
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using a flexible bis(pyrazol) ligand to construct four new Keggin-based compounds: syntheses, structures and properties†
Aixiang Tian,
Huaiping Ni,
Xuebin Ji,
Yan Tian,
Guocheng Liu and
Jun Ying*
Department of Chemistry, Bohai University, Jinzhou, 121000, P. R. China. E-mail: tian@bhu.edu.cn; ying@bhu.edu.cn
First published on 19th January 2017
Abstract
Using a flexible bis(pyrazol) ligand, four new compounds based on Keggin anions, namely [Cu(H2bdpm)2(H2O)2(HPW12O40)] (1), [Cu(H2bdpm)2(H2PW12O40)]2·2H2O (2), [Cu2(H2bdpm)3(H2O)2(SiW12O40)]·2H2O (3), and [Cu2(H2bdpm)4(H2O) (SiW12O40)]·16H2O (4) (H2bdpm = bis(3,5-dimethyl-1H-pyrazol-4-yl)methane), have been synthesized under hydrothermal conditions. Compound 1 is a zero dimension (0D) structure, showing a [Cu(H2bdpm)2]2+ mono-supporting [PW12O40]3− anion. Compound 2 shows a 1D supramolecular chain, with bi-supporting anions lined by discrete [PW12O40]3− anions. Compound 3 contains bi-nuclear Cu2+ cycles, which are linked by O4W to form a cycle-connecting-cycle chain. The [SiW12O40]4− anions connect adjacent chains to construct a 2D structure. Compound 4 contains a nested cycle subunit [Cu4(H2bdpm)6]8+, with two bi-nuclear cycles [Cu2(H2bdpm)2]4+ linked by two H2bdpm ligands. The nested cycle subunits are connected by H2bdpm molecules to construct a 2D metal–organic layer. The Keggin anions link adjacent layers to form a 3D framework. The electrochemical and photocatalytic properties of compounds 1–4 are studied.
Introduction
Recently, polyoxometalates (POMs) have attracted attention as a branch of inorganic chemistry, not only due to their diverse structures,1 but also for their extensive applications in catalysis, medicine, magnetism, photochemistry and other fields.2 POMs can provide abundant O donors to connect transition metal complexes (TMCs) to construct fascinating structures,3 which has become a booming field of POMs.Among a large number of POM-TMCs, using cycle-connecting-cycle and nested cycle subunits to modify POMs is relatively rare.4 In the synthetic strategy of forming this series, the choice of appropriate organic ligands is essential and important. Observed from reports, flexible organic ligands can construct molecular cycles with ease, containing cycle-connecting-cycle and nested cycle subunits.5 The flexible ligands own conformational freedom and can rotate in some extent, conducing to fuse TM ions and construct cycle structures. In our previous work, we have utilized a series of symmetric flexible bis(triazole) ligands to build POM-based cycle structures.6 For example, by using 1,3-bis(1,2,4-triazol-1-y1)propane (btp) we have obtained a mono-substituted Keggin-based tetra-nuclear metallamacrocycle structure.7 Thus, the usage of symmetric flexible ligands to build POM-based cycle structures is rational and challenging. In this work, we utilize a flexible bis(pyrazole) ligand bis(3,5-dimethyl-1H-pyrazol-4-yl)methane (H2bdpm) instead of bis(triazole) ligands to explore whether H2bdpm can still build POM-based cycle subunits. The selection of H2bdpm owns three advantages: (i) it has four N donors to link TM ions, inducing strong coordination capacity. (ii) The –CH2– group increases flexibility of ligand skeleton, conducing to rotate and fuse TM ions. (iii) It has four –CH3 groups, which can increase steric hindrance to avoid interpenetrating structures and induce cycle structures with ease.8 Thus, the selection of H2bdpm is a reasonable method to build metal–organic cycle modified POM compounds.
Hydrothermal technique has been proven an effective method to construct POM-TMC compounds.9 Commonly, in one-pot under hydrothermal conditions only one type of crystal could be obtained. However, two or more types of crystals obtained in one-pot are rarely observed.10 Namely, high efficient usage of hydrothermal technique to construct two or more POM-based compounds is challenging and appealing. In this work, we try to tune some influencing factors under hydrothermal conditions, aiming to form more crystals in one-pot.
Herein, by introducing H2bdpm to Keggin-Cu2+ system, four new compounds were obtained, [Cu(H2bdpm)2(H2O)2(HPW12O40)] (1), [Cu(H2bdpm)2(H2PW12O40)]2·2H2O (2), [Cu2(H2bdpm)3(H2O)2(SiW12O40)]·2H2O (3) and [Cu2(H2bdpm)4(H2O) (SiW12O40)]·16H2O (4). It is worth mentioning that compounds 3 and 4 were obtained in one pot. We also studied the electrochemical and photocatalytic properties of compounds 1–4.
Experimental
Materials and methods
The reagents and solvents for syntheses were all purchased from commercial sources and used without further purification. Elemental analyses (C, H, and N) were achieved with a Perkin-Elmer 240C elemental analyzer, and the FT-IR spectra were taken on a Varian FT-IR 640 spectrometer (KBr pellets). UV-vis absorption spectra were obtained using a Lambda 750 UV/VIS/NIR spectrophotometer. Electrochemical measurements and data collection were performed with a CHI 440 electrochemical workstation. A conventional three-electrode system was used with a saturated calomel electrode (SCE) as reference electrode and a Pt wire as counter electrode. The title compounds modified carbon paste electrodes (CPEs) were used as the working electrodes.X-ray crystallography
X-ray diffraction analysis data for compounds 1–4 were collected with a Bruker Smart Apex CCD diffractometer (Bruker Corporation, Germany) with Mo-Kα (λ = 0.71073 Å) at 293 K. The structures were solved by direct methods and refined on F2 by full-matrix least squares methods using the SHELXTL package.11 For the compounds, all the hydrogen atoms attached to carbon atoms were generated geometrically. Furthermore, the hydrogen atoms attached to water molecules were not located but were included in the structure factor calculations. In compound 4, the seventeen water molecules were highly disordered and could not be modeled properly, thus the SQUEEZE routine of PLATON was applied to remove the contributions to the scattering from the solvent molecules. The reported refinements are of the guest-free structures using the *.hkp files produced by using the SQUEEZE routine.12 All crystallographic data and structural determination is provided in Table 1. The data of selected bond lengths and angles of the title compounds are listed in Table S1.† Crystallographic data for the structures reported in this paper have been deposited in the Cambridge Crystallographic Data Center with CCDC Numbers of 1486618, 1486619, 1486620 and 1509083 for 1–4.| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | ||||
|---|---|---|---|---|
| Compounds | 1 | 2 | 3 | 4 |
| Formula | C22H37CuN8O42PW12 | C44H72Cu2N16O82P2W24 | C33H56Cu2N12O44SiW12 | C44H98Cu2N16O57SiW12 |
| Fw | 3386 | 6738 | 3686 | 4124 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | C2/c |
| a (Å) | 11.3926(10) | 12.444(2) | 15.2377(13) | 35.318(5) |
| b (Å) | 28.008(2) | 14.557(3) | 18.6736(16) | 23.532(5) |
| c (Å) | 17.0586(14) | 17.393(3) | 25.291(2) | 23.250(5) |
| β (°) | 95.775(2) | 105.644(4) | 97.928(2) | 117.176(5) |
| V (Å3) | 5415.4(8) | 2947.5(10) | 7127.7(11) | 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 190(6) 190(6) |
| Z | 4 | 1 | 4 | 8 |
| Dc (g cm−3) | 4.147 | 3.792 | 3.426 | 2.945 |
| μ (mm−1) | 25.895 | 23.787 | 19.975 | 16.569 |
| F(000) | 5952 | 2960 | 6560 | 13696 |
| R1a [I > 2σ(I)] | 0.0435 | 0.0705 | 0.0402 | 0.0487 |
| wR2b (all data) | 0.0896 | 0.2172 | 0.0806 | 0.1284 |
| GOF on F2 | 0.994 | 1.015 | 0.987 | 1.016 |
Preparation of compounds 1–4
Results and discussion
Syntheses
Under hydrothermal conditions, many factors can influence the final structures, such as temperature, reaction time, reactants and pH.9 In this work, we try to explore the effect of reactants. In PW12-Cu2+-H2bdpm system, we obtained a discrete mono-supporting structure of 1 and a supramolecular structure of 2. However, when we changed POM precursor PW12 to SiW12, two distinct compounds 3 and 4 were synthesized in one pot. So the POM precursors own influence on the structures. Furthermore, highly efficient usage of hydrothermal technique is interesting to obtain two or more crystals in one pot. Compounds 3 and 4 were obtained in one pot, which can support informative examples for highly efficient usage of hydrothermal technique.Structural description
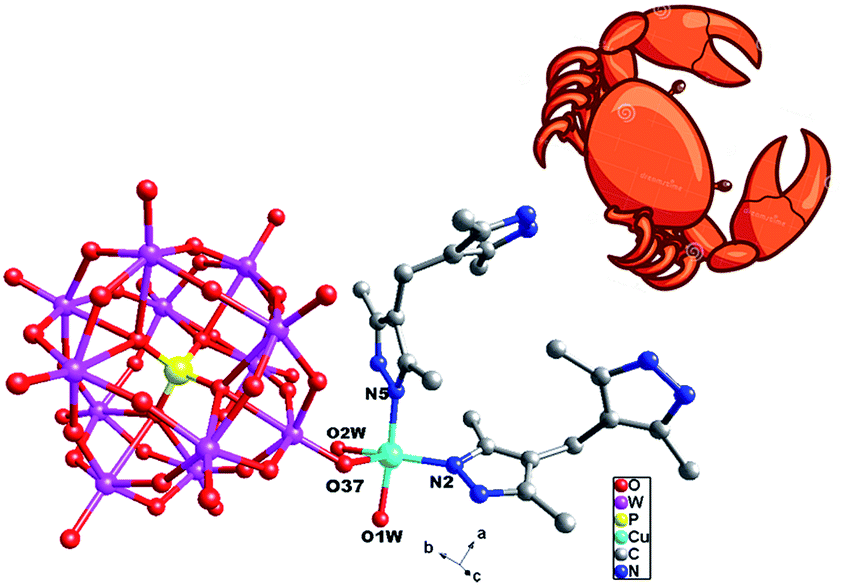 | ||
| Fig. 1 Stick/ball view of the asymmetric unit of 1. All H atoms are omitted for clarity. | ||
Compound 1 owns only one crystallographically independent Cu ion. The Cu1 is coordinated by two N donors (N2 and N5) from two H2bdpm ligands, one O atom from a PW12 anion and two coordinated water molecules (O1W and O2W). The Cu–O distances are in the range of 1.942(9)–2.553(10) Å, while the Cu–N distances range from 1.924(11) to 1.955(12) Å. The O–Cu–N angles are in the range of 90.7(4)–174.5(5)°, while the N–N–Cu angles are from 123.8(9) to 125.3(9)°.
In compound 1, the H2bdpm ligand only offers its one N atom to coordinate with one Cu2+ ion. Two H2bdpm molecules are fused by one Cu1 atom to form a [Cu(H2bdpm)2]2+ subunit, which links a PW12 anion through Cu1–O37 bond to generate a mono-supporting Keggin structure (Fig. 1). This mono-supporting anion is just like a “crab” and two H2bdpm is just like its “pincers”. The adjacent mono-supporting anions further connect each other through hydrogen bonding interactions (O31⋯O34 = 3.238 Å) to form a 1D supramolecular chain (Fig. S1†). The adjacent 1D supramolecular chains are still further linked by hydrogen bonding interactions (O1W⋯O24 = 2.80 Å) to construct a 2D supramolecular layer (Fig. 2).
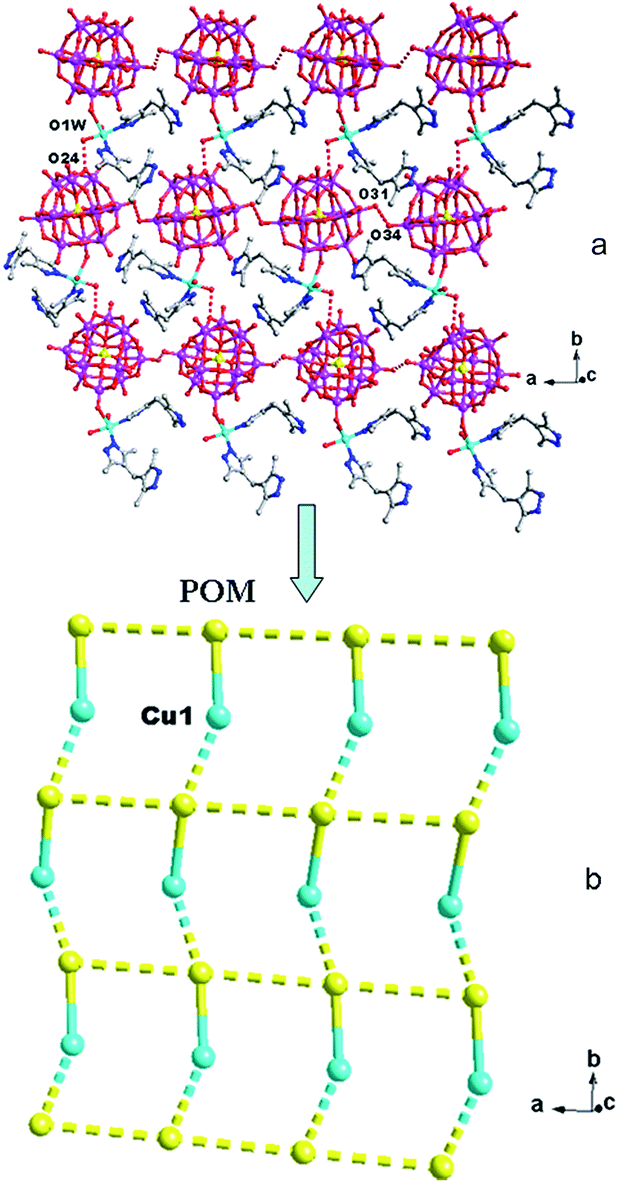 | ||
| Fig. 2 The 2D supramolecular layer (a) and its schematic view (b) of 1. | ||
 | ||
| Fig. 3 Ball/stick view of the asymmetric unit of compound 2. The hydrogen atoms and lattice water molecules are omitted for clarity. | ||
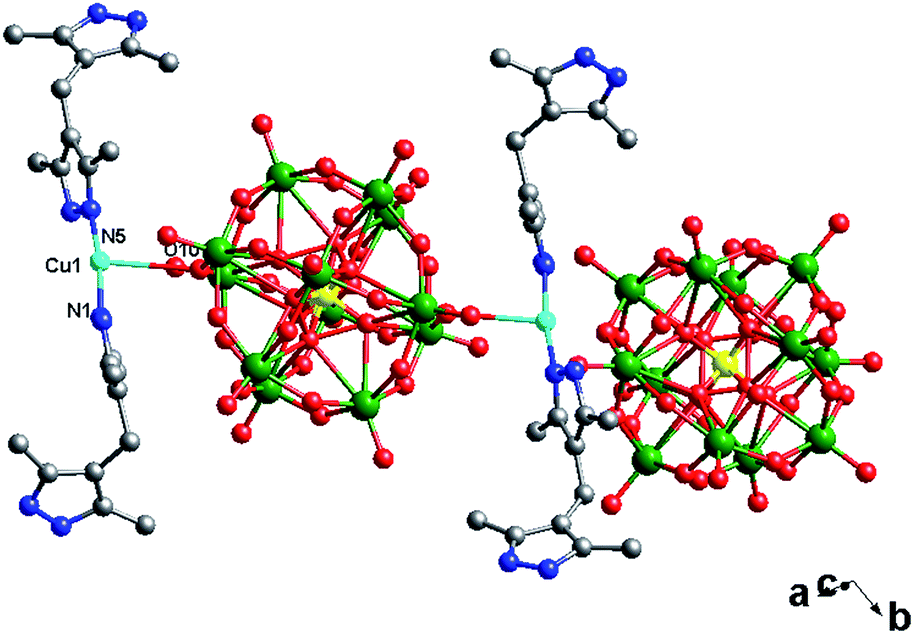
In compound 2, there is only one crystallographically independent Cu1 ion. Considering the long-range coordinative Cu–O bonds, the Cu1 atom adopts a “T”-type geometry, coordinated by two N atoms (N1 and N5) from two H2bdpm ligands and one O atom from one POM1. The Cu–N distances are in the range of 1.86(2)–1.87(2) Å, while the Cu–O distance is 2.93(2) Å. The N–Cu–N angles are in the range of 123.1(18)–124.0(16)°, while the O–Cu–O angles are from 54.5(6)–146.1(6)° (Table S1†).
In compound 2, there are two different building blocks: (i) a POM1 anion links two [Cu(H2bdpm)2] segments to form a bi-supporting anion, like alphabet “H”. (ii) A discrete anion POM2. The two building blocks connect each other through hydrogen bonding interactions (N2⋯O42 = 3.216 Å) to construct a 1D chain, as shown in Fig. 4.
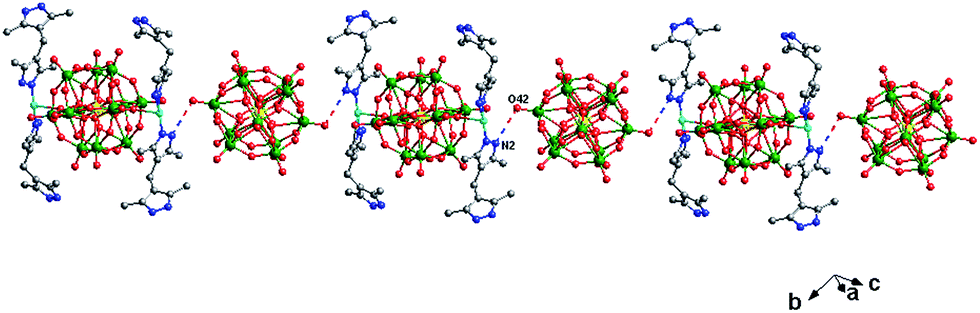 | ||
| Fig. 4 The 1D supramolecular chain of 2. | ||
 | ||
| Fig. 5 Ball/stick view of the asymmetric unit of compound 3. The hydrogen atoms and lattice water molecules are omitted for clarity. | ||
In compound 3, there are two crystallographically independent Cu1 and Cu2 ions. Both Cu1 and Cu2 ions are five-coordinated with distorted square pyramid geometry, having τ value of 0.34 for Cu1 and 0.12 for Cu2 (τ = (β − α)/60). For Cu1 the angles are N2–Cu1–O3W = 178° (β) and N12–Cu1–O4W = 156° (α), while the angles for Cu2 are N3–Cu2–O4W = 177° (β) and N7–Cu2–N9 = 170° (α). The Cu1 ion is coordinated by two nitrogen atoms (N2 and N12) from two H2bdpm molecules, one terminal O37 atom from a SiW12 anion and two coordinated water molecules (O3W and O4W). The Cu2 ion is coordinated by three N atoms (N3, N7 and N9) from three H2bdpm ligands, one terminal O35 atom from a SiW12 anion and one coordinated water molecule O4W. The Cu–O distances are in the range of 1.911(8)–2.451(10) Å, while the Cu–N distances range from 1.960(10) to 2.029(10) Å. The O–Cu–N angles are in the range of 87.4(4)–178.0(4)°, while the N–Cu–N angles are from 89.8(4) to 170.9(4)°.
In compound 3, the H2bdpm shows two kinds of coordination modes: offering only one N donor to link one Cu2+ ion (type-I) and providing two N atoms from two dimethylpyrazole groups respectively to connect two Cu2+ ions (type-II). Two type-II H2bdpm are fused by two Cu2+ ions and a bi-nuclear [Cu2(H2bdpm)2]4+ cycle is built. The adjacent bi-nuclear cycles share the same O4W to construct a cycle-connecting-cycle chain (Fig. 6b). Furthermore, the type-I H2bdpm hang this chain up and down by linking Cu2 ions (Fig. S2†). There also exists an inorganic chain constructed from SiW12 anions linked by Cu1–O4W–Cu2 units (Fig. 6a). The cycle-connecting-cycle metal–organic chains and POM-based inorganic connect each other alternately by sharing Cu ions to generate a 2D grid-like layer (Fig. 6c and d).
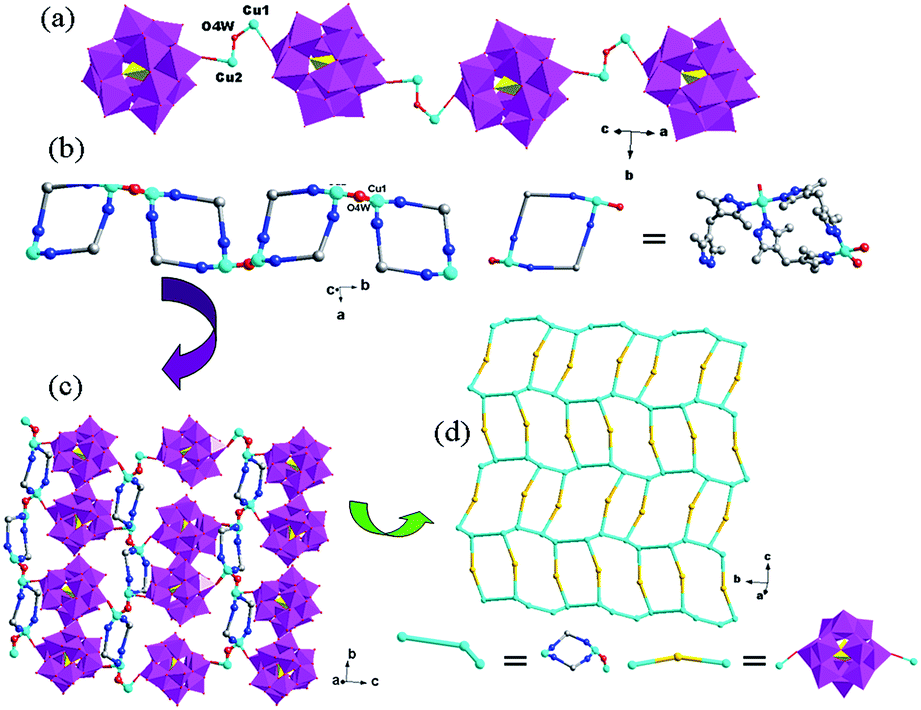 | ||
| Fig. 6 (a) The 1D inorganic chain in 3 with SiW12 anions linked by Cu1–O4W–Cu2 units. (b) The 1D cycle-connecting-cycle metal–organic chain (some C atoms are omitted for clarity). (c) The 2D grid-like layer with inorganic chains and cycle-connecting-cycle metal–organic chains linking each other. (d) The schematic diagram of the 2D layer. | ||
 | ||
| Fig. 7 Ball/stick view of the symmetric unit of compound 4. The hydrogen atoms are omitted for clarity. | ||
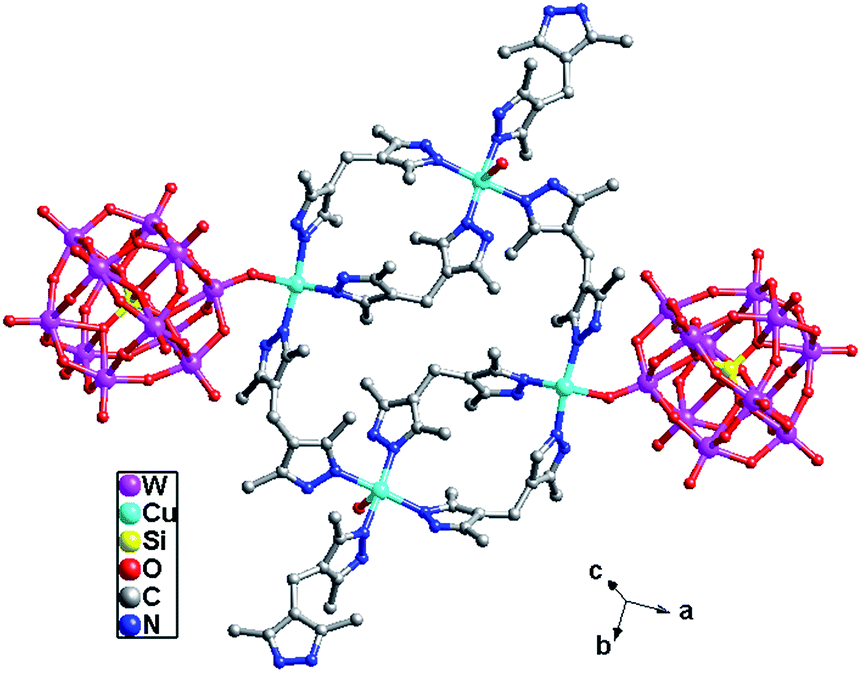
In compound 4, the H2bdpm provides two N atoms from two dimethylpyrazole groups respectively to link two Cu2+ ions, which shows two functions: linking mode (type-A) and cyclization mode (type-B) (Fig. S3†). Two type-B H2bdpm molecules are fused by two Cu2+ ions to form a bi-nuclear [Cu2(H2bdpm)2]4+ cycle. Two bi-nuclear cycles are further linked by two another type-B H2bdpm molecules and a new tetra-nuclear cycle is formed (Fig. S4†). The [Cu4(H2bdpm)6]8+ can be viewed as a nested cycle subunit. Furthermore, the type-A H2bdpm offers two N donors to connect adjacent nested cycle subunits and a 2D metal–organic layer is generated (Fig. 8a and b). The POM anions link adjacent layers covalently to construct a 3D framework of 4 (Fig. 8c and d).
 | ||
| Fig. 8 (a) View of the 2D metal–organics layer of 4 with tetra-nuclear cycles linked by type-A H2bdpm. (b) The schematic view of the 2D layer. (c) The details of the POM linking mode. (d) The 3D framework with adjacent 2D layers connected by anions. | ||
FT-IR spectra and powder X-ray diffractions
The IR spectra of compounds 1–4 are shown in Fig. S5.† The characteristic bands at 970(m), 898(m), 809(s) and 1072(m) cm−1 for 1 are attributed to ν(W–Od), ν(W–Ob–W), ν(W–Oc–W) and ν(P–O).14 The characteristic bands at 973(m), 806(s), 896(m) and 1081(m) cm−1 for 2 are attributed to ν(W–Od), ν(W–Ob–W), ν(W–Oc–W) and ν(P–O). In the spectra of 3 and 4, characteristic bands 970(m), 919(s), 792(m), 528(m), cm−1 for 3, 970(m), 919(s), 798(s), 526(m) cm−1 for 4, are attributed to ν(W–Od), ν(Si–O) and ν(W–Oc–W).15 The bands in the region of 1633–1176 cm−1 for 1, 1731–1247 cm−1 for 2, 1731–1078 cm−1 for 3, 1731–1079 cm−1 for 4 can be attributed to the H2bdpm ligand.16Fig. S6† shows the powder X-ray diffraction patterns for compounds 1–4. The diffraction peaks of both simulated and experimental patterns match well in positions, thus showing that the phase purities of the compounds 1–4 are good.
Electrochemical properties
We have investigated the electrochemical properties of compounds 1–4. Owing to the similar electrochemical behaviors of compounds 1 and 2, 3 and 4 modified carbon paste electrodes, the 1- and 3-CPEs have been taken as examples to study their electrochemical properties. The cyclic voltammograms for 1- and 3-CPEs in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates are presented in Fig. 9. In the range of −850 to +800 mV for 1-CPE, there are three reversible redox peaks II–II′, III–III′ and IV–IV′, with the half-wave potentials at −171(II–II′), −464(III–III′) and −756(IV–IV′) mV (scan rate: 100 mV s−1). These three redox peaks should be ascribed to two consecutive one-electron and one two-electron process of PW12.17 There exists one irreversible anodic peak I′ with the potential of +179 mV assigned to the oxidation of the copper centers.6a The 3-CPE shows three reversible redox peaks II–II′, III–III′ and IV–IV′ in the potential range of −850 to +800 mV, with the half-wave potentials at −200(II–II′), −463(III–III′), −754(IV–IV′) mV (scan rate: 100 mV s−1). Redox peaks II–II′ and III–III′ correspond to two consecutive one-electron processes of W centers, while IV–IV′ corresponds to a two-electron process.6a The irreversible anodic peak I′ with the potential of +85 mV is also assigned to the oxidation of the copper centers. With the scan rates from 40 to 500 mV s−1, the peak potentials change gradually: the cathodic peak potentials shift towards the negative direction and the corresponding anodic peak potentials to the positive direction with increasing scan rates. Up to 500 mV s−1, the peak currents are proportional to the scan rates (Fig. S7†), indicating that the redox processes of 1- and 3-CPEs are surface-confined.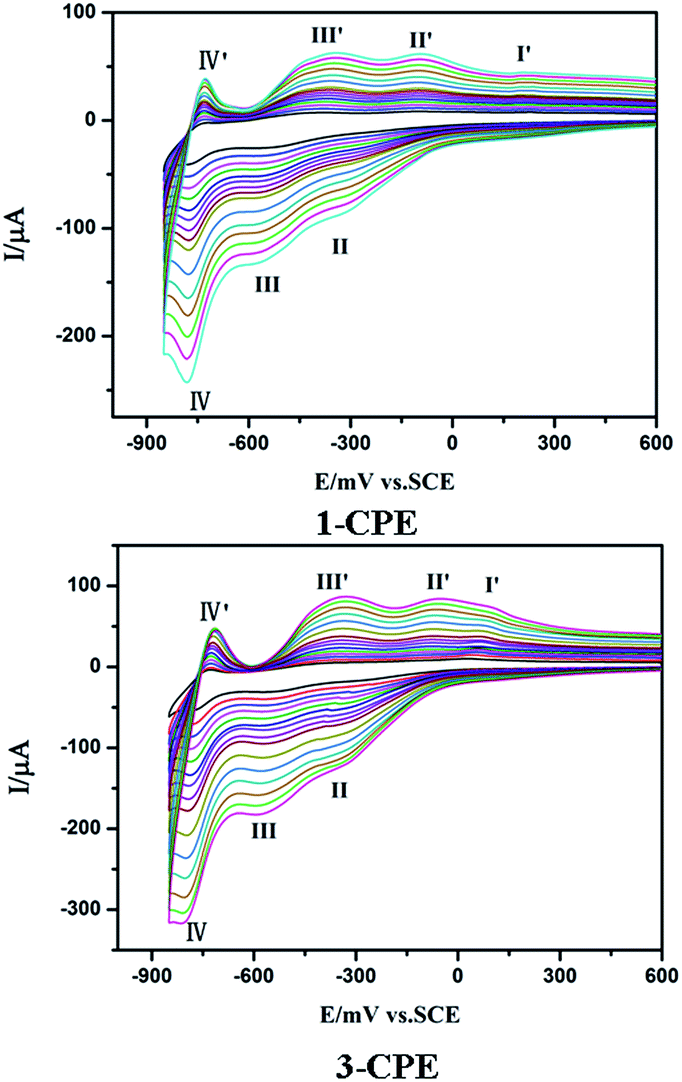 | ||
| Fig. 9 The cyclic voltammograms of the 1- and 3-CPEs in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution at different scan rates (from inner to outer: 40, 60, 80, 100, 120, 140, 160, 180, 200, 250, 300, 350, 400, 450 and 500 mV s−1, respectively.). | ||
Fig. 10 shows cyclic voltammograms for the electrocatalytic reduction of nitrite and bromate at 1- and 3-CPE in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution. With the addition of bromate, the second and third reduction peak currents increase gradually, while the corresponding oxidation peak currents gradually decrease. However, the first redox peak remains almost unchanged. Furthermore, it can be clearly seen that with adding nitrite, all three reduction peak currents gradually increased and the corresponding oxidation peak currents visibly decreased. In a word, the 1- and 3-CPE exhibit good electrocatalytic activity for the reduction of bromate and nitrite.
 | ||
| Fig. 10 Cyclic voltammograms of the 1- and 3-CPEs in 0.1 M H2SO4 + 0.5 M Na2SO4 aqueous solution containing 0.0–8.0 mM KBrO3 and KNO2, respectively. Scan rate: 100 mV s−1. | ||
Photocatalytic activity
As is known, the POM-based hybrids can exhibit good effect in the degradation of some organic dyes.18 We selected two organic dyes methylene blue (MB) and Rhodamine B (RhB) as model pollutants in aqueous media to evaluate the photocatalytic effect of compounds 1–4 under UV irradiation. In the process of photocatalysis, 100 mg compound was suspended in 90 mL of 0.02 mmol L−1 aqueous solution of MB or RhB. Furthermore, under dark conditions the suspension was magnetically stirred for about 10 min to ensure the equilibrium. Every interval (10 min for MB and 25 min for RhB) 5.0 mL samples were taken out for analysis by UV-vis spectrophotometer. As shown in Fig. 11a–c, with increasing reaction time we can clearly see that the percentage of MB degradation photocatalyzed by 1, 2 and 4 increased obviously. The conversions of MB are nearly 100% for 1 and 2, 76% for 4 (Fig. S8a†). However, the absorption peaks of MB photocatalyzed by compound 3 almost show no photocatalytic effect. Fig. 11d–f shows the photocatalytic degradation of RhB with the conversions of 58% for 2, 46% for 3 and 77% for 4 after 175 min (Fig. S8b†). But the absorption peaks of RhB with compound 1 as the catalyst shows tiny change. This result shows that compounds 1, 2 and 4 own good photocatalytic activities for the degradation of MB and compounds 2, 3 and 4 can act as good photocatalysts for the degradation of RhB. In order to confirm whether compounds 1–4 as catalysts were stable during the photocatalytic process, the IR spectra and XRD diffraction patterns of compounds 1–4 catalyst before and after catalytic reactions for MB and RhB were measure. The IR spectra (Fig. S9†) show that the characteristic bands of POMs and ligands for 1–4 catalyst after catalytic reactions for MB and RhB are maintained. The XRD diffraction patterns (Fig. S6†) of the catalyst before and after catalytic reactions show the diffraction peaks of experimental and recycled patterns mach well in positions, proving that compounds 1–4 were stable during photocatalytic process.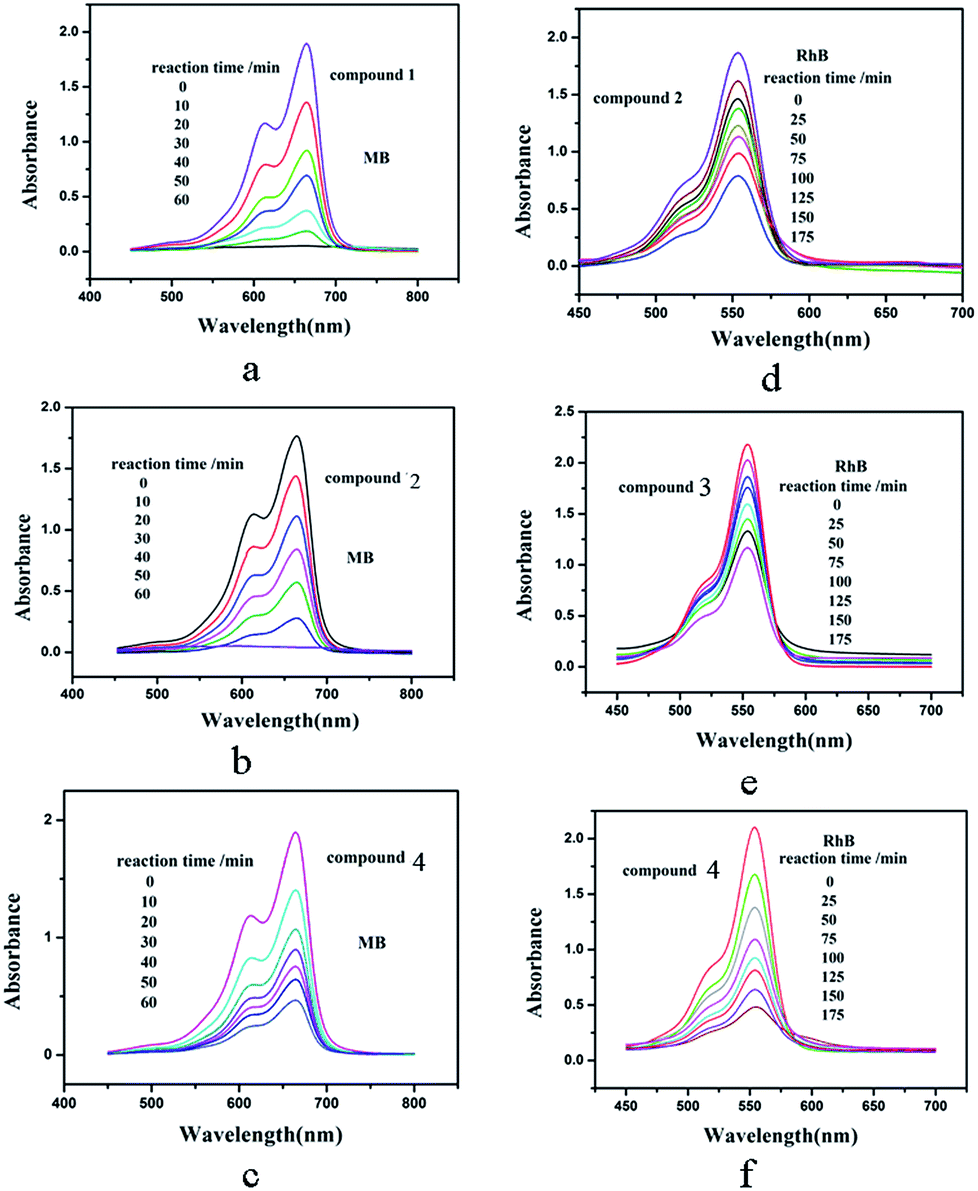 | ||
| Fig. 11 Absorption spectra of the MB (a, b and c) and RhB (d, e and f) solution during the decomposition reaction under UV irradiation at the presence of title compounds 1–4. | ||
Thermal gravimetric analyses
The TGA experiments were performed under N2 atmosphere with a heating rate of 10 °C min−1 in the temperature range of 50–850 °C, as shown in Fig. S10.† TG analyses of the title compounds all indicate two weight loss steps. The first weight loss below 350 °C may be due to the release of water molecules. The second weight loss step in 400–850 °C ascribes to the loss of organic H2bdpm molecules 14.43% (calc. 12.0%) for 1, 13.97% (calc. 12.05%) for 2, 16.61% (calc. 16.52%) for 3, 21.12% (calc. 21.17%) for 4.Conclusions
In summary, three new Keggin-based compounds have been constructed through using flexible H2bdpm ligand. The H2bdpm induces cycle subunits of the title compounds. Compound 1 shows a mono-supporting Keggin structure. Compound 2 shows a supramolecular structure. Compound 3 owns cycle-connecting-cycle chains, which are further linked by SiW12 anions to form a 2D layer. Compound 4 contains nested cycle subunits, which are linked by linking H2bdpm and anions to build a 3D framework. Further study on H2bdpm to generate new POM-based cycle structures is underway.Acknowledgements
Financial supports of this research by the National Natural Science Foundation of China (No. 21571023 and 21401010) and Program of Innovative Research Team in University of Liaoning Province (LT2012020).Notes and references
- (a) R. Yu, X. F. Kuang, X. Y. Wu, C. Z. Lu and J. P. Donahue, Coord. Chem. Rev., 2009, 253, 2872 CrossRef CAS; (b) J. W. Zhao, Y. Z. Li, L. J. Chen and G. Y. Yang, Chem. Commun., 2016, 52, 4418 RSC; (c) A. Rubinstein, P. J. Lozanao, J. J. Carbó, J. M. Poblet and R. Neumann, J. Am. Chem. Soc., 2014, 136, 10941 CrossRef CAS PubMed.
- (a) J. J. Walsh, A. M. Bond, R. J. Forster and T. E. Keyes, Coord. Chem. Rev., 2016, 306, 217 CrossRef CAS; (b) Z. J. Liu, X. L. Wang, C. Qin, Z. M. Zhang, Y. G. Li, W. L. Chen and E. B. Wang, Coord. Chem. Rev., 2016, 313, 94 CrossRef CAS; (c) T. Hirano, K. Uehara, K. Kamata and N. Mizuno, J. Am. Chem. Soc., 2012, 134, 6425 CrossRef CAS PubMed.
- J. Zhou, J. W. Zhao, Q. Wei, J. Zhang and G. Y. Yang, J. Am. Chem. Soc., 2014, 136, 5065 CrossRef CAS PubMed.
- X. L. Wang, Y. F. Bi, B. K. Chen, H. Y. Lin and G. C. Liu, Inorg. Chem., 2008, 47, 2442 CrossRef CAS PubMed.
- B. X. Dong, J. Peng, C. J. G. Garcıá, S. Benmansour, H. Q. Jia and N. H. Hu, Inorg. Chem., 2007, 46, 5933 CrossRef CAS PubMed.
- (a) A. X. Tian, J. Ying, J. Peng, J. Q. Sha, H. J. Pang, P. P. Zhang, Y. Chen, M. Zhu and Z. M. Su, Inorg. Chem., 2009, 48, 100 CrossRef CAS PubMed; (b) J. Ying, X. J. Liu, A. X. Tian and X. L. Wang, Z. Anorg. Allg. Chem., 2011, 637, 613 CrossRef CAS; (c) A. X. Tian, X. J. Liu, J. Ying, D. X. Zhu, X. L. Wang and J. Peng, CrystEngComm, 2011, 13, 6680 RSC.
- A. X. Tian, X. J. Liu, J. Ying, D. X. Zhu, X. L. Wang and J. Peng, Inorg. Chem. Commun., 2011, 14, 697 CrossRef CAS.
- A. X. Tian, Y. Yang, J. Ying, N. Li, X. L. Lin, J. W. Zhang and X. L. Wang, Dalton Trans., 2014, 43, 8405 RSC.
- (a) K. Pavani and A. Ramanan, Eur. J. Inorg. Chem., 2005, 3080 CrossRef CAS; (b) K. Pavani, S. E. Lofland, K. V. Ramanujachary and A. Ramanan, Eur. J. Inorg. Chem., 2007, 568 CrossRef CAS.
- (a) A. X. Tian, Y. L. Ning, J. Ying, J. W. Zhang, X. Hou, T. J. Li and X. L. Wang, Dalton Trans., 2015, 44, 10499 RSC; (b) J. W. Zhang, W. Zhao, Q. L. Lu, J. Luan, Y. Qu and X. L. Wang, J. Solid State Chem., 2014, 212, 151 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97, 1997 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 244 CrossRef.
- (a) A. X. Tian, Y. L. Ning, J. Ying, G. C. Liu, X. Hou, T. J. Li and X. L. Wang, CrystEngComm, 2015, 17, 5569 RSC; (b) A. X. Tian, Y. L. Ning, J. Ying, X. Hou, T. J. Li and X. L. Wang, Dalton Trans., 2015, 44, 386 RSC.
- (a) X. L. Wang, R. Zhang, X. Wang, H. Y. Lin and G. C. Liu, Inorg. Chem., 2016, 55, 6384 CrossRef CAS PubMed; (b) Z. Y. Shi, Z. Y. Zhang, J. Peng, X. Yu and X. Wang, CrystEngComm, 2013, 15, 7199 RSC.
- A. X. Tian, Y. Yang, J. Ying, N. Li, X. L. Lin, J. W. Zhang and X. L. Wang, Dalton Trans., 2014, 43, 8405 RSC.
- M. Sadakane and E. Steckhan, Chem. Rev., 1998, 98, 219 CrossRef CAS PubMed.
- X. L. Wang, X. Rong, H. Y. Lin, D. N. Liu, X. Wang and G. C. Liu, Inorg. Chem. Commun., 2016, 68, 4 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR Spectra, CV data and additional figures. CCDC 1486618, 1486619, 1486620 and 1509083. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra25156k |
| This journal is © The Royal Society of Chemistry 2017 |