
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZinc triflate-mediated cyclopropanation of oxindoles with vinyl diphenyl sulfonium triflate: a mild reaction with broad functional group compatibility†
Mingwei Zhouab,
Ke Ena,
Yimin Hu*b,
Yufang Xua,
Hong C. Shenb and
Xuhong Qian*a
aShanghai Key Laboratory of Chemical Biology, East China University of Science and Technology, Shanghai 200237, China. E-mail: xhqian@ecust.edu.cn
bRoche Pharmaceutical Research and Early Development, Roche Innovation Center Shanghai, Shanghai 201203, China. E-mail: yimin.hu@roche.com
First published on 13th January 2017
Abstract
The first use of zinc triflate for the cyclopropanation of unprotected oxindoles with vinyl diphenyl sulfonium triflate salt is reported. The reaction proceeded under ambient conditions and consistently provided high yields with broad functional group tolerability. The utility for the late-stage functionalization (LSF) of complex molecules is demonstrated.
Oxindole is a common motif in numerous molecules of great biological and medicinal interest (Fig. 1). Functionalization at the 3-position of oxindoles is a key lead derivatization strategy in medicinal chemistry. In particular, spirooxindoles have demonstrated many applications in drug discovery.1 Specifically, spirocyclopropyl oxindoles (Fig. 1: compound 4, 5, and 6) are important because such designs in drug discovery could introduce potentially favorable conformational restraint with marginal addition of molecular weight and lipophilicity. In the literature, different cyclopropanation strategies can provide access to spirocyclopropyl oxindoles.2 However, the potential of applying such strategies in the context of the late-stage functionalization3 (LSF) remains to be fully explored. There are limited choices of mild and selective conditions to tolerate aqueous environment, to be compatible with unprotected and sensitive functional groups such as amines, hydroxyl and carboxyl groups, and boronic acids, and to be suitable for functionalization of complex structures.4
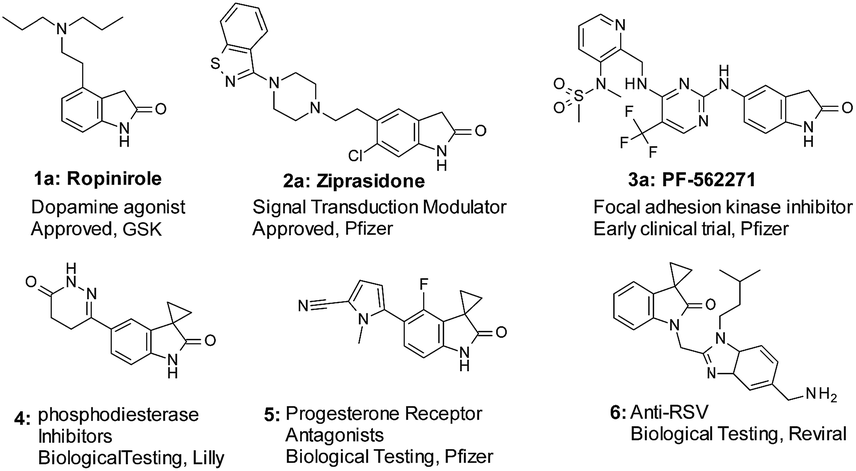 | ||
| Fig. 1 Oxindoles and spirocyclopropyl oxindoles of medicinal interests. | ||
One common cyclopropanation method involves the use of 1,2-dibromoethane. In general, a strong base, such as NaH or LiHMDS, is needed for this transformation.5 Application of such method often requires protection on oxindole nitrogen as well as other acidic functional groups. With unprotected oxindoles, over-alkylation on the nitrogen is a common side reaction. Over-alkylation can be used as a protection strategy. With unprotected oxindole 7a, Robertson obtained over-alkylated product 8a with excess reagents first.6 Subsequent treatment with magnesium achieved debromoethylation to afford cyclopropyl oxindole 9a, with an overall poor yield (Scheme 1). Recently, Marini reported a vinyl selenone-based approach for the synthesis of spirocyclopropyl oxindoles in aqueous sodium hydroxide solution.2e Limited choices of substitutions on the oxindole were reported. A strong base (NaOH) along with surfactants is necessary for optimal yields.
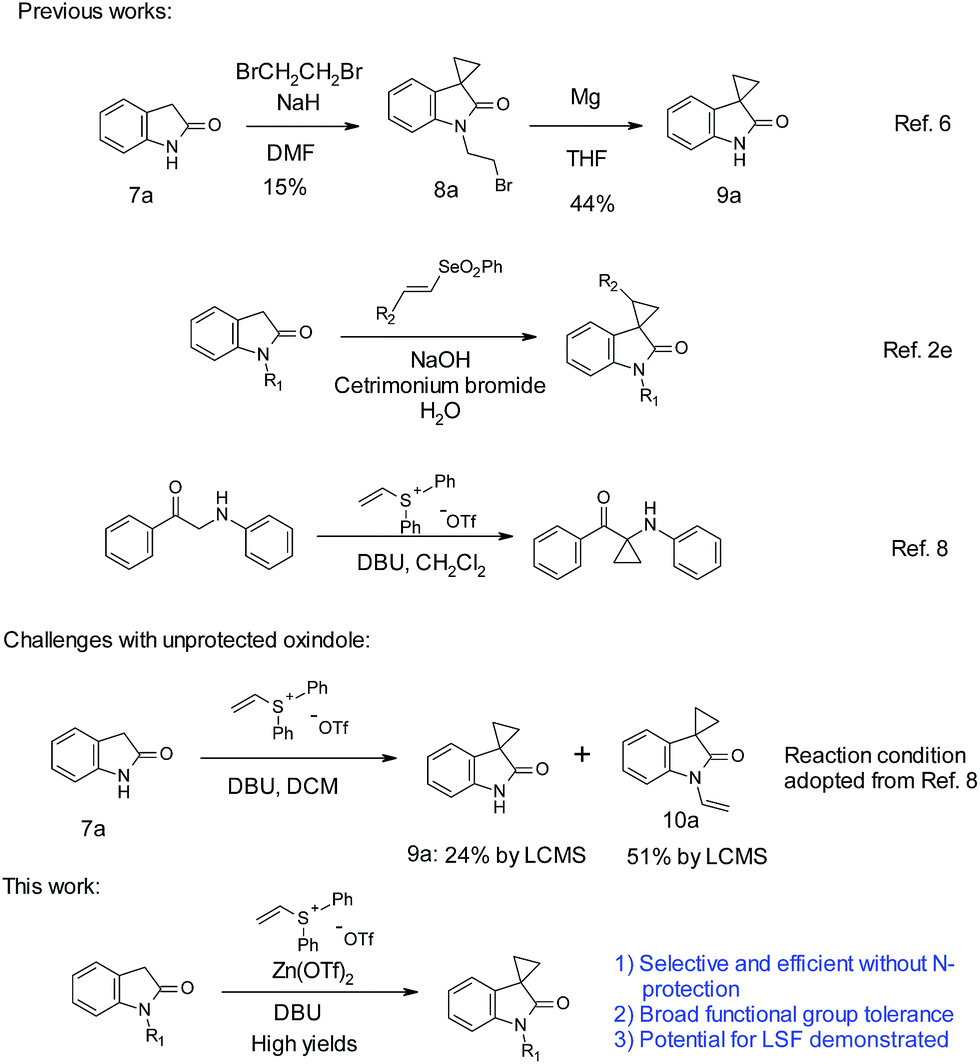 | ||
| Scheme 1 Cyclopropanation of unprotected oxindoles. | ||
On the other hand, vinyl sulfonium salts can be easily prepared with robust and safe protocols, and have demonstrated diverse applications in the construction of different carbo- and hetero-cycles.7 In 2012, Lin reported the use of vinyl sulfonium salts, with DBU as the base in dichloromethane, for the cyclopropanation of α-amino aryl ketones.8 In the case of unprotected oxindoles, due to the similar pKa of C3–H and N–H (∼18.2),9 selective C-3 functionalization could be more challenging than the case of α-amino aryl ketones. Indeed simple application of Lin's conditions with oxindole 7a did not produce satisfactory results. An incomplete conversion and more than 50% of over-alkylated product 10a were observed. During our optimization, we found that addition of Zn(OTf)2 promoted C-3 alkylation over N-alkylation. Herein, we report an efficient cyclopropanation for both N-nonsubstituted and N-substituted oxindoles with vinyl diphenylsulfonium triflate salt mediated by zinc triflate. The ambient reaction conditions are simple, mild, and devoid of alkali bases. We were pleased to find that this cyclopropanation could proceed efficiently with high yields, tolerating a variety of functional groups such as carboxylic acid, hydroxyl, amine, boronic acid, etc. The utility in LSF has also been demonstrated with three examples.
Optimization efforts with unprotected indole 7a are summarized in Table 1. As stated above, partial conversion and a 32% isolated yield of product 9a could be obtained by applying previously reported conditions, with DBU as the base and DCM as the solvent (entry 1). Screening of different bases and solvents did not produce any improvement on regioselectivity over N-alkylation (entries 2–5). A marginally improved yield of 38% was observed with a polar solvent such as DMF (entry 6). To our delight, addition of 2 equivalents of Mg(OTf)2 promoted the C-3 alkylation with a 56% yield of 9a (entry 7). Encouraged by this finding, various metal salts were screened. Zn(OTf)2 and Ca(OTf)2 gave the best yields (entry 8–12). Further refinement of conditions focused on adjusting the amounts and ratio of DBU and Zn(OTf)2 (entries 13–16). We found that reducing the amount of Zn(OTf)2 from 2 to 1 equivalent (entry 15 vs. 12) gave a clean reaction with full conversion. Over 90% of the desired product 9a was isolated with almost no N-alkylation product observed by LC/MS. The use of Zn(OAc)2 resulted in poorer yields of 9a. Different from Zn(OTf)2, reducing the equivalents of Ca(OTf)2 from 2 to 1 gave only a 50% isolated yield (entry 18 vs. 8).
|
||||
|---|---|---|---|---|
| Entry | Base (equiv.) | Additive (equiv.) | Solvent | Yield (%) |
| a Reaction conditions: 7a (0.2 mmol), vinyl diphenyl sulfonium triflate (0.24 mmol), with indicated amount of base and additive, 1 mL solvent, at room temperature (ca. 21 °C), under air, stirring for 4 h. Isolated yields are shown. | ||||
| 1 | DBU (3.0) | — | DCM | 32 |
| 2 | TEA (3.0) | — | DCM | 30 |
| 3 | DIPEA (3.0) | — | DCM | 27 |
| 4 | Pyridine (3.0) | — | DCM | 0 |
| 5 | Cs2CO3 (3.0) | — | DCM | <5 |
| 6 | DBU (3.0) | — | DMF | 38 |
| 7 | DBU (3.0) | Mg(OTf)2 (2.0) | DMF | 56 |
| 8 | DBU (3.0) | Ca(OTf)2 (2.0) | DMF | 68 |
| 9 | DBU (3.0) | Cu(OTf)2 (2.0.) | DMF | <5 |
| 10 | DBU (3.0) | Ag(OTf) (2.0) | DMF | <5 |
| 11 | DBU (3.0) | CuCl2 (2.0) | DMF | 55 |
| 12 | DBU (3.0) | Zn(OTf)2 (2.0) | DMF | 70 |
| 13 | DBU (2.0) | Zn(OTf)2 (1.0) | DMF | 78 |
| 14 | DBU (4.0) | Zn(OTf)2 (2.0) | DMF | 90 |
| 15 | DBU (3.0) | Zn(OTf)2 (1.0) | DMF | 91 |
| 16 | DBU (3.0) | Zn(OTf)2 (0.5) | DMF | 68 |
| 17 | DBU (3.0) | Zn(OAc)2 (1.0) | DMF | 46 |
| 18 | DBU (3.0) | Ca(OTf)2 (1.0) | DMF | 50 |
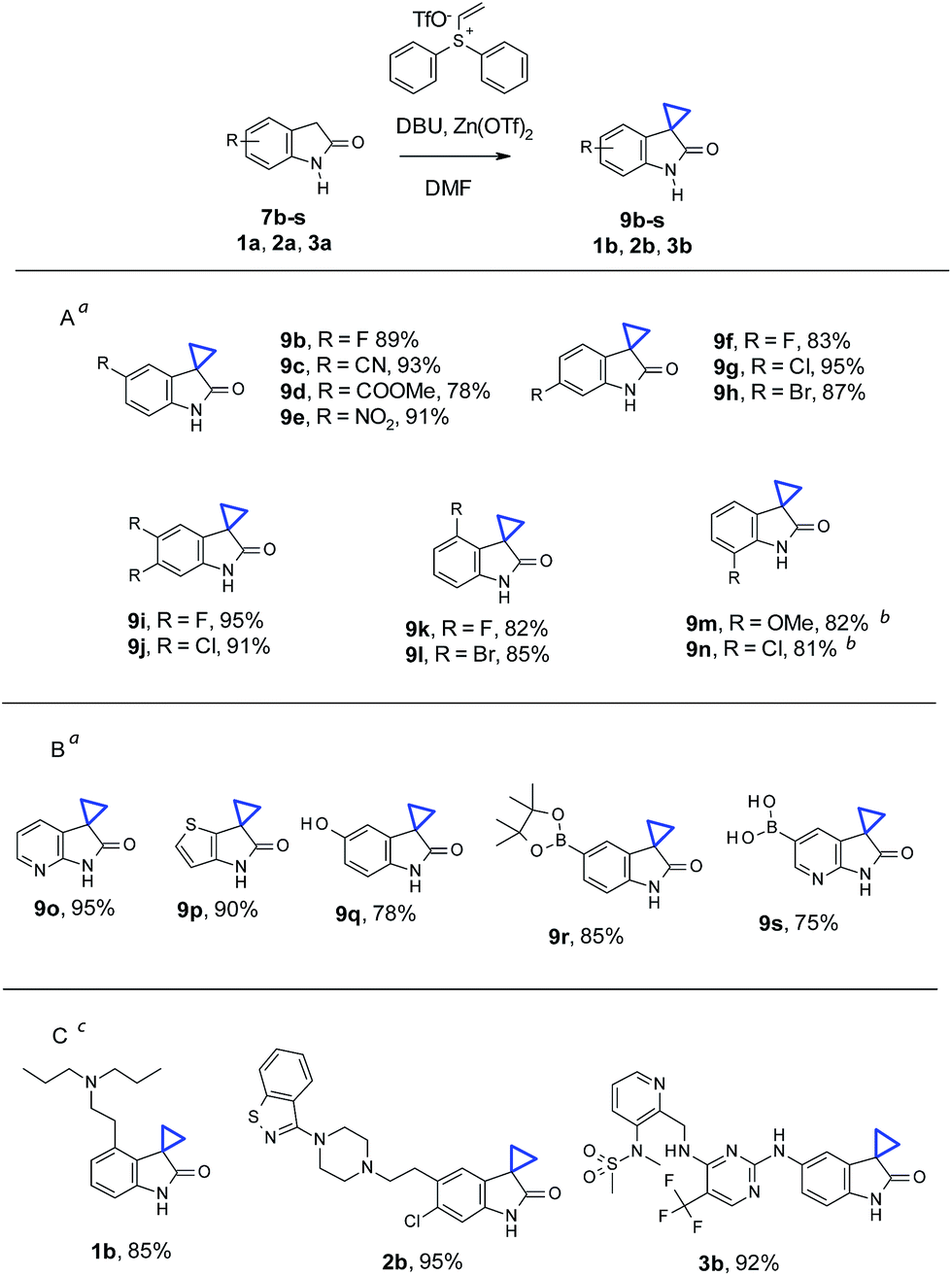
With the optimized conditions in hand, we then examined the scope of oxindole substrates (Table 2). As shown in Table 2A, reactions of substrates containing electron-rich or electron-withdrawing substituents afforded high yields of desired products. The substitution position of the oxindole had minimal influence on the reaction yields either, with the exception of 7m and 7n. 1.5 equivalents of Zn(OTf)2 were necessary to obtain full conversion and high yields of 9m and 9n. A broader substrate scope was demonstrated in Table 2B. Both electron deficient pyridine 9o and electron rich thiophene 9p were obtained in high yields. Remarkably, phenol 7q, boronic ester 7r, and even pyridyl boronic acid 7s, all underwent cyclopropanation to give the corresponding products 9q–s in good yields. Cyclopropanation on medicinally relevant and complex compounds was also performed to demonstrate the utility in LSF (Table 2C). For example, cyclopropanation of Ropinirole (1a), a dopamine agonist used in the treatment of Parkinson's disease and restless legs syndrome (RLS), and bearing a basic tertiary amine, proceeded under our standard conditions to form 1b in 85% yield. Ziprasidone (2a), an atypical antipsychotic containing a benzisothiazole found in many signal transduction modulators, also underwent a smooth cyclopropanation to give 2b in 95% yield. Finally, cyclopropanation of PF562271 (3a), a focal adhesion kinase inhibitor once in early clinical trials for treatment of cancer, with 1 active methylene group and 2 more unprotected NHs in addition to oxindole NH, reacted well to afford 3b in 92% yield. The above results, particularly in Tables 2B and C, demonstrated that chemistry described herein can be applicable for late stage cyclopropanation of important building blocks and complex drug-like molecules.
| a Reaction conditions for 7b–l, and 7o–s: oxindole (0.2 mmol), vinyl diphenyl sulfonium triflate (0.24 mmol), Zn(OTf)2 (0.2 mmol), DBU (0.6 mmol), in 1 mL DMF, at room temperature (ca. 21 °C), under air, stirring for 4 h.b For 7m and 7n: condition similar to 7b–l with 0.3 mmol of Zn(OTf)2 being used.c For 1a, 2a, and 3a: conditions similar to 7b–l with reaction time extended to 12 h. Isolated yields for all substrates described in the figure. |
|---|
 |
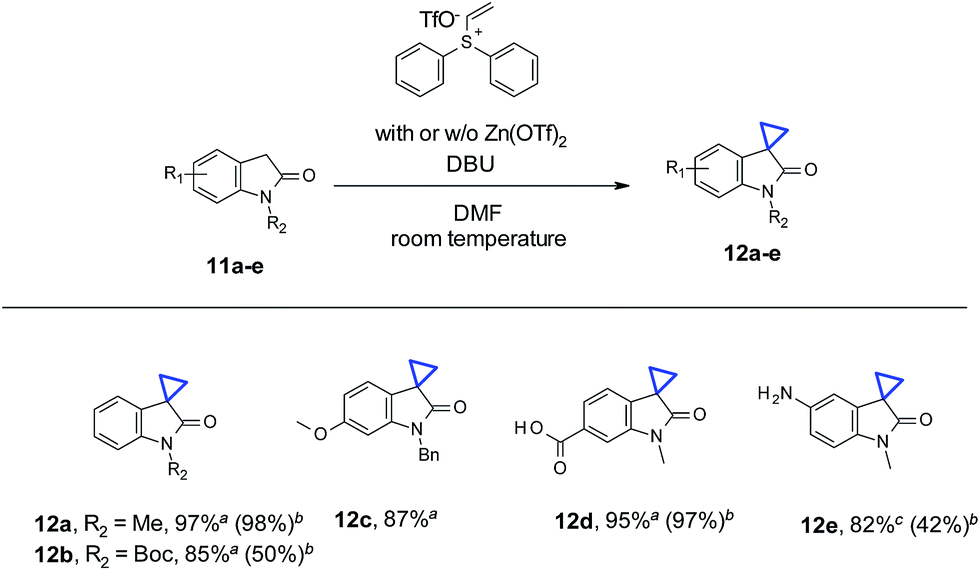
In addition to N-nonsubstituted oxindoles, we also examined cyclopropanation with N-substitued oxindole substrates (Table 3). Addition of Zn(OTf)2 is not necessary for high yields, due to the presence of “N-protecting” substitutions (11a–c). Results are comparable with or without Zn(OTf)2 (11a and 11d). In the case of Boc-protected oxindole 11b, the yield with Zn(OTf)2 was lower than the conditions without Zn(OTf)2 (50% vs. 85%). About 30% of de-Boc product was observed, presumably due to the Lewis acidity of Zn(OTf)2. Functional groups such as carboxyl acid (12d) and primary amine (12e) were well tolerated for cyclopropanation. No O-alkylation by-products were observed in the case of carboxyl acid (11d). For unprotected aniline containing substrate 11e, the reaction was sluggish under standard conditions with Zn(OTf)2. Partial conversion and N-alkylation were observed with only 42% product (12e) isolated. In the absence of Zn(OTf)2, at higher temperature (130 °C) under microwave for 1 hour, 11e did undergo cyclopropanation smoothly with full conversion, leading to 12e in 82% yield. Only minor amount (<10% by LC/MS) of the N-alkylation byproduct was observed.
| a Reaction conditions: oxindole (0.2 mmol), vinyl diphenyl sulfonium triflate (0.24 mmol), DBU (0.6 mmol), in 1 mL DMF, at room temperature (21 °C), under air, stirring for 12 h.b Yields in parentheses correspond to similar conditions with 0.2 mmol of Zn(OTf)2 being used.c Reaction conditions: oxindole (0.2 mmol), vinyl diphenyl sulfonium triflate (0.21 mmol), in 1 mL DMSO, 130 °C, microwave, 1 h. Isolated yields for all substrates described in the table. |
|---|
 |
To our delight, for substrates with sufficient aqueous solubility, cyclopropanation with vinyl diphenyl sulfonium triflate also proceeded well at room temperature (Scheme 2). Different from reported vinyl selenone chemistry,2e neither a strong base (NaOH) nor a surfactant (cetyltrimethyl ammonium bromide) was needed in our case. Oxindole 11d demonstrated adequate solubility with DBU in water and reacted with vinyl diphenyl sulfonium triflate smoothly to afford 12d in 93% yield. For substrates with less optimal aqueous solubility, such as 11a, a co-solvent system was sufficient to obtain a satisfactory yield (90% for 12a).
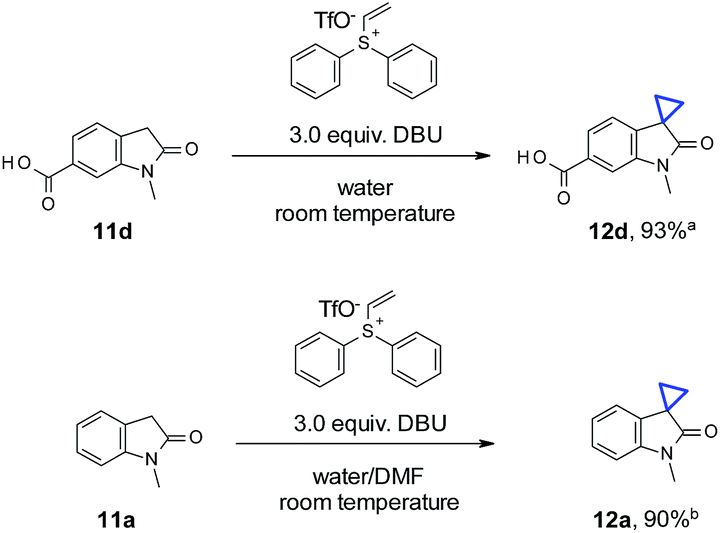 | ||
| Scheme 2 The use of water as the solvent/co-solvent areaction conditions: oxindole (0.2 mmol), vinyl diphenyl sulfonium triflate (0.24 mmol), DBU (0.6 mmol), in 1 mL H2O, room temperature (21 °C), stirring for 4 h. Isolated yield; bconditions similar to above with 0.65 mL H2O and 0.35 mL DMF as co-solvent. Isolated yield. | ||
A series of experiments were performed to understand the effect of Zn(OTf)2. We initially thought that N-alkylation could be reversible under Zn(OTf)2-mediated conditions, while the C–C bond formation may be irreversible. Furthermore, the N-alkylation product could potentially be used to provide a “vinyl intermediate” for another desirable cyclopropanation in the presence of Zn(OTf)2. In order to test this hypothesis, as shown in Scheme 3A, with 3.0 equivalents of DBU and 1.0 equivalent of vinyl diphenyl sulfonium triflate, oxindole 7a underwent a partial conversion to 8a along with significant amount of N-alkylated 10a (cannot be isolated), as suggested by the LC/MS analysis. We then added 1.0 equivalent of Zn(OTf)2 and a different oxindole 11a, in which the N-alkylation was impossible to occur. No cyclopropanation product 12a could be detected up to 12 hours. In Scheme 3B, a similar reaction with oxindole 7a was conducted with Et3N as the base. Interestingly, a different N-alkylation product 13a was observed, and 13a could be even isolated. The reaction of isolated 13a in the presence of 1.0 equivalent of oxindole 11a, 1 equivalent of Zn(OTf)2, and 3.0 equivalents of DBU in DMF gave no spirocyclopropyl oxindole 12a. Conversion of 13a to 10a was detected by LC/MS. The above results indicated that there was no transfer of vinyl group (10a) or vinyl group precursor (13a) to 11a. This suggested that the role of Zn(OTf)2 was not to promote reversibility of N-alkylation, thereby offering a vinyl intermediate for subsequent cyclopropanation reaction leading to more desired cyclopropyl products.
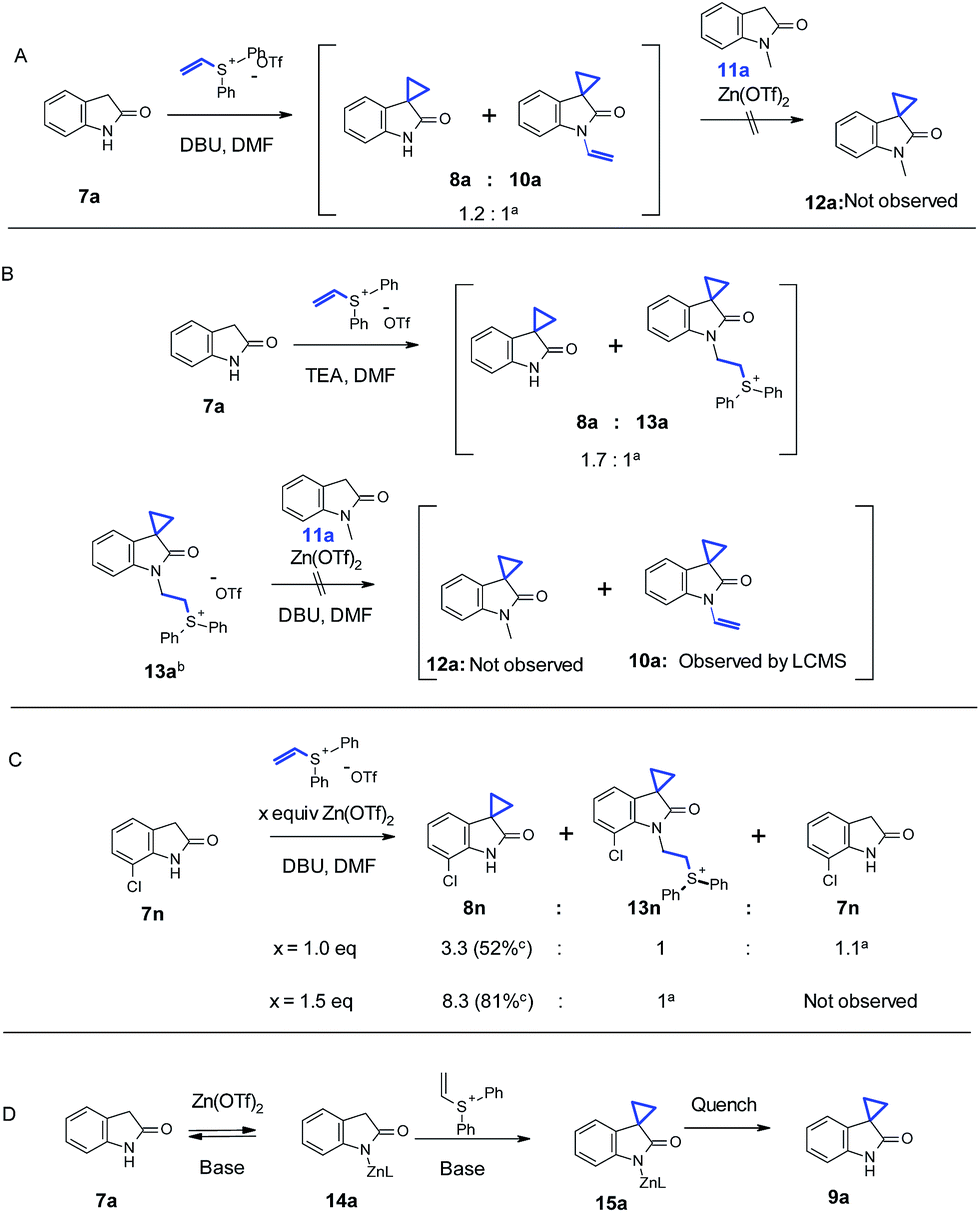 | ||
| Scheme 3 Investigation of the reaction mechanism. aRatio determined by LC/MS; bisolated by crystalization; cisolated by silica gel chromatography. | ||
In Scheme 3C, we studied the reaction in more detail with oxindole 7n. Substitution close to the oxindole nitrogen showed negative influence on the effect of Zn(OTf)2. For example, 1.5 equivalents of Zn(OTf)2 were necessary to block the N-alkylation, suggesting that the adjacent Cl could hinder the complexation of nitrogen with Zn, which in turn rendered the undesired N-alkylation product. A similar result was also observed with 7m. Thus we propose that the Zn coordination to the oxindole nitrogen could account for the observed regioselectivity under our Zn-mediated conditions. Furthermore, the proposed zinc complex 14a could enhance the acidity of the benzylic proton which is adjacent to the lactam. Therefore, weaker bases could be applied to significantly boost functional group compatibility. As shown in Scheme 3D, the Zn complexation on the nitrogen could directly block the N-alkylation pathway. Results with oxindole 11e in Table 3 under the Zn-medicated conditions also suggest that an extra coordinating group, such as the amino group of aniline, could presumably complete with the complexation of Zn(OTf)2 to the oxindole nitrogen thereby slowing down the reaction. This observation further corroborates the depicted mechanism in Scheme 3D.
In summary, we have developed an efficient cyclopropanation method for both N-nonsubstituted and N-substituted oxindoles with the commercially readily available vinyl diphenylsulfonium triflate. The conditions are convenient and mild, with no requirement of inert atmosphere or alkalis bases. A broad reaction scope is observed, with applications on late stage cyclopropanation of three medicinally interesting compounds. We envision that this synthetic methodology could be widely adopted by the medicinal chemistry community. Particularly, the employment of zinc triflate can block the N-alkylation byproduct using N-nonsubstituted oxindoles. It is conceivable that such concept could be applied in other cases where regioselectivity issue exists.
Acknowledgements
We thank Professor Guangbin Dong (University of Chicago) and Professor Xiaoguang Lei (Peking University) for helpful discussions. We also thank Mr Sheng Zhong (Roche Innovation Center Shanghai) for mass spectrometry.Notes and references
- (a) B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673 CrossRef CAS PubMed; (b) N. Ye, H. Chen, E. A. Wold, P.-Y. Shi and J. Zhou, J. Infect. Dis., 2016, 2, 382 CAS.
- (a) R. Zhou, C. Yang, Y. Liu, R. Li and Z. He, J. Org. Chem., 2014, 79, 10709 CrossRef CAS PubMed; (b) Z.-Y. Cao and J. Zhou, Org. Chem. Front., 2015, 2, 849 RSCJ.-H. Li, T.-F. Feng and D.-M. Du, J. Org. Chem., 2015, 80, 11369 CrossRef CAS PubMed; (c) M.-H. Shen, K. Xu, C.-H. Sun and H.-D. Xu, Org. Biomol. Chem., 2016, 14, 1272 RSC; (d) J. Guo, Y. Liu, X. Li, X. Liu, L. Lin and X. Feng, Chem. Sci., 2016, 7, 2717 RSC; (e) M. Palomba, L. Rossi, L. Sancineto, E. Tramontano, A. Corona, L. Bagnoli, C. Santi, C. Pannecouque, O. Tabarrini and F. Marini, Org. Biomol. Chem., 2016, 14, 2015 RSC; (f) P. B. Sampson, Y. Liu, N. K. Patel, M. Feher, B. Forrest, S.-W. Li, L. Edwards, R. Laufer, Y. Lang, F. Ban, D. E. Awrey, G. Mao, O. Plotnikova, G. Leung, R. Hodgson, J. Mason, X. Wei, R. Kiarash, E. Green, W. Qiu, N. Y. Chirgadze, T. W. Mak, G. Pan and H. W. Pauls, J. Med. Chem., 2015, 58, 130 CrossRef CAS PubMed.
- (a) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS PubMed; (b) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS PubMed; (c) S. Song, X. Sun, X. Li, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2886 CrossRef CAS PubMed; (d) K. J. Frankowski, R. Liu, G. L. Milligan, K. D. Moeller and J. Aubé, Angew. Chem., Int. Ed., 2015, 54, 10555 CrossRef CAS PubMed; (e) F. Shen, S. Tyagarajan, D. Perera, S. W. Krska, P. E. Maligres, M. R. Smith III and R. E. Maleczka Jr, Org. Lett., 2016, 18, 1554 CrossRef CAS PubMed.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC.
- (a) A. Lerchner and E. M. Carreira, J. Am. Chem. Soc., 2002, 124, 14826 CrossRef CAS PubMed; (b) J. X. Qiao, T. C. Wang, R. Ruel, C. Thibeault, A. L'Heureux, W. A. Schumacher, S. A. Spronk, S. Hiebert, G. Bouthillier, J. Lloyd, Z. Pi, D. M. Schnur, L. M. Abell, J. Hua, L. A. Price, E. Liu, Q. Wu, T. E. Steinbacher, J. S. Bostwick, M. Chang, J. Zheng, Q. Gao, B. Ma, P. A. McDonnell, C. S. Huang, R. Rehfuss, R. R. Wexler and P. Y. S. Lam, J. Med. Chem., 2013, 56, 9275 CrossRef CAS PubMed.
- D. W. Robertson, J. H. Krushinski, G. D. Pollock, H. Wilson, R. F. Kauffman and J. S. Hayes, J. Med. Chem., 1987, 30, 824 CrossRef CAS PubMed.
- (a) J. Gosselck, L. Béress and H. Schenk, Angew. Chem., Int. Ed., 1966, 5, 596 CrossRef CAS; (b) M. G. Unthank, N. Hussain and V. K. Aggarwal, Angew. Chem., Int. Ed., 2006, 45, 7066 CrossRef CAS PubMed; (c) B. Unthank, B. Tavassoli and V. K. Aggarwal, Org. Lett., 2008, 10, 1501 CrossRef PubMed.
- Z. Mao, H. Qu, Y. Zhao and X. Lin, Chem. Commun., 2012, 48, 9927 RSC.
- F. G. Bordwell and H. E. Fried, J. Org. Chem., 1991, 56, 4218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24985j |
| This journal is © The Royal Society of Chemistry 2017 |