
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effective conversion of biomass into bromomethylfurfural, furfural, and depolymerized lignin in lithium bromide molten salt hydrate of a biphasic system†
Chang Geun Yoo,
Shuting Zhang and
Xuejun Pan *
*
Department of Biological Systems Engineering, University of Wisconsin-Madison, 460 Henry Mall, Madison, WI 53706, USA. E-mail: xpan@wisc.edu
First published on 23rd December 2016
Abstract
A novel approach using a biphasic system consisting of a molten lithium bromide hydrate solution (LiBr·3H2O) and an organic solvent was developed to efficiently produce furan-based chemicals from cellulose and lignocellulosic biomass. At 125 °C for 2 h, the yield of bromomethylfurfural (BMF) from cellulose reached >90% (molar yield), and the yields of furfural (FF) and BMF from real biomass (herbage, hardwood, and softwood) were ∼70% and ∼85%, respectively. The reaction mechanisms of the polysaccharides and lignin and the role of the molten salt hydrate were investigated and elucidated. In the biphasic system, hemicelluloses and cellulose of the biomass were dissolved, hydrolyzed, dehydrated and brominated to FF and BMF, respectively, in the aqueous phase, and the furan products were simultaneously extracted into and cumulated in the organic phase. Meanwhile, lignin in the biomass was significantly depolymerized through the cleavage of β-aryl ether linkages and separated with high purity for potential coproducts.
1. Introduction
Biomass is considered as an alternative resource to petroleum-based fuels and platform chemicals because of its abundance, renewability, and environmentally-friendly and non-edible nature.1,2 In recent decades, remarkable progress has been made in developing technologies for converting biomass to fuels and chemicals.3 For instance, liquid transportation fuels such as fuel-grade alcohols and drop-in hydrocarbons have been produced from biomass through biochemical and thermochemical platforms.4–8 Many platform chemicals such as propanediol, succinic acid, hydroxymethylfurfural (HMF), furfural (FF), lactic acid, levulinic acid, glutamic acid, sorbitol, xylitol, and aromatic compounds derived from biomass have been studied.9–16Furan-based chemicals including FF, HMF, and their derivatives, are a group of products derived from the carbohydrate fractions (cellulose and hemicelluloses) of the biomass. They have wide applications in the production of liquid fuels,4,17–20 polymeric materials,21,22 pharmaceuticals,21,23 and fine chemicals.19,23–25 HMF and FF are typically produced from acid-induced dehydration of hexoses and pentoses, respectively, using homogeneous mineral acids or heterogeneous acids as catalysts in aqueous or organic solvents.17,26–29 However, the production of these furans remains challenging due to low yield, poor selectivity, and environmental issues of the associated processes.5 Currently, the annual FF production is more than 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons worldwide, but the current yield is only about 50% of the theoretical yield due to the aforementioned side reactions.30 Recently, halomethylfurfurals such as 5-bromomethylfurfural (BMF) and 5-chloromethylfurfural (CMF) have been investigated as platform chemicals for the production of solvents, polymers, and liquid hydrocarbon fuels.31–35 Diverse approaches were explored, but these processes had some limitations such as high acid loading or concentration, unsatisfactory product yield, and insufficient utilization of hemicelluloses and lignin.26,27,29,31,32
000 tons worldwide, but the current yield is only about 50% of the theoretical yield due to the aforementioned side reactions.30 Recently, halomethylfurfurals such as 5-bromomethylfurfural (BMF) and 5-chloromethylfurfural (CMF) have been investigated as platform chemicals for the production of solvents, polymers, and liquid hydrocarbon fuels.31–35 Diverse approaches were explored, but these processes had some limitations such as high acid loading or concentration, unsatisfactory product yield, and insufficient utilization of hemicelluloses and lignin.26,27,29,31,32
For effective conversion of biomass to the furans, selection of a proper reaction medium is crucial. In the previous studies, water with acid catalyst was the most common medium, but low yield and poor selectivity are the major issues due to the rehydration of HMF to levulinic acid and formic acid and/or the condensation of the furans to humins.17,36–38 It was found that ionic liquids (ILs) and high boiling point organic solvents, such as dimethylsulfoxide and N,N-dimethylformide, were able to improve the furan yield; however, these processes are economically unfavorable because of the high costs for solvent and downstream processing.17,27
In this study, we developed a simple and effective biphasic system involving molten salt hydrate to convert biomass into furans (FF and BMF) under mild conditions. Different molten halide salt hydrates for the aqueous phase and varying solvents for the organic phase were compared. The effects of the salt concentration and solvent volume on the yields of furans were also examined. The role of the molten salt hydrate and the reaction mechanism of cellulose conversion to BMF were investigated. Separated lignin was characterized by means of wet-chemistry analysis, GPC, and NMR to elucidate the lignin reactions and evaluate the potential for lignin co-products.
2. Experimental section
2.1. Materials
D(+)-Glucose, D-fructose, and D(+)-cellobiose were purchased from Fisher Scientific (Pittsburgh, PA), and microcrystalline cellulose was purchased from Sigma-Aldrich (St. Louis, MO). Lithium bromide (LiBr) and calcium bromide (CaBr2) were from Alfa Aesar, and lithium chloride (LiCl) was from Fisher Scientific. Hydrochloric acid (37 wt%), hydrobromic acid (48 wt%), and solvents including dichloromethane (DCM), toluene, benzene, and cyclohexane were purchased from Fisher Scientific. Lignocellulosic biomass including corn stover, switchgrass, poplar, aspen, and Douglas fir was air-dried at ambient temperature, ground with a Wiley mill, and the fraction between 10 and 50 mesh was used in this study.2.2. Experimental set-up
All the reactions were conducted in a stainless steel reactor with Teflon liner (50 mL of internal volume), as shown in Fig. S1.† For uniform reaction, a magnetic bar was used to stir at 500 rpm. A glycerol bath was used for heating, controlled by a hotplate (CG-1994-50, Chemglass, NJ). In a typical experiment, 0.2–1.0 g (oven dry weight) of a feedstock were loaded into the reactor with 10 mL of aqueous solution of salt with acid co-catalyst and 10–20 mL of organic solvent. At the end of reaction, the reactor was cooled down to room temperature, and then the mixture in the reactor was transferred to a Falcon™ conical centrifuge tube (Fig. S2†). Centrifuge at 4500 rpm for 10 min and carefully collect the solvent layer (organic phase, top) for the quantitation of furan products using NMR and GC-MS analyses, as described below.2.3. Quantitation of furans with NMR and GC-MS
Transfer 1 mL of the organic phase into a 2 mL vial. The organic solvent was carefully removed using a rotary evaporator (Rotavapor R-215, Switzerland), and the remaining products were dissolved in chloroform-d (1 mL) for NMR analysis. NMR spectra were acquired at 293 K on a Bruker Instruments Avance III 500 console, 11.74 T standard bore magnet NMR spectrometer, equipped with a 5 mm DCH cryoprobe. 1H-NMR (ns = 16, ds = 2, d1 = 8 s) was used to quantitate furan-based products such as BMF, CMF, and FF (Fig. S3 in ESI†). Specifically, BMF was quantitated by comparing the quantity of the protons in the samples with the protons in the internal standard (pyrazine), as presented in Fig. S3(a) in ESI.† FF and CMF were quantitated with a similar method. The molar yields of the furan-based products were calculated based on the initial pentoses or hexoses available in the feedstock with an assumption that one mole of monosaccharide would produce one mole of furan-based product. Targeted peaks of BMF were assigned at: δ = 6.55 ppm (1H) & 112.3 ppm (13C); δ = 7.16 ppm (1H) & 122.2 ppm (13C); targeted furfural peaks were assigned at: δ = 6.63 ppm (1H) & 112.7 ppm (13C); δ = 7.22 ppm (1H) & 121.8 ppm (13C) in the NMR spectra (Fig. S4 in ESI†). 1H-NMR quantification method was verified by gas chromatography-mass spectrometry (GC-MS) (GCMS-QP 2010S, Shimadzu Co., Addison, IL) equipped with SHRXI-5MS column (30 m × 0.25 mm I.D., 0.25 μm), using furfural as standard (Fig. S5 in ESI†). The GC oven temperature was programmed as follows: 100 °C for 1 min, ramp to 310 °C at 5 °C min−1 and hold for 7 min. The flow rate of carrier gas (helium) was set to 22 mL min−1. 13C-NMR analysis was conducted to confirm the structures of BMF and CMF (Fig. S3(b) in ESI†). 13C-NMR spectra from BMF (δ = 21.8 ppm) and CMF (δ = 36.1 ppm) were used to identify and quantify the formation of BMF and CMF. This result was also confirmed with GC-MS results (Fig. S5 in ESI†).2.4. Solution-state NMR analysis of whole biomass and separated lignins
Whole biomass (Douglas fir and poplar) and separated lignins from the biomass by the biphasic process were characterized with solution-state NMR analysis. For untreated whole biomass, the dried sample was pre-ground passing 50 mesh and then Soxhlet-extracted with water and ethanol for 24 h. The extractives-free sample was air-dried and further ball-milled using a PULVERISETTE 7 ball mill at 600 rpm in a ZrO2 vessel (50 mL) containing ZrO2 ball bearings (10 × 10 mm). Each sample (200 mg) was milled for 1–2 h in 10 min intervals with 5 min interval breaks to avoid excessive heating. For the separated lignin, no pretreatment was needed. To prepare sample for solution-state NMR, the ball-milled biomass or the separated lignin (50 mg) was transferred into a 5 mm NMR tube, mixed with DMSO-d6/pyridine-d5 (4:1, v/v, 0.5 mL).
Solution-state NMR spectra were acquired on a Bruker Instruments Advance III 500 console equipped with a cryoprobe. The central DMSO solvent peak was used as internal reference (δC 39.5, δH 2.49 ppm). HSQC experiments were carried out using following parameters: acquired from 11 to 1 ppm in F2 (1H) with 100 ms acquisition time, 220 to 20 ppm in F1 (13C) 8 ms acquisition time; the d1 was set 1 s and number of scans was 32. Volume integration of contours in HSQC plots used Bruker's TopSpin software.
2.5. GPC analysis of separated lignins
The weight-average molecular weight (Mw) and number-average molecular weight (Mn) of the separated lignins from three species of biomass including switchgrass, poplar, and Douglas fir were estimated by gel-permeation chromatography (GPC) on a HPLC system (ICS 3000, Dionex) with a UV detector. One guard column (Phenomenex Cat# 03B-2088-K0) and three analytical Phenogel (5 μm) columns with pore size of 10000 Å (Phenomenex Cat# 00H-0445-K0), 500 Å (Phenomenex Cat# 00H-0443-K0), and 50 Å (Phenomenex Cat# 00H-0441-K0) were connected in series, and the column temperature was maintained using a column heater (CH-460, Eppendorf) with a temperature controller (TC-50, Eppendorf). The lignin was acetylated in pyridine/acetic anhydride (1:1, v/v) to improve the solubility in tetrahydrofuran (THF). The lignin (10 mg) was suspended in 2 mL pyridine/acetic anhydride mixture and kept in the dark for 72 h. The mixture was poured into cold acidic water and stirred for 1 h. The precipitate was collected, washed with deionized water, and then dried in a freeze dryer. The acetylate lignin was dissolved in THF and filtered by a syringe filter. The THF solution (10 μL) was injected onto the GPC columns with an eluent (THF) flow rate of 1.0 mL min−1 at 30 °C. A set of polystyrene standards and aromatic compounds were used for calibration.
2.6. Experimental design and software
The conversion of cellulose to BMF in the biphasic system was optimized using response surface methodology (RSM) following the quadratic central composite design (CCD). Twenty points were tested with three variables (reaction temperature, reaction time, and hydrobromic acid (HBr) loading), and each variable varied at three levels (α = 1.0). BMF yield was the response (dependent variable). Each reaction point was conducted in duplicate. Design Expert software (version 7.1.1, Stat-Ease, Inc., Minneapolis, MN) was used for the experiment design and statistic analysis of data.3. Results and discussion
3.1. Description of the proposed biphasic system
The biphasic system used in this study is presented in Fig. 1, including an aqueous phase (bottom layer) of molten salt hydrate (concentrated LiBr solution) with a small quantity of acid as a co-catalyst and an organic phase (top layer) of extraction solvent. In the aqueous phase, cellulose and hemicelluloses are first dissolved, then hydrolysed into hexoses and pentoses, and further dehydrated or derived to BMF and FF, respectively. Meanwhile, lignin is depolymerized and left over as a solid residue. As FF and BMF have better solubility (partition) in the organic phase than in the aqueous phase, they are immediately extracted into the organic phase when formed, which prevents them from further decomposition and condensation. Lignin is cumulated between the two phases because its density is lower than that of the molten salt hydrate but higher than that of the organic solvent. The solid lignin can be easily separated from the system by filtration, while the solvent can be recycled for the following batch after FF and BMF are recovered. The aqueous phase can be reused after necessary regeneration such as adjustment of the salt and acid concentrations. | ||
| Fig. 1 Biphasic system consisting of an aqueous phase of lithium bromide solution and an organic phase of solvent for converting lignocellulose to FF and BMF and depolymerized lignin. | ||
A molten salt hydrate is basically a concentrated aqueous solution of an inorganic salt. The concentration of the molten salt hydrate varies from salt to salt, dependent on the coordination number of the salt cation.39 Molten salt hydrates have similar properties with ionic liquids, such as low vapor pressure, high boiling point, and ability to swell and dissolve cellulose, but the former is less expensive, more environment-friendly, easier to recycle, and less viscous than the latter. For example, LiBr has very high solubility in water (166.7 and 266.0 g/100 mL at 20 and 100 °C, respectively). LiBr·3H2O (equivalent to 61.7%, w/w) is a liquid at room temperature with very low viscosity (∼3 mPa S). These unique properties make the molten LiBr hydrate an attractive reaction medium for the conversion of lignocellulosic biomass.
In a perfect molten salt hydrate, all water molecules are tightly bound to the inner coordination sphere of the cation, leaving the anion naked in the system.40 For example, in the molten salt hydrate of lithium bromide (LiBr·3H2O) used in this study, lithium cation (Li+) has octahedral coordination geometry and needs 6 oxygen atoms to coordinate. Since the oxygen of each water molecule is able to coordinate with two lithium ions, the molar ratio of LiBr to H2O is 1:3 in the molten salt hydrate of LiBr. While Li+ is surrounded by water molecules, Br− is naked and free in the solution. The free bromide anion (Br−) tends to associate with the H of cellulose hydroxyl groups and form hydrogen bonds. On the other hand, Li+ is very oxophilic and able to interact with the O of cellulose hydroxyl groups when dissociated from the coordinated water molecules by heating and, in particular, when part of the water in the system is consumed for hydrolysis of cellulose and hemicelluloses. The cellulose–Br− and cellulose–Li+ associations or interactions disrupt the inter- and intra-molecular hydrogen bonding of cellulose, thereby breaking the tight crystalline matrix of cellulose, thus leading to the swelling and dissolution of cellulose. The dissolution generates a homogeneous reaction medium and facilitates the hydrolysis of cellulose and hemicelluloses. In addition, the enhanced acidity in molten salt hydrate,41 compared with that in water at the same acid concentration, promotes the hydrolysis of the polysaccharides to monomeric saccharides and the following dehydration of the monomeric saccharides to furans. Furthermore, the free bromide anions in the solution are strong nucleophiles, which are able to catalyze the dehydration of saccharides to furans and likely the cleavage of β-O-4 ether bonds of lignin resulting in the depolymerization of lignin,22,27,30,31 which will be further discussed later. Finally, the salting-out effect of the inorganic salt (LiBr) can enhance the partition coefficient of BMF and FF in the extraction solvent and thereby facilitate effective extraction of the furans from the aqueous phase to the organic phase.17,42
It is known that a biphasic system consisting of aqueous and organic phases was able to prevent or reduce undesired decomposition and condensation of furan products accumulated in the aqueous phase. In the previous studies, different biphasic systems such as HCl–H3PO4/CHCl3, ethyl acetate/ILs, acid/alkylphenol, HCl/chlorinated hydrocarbons, and AlCl3·6H2O–NaCl/THF were investigated for the production of furan-based products.18,32–34,43–46 Considering the unique properties of the molten salt hydrate, the biphasic system consisting of extraction solvent and molten salt hydrate was expected to enhance the yield of the furan products from lignocellulosic biomass.
3.2. Transformation of cellulose to BMF in the biphasic system
First, the transformation of pure cellulose to halide-methyl furfural (BMF or CMF) in the proposed biphasic system was investigated. Different molten salt hydrates, including LiBr, LiCl, and CaBr2, were tested and compared. Reaction temperature, time, and HBr (co-catalyst) loading were optimized through preliminary tests designed by RSM (Tables S1–S3 and Fig. S6†). From the RSM optimization, suggested optimal reaction conditions were 125 °C, 0.181 mL HBr loading (equivalent to 0.16 M of HBr in aqueous phase), and 126 min. As summarized in Table 1, LiBr with HBr as a co-catalyst gave the highest yield (90.3%). Under the same reaction conditions (125 °C and 126 min) and molar salt concentration (LiBr·3H2O and LiCl·3H2O), the product yield of the LiBr system (90.3% BMF) was much higher than that of the LiCl system (72.9% CMF). This observation suggests that LiBr was more efficient than LiCl at the conversion of cellulose into furans (BMF or CMF), which could be attributed to the differences in size, charge density, and electronegativity of Br− and Cl−. Br− has larger radius and higher nucleophilicity, which make it more efficient in swelling and dissolving cellulose than Cl−. Calcium bromide (CaBr2·6H2O) gave a lower BMF yield than lithium bromide (LiBr·3H2O), implying that the cation of a molten salt hydrate was also an important factor affecting the conversion of cellulose to BMF. The performance difference between Ca2+ and Li+ was likely related to their size, coordination geometry and structure, and concentration and solubility in water, which affect their interactions with Br−, water, and cellulose. Lu et al. recently reported that the structure and size of the cation affected the dissolution of cellulose.47 At the same molar concentration (0.16 M) of proton (1.29 w/v% HBr and 0.58 w/v% HCl, respectively) in the aqueous phase, HCl gave lower product yield than HBr (77.3% vs. 90.3% in Table 1).| Halide salt | Organic solvent | Acids | BMF/CMF yield [%] |
|---|---|---|---|
| a Note: reaction conditions: 2% (w/v) (200 mg) of biomass substrates loading with 0.16 M of HBr in 10 mL of aqueous phase, 20 mL of different organic solvents in organic phase, 125 °C, 500 rpm for stirring, 126 min. | |||
| LiBr | DCM | HBr | 90.3 ± 2.0 |
| CaBr2 | DCM | HBr | 70.3 ± 3.9 |
| LiCl | DCM | HBr | 72.9 ± 2.8 |
| LiBr | Toluene | HBr | 64.8 ± 4.6 |
| LiBr | Cyclohexane | HBr | 26.7 ± 5.2 |
| LiBr | Benzene | HBr | 76.5 ± 0.7 |
| LiBr | DCM | HCl | 77.3 ± 0.8 |
Efficient recovery of the furans is as important as conversion itself; therefore, selection of extraction solvent for the organic phase in this biphasic system is crucial. A good solvent should be immiscible with the aqueous phase, stable under the reaction conditions, and easy to separate from the products for reuse. In addition, FF and BMF, but not the salt and acid catalyst, should have high partition coefficient in the solvent. The results shown in Table 1 indicate that DCM gave the highest BMF yield, but it needs to be replaced with greener solvent. Toluene was reported to be an effective extraction solvent of FF and HMF in previous studies,15,34 but its performance in the present biphasic system was poorer than DCM, giving a BMF yield of only 64.8%. Cyclohexane turned out to be not a good solvent because only 26.7% BMF was recovered under the same conditions. Benzene showed the potential as an extraction solvent (76.5% BMF yield), although its performance needs to be improved by further investigation.
It was observed that the volume ratio of organic phase to aqueous phase was also a critical factor that affected not only the load of the solvent but also the total yield of BMF. As shown in Fig. 2, under the same reaction conditions, when the volume ratio of organic phase to aqueous phase increased from 1.0 to 2.0, the recovery yield of BMF increased from 73.9% to 90.3%. The results suggest that sufficient organic solvent be important for satisfactory BMF recovery. To investigate how much the generated BMF was extracted to the solvent layer at the ratio of 1.0, the aqueous phase was further extracted after the reaction with fresh DCM, and the combined ratio of solvent to aqueous phase was 2. Additional 5.1% BMF was recovered (Fig. 2), and the total HMF yield ended up to 79.0%, which, however, was still 11.3% lower than that (90.3%) from the experiment with the ratio of 2. The results indicate that sufficient solvent is crucial to extract the newly formed BMF and prevent it from condensation or further degradation. Continuous extraction of the product during the reaction might be able to further reduce solvent use and improve BMF yield, which needs further investigation.
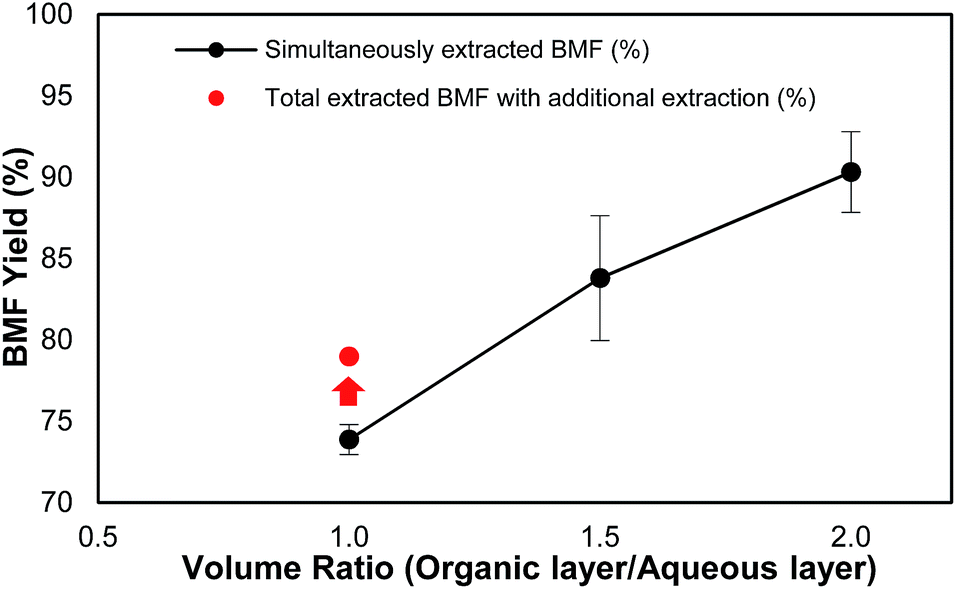 | ||
| Fig. 2 Effect of volume ratio of organic phase to aqueous phase on BMF recovery. Note: reaction conditions: 2% (w/v) (200 mg) of cellulose loading with 0.16 M of HBr in 10 mL of LiBr hydrate (61.6%), 10–20 mL of organic phase (DCM), 125 °C, 500 rpm for stirring, 126 min. | ||
The effect of LiBr concentration on BMF production from cellulose was investigated to demonstrate the importance of molten salt hydrate. As shown in Fig. 3, the investigated LiBr concentrations include 20.2, 40.8, 44.6, 49.1, 54.7, 61.6, and 70.7% (w/w), which correspond to the molar ratios of H2O to LiBr of 19, 7, 6, 5, 4, 3, and 2, respectively. The results clearly indicated the cruciality of LiBr concentration. No BMF was detected at a concentration of 20.2% LiBr (LiBr·19H2O), and no significant BMF was formed until the LiBr concentration reached 49.1% (LiBr·5H2O). The highest BMF yield (90.3%) was observed at 61.6% concentration (LiBr·3H2O), at which the molten salt hydrate of LiBr is formed. Further increasing LiBr concentration to 70.7% (LiBr·2H2O) did not enhance but reduced BMF yield (Fig. 3), which was likely because the water deficient condition at high LiBr concentration depressed the cellulose hydrolysis. The results verified the hypothesis that the molten salt hydrate system is crucial for the conversion of cellulose to BMF.
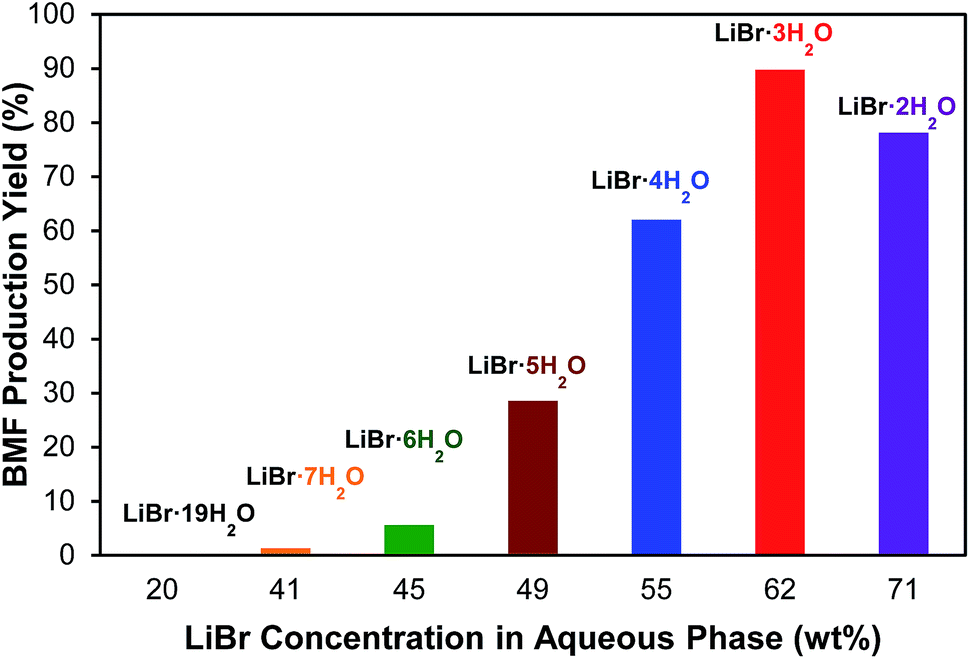 | ||
| Fig. 3 Production of BMF from cellulose at different LiBr concentrations. Note: reaction conditions: 2% (w/v) (200 mg) of cellulose loading with 0.16 M of HBr in 10 mL of different concentration of LiBr hydrates, 20 mL of organic phase (DCM), 125 °C, 500 rpm for stirring, 126 min. | ||
3.3. Proposed reaction mechanism of cellulose to BMF
To understand the pathway from cellulose to BMF, the conversion of glucose, fructose, cellobiose, and cellulose to BMF was investigated. Cellobiose, glucose, and fructose were selected because they are possible intermediates of the conversion from cellulose to BMF. As shown in Fig. 4, 94.3% of fructose was converted to BMF within the first 30 min; the BMF yield decreased slightly when the reaction was extended to 2 h, probably, in part, due to the condensation/decomposition of BMF. Since glucose and cellobiose are the hydrolysis products of cellulose, they were expected to have faster reactions and higher BMF yield than cellulose; however, under the same conditions, the conversion of glucose, cellobiose, and cellulose to BMF followed a similar trend with regarding to reaction rate and product yield. The conversion of glucose, cellobiose, and cellulose was slower and resulted in lower BMF yield than that of fructose. As fructose was quickly converted to BMF with high yield (Fig. 4), it is reasonable to hypothesize that the hydrolysis of cellulose to glucose was probably not the rate-limiting reaction, but rather the isomerization of glucose to fructose likely controlled the overall reaction rate.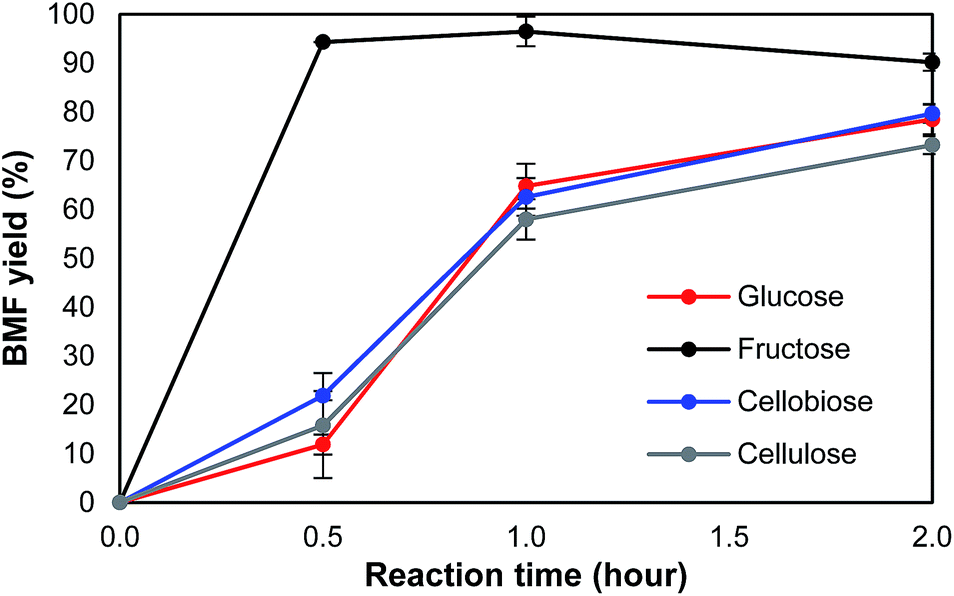 | ||
| Fig. 4 Conversion of different carbohydrates into BMF. Note: reaction conditions: 2% (w/v) (200 mg) of carbohydrates loading with 0.16 M of HBr in 10 mL of LiBr hydrate (61.6%), 20 mL of organic phase (DCM), 125 °C, 500 rpm for stirring, 0–120 min. | ||
Based on the results and discussions above, the pathway from cellulose to BMF in molten salt hydrate (LiBr·3H2O) is proposed in Fig. 5, which includes the steps of cellulose dissolution, cellulose hydrolysis to glucose, glucose isomerization to fructose, fructose dehydration to HMF, and HMF bromination to BMF. First of all, cellulose is swelled and dissolved in the molten salt hydrate because of the disruption of intermolecular and intramolecular hydrogen bonds of cellulose by the formation of Br–H⋯O and Li–O⋯H interactions, as discussed above. The dissolved cellulose is more readily and quickly hydrolyzed into glucose by acid than solid and crystalline cellulose. As mentioned above (Fig. 4), dissolution and hydrolysis of cellulose seem to not be the rate-limiting steps of cellulose conversion to BMF. The following step is the isomerization of glucose to fructose. Because of the presence of H+ and Li+ in the system, there are two possible isomerization pathways catalyzed by them, respectively. In path 1, glucose is isomerized to fructose through the acid-catalyzed intramolecular hydrogen transfer from C-2 to C-1, initiated by protonation of the carbonyl oxygen atom on C-1.48,49 In path 2, the isomerization is achieved by a metal-catalyzed hydrogen transfer between C-2 and C-1 via a hydride shift.50–52 Li+ attacks the O of the hydroxyl group on C-2 and leads to the formation of intermediate, a 5-member complex involving Li+, which then rearranges to fructose. In the third step, fructose is dehydrated to HMF, catalyzed by either Br− 53 or H+ 54 in the acidic molten salt hydrate system. The last step of cellulose-to-BMF is the bromination of HMF. The hydroxyl group of HMF is protonated to form a good leaving group (water), which is then displaced by bromide through a SN2 mechanism.
 | ||
| Fig. 5 Proposed conversion pathways from cellulose to BMF. | ||
3.4. Transformation of real biomass in the biphasic system
The biphasic system was applied to different types of real biomass including switchgrass (energy crop), corn stover (agricultural residue), poplar and aspen (hardwood), and Douglas fir (softwood). As summarized in Table 2, the process was able to convert cellulose and hemicelluloses in the biomass simultaneously to BMF and FF, respectively, with high yields. The molar yields of BMF and FF were calculated based on original hexoses and pentoses in the biomass (Table S4†), respectively. Under the same conditions, aspen had the highest BMF yield (87.1%), while corn stover, switchgrass, and poplar gave similar yields of approximately 70%. Softwood Douglas fir had the lowest BMF yield, which was presumably attributed to its different physical structure and chemical composition, in particular high lignin content. The yield of FF varied from biomass to biomass within the range of 51–84%, dependent on the species.| Biomass | BMF yield [%] | Furfural yield [%] |
|---|---|---|
| a Note: reaction conditions: 2% (w/v) (200 mg) of different biomass loading with 0.16 M of HBr in 10 mL of LiBr hydrate (61.6%), 20 mL of organic phase (DCM), 125 °C, 500 rpm for stirring, 126 min. | ||
| Corn stover | 71.3 ± 1.6 | 69.4 ± 5.6 |
| Switchgrass | 70.2 ± 0.7 | 51.2 ± 2.0 |
| Poplar | 72.2 ± 2.8 | 61.9 ± 1.3 |
| Aspen | 87.1 ± 1.5 | 83.9 ± 5.9 |
| Douglas fir | 63.9 ± 2.3 | 57.3 ± 0.0 |
3.5. Transformation of biomass at high solid loading
It is apparent that increasing solid loading could improve the efficiency and productivity of biomass conversion processes. As all the tests above (Table 2) were conducted at low solid loading (2 wt% in aqueous phase), higher solid loading was explored. As shown in Fig. 6, the yields of BMF and FF from aspen were 87% and 84%, respectively, at 2 wt% solid loading, and slightly decreased to 80% and 78%, respectively, at 5% aspen loading; however, the yields significantly dropped when solid loading was further increased to 10%. In particular, the BMF yield sharply dropped to 44%, which was probably due to the insufficient swelling and hydrolysis of cellulose at high solid loading. Extending reaction time from 126 to 189 min brought FF yield back to 85% (solid red triangle in Fig. 6), but BMF yield was only slightly improved (solid red circle in Fig. 6), implying that extending reaction time did not substantially promote the conversion of cellulose. Differently, increasing catalyst (HBr) loading by 3 times (to 0.48 M in aqueous phase) elevated the BMF yield to 67% (solid blue circle in Fig. 6), but FF yield was not improved, which was probably due to the enhanced FF condensation by the additional acid. These results suggest that additional acid catalyst and longer reaction time are necessary to reach satisfactory FF and BMF yields at high solid loading.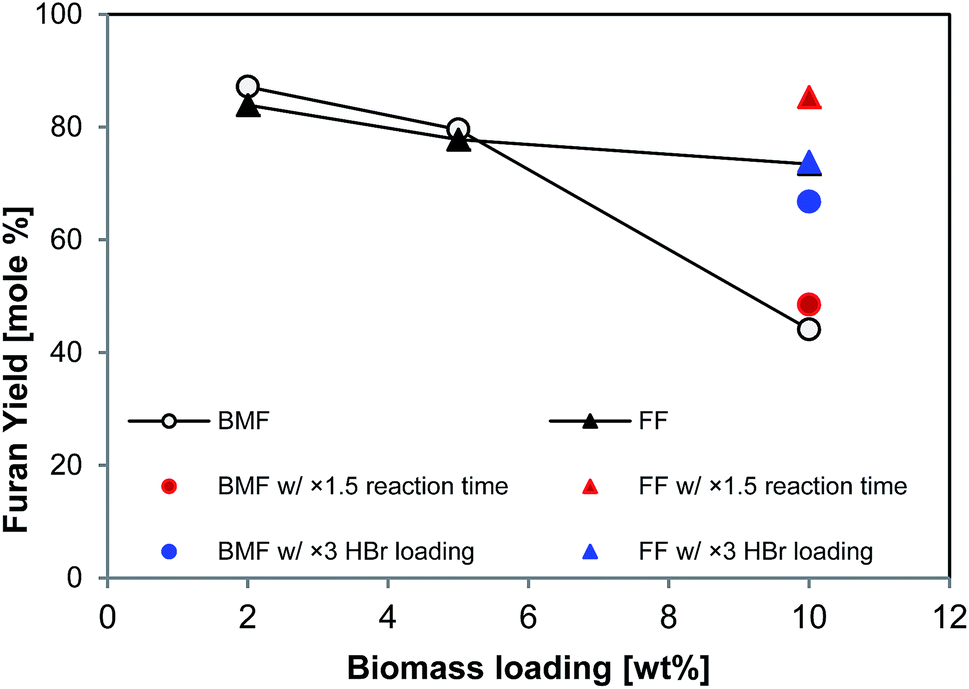 | ||
| Fig. 6 BMF and FF production from Aspen with different substrates loading. Note: reaction conditions: 2–10% (w/v) of aspen loading with 0.16 M of HBr in 10 mL of LiBr hydrate (61.6%), 20 mL of organic phase (DCM), 125 °C, 500 rpm for stirring, 126 min. | ||
3.6. Characterization of separated lignin
When cellulose and hemicelluloses of the biomass were converted to BMF and FF, respectively, lignin was left over as an insoluble residue between the two phases, as shown in Fig. 1 and S2.† The separated lignin was preliminarily investigated to elucidate the change of lignin and evaluate the potential for lignin co-products. Firstly, purity of three lignin samples from switchgrass, poplar, and Douglas fir was analyzed. It was found that 97% of the solid residues were lignin, and trace or no residual carbohydrates were detected (Table S5†). This observation indicates that the present biphasic system not only completely convert cellulose and hemicelluloses into BMF and FF, respectively, but produce high-purity lignin.The molecular weight of the lignin samples was estimated by GPC after acetylation. It was found that the acetylated lignins were not completely soluble in THF, which was used as the eluent for the GPC analysis. The weight average molecular weights of the THF-soluble fractions of the lignins are summarized in Table 3. They fell between 1000 and 1300 g mol−1, which was much lower than those of native lignins (milled wood lignins) (5900–23500 g mol−1) and separated lignins from other processes (2000–8700 g mol−1).55 The results suggest that the THF-soluble lignin was extensively depolymerized during the reaction. The THF-insoluble lignin represented the fraction that was either insufficiently depolymerized or repolymerized (condensed) during the reaction.
| Lignin | PDI | Mn (g mol−1) | Mw (g mol−1) |
|---|---|---|---|
| a Note: reaction conditions: 2% (w/v) of poplar/switchgrass/Douglas fir loading with 0.16 M of HBr in 10 mL of LiBr hydrate (61.6%), 20 mL of organic phase (DCM), 125 °C, 500 rpm for stirring, 126 min.b PDI – polydispersity index (= Mw/Mn). | |||
| Poplar lignin | 2.60 | 500 | 1300 |
| Switchgrass lignin | 2.50 | 400 | 1000 |
| Douglas fir lignin | 2.60 | 500 | 1300 |
The separated lignins from softwood Douglas fir and hardwood poplar were further characterized using NMR and compared with the native lignins in the original woods to understand the changes in the chemical structure of lignins during the transformation of cellulose and hemicelluloses. In the aliphatic (lignin side-chain and polysaccharides) regions (top row in Fig. 7) of the NMR spectra, almost all of the peaks for polysaccharides in both Douglas fir and poplar were removed after the reaction, verifying that the separated lignins were carbohydrate-free, which is consistent with the lignin purity analysis. In addition, major inter-linkage (β-aryl ether) in lignin was clearly removed, which is the direct evidence of lignin depolymerization. Aromatic regions (bottom row in Fig. 7) also indicate that the separated lignins were significantly different from the native lignins in Douglas fir and poplar, respectively. In particular, the G2 and G6 correlation peaks in both softwood and hardwood lignins were decreased and shifted after the reaction in the separated lignins. Similarly, the S2/6 correlation peak in hardwood lignin diminished significantly after the reaction. This might result either from the deconstruction of aromatic rings or from the condensations of lignin at these sites on benzene rings (C2 and C6) with the α-C of other structural units under acidic condition.56–58
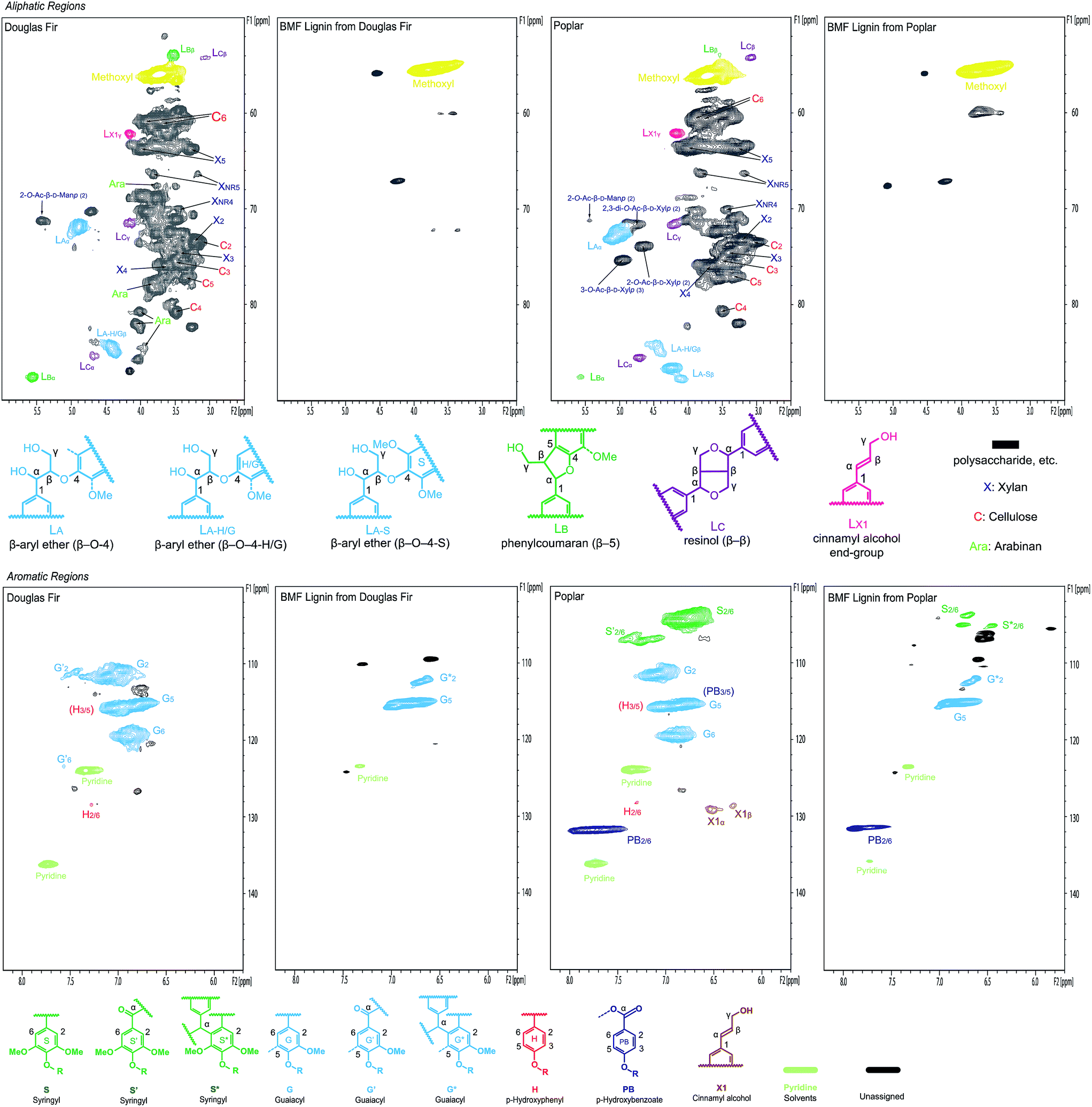 | ||
| Fig. 7 2D NMR spectra of biomass (Douglas fir and poplar) whole cell wall and separated lignins in DMSO-d6/pyridine-d5 (4:1). | ||
4. Conclusion
In summary, the biphasic process including acidic molten salt hydrate (LiBr·3H2O) investigated in this study could effectively convert cellulose and hemicelluloses of lignocellulosic biomass into BMF and FF, respectively, with high yields at moderate temperature. With unique properties, the molten salt hydrate of lithium bromide was able to swell and dissolve cellulose, enhance the hydrolysis of cellulose to glucose, catalyze the isomerization of glucose to fructose and the dehydration of fructose to HMF, and brominate HMF to BMF. Simultaneously, lignin of the biomass was extensively depolymerized and separated for co-products.Acknowledgements
This research was supported by National Science Foundation (CBET-1159561 and CBET 1236562), USDA McIntire Stennis project (WIS01861), and Wisconsin Alumni Research Foundation (WARF) Accelerator Program. The authors thank Dr Hoon Kim for his kind assistance with whole cell wall gel NMR and discussion of NMR spectra.Notes and references
- N. Sathitsuksanoh, A. George and Y. H. P. Zhang, J. Chem. Technol. Biotechnol., 2013, 88, 169–180 CrossRef CAS.
- J. Serrano-Ruiz and J. Dumesic, in Catalysis for Alternative Energy Generation, ed. L. Guczi and A. Erdôhelyi, Springer, New York, 2012, ch. 2, pp. 29–56 Search PubMed.
- V. B. Agbor, N. Cicek, R. Sparling, A. Berlin and D. B. Levin, Biotechnol. Adv., 2011, 29, 675–685 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- B. Joshi, M. R. Bhatt, D. Sharma, J. Jarina, R. Malla and L. Sreerama, Biotechnol. Mol. Biol. Rev., 2011, 6, 172–182 CAS.
- C. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- J. Hill, E. Nelson, D. Tilman, S. Polasky and D. Tiffany, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11206–11210 CrossRef CAS PubMed.
- J. Akhtar, A. Idris and R. Abd Aziz, Appl. Microbiol. Biotechnol., 2014, 98, 987–1000 CrossRef CAS PubMed.
- C. Gao, C. Ma and P. Xu, Biotechnol. Adv., 2011, 29, 930–939 CrossRef CAS PubMed.
- R. D. Cortright, M. Sanchez-Castillo and J. A. Dumesic, Appl. Catal., B, 2002, 39, 353–359 CrossRef CAS.
- G. Marcotullio and W. d. Jong, Carbohydr. Res., 2011, 346, 1291–1293 CrossRef CAS PubMed.
- J. M. Cruz, J. M. Domínguez, H. Domínguez and J. C. Parajó, Biotechnol. Lett., 2000, 22, 1895–1898 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- M. Mascal and E. B. Nikitin, Green Chem., 2010, 12, 370–373 RSC.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef PubMed.
- L. Hu, G. Zhao, W. Hao, X. Tang, Y. Sun, L. Lin and S. Liu, RSC Adv., 2012, 2, 11184–11206 RSC.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS.
- X. Tong, Y. Ma and Y. Li, Appl. Catal., A, 2010, 385, 1–13 CrossRef CAS.
- A. Gandini and M. Belgacem, J. Polym. Environ., 2002, 10, 105–114 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- C. Li, Z. K. Zhao, A. Wang, M. Zheng and T. Zhang, Carbohydr. Res., 2010, 345, 1846–1850 CrossRef CAS PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 Search PubMed.
- M. E. Zakrzewska, E. Bogel-łukasik and R. Bogel-łukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS PubMed.
- S. Dutta, S. De, B. Saha and M. I. Alam, Catal. Sci. Technol., 2012, 2, 2025–2036 CAS.
- L. Wang, F. Yuan, X. Niu, C. Kang, P. Li, Z. Li and Y. Zhu, RSC Adv., 2016, 6, 40175–40184 RSC.
- Y. Xing, B. Yan, Z. Yuan and K. Sun, RSC Adv., 2016, 6, 59081–59090 RSC.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268–1272 CrossRef CAS PubMed.
- N. Kumari, J. K. Olesen, C. M. Pedersen and M. Bols, Eur. J. Org. Chem., 2011, 1266–1270 CrossRef CAS.
- W. Gao, Y. Li, Z. Xiang, K. Chen, R. Yang and D. S. Argyropoulos, Molecules, 2013, 18, 7675–7685 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, ChemSusChem, 2009, 2, 859–861 CrossRef CAS PubMed.
- M. Mascal and E. B. Nikitin, Angew. Chem., Int. Ed., 2008, 120, 8042–8044 CrossRef.
- M. Mascal and S. Dutta, in Selective Catalysis for Renewable Feedstocks and Chemicals, Springer, 2014, pp. 41–83 Search PubMed.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2006, 45, 2163–2173 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 7703–7710 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and J. R. L. Smith, Green Chem., 2009, 11, 1273–1484 RSC.
- H. Leipner, S. Fischer, E. Brendler and W. Voigt, Macromol. Chem. Phys., 2000, 201, 2041–2049 CrossRef CAS.
- S. Sen, J. D. Martin and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2013, 1, 858–870 CrossRef CAS.
- J. A. Duffy and M. D. Ingram, Inorg. Chem., 1978, 17, 2798–2802 CrossRef CAS.
- T. Tan and S. Aravinth, Fluid Phase Equilib., 1999, 163, 243–257 CrossRef CAS.
- M. Brasholz, K. von Känel, C. H. Hornung, S. Saubern and J. Tsanaktsidis, Green Chem., 2011, 13, 1114–1117 RSC.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef PubMed.
- Y. Yang, C.-W. Hu and M. M. Abu-Omar, Green Chem., 2012, 14, 509–513 RSC.
- B. R. Caes and R. T. Raines, ChemSusChem, 2011, 4, 353–356 CrossRef CAS PubMed.
- B. Lu, A. Xu and J. Wang, Green Chem., 2014, 16, 1326–1335 RSC.
- D. W. Harris and M. S. Feather, Carbohydr. Res., 1973, 30, 359–365 CrossRef CAS.
- N. Shi, Q. Liu, L. Ma, T. Wang, Q. Zhang, Q. Zhang and Y. Liao, RSC Adv., 2014, 4, 4978–4984 RSC.
- C. A. Collyer, K. Henrick and D. M. Blow, J. Mol. Biol., 1990, 212, 211–235 CrossRef CAS PubMed.
- R. W. Nagorski and J. P. Richard, J. Am. Chem. Soc., 2001, 123, 794–802 CrossRef CAS PubMed.
- M. Moliner, Y. Román-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS PubMed.
- Y.-N. Li, J.-Q. Wang, L.-N. He, Z.-Z. Yang, A.-H. Liu, B. Yu and C.-R. Luan, Green Chem., 2012, 14, 2752–2758 RSC.
- S. Caratzoulas and D. G. Vlachos, Carbohydr. Res., 2011, 346, 664–672 CrossRef CAS PubMed.
- A. Tolbert, H. Akinosho, R. Khunsupat, A. K. Naskar and A. J. Ragauskas, Biofuels, Bioprod. Biorefin., 2014, 8, 836–856 CrossRef CAS.
- K. Shimada, S. Hosaya and T. Ikeda, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef CAS.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS PubMed.
- X. Yang, N. Li, X. Lin, X. Pan and Y. Zhou, J. Agric. Food Chem., 2016, 64, 8379–8387 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, optimization of reaction conditions, and NMR data for product quantification and lignin characterization. See DOI: 10.1039/c6ra25025d |
| This journal is © The Royal Society of Chemistry 2017 |