
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-performance carbon-coated mesoporous LiMn2O4 cathode materials synthesized from a novel hydrated layered-spinel lithium manganate composite†
Caihua Jianga,
Zilong Tang*a,
Shiqing Dengab,
Ye Honga,
Shitong Wanga and
Zhongtai Zhanga
aState Key Laboratory of New Ceramics and Fine Processing, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, P. R. China. E-mail: tzl@tsinghua.edu.cn
bNational Center for Electron Microscopy in Beijing, School of Materials Science and Engineering, The State Key Laboratory of New Ceramics and Fine Processing, Key Laboratory of Advanced Materials (MOE), Tsinghua University, Beijing 100084, P. R. China
First published on 17th January 2017
Abstract
Carbon-coated mesoporous spinel LiMn2O4 has been synthesized from a novel hydrated layered-spinel lithium manganate composite through a facile hydrothermal process and subsequent thermal treatment. Benefiting from the carbon coating and mesoporous structure, the LiMn2O4 material exhibits superior high-rate capability and long-life cycling stability, delivering the initial discharge capacity of 117.8 mA h g−1 at 30C with over 90% capacity retention after 1500 cycles. Moreover, the innovative employment of hydrated layered Li-deficient and spinel Li-rich intermediates might provide greater inspiration for other high-performance electrode materials with multiple layer architectures and optimized phase compositions.
Introduction
Nowadays, lithium-ion batteries (LIBs) have extensive applications in electrochemical energy storage and conversion including hybrid electric vehicles (HEVs), electric vehicles (EVs), portable electronic devices and energy storage systems (ESSs). However, at the present stage, lithium-ion batteries are still facing challenges to meet the increasing market demands for higher energy and power densities, longer cycling life, better security and lower cost.1–4 Since cathode materials can severely restrict the electrochemical properties, breakthroughs in the cathodes become more essential and urgent.1,5–7 Among all the appealing cathode materials, spinel LiMn2O4 has attracted much attention by virtue of its low cost, environmental friendliness, high operating plateaus, acceptable energy density and excellent safety.8–10 Nevertheless, the electrochemical properties of LiMn2O4 seem far from satisfactory: (1) poor high-rate capability hampered by the low electronic and ionic conductivities; (2) capacity deterioration caused by the severe manganese dissolution and side reactions.9,11,12To improve the high-rate capability and cycling stability of LiMn2O4, designs of carbon coating and mesoporous structures have been extensively investigated.13–19 Carbon coating can increase the electronic conductivities by forming a continuous electron transport route along the materials' surface, while mesoporous structures can increase the ionic conductivities by reducing the pathway of Li+ diffusion and providing more reaction sites, which contribute to the rate capability.13–17 Meanwhile, upon lithium insertion/extraction, carbon coating can protect the materials from volume changes and side reactions, while the mesoporous structures can accommodate the structural strain, thus contributing to the cycling stability.13,16,18,19
As for the synthesis of LiMn2O4, various polymorphs of manganese oxides have been explored such as α-MnO2,20,21 β-MnO2,22,23 γ-MnO2,21,24 δ-MnO2.25–27 and R-MnO2.28 Among them, birnessite-type lithium manganese oxide (δ-type or Li-birnessite) possesses a hydrated layered structure containing Li ions and crystal water between the layers,29–31 contributing to the exotic properties in many fields.32–34 Moreover, the layered phase is prone to generate the lamellar nanostructure32,33 which can reduce the Li+ diffusion pathway in the thickness direction and further improve the rate performance. Unfortunately, though having been reported as precursors or immediate products for years, the Li-birnessite troubles with impurities such as Li-deficient LixMnO2 or Mn2O3 and lamellar aggregation during the transformation to spinel LiMn2O4,25,31,35 resulting in inferior electrochemical performances. Thus, how to utilize the Li-birnessite with novel structure to synthesize high-performance LiMn2O4 is worth further study.
In this work, we have employed a novel hydrated layered-spinel lithium manganese oxide composite to synthesize the carbon-coated mesoporous spinel LiMn2O4 via a facile hydrothermal process and subsequent thermal treatment. Based on the gradual transformation from layered Li-birnessite phase to spinel LiMn2O4 phase, the resultant mesoporous structure and carbon coating can effectively improve the rate capability and cycling stability. The outstanding electrochemical performance thus obtained, is of interest as a cathode material for lithium-ion batteries.
Results and discussion
As illustrated in Fig. 1, the formation mechanism of the carbon-coated mesoporous LiMn2O4 involves two steps: the hydrothermal reaction between manganese oxides (denoted as MO in Fig. 1a) and lithium hydroxide solution at 160 °C (Step I) and the thermal treatment under vacuum at 600 °C (Step II). The absorbed organic group CH3COO− (shown in Fig. 1a and b) is originated from the reduction of ethanol in the synthesis process of the initial MO. Through Step I, the obtained hydrothermal products (Fig. 1b) consist of two phases: layered Li-birnessite and spinel LMO. The blooming flower-like nanosheets at different levels were interlaced, contributing to the mesoporous structure. Then through Step II, carbon-coated mesoporous LiMn2O4 (Fig. 1c) was obtained where the residual Li-birnessite fully transformed into the spinel LMO accompanied by the surface carbonization (see more experimental details in the ESI†).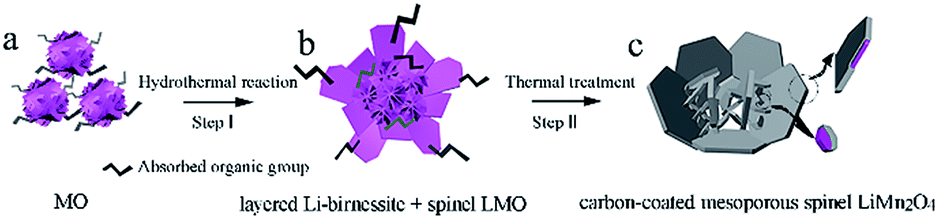 | ||
| Fig. 1 A schematic illustrating the formation mechanism of the carbon-coated mesoporous spinel LiMn2O4. | ||
The hydrothermal process of Step I is so critical through the whole synthesis process that deserves further investigation and analysis. X-ray diffraction (XRD) patterns of the prepared MO and hydrated hydrothermal products are shown in Fig. 2a. The weak peaks of MO indicate the disordered nature of the birnessite-type manganese oxides. While after the hydrothermal treatment, the layered monoclinic Li-birnessite (Li4Mn14O27·xH2O, JCPDS 50-0009) coexists with the spinel lithium manganese oxide (LMO). The layered Li-birnessite phase contains crystal water and Li-ions between layers with a basal spacing of ∼0.7 nm (presented in Fig. 2b), which corresponds to the (001) diffraction peak at around 12.8°. Typical (111) diffraction at 2θ = 18.9° confirms the appearance of Li-rich spinel phase Li1.27Mn1.73O4 (JCPDS 51-1582) which is a common hydrothermal product consistent with previous reports.36 Fig. 2c shows the crystal structure of the spinel lithium manganese oxide. The two-phase mixture (denoted as hydrated L–S) consisting of both Li-deficient layered phase (compared with Li![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mn = 1:2 (atom%) for stoichiometric LiMn2O4) and Li-rich spinel phase can be significant for the subsequent changes. XRD patterns of hydrothermal products at intermediate time are depicted in Fig. S1a and c.† After 1 h reaction, only single layered Li-birnessite phase exists and when the reaction time increased, the spinel lithium manganese oxide (LMO) comes out and coexists with the layered Li-birnessite. The above results assist to understand the phase transformation process which are based on the ‘hydrothermal-soft chemical reaction’:25,26,37–39 Li+ inserts into the interlayer spaces of Li-birnessite (soft chemical reaction); the layered structure collapses along with the squeezing of water and changes into the spinel structure (hydrothermal reaction).
:Mn = 1:2 (atom%) for stoichiometric LiMn2O4) and Li-rich spinel phase can be significant for the subsequent changes. XRD patterns of hydrothermal products at intermediate time are depicted in Fig. S1a and c.† After 1 h reaction, only single layered Li-birnessite phase exists and when the reaction time increased, the spinel lithium manganese oxide (LMO) comes out and coexists with the layered Li-birnessite. The above results assist to understand the phase transformation process which are based on the ‘hydrothermal-soft chemical reaction’:25,26,37–39 Li+ inserts into the interlayer spaces of Li-birnessite (soft chemical reaction); the layered structure collapses along with the squeezing of water and changes into the spinel structure (hydrothermal reaction).
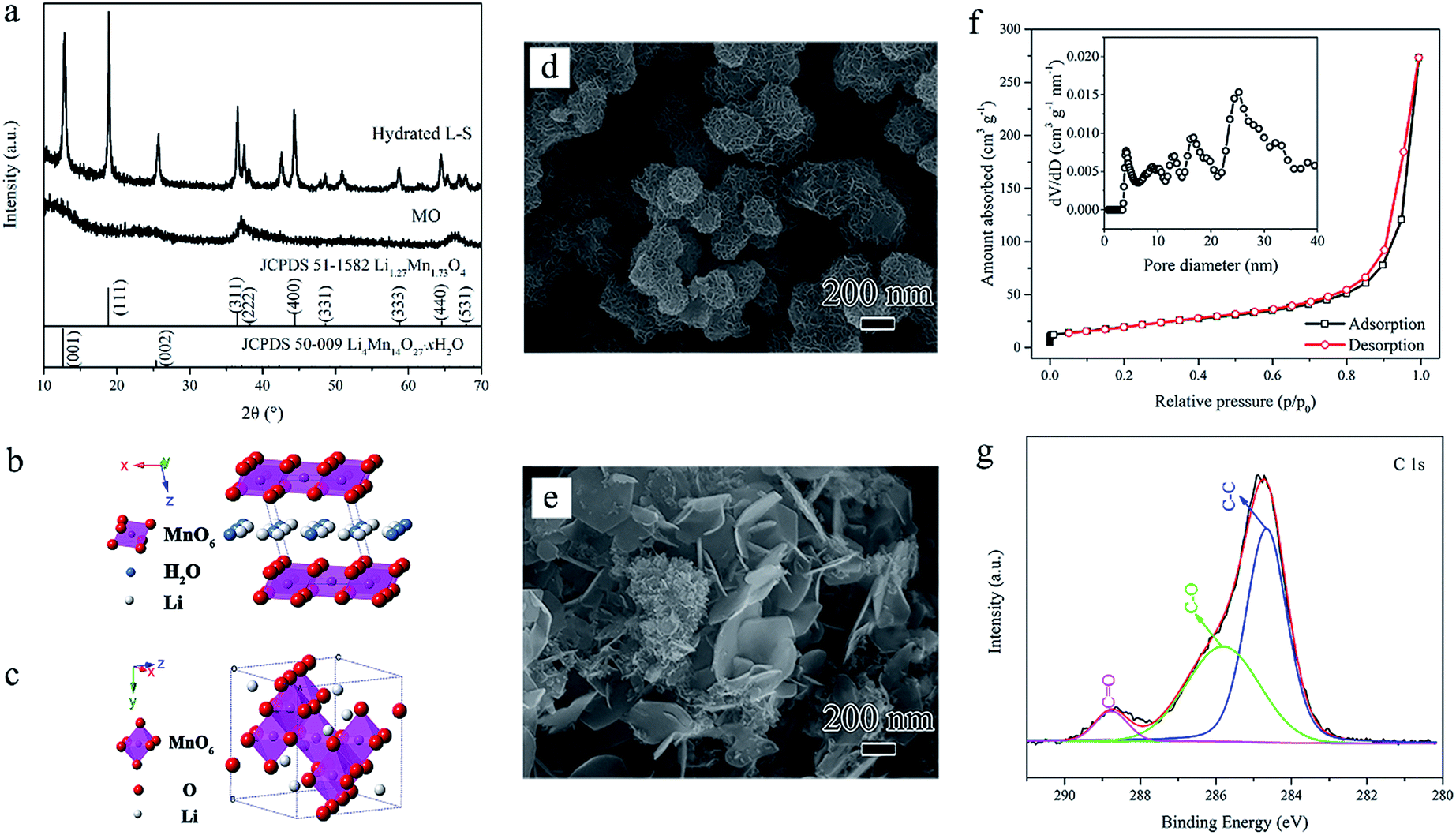 | ||
| Fig. 2 (a) XRD patterns of MO and hydrated L–S. (b and c) Crystal structure of the layered hydrated Li-birnessite (b) and spinel LMO (c). (d and e) SEM images of MO and hydrated L–S. (f) N2 adsorption–desorption isotherms and the corresponding pore size distribution of hydrated L–S. (g) XPS C1s spectrum of the initial manganese oxide (MO). | ||
Furthermore, scanning electron microscopy (SEM) images in Fig. 2d, e, S1b and d† reveal the corresponding morphology evolution. The initial MO are bud-like nanoclusters of 200–300 nm assembled by interconnected thin nanosheets (Fig. 2d) and transmission electron microscopy (TEM) images show that these nanosheets are only a few nanometers in thickness (Fig. S2a and b†). Then after hydrothermal treatment, inhomogeneous nanostructures appear which consist of both pedal-like bigger nanosheets and bud-like smaller nanoclusters (Fig. 2e). Additionally, as can be seen from Fig. S2c and d,† the bigger nanosheets are of several hundred nanometers and the nanoclusters are assembled by small nanosheets (tens of nanometers). Fig. S1b† reveals that the morphology of nanosheets and nanoclusters originated from the layered Li-birnessite phase which is more liable to generate the laminar morphology. The obtained nanosheets with different sizes and the whole morphology evolution can be interpreted as Ostwald ripening, nucleation–dissolution–recrystallization and self-assembly processes in consistent with previous reports.25,39–42 In particular, the MO nanodomains separated from the edge of original nanosheets and new MnO2 nuclei adsorbed with the CH3COO− group formed. Due to the coexistence of multi-phases in the hydrothermal system as described before and energy difference, more than one nucleus of the crystal units may be contained with different nucleation, orientation and growth rates,40,42 which can explain the inhomogeneous outward migration of crystallites. Moreover, the relatively higher concentration of Li+ facilitates dissolution of manganese oxide39,42 and thus favors the generation of those bud-like smaller nanoclusters according to the dissolution–recrystallization mechanism. Ultimately, the blooming flower-like nanosheets at different levels are self-assembled, the interconnection between which can result in the mesoporous structure. The mesoporous structure is further confirmed by the nitrogen adsorption–desorption isotherms with a type-IV curve and hysteresis loop (Fig. 2f). The pore size distribution curve calculated by using the density functional theory (DFT) shows a multimodal feature with peaks ranging from 3–30 nm. More importantly, X-ray photoelectron spectroscopy (XPS) investigation of C1s in Fig. 2g reveals peak locations at 284.6 eV (C–C), 285.8 eV (C–O) and 288.6 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O),15,43,44 confirming organic groups terminated on the surface which guarantees the subsequent carbon coating.
O),15,43,44 confirming organic groups terminated on the surface which guarantees the subsequent carbon coating.
Then subsequent thermal treatment under vacuum through Step II leads to the ultimate pure phase spinel LiMn2O4 transforming from the intermediate of hydrated L–S. XRD characterization (Fig. 3a) of the heat-treated product reveals that all the reflections can be identified as spinel LiMn2O4 (JCPDS 35-0782). The Li:Mn atomic ratio of 0.505 tested by inductively coupled plasma optical emission spectrometry (ICP-OES) method confirms the nearly stoichiometric composition. To further verify the disappearance of the Li-birnessite phase, thermogravimetric-differential thermal analysis (TG-DTA) was performed (Fig. S3a†). An obvious endothermic peak with remarkable weight loss of ∼3.0% is observed at ∼150 °C, which can be ascribed to dehydration of physically absorbed water and partial interlayer water.37,41,45 XRD pattern of the sample annealed at 150 °C in Fig. S3b† also confirms the collapse of the layered Li-birnessite. The weight loss over 150 °C can be assigned to the further dehydration of OH groups, release of oxygen molecules and structural rearrangement.37,41,45 Moreover, when increasing the annealing temperature, the increasing intensity and slight left shift of (111) diffraction for the spinel structure can be observed (Fig. S3b†), implying the composition change of spinel phase. So far, the transformation from the two-phase mixture consisting of layered Li-deficient phase and spinel Li-rich phase to the single stoichiometric spinel LiMn2O4 phase has been successfully completed.
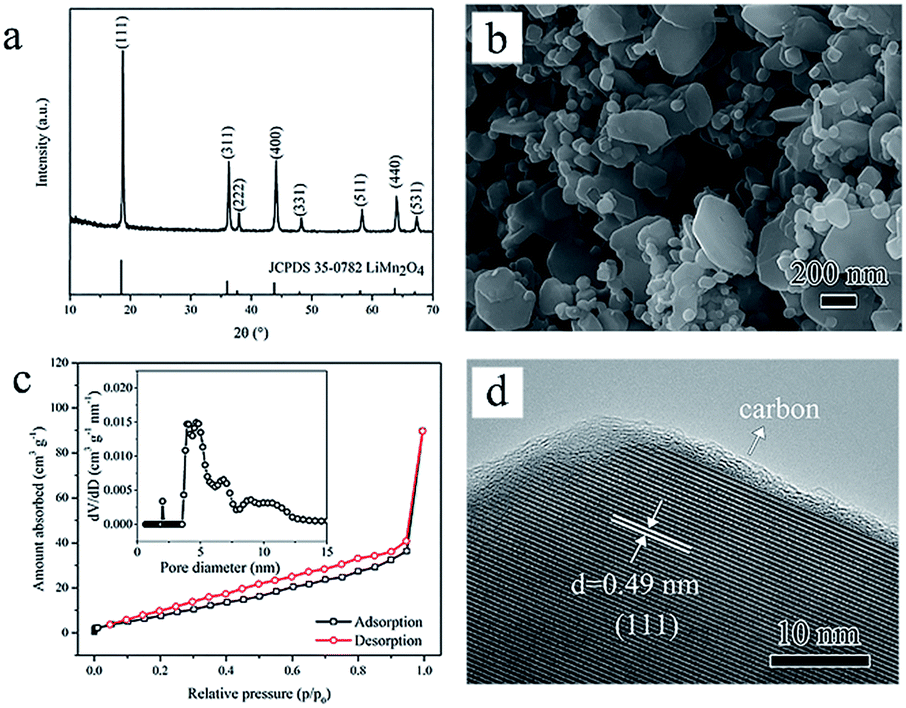 | ||
| Fig. 3 Characterizations of the as-synthesized carbon-coated mesoporous spinel LiMn2O4. (a) XRD pattern, (b) SEM image, (c) N2 adsorption–desorption isotherms together with the corresponding pore size distribution and (d) HRTEM image. | ||
Fig. 3b shows the SEM image of the as-prepared LiMn2O4. It can be observed that the heat-treated product inherits the former multiform lamellar structure but with coarsened nanoplates (200–500 nm) and distributed small nanoplatelets. The thickness of these nanoplates is only tens of nanometers and this is supposed to shorten the Li+ diffusion pathways. Noteworthily, the smaller nanoplatelets spread around the bigger nanoplates, which can prevent the mutual aggregate and provide more interspaces. Nitrogen adsorption–desorption isotherms with the hysteresis loop shown in Fig. 3c confirm the mesoporous nature of the LiMn2O4 product. Relatively smaller pore size is mainly due to the structural shrinkage during the annealing process compared with that of the hydrothermal product. Such mesoporous structure is beneficial to Li+ storage by providing more convenient contact between the electrolyte and electrode materials.13,14
The crystal structure and surface feature of the LiMn2O4 product were further studied by using high-resolution transmission electron microscopy (HRTEM) method combined with energy disperse spectroscopy (EDS) point mapping analysis. HRTEM image in Fig. 3d exhibits a clear interplanar distance of ∼0.49 nm, corresponding to the (111) plane of cubic spinel LiMn2O4. In addition, originated from surface carbonization, an apparent thin carbon layer wrapped on the surface can be distinguished. EDS point mapping results in Fig. S4† indicates the higher relative concentration of C at the edge than that in the middle zone. The carbon content is 0.27 wt% measured by an element analyzer. The amorphous carbon layer can form a 3D conductive network and prevent carbon-coated LiMn2O4 from excessive Mn dissolution during the charge/discharge cycling.16–19
Based on the above observations, the carbon-coated mesoporous spinel LiMn2O4 (denoted as LMO-CM), as the final state, can be thus reasonably confirmed. When exploring the electrochemical performances as the cathode material for LIBs, we also introduced two control groups for comparison: the no carbon-coated spinel LiMn2O4 (denoted as LMO-NC) which was annealed in air with the other experimental variables the same as those of LMO-CM and the commercial spinel lithium manganese oxide (denoted as LMO-COM). Their typical XRD patterns and SEM images are shown in Fig. S5.† Fig. 4a shows the first galvanostatic discharge–charge profiles between 3.2–4.3 V vs. Li+/Li at 0.2C (1C = 140 mA g−1). All the three samples have two voltage plateaus at around 4.0 V and 4.1 V, which can be attributed to the Mn3+/Mn4+ redox couple associated with the two-stage mechanism of Li+ insertion and extraction. The corresponding electrochemical conversion reactions can be expressed as follows: LiMn2O4 → Li0.5Mn2O4 + 0.5Li+ + 0.5e− (at low plateau); Li0.5Mn2O4 → 2λ-MnO2 + 0.5Li+ + 0.5e− (at high plateau).10,46,47 In contrast, LMO-CM has much flatter voltage plateaus, suggesting its better crystallinity property and reversibility; LMO-CM has the highest reversible discharge specific capacity of 129.7 mA h g−1 compared with 116.1 mA h g−1 and 100.7 mA h g−1 for LMO-NC and LMO-COM. These results are in accordance with the most distinguished cathodic/anodic peaks as well as the largest integral area surrounded by the relevant cyclic voltammetry (CV) plot (Fig. S6†).
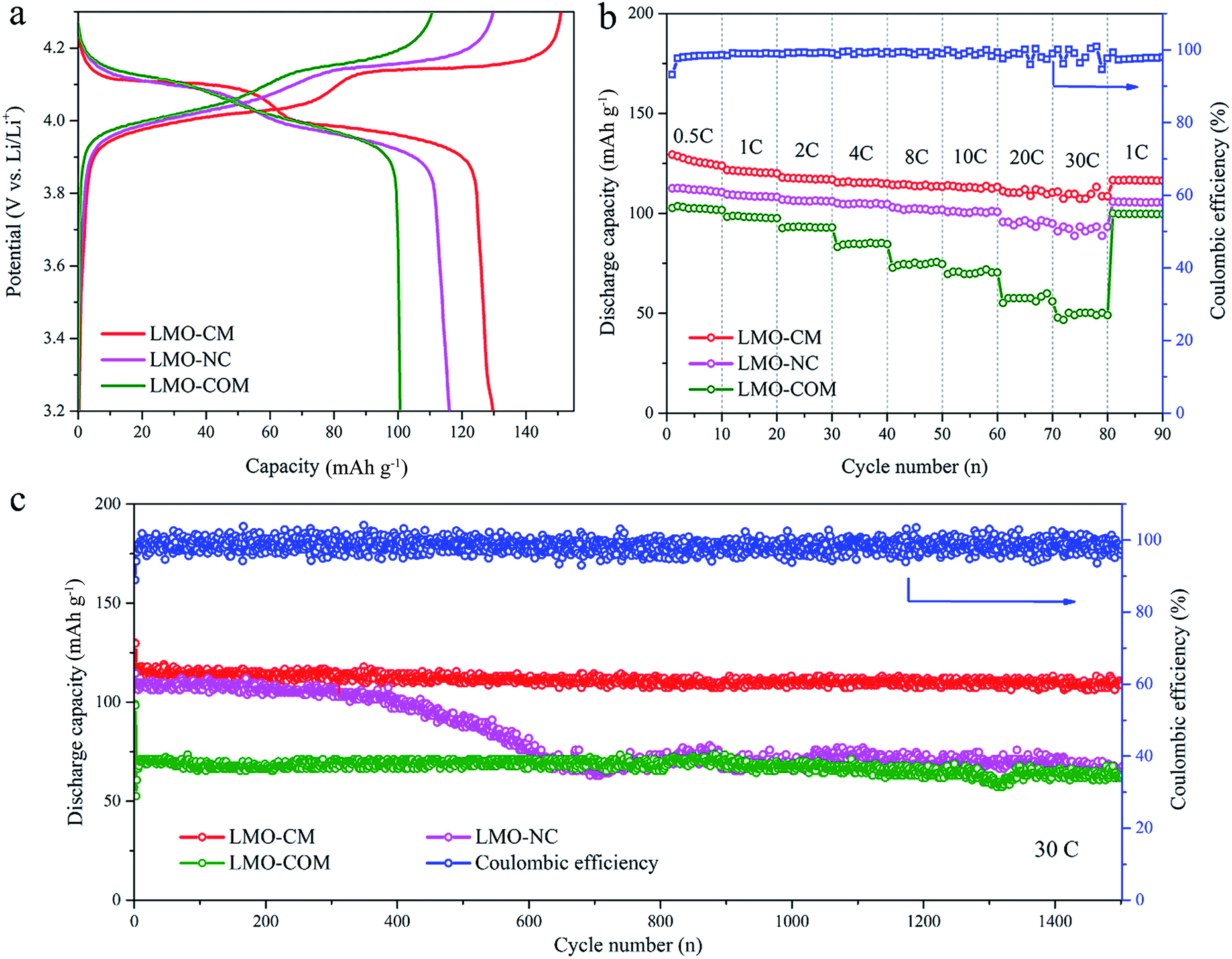 | ||
| Fig. 4 Comparison of electrochemical performances for carbon-coated mesoporous LMO-CM, no carbon-coated LMO-NC and commercial LMO-COM. (a) The first galvanostatic discharge–charge profiles at 0.2C. (b) Discharge capacity with cycle number at different rates ranging from 0.5C to 30C. (c) Long-term cycling performance at 30C for all the three samples. Note that the given coulombic efficiency in (b) and (c) is only for LMO-CM. | ||
Notably, LMO-CM exhibits superior rate capability as shown in Fig. 4b. It delivers high reversible capacities of 129.4 mA h g−1, 121.7 mA h g−1, 115.4 mA h g−1, 114 mA h g−1 and 110.8 mA h g−1 when cycled at 0.5C, 1C, 4C, 10C, 30C for each 10 cycles, respectively. Also, the capacity can be well recovered when cycled back to 1C. While LMO-NC and LMO-COM only remain 91.0 mA h g−1 and 47.8 mA h g−1 at 30C, much lower than that of LMO-CM, besides, their corresponding capacity retention is ∼83% and ∼49% (versus capacity at 1C), inferior to ∼91% for LMO-CM. Rate performance of the as-synthesized LMO-CM was also compared with that of other typical high-rate LMO/C materials reported previously as displayed in Fig. S7,† showing the superior rate capability of our work especially at rates higher than 20C.
To investigate cycle performance at high rates, the three samples were cycled at 30C for 1500 cycles as presented in Fig. 4c. When charged and discharged at the same rate of 30C, the initial discharge capacity of LMO-CM is 117.8 mA h g−1 (nearly 80% of the theoretical specific capacity of 148 mA h g−1), higher than that of LMO-NC (108.5 mA h g−1) and LMO-COM (52.5 mA h g−1). What's more, LMO-CM can retain ∼92% capacity (108.5 mA h g−1) after 1500 cycles with the coulombic efficiency retaining nearly 100%, whereas LMO-NC has a drastic capacity decay after about 500 cycles and ultimately retains ∼61% capacity (66.5 mA h g−1). As given in Fig. S8,† remarkably, at 60C (charged within 1 min actually), over 90% stable capacity retention is maintained with an initial capacity of 102.7 mA h g−1. Even cycled at 80C (∼34 s) and 120C (∼28 s), LMO-CM can still yield a discharge capacity as high as 102.5 mA h g−1 and 88.6 mA h g−1, showing the unprecedented high-rate and long-life cycling stability. Though mainly confined to the laboratory level, the current results are still encouraging.
As schematically illustrated in Fig. 5, the outstanding electrochemical performance of the as-synthesized carbon-coated mesoporous LiMn2O4 can be ascribed to three main factors. Firstly, the thin carbon layer decorated on the surface can not only reduce the interface resistance by forming a three dimensional electro-conductive network, but also improve the structural integrity by suppressing volume change and side reactions during cycling operations.16–18,48,49 Electrochemical impedance spectroscopy (EIS) analysis in Fig. S9† suggests a smaller charge-transfer resistance of LMO-CM. Secondly, the novel mesoporous structure constructed by bigger nanoplates and distributed smaller nanoplates can ensure intimate contact between the electrolyte and electrode materials, thus providing more sites for Li+ storage.13,14,50,51 Finally, the thickness of all existing nanoplates is only tens of nanometers and such small dimension can support to shorten the pathways for Li+ diffusion.52–54 Therefore, all the above factors enable the obtained LiMn2O4 material with superior rate capability and remarkable cycling stability, which provides a perspective on the potential applications in high-power electric vehicles and smart grids.
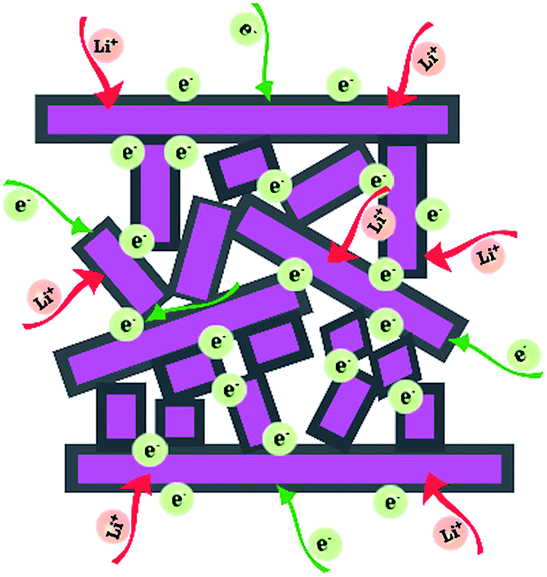 | ||
| Fig. 5 Schematic illustration of electron conduction and Li+ storage in LMO-CM. | ||
Conclusions
In summary, by employing the hydrated layered Li-deficient and spinel Li-rich composite, we have successfully synthesized a carbon-coated mesoporous LiMn2O4 via a facile hydrothermal process and subsequent thermal treatment. Remarkably, the as-prepared LiMn2O4 exhibits superior high-rate capability and long-life cycling stability, delivering the initial discharge capacity of 117.8 mA h g−1 at 30C with over 90% capacity retention after 1500 cycles. Additionally, this work paves a fresh route to obtain pure and high-performance LiMn2O4 with mesoporous structure by utilizing layered birnessite-type manganese oxides. The innovative employment of hydrated layered Li-deficient and spinel Li-rich intermediate product might provide inspiration to high-performance LiMn2O4 synthesized from other methods or other high-performance electrode materials with multiple layer architecture and optimized phase composition.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 51472137) and Ministry of Education of China (no. 20120002110007).Notes and references
- R. Chen, T. Zhao, X. Zhang, L. Li and F. Wu, Nanoscale Horiz., 2016, 1, 423–444 RSC.
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- S. Myung, K. Amine and Y. Sun, J. Power Sources, 2015, 283, 219–236 CrossRef CAS.
- R. Pitchai, V. Thavasi, S. G. Mhaisalkar and S. Ramakrishna, J. Mater. Chem., 2011, 21, 11040 RSC.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51–65 CrossRef CAS.
- F. Mao, W. Guo and J. Ma, RSC Adv., 2015, 5, 105248 RSC.
- S. Lee, Y. Cho, H. Song, K. T. Lee and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 8748–8752 CrossRef CAS PubMed.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- G. Xu, Z. Liu, C. Zhang, G. Cui and L. Chen, J. Mater. Chem. A, 2015, 3, 4092 CAS.
- O. K. Park, Y. Cho, S. Lee, H.-C. Yoo, H.-K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621–1633 CAS.
- H. Lin, J. Hu, H. Rong, Y. Zhang, S. Mai, L. Xing, M. Xu, X. Li and W. Li, J. Mater. Chem. A, 2014, 2, 9272 CAS.
- J. Luo, Y. Wang, H. Xiong and Y. Xia, Chem. Mater., 2007, 19, 4791–4795 CrossRef CAS.
- W. Sun, H. Liu, Y. Liu, G. Bai, W. Liu, S. Guo and X. Zhao, Nanoscale, 2015, 7, 13173–13180 RSC.
- H. Zhang, Z. Li, S. Yu, Q. Xiao, G. Lei and Y. Ding, J. Power Sources, 2016, 301, 376–385 CrossRef CAS.
- H. Li and H. Zhou, Chem. Commun., 2012, 48, 1201–1217 RSC.
- A. R. Han, T. W. Kim, D. H. Park, S.-J. Hwang and J.-H. Choy, J. Phys. Chem. C, 2007, 111, 11347–11352 CAS.
- T. Yi, Y. Zhu, X. Zhu, J. Shu, C. Yue and A. Zhou, Ionics, 2009, 15, 779–784 CrossRef CAS.
- H.-W. Lee, P. Muralidharan, R. Ruffo, C. M. Mari, Y. Cui and D. K. Kim, Nano Lett., 2010, 10, 3852–3856 CrossRef CAS PubMed.
- J. Luo, H. Xiong and Y. Xia, J. Phys. Chem. C, 2008, 112, 12051–12057 CAS.
- Y. Ding, J. Xie, G. Cao, T. Zhu, H. Yu and X. Zhao, Adv. Funct. Mater., 2011, 21, 348–355 CrossRef CAS.
- J. Lu, X. Fan, C. Zhou, Z. Liu, F. Zheng, K. S. Lee and L. Lu, J. Electrochem. Soc., 2016, 163, 197–202 CrossRef.
- L. He, S. Zhang, X. Wei, Z. Du, G. Liu and Y. Xing, J. Power Sources, 2012, 220, 228–235 CrossRef CAS.
- F. v. Bülow, H. Zhang and D. E. Morse, Adv. Energy Mater., 2012, 2, 309–315 CrossRef.
- X. Lv, S. Chen, C. Chen, L. Liu, F. Liu and G. Qiu, Solid State Sci., 2014, 31, 16–23 CrossRef CAS.
- M. Tang, A. Yuan and J. Xu, Electrochim. Acta, 2015, 166, 244–252 CrossRef CAS.
- B. Zou, X. Ma, Z. Tang, C. Ding, Z. Wen and C. Chen, J. Power Sources, 2014, 268, 491–497 CrossRef CAS.
- R. M. Mckenzie, Mineral. Mag., 1971, 38, 493–502 CAS.
- V. V. Vol’Khin, O. A. Pogodina and G. V. Leont’eva, Russ. J. Gen. Chem., 2002, 72, 173–177 CrossRef.
- X. Yang, W. Tang, Z. Liu, Y. Makita and K. Ooi, J. Mater. Chem., 2002, 12, 489–495 RSC.
- L. Dang, C. Wei, H. Ma, Q. Lu and F. Gao, Nanoscale, 2015, 7, 8101 RSC.
- M. Qin, H. Zhao, W. Yang, Y. Zhou and F. Li, RSC Adv., 2016, 6, 23905 RSC.
- A. K. Thapa, B. Pandit, R. Thapa, T. Luitel, H. S. Paudel, G. Sumanasekera, M. K. Sunkara, N. Gunawardhana, T. Ishihara and M. Yoshio, Electrochim. Acta, 2014, 116, 188–193 CrossRef CAS.
- T. L. Christiansen, E. D. Bøjesen, M. Søndergaard, S. Birgisson, J. Becker and B. B. Iversen, CrystEngComm, 2016, 18, 1996–2004 RSC.
- B. J. Liddle, S. M. Collins and B. M. Bartlett, Energy Environ. Sci., 2010, 3, 1339 CAS.
- L. Liu, Q. Feng, K. Yanagisawa, G. Bignall and T. Hashida, J. Mater. Sci., 2002, 37, 1315–1320 CrossRef CAS.
- Q. Feng, K. Yanagisawa and N. Yamasaki, Chem. Commun., 1996, 14, 1607–1608 RSC.
- X. Huang, Q. Zhang, J. Gan, H. Chang and Y. Yang, J. Electrochem. Soc., 2011, 158, 139–145 CrossRef.
- K. A. M. Ahmed and K. Huang, Solid State Sci., 2014, 30, 11–16 CrossRef CAS.
- X. Zhang, P. Yu, H. Zhang, D. Zhang, X. Sun and Y. Ma, Electrochim. Acta, 2013, 89, 523–529 CrossRef CAS.
- Z. Zhang, G. Hu, Y. Cao, J. Duan, K. Du and Z. Peng, RSC Adv., 2015, 5, 81461–81467 RSC.
- J. W. Lee, A. S. Hall, J.-D. Kim and T. E. Mallouk, Chem. Mater., 2012, 24, 1158–1164 CrossRef CAS.
- H. Luo, Y. Sun, F. Zhang, D. Zhang, Y. Yang and J. Zhang, Electrochim. Acta, 2014, 148, 228–236 CrossRef CAS.
- Q. Feng, K. Yanagisawa and N. Yamasaki, J. Porous Mater., 1998, 5, 153–161 CrossRef CAS.
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769–775 CrossRef CAS.
- K. K. A. G. W. Liu, J. Electrochem. Soc., 1998, 145, 459–465 CrossRef.
- J. Wang, J. Yang, Y. Tang, R. Li, G. Liang, T.-K. Sham and X. Sun, J. Mater. Chem. A, 2013, 1, 1579 CAS.
- S. Xin, Y. Guo and L. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS PubMed.
- J. Wang, W. Liu, S. Liu, J. Chen, H. Wang and S. Zhao, Electrochim. Acta, 2016, 188, 645–652 CrossRef CAS.
- C. Zhu, G. Saito and T. Akiyama, J. Mater. Chem. A, 2013, 1, 7077 CAS.
- Y. Chen, K. Xie, Y. Pan and C. Zheng, J. Power Sources, 2011, 196, 6493–6497 CrossRef CAS.
- M. Okubo, Y. Mizuno, H. Yamada, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, ACS Nano, 2010, 4, 741–752 CrossRef CAS PubMed.
- K. M. Shaju and P. G. Bruce, Chem. Mater., 2008, 20, 5557–5562 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25802f |
| This journal is © The Royal Society of Chemistry 2017 |