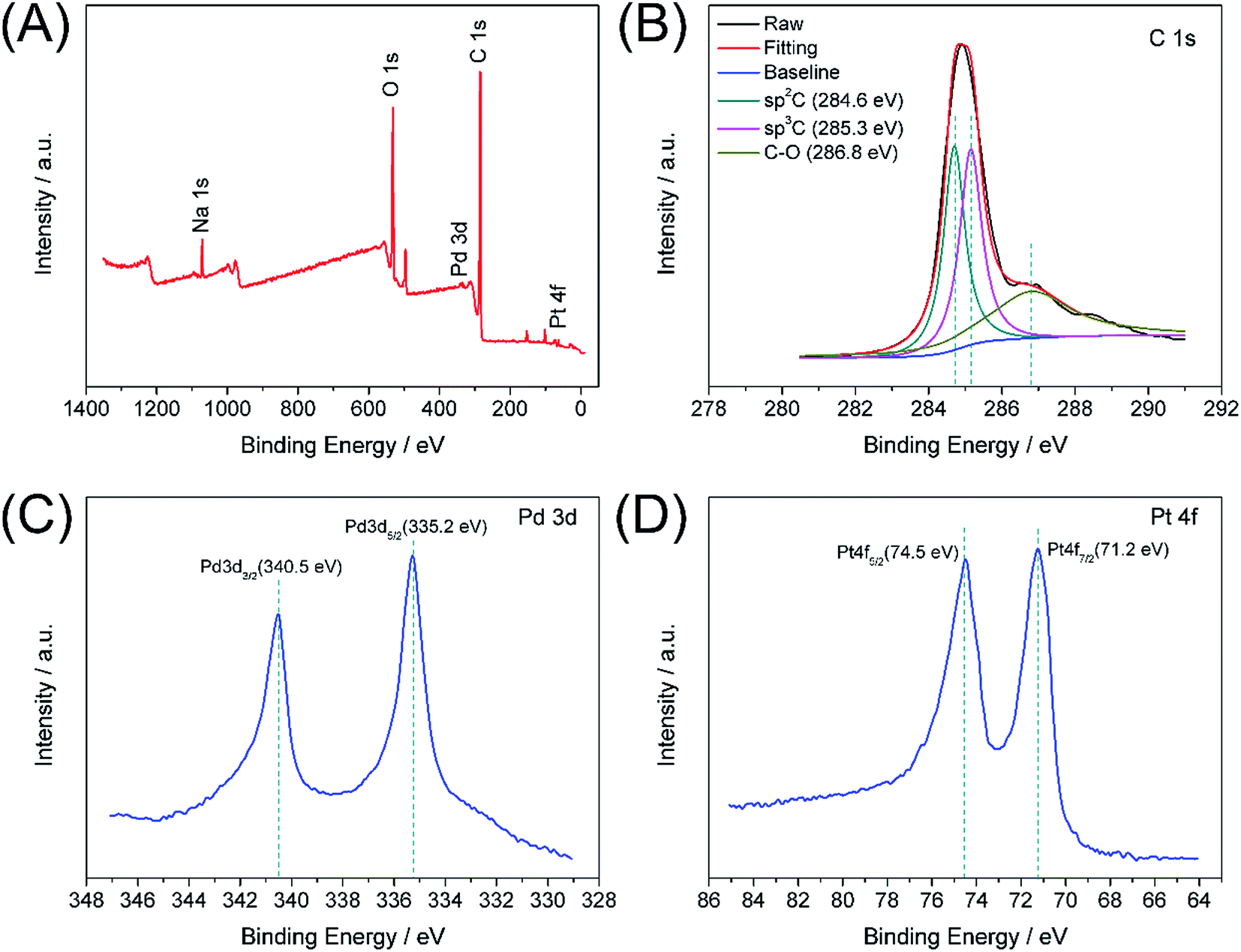 | ||
| Fig. 4 XPS survey spectrum of Pd–Pt@β-CD-RGO (A); the narrow spectra of C 1s (B), Pd 3d (C), and Pt 4f (D). | ||
The electrochemical active surface areas (ECSAs) of the Pd–Pt@β-CD-RGO, Pd–Pt@RGO, Pd@β-CD-RGO, Pt@β-CD-RGO, and commercial Pd/C catalysts were determined by the CV measurements in the N2 saturated electrolyte of 0.5 M H2SO4 at a scanning rate of 50 mV s−1. As shown in Fig. S3,† all the CV curves exhibit obvious multiple peaks in the lower potential range of −0.3 to 0 V associated with hydrogen adsorption/desorption and Pt/Pd oxide formation/reduction. The anodic peaks at the higher potential approximately 0.6 V and its cathodic counterparts in the potential range of 0.45 to 0.33 V are due to the Pt/Pd oxide formation/reduction. The ECSAs were estimated by the integrated charge (QH) in the hydrogen desorption region after deduction of the double-layer region. On the basis of the assumption that the charge density for the formation of a fully covered Pd(OH)2 monolayer was 430 μC cm−2, according to the equation ECSA = QH/(430 mC cm−2 × Pd loading), the ECSA for Pd/C and Pd@β-CD-RGO were 27.9 and 38.0 m2 g−1, respectively. According to the equation ECSA = QH/(210 mC cm−2 × Pt loading), the ECSA of Pt@β-CD-RGO was 21.4 m2 g−1. According to the equation ECSA = QH/(210 mC cm−2 × Pt + Pd loading), the ECSA of Pd–Pt@β-CD-RGO and Pd–Pt@RGO were 51.0 and 41.3 m2 g−1, respectively. The higher ECSA of Pd–Pt@β-CD-RGO is favorable to the electrocatalytic activity for methanol or ethanol oxidation.
The catalytic performance of the Pd–Pt@β-CD-RGO toward methanol oxidation reaction (MOR) was investigated with three-electrode system in the N2 saturated electrolytes of 1.0 M KOH and 1.0 M CH3OH at a scan rate of 50 mV s−1 at room temperature. For comparison, the catalytic performance of the Pd–Pt@RGO, Pd@β-CD-RGO, Pt@β-CD-RGO, and commercial Pd/C was also tested. The currents were normalized to the loading amount of Pt + Pd in order to compare the mass activity of different catalyst. As shown in Fig. 5A, the Pd–Pt@β-CD-RGO exhibits the highest catalytic activity with a mass-normalized current density of 1609 mA mg−1, which is approximately 1.5, 2.7, 3.8, and 5.3 times greater than that of Pd–Pt@RGO (1077 mA mg−1), Pd@β-CD-RGO (595 mA mg−1), Pt@β-CD-RGO (426 mA mg−1), and commercial Pd/C (305 mA mg−1) catalyst, respectively. The detailed electrochemical parameters of the as-prepared catalysts obtained from the CV curves were listed in Table 1. It should be noted that the specific activity of Pd–Pt@β-CD-RGO (3.15 mA cm−2) was also higher than that of Pd–Pt@RGO (2.60 mA cm−2), Pd@β-CD-RGO (1.57 mA cm−2), Pt@β-CD-RGO (1.99 mA cm−2), and commercial Pd/C (1.09 mA cm−2). Moreover, at the same current density in the positive scan (as indicated by the dashed line in inset of Fig. 5A), the corresponding oxidation potential on the Pd–Pt@β-CD-RGO was the lowest, suggesting the enhancement of the methanol oxidation kinetics on the Pd–Pt@β-CD-RGO.13 The improved electrooxidation kinetics of the Pd–Pt@β-CD-RGO may originate from the bimetallic Pd–Pt composition and the too small size (2.0 nm). In addition, the ratio of the mass current density values in two sequential forward and backward sweeps (Jf/Jb), (as considered to be an important indicator of the catalyst tolerance to poisoning species14) showed significant differences for these catalysts. A higher ratio indicates more effective removal of the poisoning species on the catalyst surface.38 The Jf/Jb values were 5.8, 5.6, 2.4, 2.3, and 2.7 for Pd–Pt@β-CD-RGO, Pd–Pt@RGO, Pd@β-CD-RGO, Pt@β-CD-RGO, and commercial Pd/C, respectively. Thus, the Pd–Pt@β-CD-RGO exhibited best tolerance to the poisoning species than other and the Pd/C catalysts.
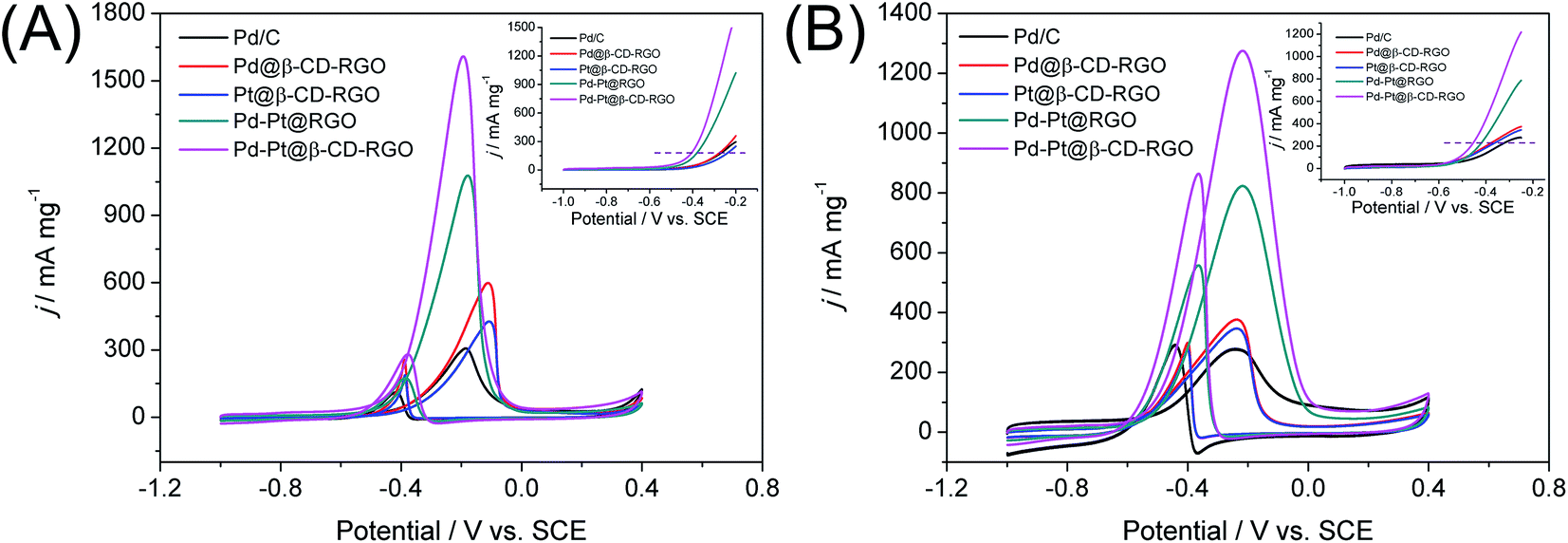 | ||
| Fig. 5 Stable CV curves for methanol (A) and ethanol (B) oxidation reactions catalyzed by Pd/C, Pd@β-CD-RGO, Pt@β-CD-RGO, Pd–Pt@RGO, and Pd–Pt@β-CD-RGO in a solution of 1.0 M KOH containing 1.0 M methanol or 1.0 M ethanol at a scan rate of 50 mV s−1. | ||
| Catalyst | Potential (V) | ECSA (m2 g−1) | Mass activity (mA mg−1) | Specific activity (mA cm−2) | |||
|---|---|---|---|---|---|---|---|
| Methanol | Ethanol | Methanol | Ethanol | Methanol | Ethanol | ||
| Pd/C | −0.20 | −0.23 | 27.9 | 305 | 272 | 1.09 | 0.97 |
| Pd@β-CD-RGO | −0.08 | −0.23 | 38.0 | 595 | 376 | 1.57 | 0.99 |
| Pt@β-CD-RGO | −0.10 | −0.23 | 21.4 | 426 | 346 | 1.99 | 1.62 |
| Pd–Pt@RGO | −0.20 | −0.24 | 41.3 | 1077 | 820 | 2.60 | 1.98 |
| Pd–Pt@β-CD-RGO | −0.24 | −0.24 | 51.0 | 1609 | 1274 | 3.15 | 2.50 |
The Pd–Pt@β-CD-RGO, Pd–Pt@RGO, Pd@β-CD-RGO, Pt@β-CD-RGO, and commercial Pd/C were further studied for ethanol oxidation reaction (EOR) in 1.0 M KOH containing 1.0 M C2H5OH under N2 protection. As illustrated in Fig. 5B, the Pd–Pt@β-CD-RGO also displayed the highest catalytic activity with a mass current density of 1274 mA mg−1, which is 1.6, 3.4, 3.7, and 4.6 times higher than that of Pd–Pt@RGO (820 mA mg−1), Pd@β-CD-RGO (376 mA mg−1), Pt@β-CD-RGO (346 mA mg−1), and Pd/C (272 mA mg−1) catalyst, respectively. The catalytic activity follows the order: Pd–Pt@β-CD-RGO > Pd–Pt@RGO > Pd@β-CD-RGO > Pt@β-CD-RGO > Pd/C, which is similar to the order of MOR. As shown in Table 1, the specific activity for EOR of Pd–Pt@β-CD-RGO (2.50 mA cm−2) was also higher than that of Pd–Pt@RGO (1.98 mA cm−2), Pd@β-CD-RGO (0.99 mA cm−2), Pt@β-CD-RGO (1.62 mA cm−2), and commercial Pd/C (0.97 mA cm−2). The onset potential (inset of Fig. 5B) of Pd–Pt@β-CD-RGO was more negative than those of other catalysts, also indicating the higher electrocatalytic performance of the Pd–Pt@β-CD-RGO toward ethanol oxidation. For the EOR, the Jf/Jb value of Pd–Pt@β-CD-RGO (1.5) was also higher that of Pd–Pt@RGO (1.4), Pd@β-CD-RGO (1.2), Pt@β-CD-RGO (1.3), and Pd/C (0.9), suggesting the better tolerance of Pd–Pt@β-CD-RGO to the poisoning species than other catalysts. In addition, the activity of the Pd–Pt@β-CD-RGO catalyst is also higher than those of recently reported Pd–Pt bimetallic catalysts as summarized in Table 2.
| Catalysts | Shapes of Pd–Pt | Potential (V) | ECSA (m2 g−1) | Specific activity (mA cm−2) | Ref. |
|---|---|---|---|---|---|
| PdPt nanoalloys | Islands | 0.60 | 21.7 | 1.08 | 20 |
| Pt–Pd | Cubes | 0.85 | — | 1.49 | 14 |
| Pt-on-Pd | Nanodendrites | 0.70 | 48.0 | 1.04 | 13 |
| Pd@Pt | Porous particles | 0.70 | 38.2 | 1.31 | 24 |
| Pt–Pd@β-CD-RGO | Clusters | −0.24 | 51.0 | 3.15 | This work |
The enhanced electrocatalytic activity of the Pd–Pt@β-CD-RGO could be attributed to three main factors: (a) the Pd–Pt clusters with very small size (2.0 nm) would offer more accessibility methanol or ethanol oxidation, thus remarkably improving the atomic utilization efficiency; (b) the excellent monodispersity and uniformity of Pd–Pt loaded on β-CD-RGO with high surface area might be favorable for improving the catalytic performance; (c) the formation of bimetallic surface atom arrangement (coexistence of surface Pd and Pt sites) and/or the synergistic effect originating from the coexistence of surface Pd and Pt atoms should also be emphasized. The TEM images of Pd/C were investigated and displayed in Fig. S4.† The size of Pd in Pd/C is approximately 7.5 ± 2.0 nm. From these results, we can conclude that decreasing the size of noble metal nanoparticles can significantly improve the catalytic activity.
The durability of Pd–Pt@β-CD-RGO and commercial Pd/C was compared in 1.0 M KOH solution containing 1.0 M methanol using chronoamperometry and CV. At a fixed potential, the intermediate products such as CO from the methanol oxidation would adsorb and accumulate on the electrode surface, which might lead to the catalysts inactivation and the rapid current attenuation.39 As shown in Fig. 6A, the current–time curves recorded at −0.2 V for 4000 s are obtained. The oxidation current on commercial Pd/C modified electrode decreases much more rapidly than that on Pd@β-CD-RGO, Pt@β-CD-RGO, Pd–Pt@RGO, and Pd–Pt@β-CD-RGO modified electrodes at the initial stage. The currents on these modified electrodes gradually decay and achieve a pseudo-steady state. Obviously, the Pd–Pt@β-CD-RGO catalyst retains the highest current than other catalysts for the whole time. This result confirms that Pd–Pt@β-CD-RGO catalyst has the higher poison tolerance and better electrochemical stability than commercial Pd/C. The electrocatalytic cycling stabilities of these catalysts and commercial Pd/C in 1.0 M KOH solution containing 1.0 M methanol have also been investigated using CV, as shown in Fig. 6B. It can be observed that the forward peak current density for methanol oxidation on Pd–Pt@β-CD-RGO modified electrode decreases by about 11.8% within the 400 scans. However, the peak current density on Pd–Pt@RGO, Pd@β-CD-RGO, Pt@β-CD-RGO, and commercial Pd/C modified electrodes declines about 17.6%, 39.8%, 32.2%, and 52.8% in the same scans. The good cycling stabilities may be attributed to the more durable nature of the {111} facet of Pd–Pt bimetallic nanoalloys and the synergetic effect of the Pd and Pt atoms,14,20 which has been reported in the MOR with Pd–Pt bimetallic electrocatalysts. The TEM image of Pd–Pt@β-CD-RGO after durability was obtained as shown in Fig. S5.† From the TEM image, we can see that part of the Pd–Pt nanoclusters have begun to aggregate. This aggregation may result in the decrease of the activity of the Pd–Pt@β-CD-RGO catalyst. This implies that the small size and uniform dispersibility are indeed very important for the catalytic activity of the catalyst. Thus, we will further study on this problem in our lab for improving the durability of such catalyst.
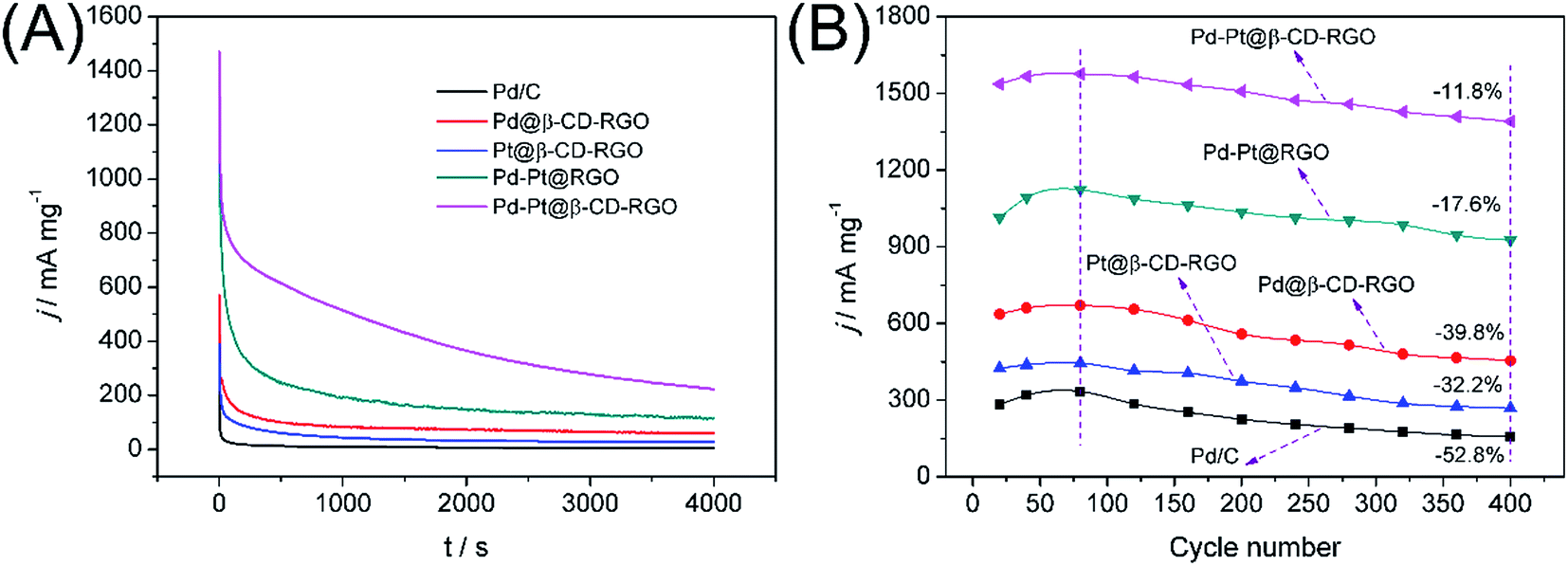 | ||
| Fig. 6 (A) Chronoamperometric curves for methanol oxidation reactions catalyzed by Pd/C, Pd@β-CD-RGO, Pt@β-CD-RGO, Pd–Pt@RGO, and Pd–Pt@β-CD-RGO recorded at −0.2 V (vs. SCE) for 4000 s in 1.0 M KOH containing 1.0 M methanol. (B) The durability performance of the Pd/C, Pd@β-CD-RGO, Pt@β-CD-RGO, Pd–Pt@RGO, and Pd–Pt@β-CD-RGO reflected by the forward peak current density based on 400 cycles in 1.0 M KOH containing 1.0 M methanol. | ||
4. Conclusions
In conclusion, we have developed a facile aqueous method to load Pd–Pt bimetallic nanoclusters with a uniform size of 2.0 nm on β-CD-RGO. The integration of β-CD and RGO was responsible for the formation of the monodispersed 2.0 nm Pd–Pt bimetallic clusters. The β-CD functionalization enhanced the affinity between supports and metal precursors, thus preventing aggregation and overgrowth of nanocrystals. Moreover, compared with commercial Pd/C catalyst, the β-CD-RGO supported Pd–Pt nanoclusters exhibited better electrocatalytic activities for methanol and ethanol oxidation. The Pd–Pt@β-CD-RGO nanocatalyst may also be used in other potential applications such as CO oxidation or hydrogenation reaction.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China under the Grant No. 51361028, 51161025, 51661033, and 31660538.Notes and references
- H.-H. Li, S.-Y. Ma, Q.-Q. Fu, X.-J. Liu, L. Wu and S.-H. Yu, J. Am. Chem. Soc., 2015, 137, 7862–7868 CrossRef CAS PubMed.
- X. M. Chen, G. H. Wu, J. M. Chen, X. Chen, Z. X. Xie and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed.
- H. T. Qi, P. Yu, Y. X. Wang, G. C. Han, H. B. Liu, Y. P. Yi, Y. L. Li and L. Q. Mao, J. Am. Chem. Soc., 2015, 137, 5260–5263 CrossRef CAS PubMed.
- B. Liu, H. Q. Yao, W. Q. Song, L. Jin, I. M. Mosa, J. F. Rusling, S. L. Suib and J. He, J. Am. Chem. Soc., 2016, 138, 4718–4721 CrossRef CAS PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. MacQuarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- Y. J. Wang, D. P. Wilkinson and J. Zhang, Chem. Rev., 2011, 111, 7625–7651 CrossRef CAS PubMed.
- H. J. Yin, H. J. Tang, D. Wang, Y. Gao and Z. Y. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS PubMed.
- H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, ACS Nano, 2012, 6, 2693–2703 CrossRef CAS PubMed.
- P. A. Khomyakov, G. Giovannetti, P. C. Rusu, G. Brocks, J. Van den Brink and P. Kelly, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 195425 CrossRef.
- Q. J. Wang and J. G. Che, Phys. Rev. Lett., 2009, 103, 066802 CrossRef CAS PubMed.
- I. Cabria, M. J. López and J. A. Alonso, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 035403 CrossRef.
- A. C. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- L. Wang, Y. Nemoto and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 9674–9677 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- S. J. Guo, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- B. Lim and Y. N. Xia, Angew. Chem., Int. Ed., 2011, 50, 76–85 CrossRef CAS PubMed.
- Q. Yuan, Z. Y. Zhou, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 1491–1493 RSC.
- X. Q. Huang, H. H. Zhang, C. Y. Guo, Z. Y. Zhou and N. F. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808–4812 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, W. Zhu, H.-S. Wu, Y.-W. Zhang and C.-H. Yan, Chem. Commun., 2012, 48, 543–545 RSC.
- D.-B. Huang, Q. Yuan, H.-H. Wang and Z.-Y. Zhou, Chem. Commun., 2014, 50, 13551–13554 RSC.
- B. Lim, J. G. Wang, P. H. C. Camargo, M. J. Jiang, M. J. Kim and Y. N. Xia, Nano Lett., 2008, 8, 2535–2540 CrossRef CAS PubMed.
- Y. Chen, J. Yang, Y. Yang, Z. Y. Peng, J. H. Li, T. Mei, J. Y. Wang, M. Hao, Y. L. Chen, W. L. Xiong, L. Zhang and X. B. Wang, Chem. Commun., 2015, 51, 10490–10493 RSC.
- W. Hong, C. S. Shang, J. Wang and E. K. Wang, Energy Environ. Sci., 2015, 8, 2910–2915 CAS.
- B. Jiang, C. L. Li, J. Henzie, T. Takei, Y. Bando and Y. Yamauchi, J. Mater. Chem. A, 2016, 4, 6465–6471 CAS.
- Y. Q. Li, C. L. Li, B. P. Bastakoti, J. Tang, B. Jiang, J. Kim, M. Shahabuddin, Y. Bando, J. H. Kim and Y. Yamauchi, J. Mater. Chem. A, 2016, 4, 9169–9176 CAS.
- B. Lim, M. J. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- Z. M. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542–7543 CrossRef CAS PubMed.
- Y. J. Guo, S. J. Guo, J. T. Ren, Y. M. Zhai, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 4001–4010 CrossRef CAS PubMed.
- G. B. Zhu, X. Zhang, P. B. Gai, X. H. Zhang and J. H. Chen, Nanoscale, 2012, 4, 5703–5709 RSC.
- Q. Zhang, Z. Y. Bai, M. Shi, L. Yang, J. L. Qiao and K. Jiang, Electrochim. Acta, 2015, 177, 113–117 CrossRef CAS.
- Q. B. Zhang, J. P. Xie, J. H. Yang and J. Y. Lee, ACS Nano, 2009, 3, 139–148 CrossRef CAS PubMed.
- Y. J. Xiong, J. M. McLellan, Y. D. Yin and Y. N. Xia, Angew. Chem., Int. Ed., 2007, 46, 790–794 CrossRef CAS PubMed.
- Z. Gao, W. L. Yang, J. Wang, H. J. Yan, Y. Yao, J. Ma, B. Wang, M. L. Zhang and L. H. Liu, Electrochim. Acta, 2013, 91, 185–194 CrossRef CAS.
- Z. Fan, W. Kai, J. Yan, T. Wei, L. Zhi, J. Feng, Y. Ren, L. Song and F. Wei, ACS Nano, 2011, 5, 191–198 CrossRef CAS PubMed.
- X. W. Mao, D. M. Tian and H. B. Li, Chem. Commun., 2012, 48, 4851–4853 RSC.
- D. H. Du, P. C. Li and J. Y. Ouyang, ACS Appl. Mater. Interfaces, 2015, 7, 26952–26958 CAS.
- G. T. Fu, L. Tao, M. Zhang, Y. Chen, Y. W. Tang, J. Lin and T. H. Lu, Nanoscale, 2013, 5, 8007–8014 RSC.
- M. Chen, Y. Meng, J. Zhou and G. W. Diao, J. Power Sources, 2014, 265, 110–117 CrossRef CAS.
- H. Huang, D. Sun and X. Wang, J. Phys. Chem. C, 2011, 115, 19405–19412 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24893d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |