
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitrogen, sulfur and phosphorus-codoped carbon with a tunable nanostructure as an efficient electrocatalyst for the oxygen reduction reaction†
Kai Chena,
Yajuan Haoa,
Meirong Zhanga,
Dongying Zhoua,
Yingjie Caoa,
Ying Wangb and
Lai Feng*a
aCollege of Physics, Optoelectronics and Energy & Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou 215006, China. E-mail: fenglai@suda.edu.cn
bTesting and Analysis Center, Soochow University, Suzhou 215006, China
First published on 4th January 2017
Abstract
Novel types of N,S,P-codoped carbons with varied nanostructures are developed as efficient and stable electrocatalysts for the oxygen reduction reaction (ORR). The carbon catalysts were facilely prepared using poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) (PZS) nanospheres as single precursors through a pyrolysis procedure in the presence or absence of melamine. The as-prepared microporous carbon nanospheres NSP-PC-1 with a high surface area (967 m2 g−1) exhibit a significantly enhanced ORR catalytic activity compared to their solely N-doped counterpart (N-PC-1), suggesting the remarkable contribution of additional S,P-doping on the ORR performance. More importantly, the as-prepared mesoporous carbon nanosheets NSP-PC-2 with a moderately high surface area (613 m2 g−1) and comparable overall NSP content show greatly improved ORR catalytic activity relative to that of NSP-PC-1, which is even slightly superior to that of commercial Pt/C catalysts. The excellent performance of NSP-PC-2 is mainly attributed to its proper N,S,P-codoping as well as advanced structural features.
1. Introduction
Fuel cells have been of continuous interest in the field of clean energy owing to their high power density and low pollutant emissions.1 Platinum (Pt) or Pt-based precious metal catalysts have been widely used for promoting the electrochemical oxygen reduction reaction (ORR) on the electrode of fuel cells.2–6 Nevertheless, though Pt-based catalysts exhibit high efficiency for ORR, their high cost and low tolerance to fuel crossover reduce their potential in practical use and hinder the large-scale commercialization of fuel cells.7–9 In recent years, various Pt-free catalysts have been developed,6 such as transition metal oxides,10,11 porous carbon,12–14 graphene15,16 and their composites.17–19 Among them, porous carbon catalysts have drawn considerable interest owing to their evident advantages such as low cost, tunable catalytic activity as well as high thermal/chemical stability. Therefore, in recent years, intensive efforts have been devoted to developing novel porous carbons with enhanced catalytic performance.It has been well established that two major considerations shall be taken into account when designing and preparing high-performance ORR carbon catalysts: (1) the catalytic nature and density of the active sites, which could be improved by proper heteroatom-doping.20,21 For instance, N-doping can introduce catalytic active sites by breaking the electroneutrality of the carbon material, thus enhancing the ORR activity.22–25 It was also suggested that the co-doping of two or multiple elements, such as N/B, N/S, N/P, N/S/P in carbon catalyst can introduce asymmetrical spin or charge density due to their synergistic effects, therefore generating more active sites and higher catalytic activity relative to that of unitary doped catalyst.26–30 (2) The accessibility to the active sites and mass transfer properties, which are closely related to the structural feature of carbon catalyst (i.e. morphology, particle size and pore structure).31,32 Thus, these two considerations have been combined to afford rational design in recent studies. A variety of heteroatom-doped carbons with either meso or microporosity and large specific surface area have been prepared and employed as ORR catalysts.33–36 Typically, one of state-of-the-art carbon catalysts was reported by Tour et al. very recently, which was highly mesoporous with proper B,N codoping and showed excellent ORR electrocatalytic activity, even much better than the commercial Pt/C catalysts.30 However, the carbon catalysts with such desirable ORR performance are very rare. Most of carbon catalysts reported so far exhibited only moderate ORR performance, which is still inferior to that of the commercial Pt/C catalysts.
Herein, we present a novel type of N,S,P-codoped carbon catalysts with either micro- or mesoporosity. These catalysts can be facilely prepared by a pyrolysis process using poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) (PZS) nanospheres as single precursor. It is demonstrated that the as-prepared carbon catalysts exhibit much better catalytic performance than the solely N-doped counterpart thanks to the contribution of additional S,P doping. More importantly, the catalyst with mesoporous structure shows an excellent ORR performance very comparable to that of commercial Pt/C in alkaline media, probably due to the synergistic effect of the proper heteroatom-doping and its structural advantages.
2. Experimental
2.1. Material preparation
Crosslinking poly(cyclotriphosphazene-co-4,4′-sulfonyldiphenol) nanospheres (PZS) were prepared according to the method as described in the literatures.37,38 Briefly, 1.6 g of phosphonitrilic chloride trimer and 3.455 g of 4,4′-sulfonyldiphenol were dissolved in a acetonitrile solution (160 mL) with ultrasonication. Then, 8 mL triethylamine was added slowly and the reaction mixture was maintained for 10 min with ultrasonication, yielding a solution with white suspensions. Then, the reaction mixture was centrifuged to wipe off the solvent and washed by using ethanol and distilled water. The product of PZS microspheres was freeze-dried in vacuum overnight.The N,S,P co-doped porous carbon denoted as NSP-PC-1 was obtained through directly pyrolyzing PZS nanospheres at 900 °C for 60 min with a heating rate of 10 °C min−1 and then cooling down to room temperature under N2 flow. Another co-doped porous carbon of NSP-PC-2 were prepared by heating the mixture of melamine and PZS nanospheres (with a mass ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in a semiclosed system under N2 atmosphere for minimizing the elution of decomposed melamine at high temperature. In a typical run, 0.5 g PZS microspheres and 5 g melamine were dispersed in a hot water solution (80 °C) and stirred for more than 2 h. The resulting mixture was freeze-dried in vacuum overnight and pyrolyzed at 900 °C for 60 min under N2 atmosphere. The final products were washed by ethanol and dried for use.
:1) in a semiclosed system under N2 atmosphere for minimizing the elution of decomposed melamine at high temperature. In a typical run, 0.5 g PZS microspheres and 5 g melamine were dispersed in a hot water solution (80 °C) and stirred for more than 2 h. The resulting mixture was freeze-dried in vacuum overnight and pyrolyzed at 900 °C for 60 min under N2 atmosphere. The final products were washed by ethanol and dried for use.
2.2. Structural characterizations
The as-prepared carbon catalysts were characterized by the field emission scanning electron microscope (FESEM, Hitachi S-4800) and transmission electron microscope (TEM, FEI Tecnai F20) for the morphology and microstructure identifications. Raman spectra were recorded on a micro-Raman spectroscopy system (Renishaw inVia-reflex, 532 nm excitation laser). X-ray photoelectron spectroscopy (XPS) studies were carried out with an ESCALAB 250 spectrometer using a monochromated Al Kα excitation source. Nitrogen adsorption–desorption isotherms at −196 °C were measured on an adsorption volumetric analyzer ASAP 2020 manufactured by Micromeritics, Inc. (Norcross, Georgia, USA). All samples were degassed at 200 °C overnight prior to adsorption measurements. A part of the N2 sorption isotherm in the P/P0 range of 0.005–0.05 was fitted to the BET equation to estimate the BET surface area.39 Total pore volume (Vtotal) was obtained at a relative pressure of 0.985. The pore size distribution was obtained using the NLDFT model in the Micromeritics ASAP 2020 software package (assuming slit pore geometry). Micropore area (Smicro) and volume (Vmicro) were calculated using the t-plot method.2.3. Electrochemical tests
All electrochemical measurements were performed at room temperature on a potentiostat (CHI 760E, CH Instrument, Shanghai, China) and a rotating ring disk electrode system (RRDE-3A, ALS Co., Ltd, Japan) with a standard three electrode cell. A Pt wire and an Hg/HgO/0.1 M OH− electrode (0.164 V vs. NHE) were used as the counter and reference electrode, respectively. A RDE with glassy carbon (GC) disk electrode (4 mm in diameter) and a rotating ring-disk electrode (RRDE, ALS Co., Ltd, Japan) with a Pt ring (5 mm inner diameter and 7 mm outer diameter) and a GC disk (4 mm diameter) are used as the substrate for the working electrodes. Before use, the GC electrodes in RDE/RRDE are polished using aqueous alumina suspension on polishing pad. All electrode potentials were reported relative to the reversible hydrogen electrode (RHE), which were converted from the Hg/HgO/0.1 M OH− electrode according to the formula: ERHE = E(Hg/HgO) + ϕ(Hg/HgO/0.1 M OH−) + 0.0591 × pH V.40To fabricate the catalyst-modified working electrode, the catalyst ink was prepared by ultrasonically dispersing the as-prepared carbon catalyst (5 mg) in a solution containing 100 μL Nafion (5 wt%) solution and 900 μL deionized water. The Pt/C catalyst (10 wt% Pt on graphitized carbon, Sigma-Aldrich) ink was prepared in the same way. The above prepared catalyst ink was casted onto the fresh surface of the GCE (a diameter of 4 mm). The catalyst loadings on RDE and RRDE were 400 μg cm−2 (40 μgPt cm−2 for the Pt/C catalyst).
ORR performance of the catalysts was investigated via cyclic voltammogram (CV) and linear sweep voltammogram (LSV) in either N2 or O2 saturated 0.1 M KOH solution. The scan rate was 50 mV s−1 for CV and 10 mV s−1 for LSV tests. Oxygen reduction current was evaluated by subtracting the background capacitive current, which was measured by scanning the electrode in a N2-saturated 0.1 M KOH solution under the same conditions. The electron transfer number (n) was calculated using Koutecky–Levich (K–L) equations:
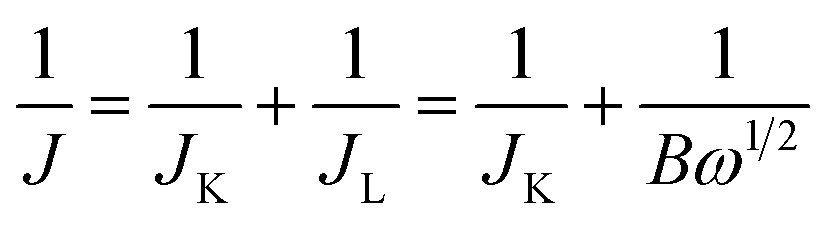 | (1) |
| B = 0.20nFC0D02/3ν−1/6 | (2) |
485 C mol−1), C0 is the bulk concentration (1.2 × 10−3 mol L−1) of O2, D0 is the diffusion coefficient (1.9 × 10−5 cm2 s−1) of O2 in the KOH solution, and ν is the kinetic viscosity (0.01 cm2 s−1) of the electrolyte.41
Rotating ring-disk electrode (RRDE) tests for ORR were measured in the O2 saturated 0.1 M KOH solution with a scan rate of 10 mV s−1, and the ring potential was set at 0.7 V for H2O2 production.40 The electron transfer number n and the production yield of HO2− (HO2−%) were determined by the following equations:
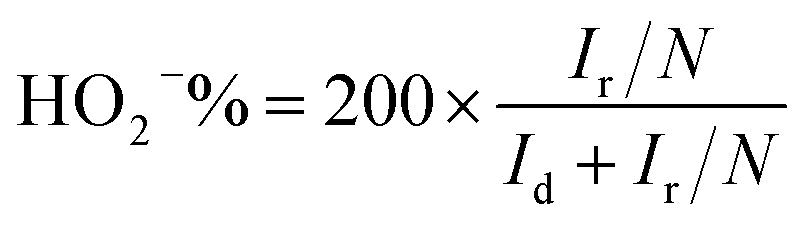 | (3) |
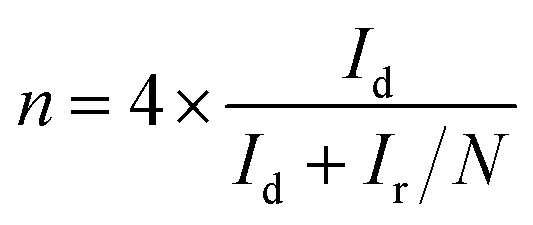 | (4) |
Durability of catalysts was evaluated from the i–t chronoamperometric responses of as-prepared carbon catalyst and Pt/C catalyst in O2-saturated 0.1 M KOH at 0.6 V (vs. RHE) for 5.5 h with a rotation rate of 400 rpm. For fuel crossover effect tests, the chronoamperometric response at 0.6 V (vs. RHE) was recorded by RDE tests with a rotation rate of 1600 rpm, and followed by the introduction of methanol (3 M) into 0.1 M KOH solution.
3. Results and discussion
The catalysts NSP-PC-1 and NSP-PC-2 were prepared through a two-step method (Scheme 1): (1) highly cross-linked PZS nanospheres were prepared according to the literature-reported method;37,38 (2) the as-prepared PZS nanospheres were carbonized at 900 °C with or without the presence of melamine under N2 atmosphere to form N,S,P co-doped carbon catalysts. Particularly, PZS nanospheres act as carbon source and N,S,P-doping sources, which might result in a uniform distribution of N, S, P heteroatoms in the carbon catalyst and simplify the preparation. In the case of NSP-PC-2, melamine was added during pyrolysis for additional N-doping and porosity adjustment. The following mechanism has been proposed: in the temperature range of 300–600 °C, melamine is decomposed into carbon nitrides,42 which diffuses into the matrix of semi-carbonized PZS. At a higher temperature (i.e., >750 °C), carbon nitrides are further decomposed into N2, NH3 and some reactive species (i.e., C2N2+, C3N2+, C3N3+),43–46 which are released from or react with the carbon matrix to contribute to the porosity adjustment or to introduce the N-containing functionalities in the carbon product.47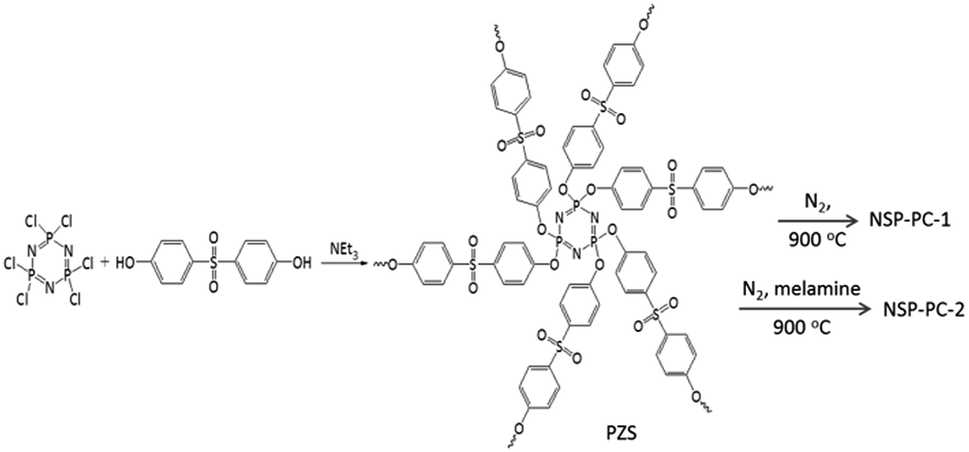 | ||
| Scheme 1 Preparations of NSP-PC-1 and NSP-PC-2. | ||
The structures and morphologies of the as-prepared catalysts were investigated by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in SEM images (Fig. 1), NSP-PC-2 has a sheet-like morphology, unlike the nanospherical morphology of NSP-PC-1 or PZS (see Fig. S1a†). Thus, the formation of the sheet-like structures of NSP-PC-2 might be attributed to the presence of melamine during pyrolysis, though the mechanism is still unknown. The TEM image in Fig. 2a also reveals the sheet-like feature of NSP-PC-2. Furthermore, a high-resolution (HR)TEM image (Fig. 2b) suggests that NSP-PC-2 is amorphous in nature. Fig. 2c shows a typical scanning transmission electron microscopy (STEM) annular dark field (ADF) image and elemental mapping of NSP-PC-2, where all the elements (C, O, N, P and S) are homogeneously distributed throughout the carbon nanosheet.
 | ||
| Fig. 1 SEM images of (a) NSP-PC-1, (b) NSP-PC-2. | ||
 | ||
| Fig. 2 (a and b) TEM and HRTEM images of NSP-PC-2. (c) HADDF image and compositional EDS mapping of NSP-PC-2 using scanning transmission electron microscopy. | ||
The X-ray photoelectron spectroscopy (XPS) analysis (Fig. 3 and S2†) further reveals the N,S,P-codoping in the catalyst NSP-PC-2 as well as NSP-PC-1. Particularly, the N 1s spectrum demonstrates three types of N species in NSP-PC-2, corresponding to graphitic-N (401.0 eV),48 pyrrolic-N (399.8 eV),49 and pyridinic-N or P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/P–N (398.2 eV). Among them, graphitic-N, pyrrolic-N and pyridinic-N or PN/P–N are all considered to be favorable for ORR.50–53 The S 2p spectrum reveals only thiophene-S in NSP-PC-2, which is also believed to play an important role in enhancing catalytic activity. The two peaks positioned at 165.1 and 163.9 eV are consistent with the S 2p1/2 and S 2p3/2 of thiophene-S, owing to their spin–orbit coupling.54,55 The P 2p spectrum can be fitted with two peaks at 132.6 and 133.6 eV, corresponding to P–C and PN/P–N bonding, respectively,51,56 indicating that P atoms were incorporated into the N-doped carbon framework. Such a doping way was found to create more active sites and trigger synergistic effects for ORR electrocatalysis.16 NSP-PC-1 also exhibits similar XPS spectra (Fig. S2†). However, the N, S, P contents of the carbon catalysts are very different (see Table S1 in ESI†). Particularly, NSP-PC-2 has 9.05 at% N, 0.6 at% S and 1 at% P, while NSP-PC-1 has 3.75 at% N, 4 at% S and 0.1 at% P. The higher N-content and lower S-content in NSP-PC-2 may be attributed to the additional precursor of melamine, which not only provides additional N source but also results in desulfurization to some extent.57 On the other hand, the higher P content in NSP-PC-2 might be related to the increase of N-doping. As suggested in P 2p spectrum (Fig. 3), the higher P-doping in NSP-PC-2 occurs with bonding configuration of C–PN/C–P–N.
N/P–N (398.2 eV). Among them, graphitic-N, pyrrolic-N and pyridinic-N or PN/P–N are all considered to be favorable for ORR.50–53 The S 2p spectrum reveals only thiophene-S in NSP-PC-2, which is also believed to play an important role in enhancing catalytic activity. The two peaks positioned at 165.1 and 163.9 eV are consistent with the S 2p1/2 and S 2p3/2 of thiophene-S, owing to their spin–orbit coupling.54,55 The P 2p spectrum can be fitted with two peaks at 132.6 and 133.6 eV, corresponding to P–C and PN/P–N bonding, respectively,51,56 indicating that P atoms were incorporated into the N-doped carbon framework. Such a doping way was found to create more active sites and trigger synergistic effects for ORR electrocatalysis.16 NSP-PC-1 also exhibits similar XPS spectra (Fig. S2†). However, the N, S, P contents of the carbon catalysts are very different (see Table S1 in ESI†). Particularly, NSP-PC-2 has 9.05 at% N, 0.6 at% S and 1 at% P, while NSP-PC-1 has 3.75 at% N, 4 at% S and 0.1 at% P. The higher N-content and lower S-content in NSP-PC-2 may be attributed to the additional precursor of melamine, which not only provides additional N source but also results in desulfurization to some extent.57 On the other hand, the higher P content in NSP-PC-2 might be related to the increase of N-doping. As suggested in P 2p spectrum (Fig. 3), the higher P-doping in NSP-PC-2 occurs with bonding configuration of C–PN/C–P–N.
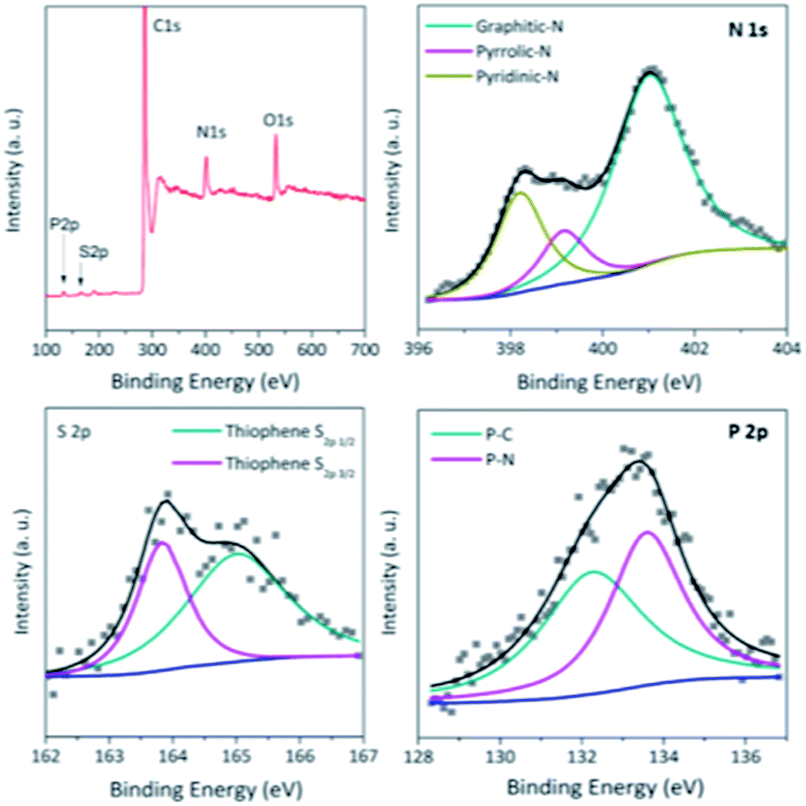 | ||
| Fig. 3 XPS spectra of NSP-PC-2. | ||
Raman spectroscopy is adopted to further investigate the structural feature of the carbon catalysts. As shown in Fig. S4 (in ESI†), the two characteristic peaks at around 1350 and 1580 cm−1 are assigned to the D band and G band, respectively, confirming the presence of disordered carbon and graphitic carbon.58 Particularly, NSP-PC-1 and NSP-PC-2 show similar ID/IG band intensity ratio (i.e., 0.925 and 0.923), indicating their similar disorder degree in spite of their different pyrolysis conditions (i.e., with or without melamine).
The porous characteristics of the catalysts NSP-PC-2 and NSP-PC-1 were investigated by N2 adsorption–desorption measurements. The textural properties were summarized in Table 1. As presented in Fig. 4a, NSP-PC-2 presents a typical type-IV isotherm according to the IUPAC classification, which shows a small N2 uptake at low relative pressure (P/P0 < 0.001), a clear hysteresis loop and an evident rise in the medium and high pressure regions (P/P0 = 0.8–1.0), indicative of mainly mesoporous structures with a moderately high BET surface area of 613 m2 g−1 and a large pore volume of 1.42 cm3 g−1. In contrast, the isotherm for NSP-PC-1 is assigned to type-I, suggesting sheer microporous structures (up to 93% microporosity) with a high BET surface area of 967 m2 g−1 and a pore volume of 0.38 cm3 g−1. The different porosities of these carbon catalysts were also demonstrated by pore size distribution (PSD) results calculated using NLDFT method. As shown in Fig. 4b, the pore volume of NSP-PC-1 is mainly contributed by the micropores (with sizes smaller than 2 nm), while the porosity of NSP-PC-2 is mainly distributed into a mesopore system with sizes ranging from 2.5 nm to 50 nm (∼90% mesoporosity). Thus, it appears that the melamine plays a significant role in achieving the mesoporosity of NSP-PC-2. This may be attributed to the generation of gaseous agents (i.e., N2 and NH3) due to the decomposition of melamine or its derivative (i.e., carbon nitride) at high temperature (i.e., 900 °C),44,45 which might swell the carbon matrix and induces the formation of mesopores.
| Catalysts | SBETa (m2 g−1) | Vtotalb (cm3 g−1) | Smicroc (m2 g−1) | Vmicroc (cm3 g−1) | Smesod (m2 g−1) | Vmesod (cm3 g−1) |
|---|---|---|---|---|---|---|
| a Surface areas (SBET) were calculated by the BET method.b The total pore volumes (Vtotal) were estimated from the adsorbed amount at a relative pressure P/P0 = 0.985.c Micropore area (Smicro) and volume (Vmicro) were calculated using the t-plot method.d Mesopore areas (Smeso) and volume (Vmeso) were calculated from the difference of the BET surface area and micropore area (Smeso = SBET − Smicro), and the total pore volume and micropore volume (Vmeso = Vtotal − Vmicro), respectively. | ||||||
| NSP-PC-1 | 967 | 0.38 | 898 | 0.346 | 69 | 0.036 |
| N-PC-1 | 845 | 0.33 | 807 | 0.307 | 38 | 0.023 |
| NSP-PC-2 | 613 | 1.42 | 49 | 0.019 | 564 | 1.40 |
| NSP-PC-3 | 648 | 1.56 | 27 | 0.009 | 621 | 1.55 |
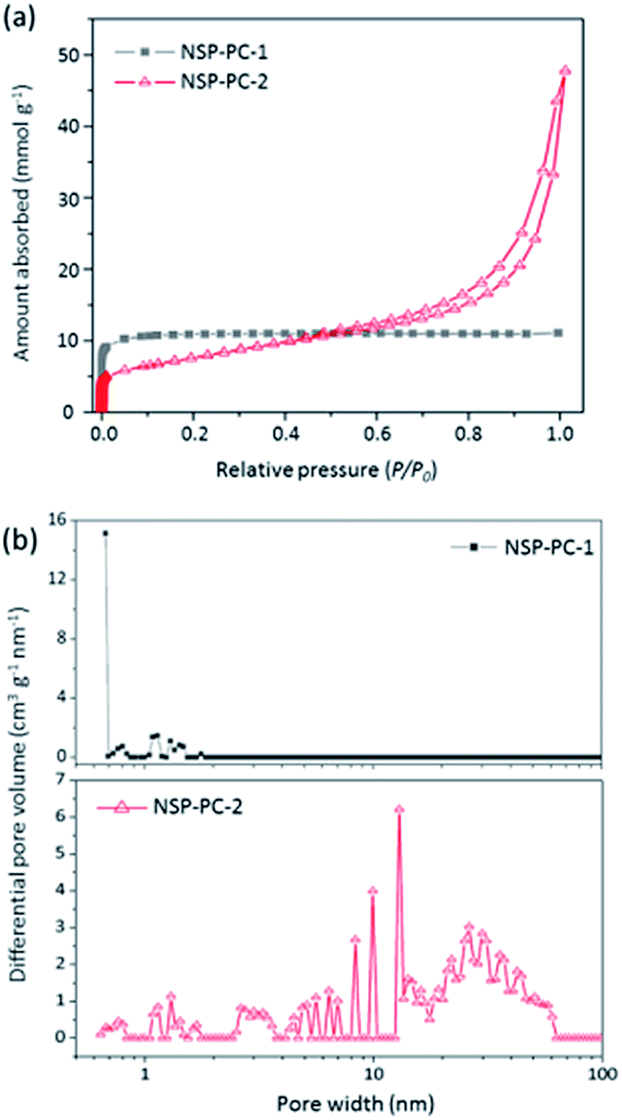 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms and (b) pore size distributions of NSP-PC-1 and NSP-PC-2 calculated using NLDFT. | ||
The ORR catalytic activity of the catalysts NSP-PC-1 and NSP-PC-2 was first assessed by means of cyclic voltammetry (CV) and linear sweep voltammetry (LSV) in 0.1 M KOH solution saturated with O2. In order to better understand the catalytic performances and reveal the role of heteroatom-doping, a control catalyst was prepared by pyrolyzing melamine-doped polymeric nanospheres,59,60 noted as N-PC-1 (see ESI† for preparation details). Though N-PC-1 was derived from the precursor different from that of NSP-PC-1, its structural features including the nanospherical morphology, microporous characteristics and BET area are very similar to those of NSP-PC-1 (see Fig. S1a, S5 in ESI† and Table 1). Moreover, as suggested by XPS analysis, N-PC-1 contains no S, P but only N species. The identified N-compositions and content in N-PC-1 are similar to those found in NSP-PC-1. (Fig. S3 and Table S1 in ESI†).
As shown in Fig. 5a, the CV results indicate that the control catalyst N-PC-1 shows a poorer ORR catalytic activity in terms of the more negative oxygen reduction peak potential as compared to that of NSP-PC-1 or NSP-PC-2. This trend is again demonstrated by the LSV results (Fig. 5b). The control catalyst N-PC-1 exhibits an apparently less enhanced catalytic activity for ORR in view of an E1/2 of 0.71 V, which is 70 and 140 mV lower than those of N,S,P co-doped catalysts NSP-PC-1 and NSP-PC-2, respectively. Considering the similar structural features of N-PC-1 and NSP-PC-1, the better catalytic activity of the latter can be safely attributed to the contribution of additional S,P-doping. It is also clear seen that the catalyst NSP-PC-2 shows the best catalytic activity among three as-prepared carbon catalysts in terms of the most positive E1/2 of 0.85 V. The improved catalytic activity of NSP-PC-2 relative to that of NSP-PC-1 might be due to its slightly higher overall NSP content and more importantly, the distinct structural feature (such as the mesoporosity), which makes the active sites very accessible for the reactants and facilitates the mass-transfer despite of the lower surface area of NSP-PC-2 relative to that of microporous NSP-PC-1.
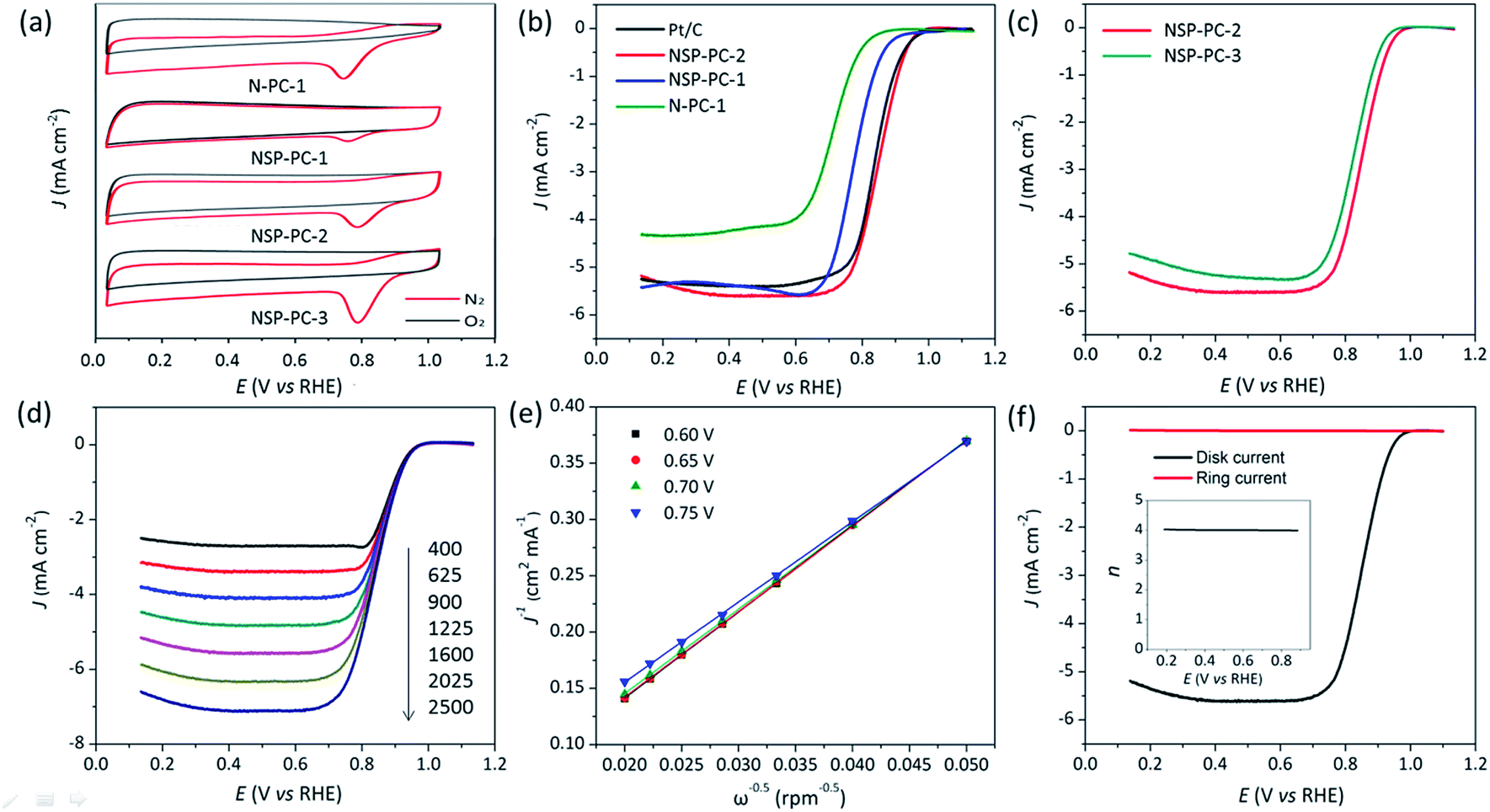 | ||
| Fig. 5 (a) CV curves of different carbon catalysts recorded in O2-saturated (red line) or N2-saturated (black line) in 0.1 M KOH. (b and c) LSV curves of NSP-PC-1 and NSP-PC-2 compared to Pt/C and control catalysts in O2-saturated 0.1 M KOH at 1600 rpm. (d) LSV curves of NSP-PC-2 at different rotation speeds (rpm) in 0.1 M KOH. (e) The Koutecky–Levich (K–L) plots for NSP-PC-2 at different potentials (see Fig. S11† for theoretical K–L plots for 2 and 4 electron-transfer, respectively). (f) RRDE test of the ORR on NSP-PC-2 in O2-saturated 0.1 M KOH at 1600 rpm. The inset shows the n value against electrode potential. The loading of all nonprecious catalysts is 400 μg cm−2. The Pt loading is 40 μgPt cm−2. | ||
To further explore the effect of the dosage of introduced heteroatoms on the electrocatalytic activity of the as-prepared carbon catalyst, another control catalyst NSP-PC-3 was prepared by the same synthetic route as NSP-PC-2 except for lower pyrolysis temperature (i.e., 800 °C). As suggested by N2 adsorption–desorption measurement, Raman and XPS analysis (see Fig. S1b, S4, S6, S7 and Table S1†), NSP-PC-3 exhibits similar structural feature as that of NSP-PC-2 but much higher overall NSP content. However, NSP-PC-3 shows a poorer catalytic activity in terms of smaller diffusion limiting current density and an E1/2 at 0.83 V, which is 20 mV lower than that of NSP-PC-2 (see Fig. 5d). Such a degraded catalytic performance might be attributed to the reduction of electrical conductivity with higher NPS content, which obstructs the ORR process in NSP-PC-3.
It is noteworthy that the E1/2 of NSP-PC-2 is even 10 mV higher than that of the commercial Pt/C catalyst (i.e., a value of 0.84 V comparable to the reference data with similar Pt loading),61–63 suggesting an excellent ORR activity for NSP-PC-2. This E1/2 value is also more positive than those of recently reported NSP-codoped porous carbons, which are remarkably lower than that of commercial Pt/C catalyst.27,28 The K–L plots (J−1 vs. ω−1/2 in Fig. 5e and S11†) derived from the LSVs of NSP-PC-2 show an excellent linearity, implying a first-order reaction in this potential range. The calculated average electron-transfer number (n) is 3.84. This value is close to that obtained from the rotating ring-disk electrode (RRDE) measurements (n = 4.02–3.99, see Fig. 5f), suggesting a one-step, four-electron oxygen reduction process for this carbon catalyst. The RRDE measurements also reveal that the production of HO2− was suppressed on the catalyst NSP-PC-2 with a yield below ∼7.3% over the potential range of 0.4–0.9 V (see Fig. S12 in ESI†), which is closely comparable to that reported for Pt/C catalyst (4–5%).30 Moreover, the Tafel slope of NSP-PC-2 was calculated to be 84.1 mV dec−1 (see Fig. S13 in ESI†), very close to that of Pt/C (83.8 mV dec−1) measured under the same condition, indicating the transfer of the first electron is probably the rate-determining step in ORR catalyzed by NSP-PC-2, similar to platinum.64 Thus, all these results suggest an excellent ORR activity for the catalyst NSP-PC-2, which is very comparable to that of commercial Pt/C catalyst.
The durability and possible fuel crossover effect are also important for cathodic catalysts in fuel cells. The durability of the NSP-PC-2 was examined via a continuous chronoamperometric measurement and compared to that of Pt/C catalyst. As shown in Fig. 6a, the Pt/C modified electrode suffers a 20% current decay after 5.5 h, while only a small current decay of 5% is observed for NSP-PC-2-modified electrode, indicating much better stability of this carbon catalyst in an alkaline environment. This result was confirmed by a repeated potentiodynamic cycling durability test. As shown in Fig. S14,† a slightly shifted LSV curve (ΔE1/2 = 10 mV) was observed for NPS-PC-2 after 5000 CV cycles between 0.6 to 1.0 V, while there was a 40 mV loss of half-wave potential for the Pt/C catalyst under the same conditions. Moreover, the current density of NPS-PC-2 is almost unchanged upon CV cycling, indicating that surface properties were maintained during potential cycling tests.65–67 In addition, the methanol crossover effect on NSP-PC-2 was evaluated using chronoamperometric measurements. As shown in Fig. 6b, upon the addition of 3 M methanol, Pt/C electrode presents a significant drop of ca. 50% in the ORR current, whereas no noticeable change is observed for the NSP-PC-2-modified electrode. This result suggests that the catalyst NSP-PC-2 has a greater tolerance to methanol crossover than the commercial Pt/C catalyst.
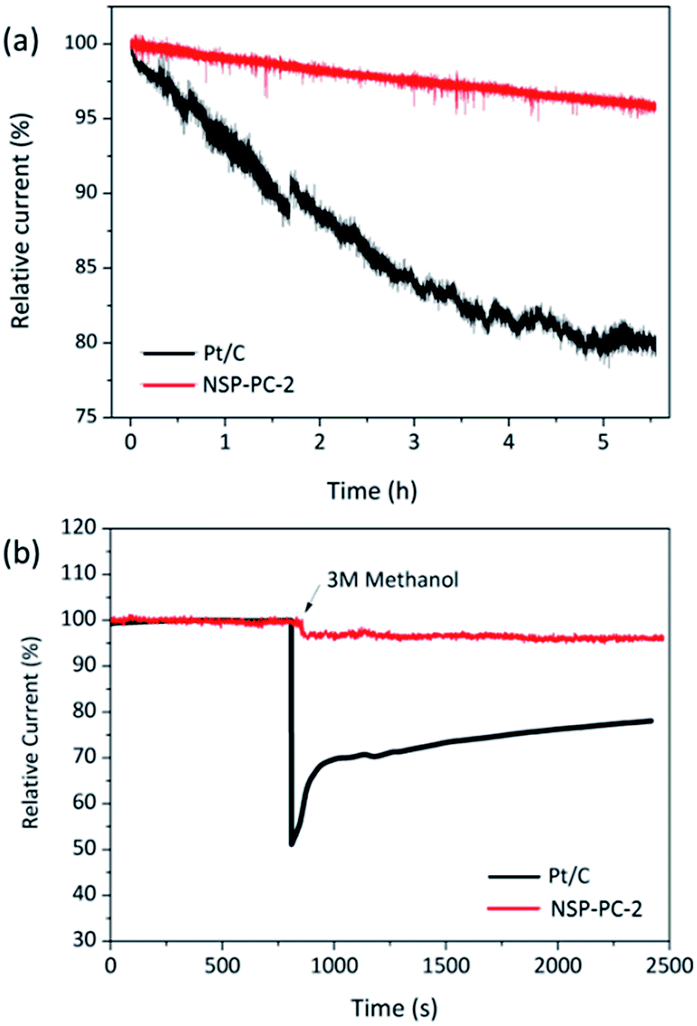 | ||
| Fig. 6 (a) The durability of the catalyst-modified electrodes for the ORR in oxygen-saturated 0.1 M KOH. (b) i–t chronoamperometric responses of the catalyst-modified electrodes at 0.6 V (vs. RHE) with a rotating speed of 1600 rpm in oxygen-saturated 0.1 M KOH. The arrow indicates the addition of methanol into the electrolytic cell. | ||
4. Conclusions
In summary, starting with PZS nanospheres as the single precursors, we have developed N,S,P-codoped carbons (i.e., NSP-PC-1 and NSP-PC-2) with either micro- or mesoporosity as efficient and stable ORR electrocatalysts. As the microporous catalyst NSP-PC-1 exhibits a significantly enhanced ORR catalytic activity compared to solely N-doped counterpart, it unambiguously suggests that additional S,P-doping can dramatically promote the ORR activity of carbon catalyst. Moreover, the mesoporous catalyst NSP-PC-2 shows an optimal catalytic activity in terms of an E1/2 at 0.85 V (vs. RHE) in alkaline media, which is closely comparable to that of Pt/C and 70 mV higher than that of microporous NSP-PC-1. The superior catalytic activity of NSP-PC-2 is mainly attributed to its proper N,S,P-codoping (∼11 at%) as well as its structural advantages such as the high mesoporosity and moderately high surface area (613 m2 g−1), which might make the active sites very accessible and facilitates the O2 mass transfer.Acknowledgements
This work is the supported in part by the Natural Science Foundation of China (51372158), Jiangsu Specially Appointed Professor Program (SR10800113), and the Project for Jiangsu Scientific and Technological Innovation Team (2013).Notes and references
- B. C. Steele and H. A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- M. Nesselberger, M. Roefzaad, R. F. Hamou, P. U. Biedermann, F. F. Schweinberger, S. Kunz, K. Schloegl, G. K. H. Wiberg, S. Ashton, U. Heiz, K. J. J. Mayrhofer and M. Arenz, Nat. Mater., 2013, 12, 919–924 CrossRef CAS PubMed.
- A. Anastasopoulos, J. C. Davies, L. Hannah, B. E. Hayden, C. E. Lee, C. Milhano, C. Mormiche and L. Offin, ChemSusChem, 2013, 6, 1973–1982 CrossRef CAS PubMed.
- J. Wu, M. Shi, X. Yin and H. Yang, ChemSusChem, 2013, 6, 1888–1892 CrossRef CAS PubMed.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruña, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165–2173 CrossRef CAS.
- N. Alonso-Vante, ChemPhysChem, 2010, 11, 2732–2744 CrossRef CAS PubMed.
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS.
- X. Tong, S. Chen, C. Guo, X. Xia and X.-Y. Guo, ACS Appl. Mater. Interfaces, 2016, 8, 28274–28282 CAS.
- J. Wu, R. Mi, S. Li, P. Guo, J. Mei, H. Liu, W. Lau and L. Liu, RSC Adv., 2015, 5, 25304–25311 RSC.
- G. Tao, L. Zhang, L. Chen, X. Cui, Z. Hua, M. Wang, J. Wang, Y. Chen and J. Shi, Carbon, 2015, 86, 108–117 CrossRef CAS.
- K.-H. Wu, D.-W. Wang, D.-S. Su and I. R. Gentle, ChemSusChem, 2015, 8, 2772–2788 CrossRef CAS PubMed.
- P. Trogadas, T. F. Fuller and P. Strasser, Carbon, 2014, 75, 5–42 CrossRef CAS.
- Y. Ito, H.-J. Qiu, T. Fujita, Y. Tanabe, K. Tanigaki and M. Chen, Adv. Mater., 2014, 26, 4145–4150 CrossRef CAS PubMed.
- R. Li, Z. Wei and X. Gou, ACS Catal., 2015, 5, 4133–4142 CrossRef CAS.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- G. Zhang, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2014, 26, 2408–2412 CrossRef CAS PubMed.
- Y. Gao, H. Zhao, D. Chen, C. Chen and F. Ciucci, Carbon, 2015, 94, 1028–1036 CrossRef CAS.
- D.-W. Wang and D. Su, Energy Environ. Sci., 2014, 7, 576–591 CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 CAS.
- W. Xiong, F. Du, Y. Liu, A. Perez Jr, M. Supp, T. S. Ramakrishnan, L. M. Dai and L. Jiang, J. Am. Chem. Soc., 2010, 132, 15839–15841 CrossRef CAS PubMed.
- Y. L. Zhu, C. Su, X. M. Xu, W. Zhou, R. Ran and Z. P. A. Shao, Chem.–Eur. J., 2014, 20, 15533–15542 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- W. Ai, Z. M. Luo, J. Jiang, J. H. Zhu, Z. Z. Du, Z. X. Fan, L. H. Xie, H. Zhang, W. Huang and T. Yu, Adv. Mater., 2014, 26, 6186–6192 CrossRef CAS PubMed.
- J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496–11500 CrossRef CAS PubMed.
- S. Gao, X. Wei, H. Liu, K. Geng, H. Wang, H. Moehwald and D. Shchukin, J. Mater. Chem. A, 2015, 3, 23376–23384 CAS.
- J. Wu, X. Zheng, C. Jin, J. Tian and R. Yang, Carbon, 2015, 92, 327–338 CrossRef CAS.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084–7091 CrossRef CAS PubMed.
- Y. Gong, H. Fei, X. Zou, W. Zhou, S. Yang, G. Ye, Z. Liu, Z. Peng, J. Lou, R. Vajtai, B. I. Yakobson, J. M. Tour and P. M. Ajayan, Chem. Mater., 2015, 27, 1181–1186 CrossRef CAS.
- D. Eisenberg, W. Stroek, N. J. Geels, C. S. Sandu, A. Heller, N. Yan and G. A. Rothenberg, Chem.–Eur. J., 2016, 22, 501–505 CrossRef CAS PubMed.
- G. A. Ferrero, K. Preuss, A. B. Fuertes, M. Sevilla and M.-M. Titirici, J. Mater. Chem. A, 2016, 4, 2581–2589 CAS.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 CAS.
- X. Zhao, H. Zhao, T. Zhang, X. Yan, Y. Yuan, H. Zhang, H. Zhao, D. Zhang, G. Zhu and X. Yao, J. Mater. Chem. A, 2014, 2, 11666–11671 CAS.
- J.-Y. Choi, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2010, 114, 8048–8053 CAS.
- Y. He, X. Han, Y. Du, B. Song, P. Xu and B. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 3601–3608 CAS.
- J. Zhou, L. Meng, X. Feng, X. Zhang and Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 8476–8479 CrossRef CAS PubMed.
- S. Yang, L. Peng, P. Huang, X. Wang, Y. Sun, C. Cao and W. Song, Angew. Chem., Int. Ed., 2016, 55, 647–648 Search PubMed.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS PubMed.
- J. Guo, A. Hsu, D. Chu and R. Chen, J. Phys. Chem. C, 2010, 114, 4324–4330 CAS.
- S. Yang, X. Feng, X. Wang and K. Müllen, Angew. Chem., Int. Ed., 2011, 50, 5339–5343 CrossRef CAS PubMed.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- S. Trasobares, C. Kolczewski, R. Raty, N. Borglund, A. Bassan, G. Hug, C. Colliex, S. Csillag and L. G. M. Pettersson, J. Phys. Chem. A, 2003, 107, 228–235 CrossRef CAS.
- S. Ju, C. Han, C. Wu, M. Mebel and Y. Chen, J. Phys. Chem. B, 1999, 103, 582–596 CrossRef CAS.
- A. B. Fuertes, G. A. Ferrero and M. Sevilla, J. Mater. Chem. A, 2014, 2, 14439–14448 CAS.
- W.-H. Lee and J. H. Moon, ACS Appl. Mater. Interfaces, 2014, 6, 13968–13976 CAS.
- H. L. Peng, Z. Y. Mo, S. J. Liao, H. G. Liang, L. J. Yang and F. Luo, Sci. Rep., 2013, 3, 1765 Search PubMed.
- R. L. Liu, D. Q. Wu, X. L. Feng and K. Mullen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- C. Wang, L. Sun, Y. Zhou, P. Wan, X. Zhang and J. Qiu, Carbon, 2013, 59, 537–546 CrossRef CAS.
- F. Razmjooei, K. P. Singh, M. Y. Song and J.-S. Yu, Carbon, 2014, 78, 257–267 CrossRef CAS.
- D. S. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. S. Yu, J. Am. Chem. Soc., 2012, 134, 16127–16130 CrossRef CAS PubMed.
- T. Y. Ma, J. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- J. Xu, G. Dong, C. Jin, M. Huang and L. Guan, ChemSusChem, 2013, 6, 493–499 CrossRef CAS PubMed.
- H. Jiang, Y. Zhu, Q. Feng, Y. Su, X. Yang and C. Li, Chem.–Eur. J., 2014, 20, 3106–3112 CrossRef CAS PubMed.
- J. Xiao, Y. Zhang, Q. Zhong, F. Li, J. Huang and B. Wang, Energy Fuels, 2016, 30, 3385–3391 CrossRef CAS.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- H. Cong, M. Zhang, Y. Chen, K. Chen, Y. Hao, Y. Zhao and L. Feng, Carbon, 2015, 92, 297–304 CrossRef CAS.
- H. Zhou, S. Xu, H. Su, M. Wang, W. Qiao and L. Ling, Chem. Commun., 2013, 49, 3763–3765 RSC.
- H. Ba, Y. Liu, L. Truong-Phuoc, C. Duong-Viet, J.-M. Nhut, D. L. Nguyen, O. Ersen, G. Tuci, G. Giambastiani and C. Pham-Huu, ACS Catal., 2016, 6, 1408–1419 CrossRef CAS.
- C. Guo, W. Liao, Z. Li, L. Sun and C. Chen, Nanoscale, 2015, 7, 15990–15998 RSC.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006–15013 CrossRef.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- J. Sanetuntikul, T. Hang and S. Shanmugam, Chem. Commun., 2014, 50, 9473–9476 RSC.
- J. Sanetuntikul, K. Ketpang and S. Shanmugam, ACS Catal., 2015, 5, 7321–7327 CrossRef CAS.
- J. Sanetuntikul, C. Chuaicham, Y.-W. Choi and S. Shanmugam, J. Mater. Chem. A, 2015, 3, 15473–15481 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21540h |
| This journal is © The Royal Society of Chemistry 2017 |