
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A green approach to the synthesis of a nano catalyst and the role of basicity, calcination, catalytic activity and aging in the green synthesis of 2-aryl bezimidazoles, benzothiazoles and benzoxazoles
Pramod K. Sahu*
School of Studies in Chemistry, Jiwaji University, Gwalior-474011, Madhya Pradesh, India. E-mail: sahu.chemistry@gmail.com; researchdata6@gmail.com
First published on 30th August 2017
Abstract
A green synthesis of hydrotalcite (a double layered catalyst) by a grinding method using Al/Mg molar ratios of 1.0–3.0 at room temperature is described. The prepared double layered catalyst (hydrotalcite) has been characterized by TG, FT-IR, SEM, XRD and Hammett titration methods. Different factors such as the effect of molar ratios, catalyst loading, reaction time, aging time, and basicity have been investigated for the facile, efficient and green synthesis of 2-arylbenzimidazoles, benzothiazoles and benzoxazoles under solvent free conditions. The influence of catalyst loading on reactivity was studied and a catalyst loading of 20 mg was found to be optimal, giving the best yield with minimum time as compared to other catalysts. The present methodology reports, herein, a rapid and cost effective synthesis of hydrotalcite and its versatile applications in the synthesis of 2-arylbenzimidazoles, benzothiazoles and benzoxazoles.
Introduction
The benzimidazole, benzoxazole and benzothiazole skeletons may be found in numerous pharmaceutical agents with a diverse spectrum of biological activities.1 These are important structural intermediates in the synthesis of a variety of pharmaceutical, natural, and agrochemical compounds (Fig. 1). However various methods have been reported for the synthesis of benzimidazoles,2 benzoxazoles,3 and benzothiazoles,4 but a real need exists for new and simple procedures that support many kinds of structural diversity and various substitution patterns in the target library. A variety of catalysts was used for the synthesis of 1,2-disubstituted benzimidazoles.5,6 Similarly the substituted benzothiazoles and benzoxazoles were also synthesized by several methods.7 Unfortunately, many of these reported methods suffer from one or other limitations such as drastic reaction conditions, long reaction time with poor yield, side product formation, strong oxidizing agents, use of toxic reagents, hazardous solvents, use of expensive catalyst, more catalyst loading, and tedious workup procedure. Most of the reported catalysts are homogeneous having no recyclability whereas the reactions carried out with heterogeneous catalysts required non green solvents and higher temperatures. | ||
| Fig. 1 Some benzimidazoles, benzothiazoles, and benzoxazoles. | ||
Layered double hydroxides (LDHs), which are referred to anionic clays in comparison with cationic clays and also as hydrotalcite like compounds (HT) are an important class of ionic lamellar solids. The main property of hydrotalcites is their anion exchange capacity, which makes them unique inorganic materials to intercalate organic or inorganic anion.8 Hydrotalcites are increasingly regarded as a good alternative to the traditional homogenous base catalysts such as NaOH and KOH for several base-catalyzed reactions that are important for the pharmaceutical and fragrance industries,9 electrode modifiers and catalyst supports.10 A well-documented example is the isomerization of eugenol and safrole.11 The structure consists of positively charged brucite (magnesium hydroxide)-like layers with interlayer space containing charge compensating anions and water molecules.12 As far as green chemistry is concerned, hydrotalcites offer several advantages over corrosive and dissolved catalysts; easy separation from the reaction mixture, recycling possibilities, decreased corrosion of the reactor.13 Hydrotalcites can be implicated in the preparation of catalysts dedicated to the production of H2,14 wide range of organic compounds,15 and production of biodiesel by trans-esterification of triglycerides with methanol.16 In addition to the above numerous experimental investigations have been published on the use of hydrotalcites for catalytic applications.17
Considering all above issues, there is a need to develop environmental benign green route and search new catalyst for the synthesis of benzimidazoles, benzothiazoles, and benzoxazoles. Hydrotalcite have higher catalytic activity due to high surface area than their bulk counterparts and due to this they have attracted considerable attention in organic synthesis. Therefore, as a part of our ongoing research aimed at the development of new catalysts and their application in synthesis of hetrocycles,18 we report herein, a rapid and cost effective method (grinding method) for synthesis of hydrotalcite and used in synthesis of benzimidazoles, benzothiazoles, and benzoxazoles under solvent free conditions (Fig. 2).
 | ||
| Fig. 2 Reaction of 1,2-phenylenediamine, 2-aminothiophenol, and 2-aminophenol with aldehyde respectively using hydrotalcite. | ||
Results and discussion
Characterization of hydrotalcite
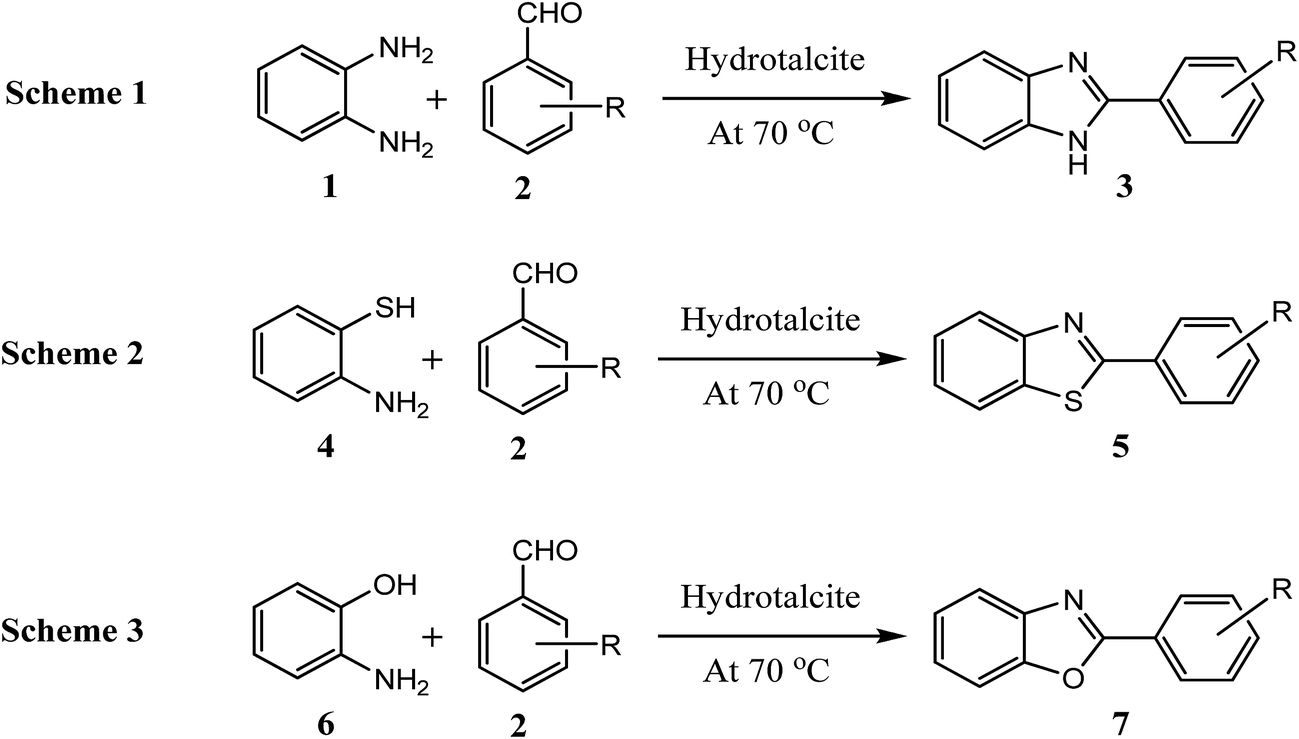
Absorption band at higher wave numbers 3695 and 3471 cm−1 in IR spectra (Fig. 3) assigned to the O–H stretching vibrations of water bonded to M3OH units. The shorter O–H bonds existing in hydrotalcite causes an increase in electrostatic attraction within the hydrotalcite layers.19 The weak bands at 2930 cm−1 and 2880 cm−1 are suggested to the strongly hydrogen bonded water molecules to interlayer anions such as carbonate.20 Strong water deformation modes at 1631 cm−1 found due to hydrotalcites containing physically adsorbed water.19 Carbonate which is bonded to hydroxyl surface of hydrotalcite shown a band at 1364 cm−1.20 The IR absorption band at 1024 cm−1 has been assigned to symmetric stretch of free carbonate canions.19 Bending vibration of free carbonate anions give a band at 777 cm−1. The IR absorption band at 443 cm−1 has been assigned to Al–O bonds.21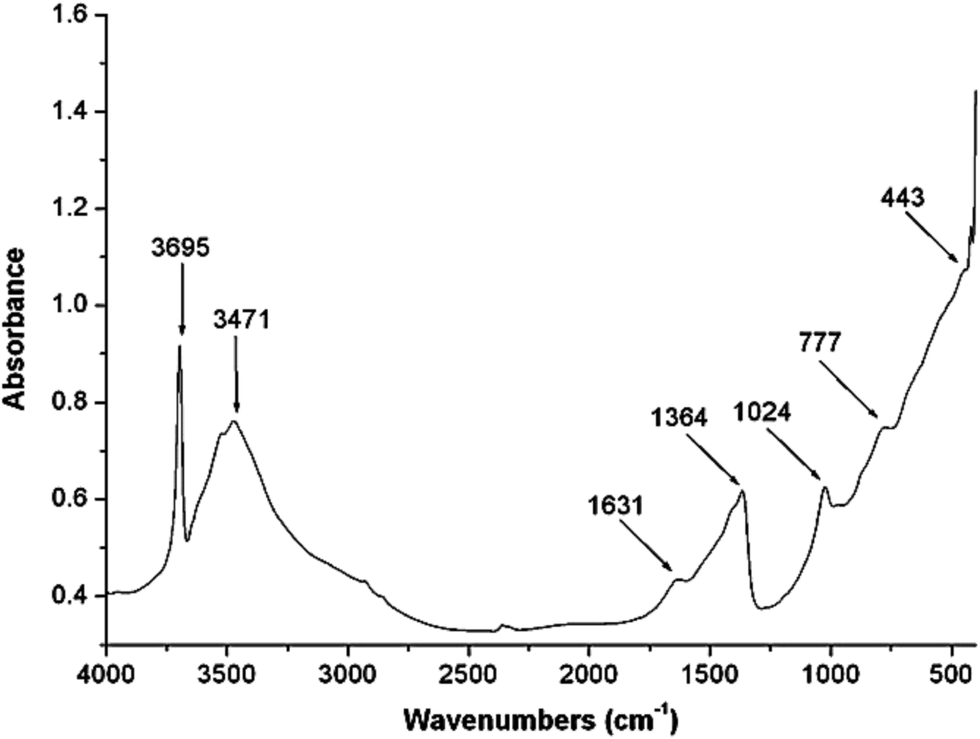 | ||
| Fig. 3 FT-IR spectra of the hydrotalcite (Mg–Al–CO3). | ||
Mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al atomic ratio was measured using X-ray microanalysis and found 3.16, which is in good agreement with the metallic ratio (3.0) taken in solution. The value of x [x = MIII/MII + MIII] was found to 0.24, which suggest the purity of hydrotalcite.22 Powder X-ray diffraction (P-XRD) pattern for sample Mg–Al–CO3 is shown in Fig. 4. The presence of CO32− anion in the interlayer gallery of the hydrotalcite is confirmed by the characteristic basal spacing d003 = 7.76 Å. This indicates a gallery height of 2.96 Å (assuming a thickness of 4.8 Å for the cationic sheets). The material is reasonably crystalline and suggests a relatively well-ordered sheet arrangement.23 The crystallite size of this sample was found 24.87 nm as calculated using Scherrer formula.24 More intensive and sharper reflections of the (003) and (006) planes has found at low 2θ values (11–23°). A typical SEM image of Mg–Al–CO3 hydrotalcite is shown in Fig. 4. This figure indicates the existence of lamellar particles looks like rounded hexagonal shape and typical of hydrotalcite like material. The material was found mesoporous with the surface area 90 m2 g−1.
:Al atomic ratio was measured using X-ray microanalysis and found 3.16, which is in good agreement with the metallic ratio (3.0) taken in solution. The value of x [x = MIII/MII + MIII] was found to 0.24, which suggest the purity of hydrotalcite.22 Powder X-ray diffraction (P-XRD) pattern for sample Mg–Al–CO3 is shown in Fig. 4. The presence of CO32− anion in the interlayer gallery of the hydrotalcite is confirmed by the characteristic basal spacing d003 = 7.76 Å. This indicates a gallery height of 2.96 Å (assuming a thickness of 4.8 Å for the cationic sheets). The material is reasonably crystalline and suggests a relatively well-ordered sheet arrangement.23 The crystallite size of this sample was found 24.87 nm as calculated using Scherrer formula.24 More intensive and sharper reflections of the (003) and (006) planes has found at low 2θ values (11–23°). A typical SEM image of Mg–Al–CO3 hydrotalcite is shown in Fig. 4. This figure indicates the existence of lamellar particles looks like rounded hexagonal shape and typical of hydrotalcite like material. The material was found mesoporous with the surface area 90 m2 g−1.
 | ||
| Fig. 4 XRD pattern and SEM image of hydrotalcite (Mg–Al, 3:1). | ||
A total mass 38.26% at 330–570 °C in TG graph (Fig. 5) of hydrotalcite (Al:Mg:CO3) and other mass loss at 60–230 °C, 230–330 °C, and 570–670 °C respectively. The TG results point out that the hydrotalcite (Al:Mg:CO3) is thermally stable up to 230 °C.25 The second mass loss between 223 and 330 °C has been ascribed to the dehydroxylation of the brucite like layers along with anion decomposition leaving a Mg, Al oxo-hydroxide up to 330 °C. Finally third mass loss has been assigned to progressive elimination of hydroxyl ions and produce metal oxides and spinel structure.
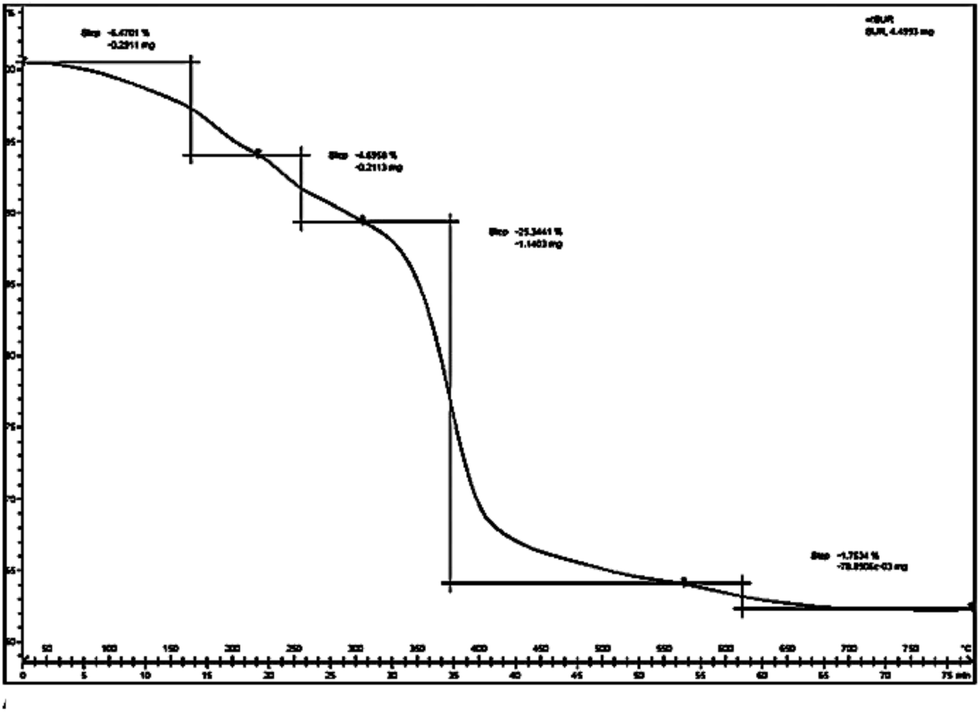 | ||
| Fig. 5 DTG curve of hydrotalcite (Mg–Al; 3:1). | ||
The Ca:Al metal ratio was found in good agreement with the initially taken metallic ratio. The observed value for Ca–Al–CO3 was 3:2:1. The P-XRD patterns for the LDHs Ca–Al–CO3 exhibits features commonly shown by layered materials. There are narrow, symmetric, strong lines at low 2θ values and weaker, less symmetric lines at high 2θ value (Fig. 6).26 Presence of CO32− anion in the interlayer gallery of the hydrotalcite is confirmed by the characteristic basal spacing d003 = 3.38 Å. Sharp intense peaks at low diffraction angles (peaks close to 2θ = 11°, 24°, and 35°; ascribed to diffraction by basal planes (003), (006), and (009), respectively) and broad, less intense peaks at higher angles (peaks close to 2θ = 38°, 46°, and 60°) ascribed to diffraction by (105), (108) and (110) planes confirm the presence of hydrotalcite. The crystallite size of this sample was found 47.025 nm. SEM (Fig. 6) of the material shows high crystallinity. The particles of hydrotalcite Ca–Al–CO3 clearly exhibit the hexagonal shape; however, big needle shape particles are also visible.
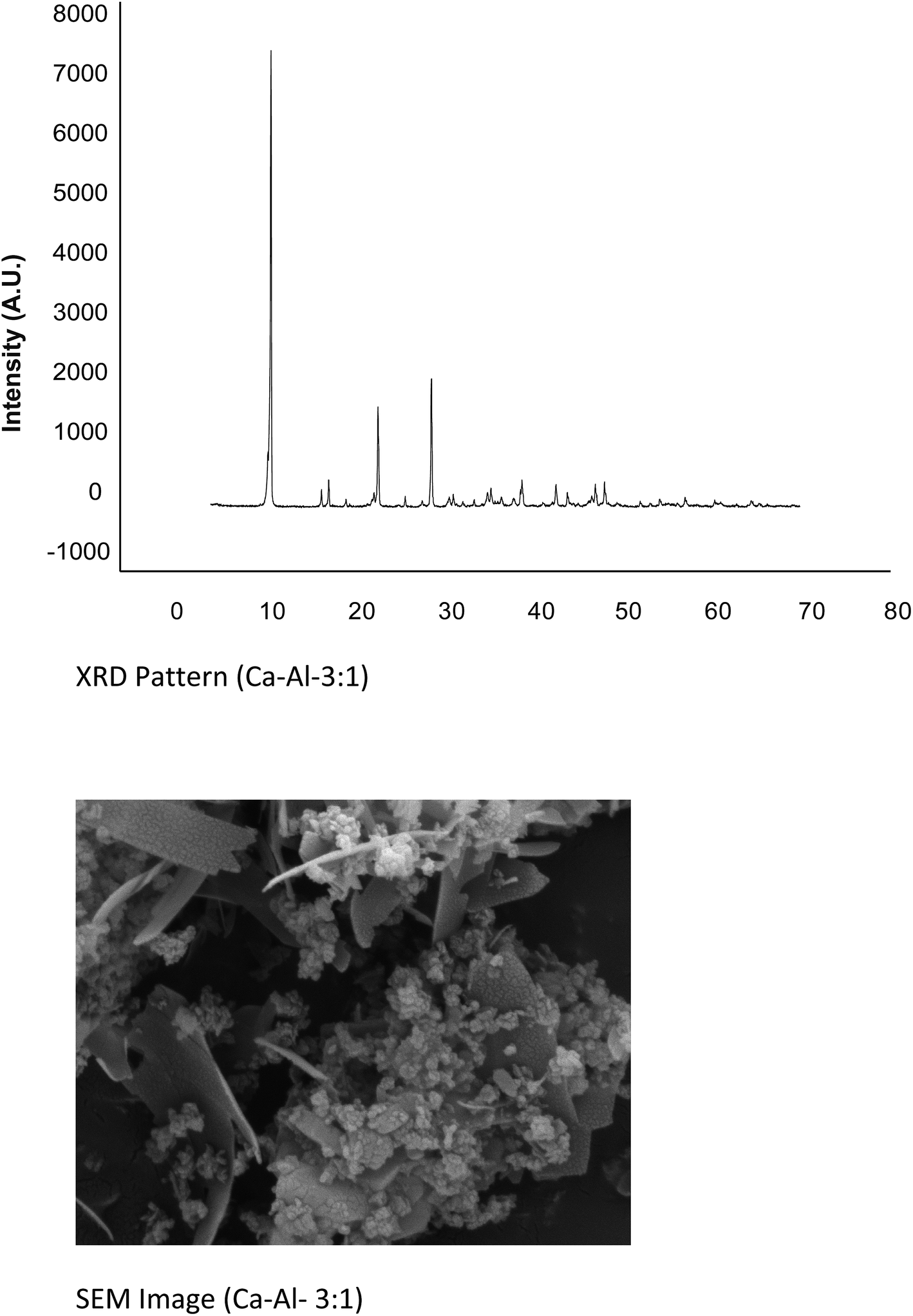 | ||
| Fig. 6 P-XRD and pattern and SEM image of hydrotalcite (Ca–Al, 3:1). | ||
Chemistry
To optimize the reaction conditions, a model reaction of 4-hydroxy coumarin, benzaldehyde and 2-aminobenzothiazole was carried under solvent free conditions. Under room temperature (Table 1, entry 1), reaction was slow and yield of target product was too low, even reaction has performed at 90 and 120 °C (Table 1, entries 4 and 5). Best yield was obtained at 70 °C with minimum time (3.0 h). So, 70 °C temperature was choose for further study. As shown in Fig. 2, the reactions of 1,2-phenylenediamine with benzaldehyde (Scheme 1), 2-aminothiophenol with benzaldehyde (Scheme 2), and 2-aminophenol with benzaldehyde (Scheme 3) were selected as model reactions to investigate catalytic activity of hydrotalcite under solvent free conditions. To optimize the reaction conditions, variety of catalysts were screened in a model reaction of o-phenylenediamine and benzaldehyde and results are summarized in Table 2. This clearly indicates that in the absence of catalyst, reaction did not work beneficially (Table 2, entry 1). From Table 2, it has been found that best yield was obtained with hydrotalcite (HT, Mg–Al–CO3) with lower time as compared to other catalysts. Further to identify the catalyst loading, different amount of loading (Table 2, entries 2–5) was used and 20 mg of catalyst (HT) was suitable to promote the reaction efficiently (Table 2, entry 4). An increment in catalyst concentration more than 20 mg did not show effective improvement in product yield (Table 2, entry 5). Slightly lower yield was obtained with Ca–Al–CO3 hydrotalcite (Table 2, entry 6) as compared to Mg–Al–CO3 hydrotalcite. Other metal oxide, hydroxide and chlorides give moderate yield of final product (Table 2, entries 7–13).
|
|||
|---|---|---|---|
| Entry | Catalyst (in grams) | Time (h) | Yield% of 3ab |
| a Reaction conditions: benzaldehyde (1 mmol), o-phenylenediamine (1 mmol), hydrotalcite (20 mg), at 70 °C.b Isolated yield. | |||
| 1 | No catalyst | 6.0 | 25 |
| 2 | Mg–Al–CO3, HT (0.005) | 6.0 | 75 |
| 3 | Mg–Al–CO3, HT (0.01) | 4.0 | 88 |
| 4 | Mg–Al–CO3, HT (0.02) | 3.0 | 95 |
| 5 | Mg–Al–CO3, HT (0.05) | 3.0 | 95 |
| 6 | Ca–Al–CO3, HT (0.02) | 2.0 | 89 |
| 7 | AlCl3 (0.02) | 5.0 | 59 |
| 8 | SiO2 (0.02) | 4.0 | 72 |
| 9 | MnO2 (0.02) | 4.0 | 61 |
| 10 | Al(OH)3 (0.02) | 4.5 | 50 |
| 11 | Ca(OH)2 (0.02) | 4.0 | 51 |
| 12 | Al2O3 (0.02) | 4.5 | 60 |
| 13 | Mg(OH)2 (0.02) | 4.5 | 42 |
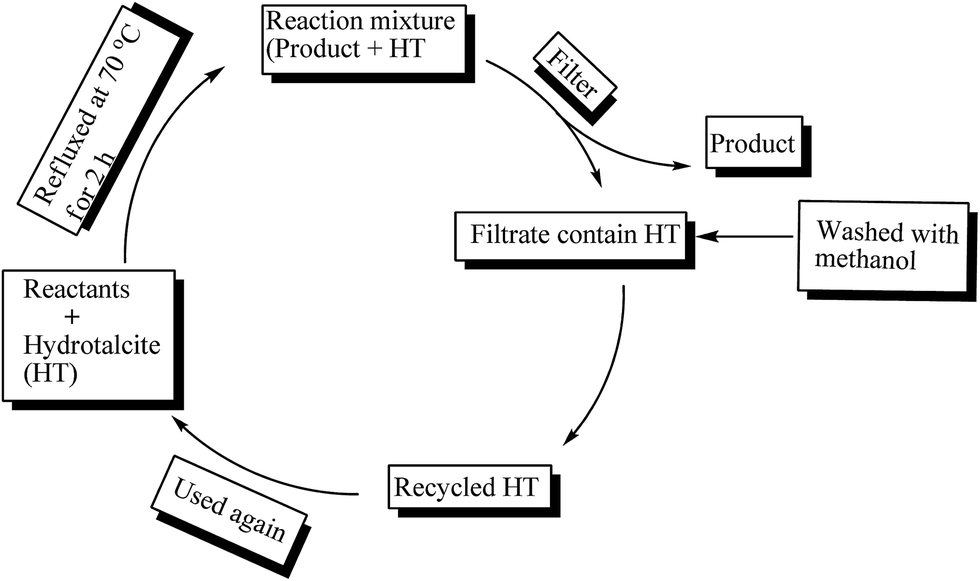
Recyclability of the catalyst is an important task in industrial applications. Therefore reusability of hydrotalcite (Mg–Al–CO3) was investigated for eight runs (Table 3, entries 2–9) in a model reaction of benzaldehyde and o-phenylenediamine by incorporating 20 mg of hydrotalcite as catalyst (Fig. 7). After completion of reaction, the contents were filtered to recycle the hydrotalcite catalyst. Recycled hydrotalcite washed with methanol to remove organic impurities. MgII hydrotalcite catalyst can be readily recovered and reused for eight runs. It was found that the reactivity of the catalyst decreases marginally (approx. 9–10%) for the eight runs.
 | ||
| Fig. 7 Recyclability and reusability of hydrotalcite catalyst. | ||
To generalize the applicability and scope of this protocol in the formation of functionalized heterocycles were investigated by using various substituted aldehydes. Different electron donating and electron withdrawing aldehydes were used and as expected it give good yield of target products, it can be seen that electron donating and electron withdrawing groups does not show any significant difference on the reaction yields (Tables 4–6). This indicated that present catalytic system efficiently makes the condensation reaction much faster with increased yields.
|
||||||
|---|---|---|---|---|---|---|
| R | Product | Yield (%) | Time (h) | MP (°C) | ||
| Found | Reported | Ref. | ||||
| a Reaction conditions: aldehydes (1 mmol), o-phenylenediamine (1 mmol), hydrotalcite (20 mg), at 70 °C temp. | ||||||
| H | 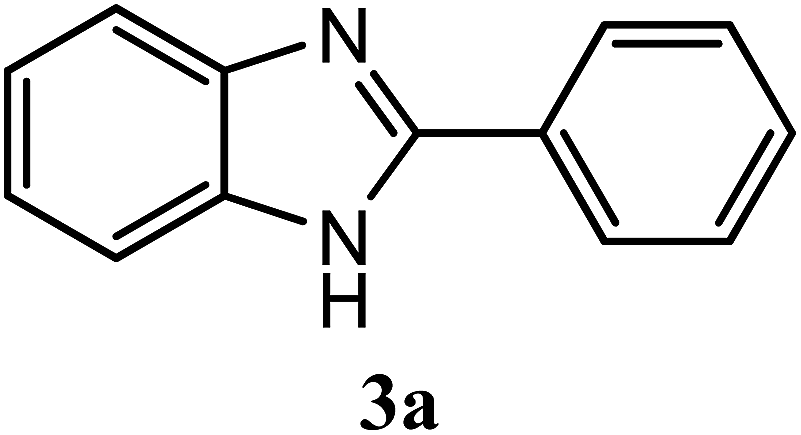 |
95 | 3.0 | 290–291 | 290–293 | 7f |
| 4-NO2 | 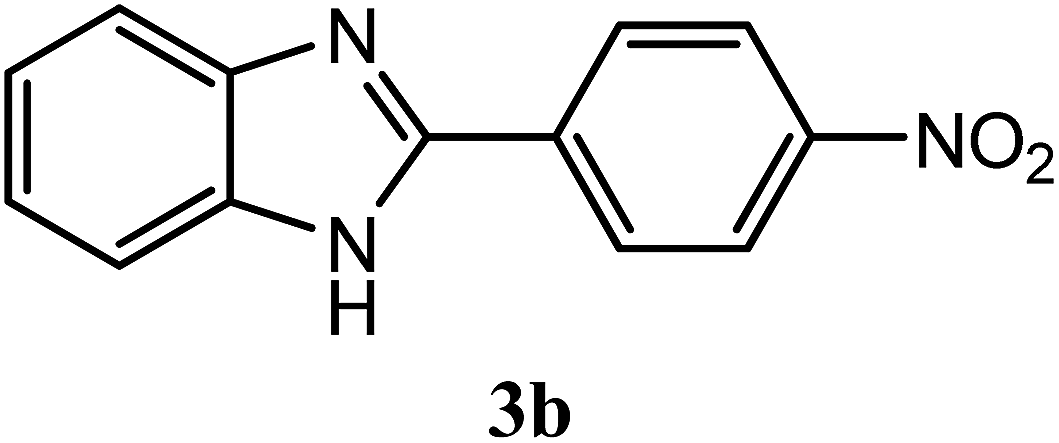 |
91 | 2.5 | 321–323 | 322–323 | 7f |
| 4-OH | 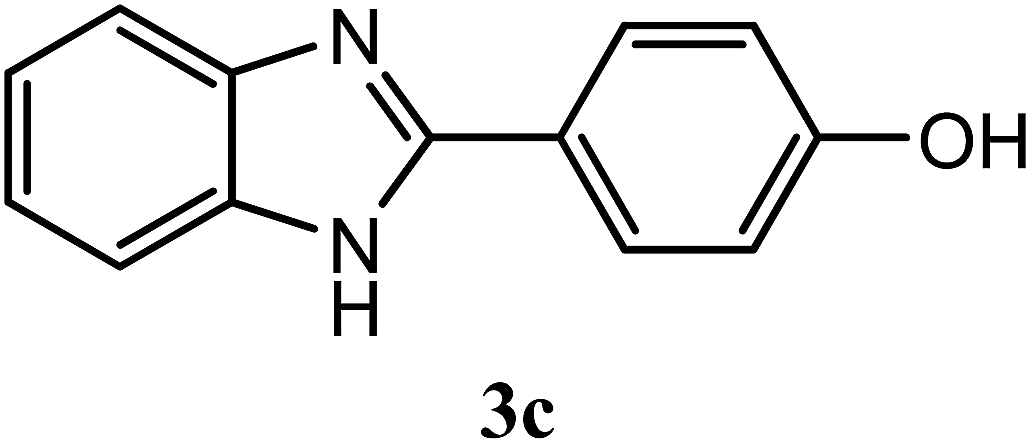 |
90 | 2.0 | 277–279 | 265–267 | 7h |
| 2-OH | 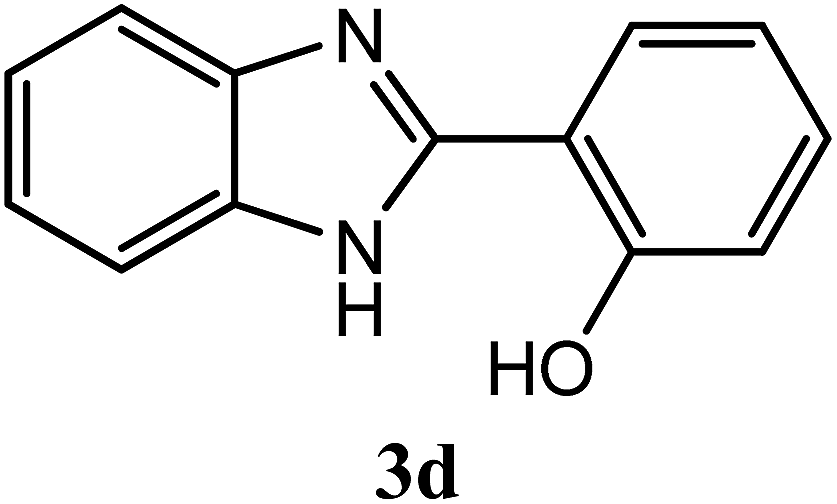 |
93 | 2.5 | 222–224 | 236–237 | 7n |
| 4-Cl | 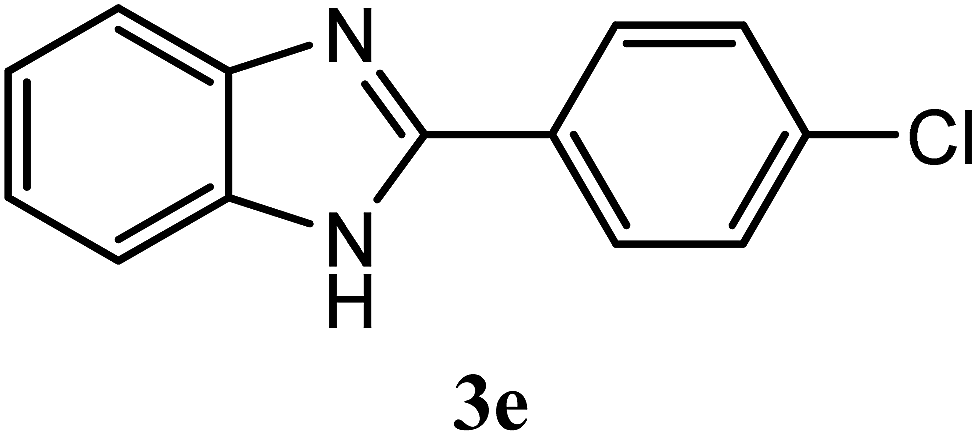 |
92 | 2.0 | 288–291 | 288–291 | 7g |
| 4-OCH3 | 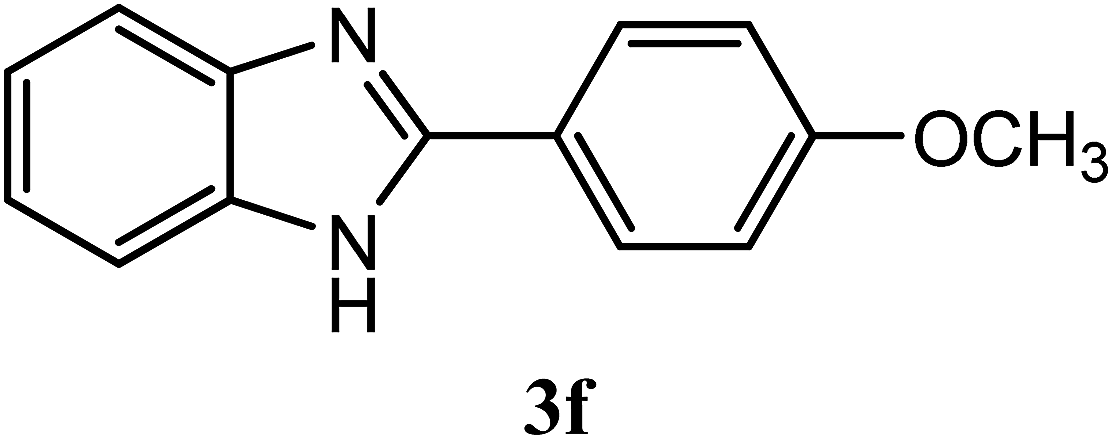 |
96 | 2.0 | 221–223 | 180–182 | 7i |
| 4-CH3 | 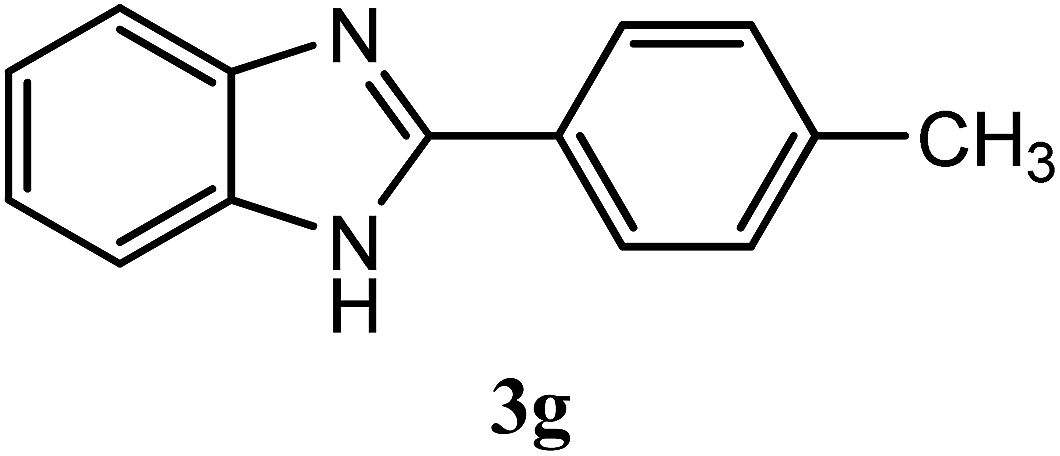 |
95 | 2.0 | 260–262 | 264–265 | 7f |
|
||||||
|---|---|---|---|---|---|---|
| R | Product | Yield (%) | Time (h) | MP (°C) | ||
| Found | Reported | Ref. | ||||
| a Reaction conditions: aldehyde (1 mmol), 2-aminothiophenol (1 mmol), hydrotalcite (20 mg), at 70 °C temp. | ||||||
| H |  |
96 | 2.0 | 112–114 | 112–114 | 7k |
| 4-Cl |  |
93 | 2.5 | 104–106 | 103–104 | 7l |
| 4-Br |  |
92 | 2.0 | 106–108 | 102–104 | 7p |
| 4-NO2 | 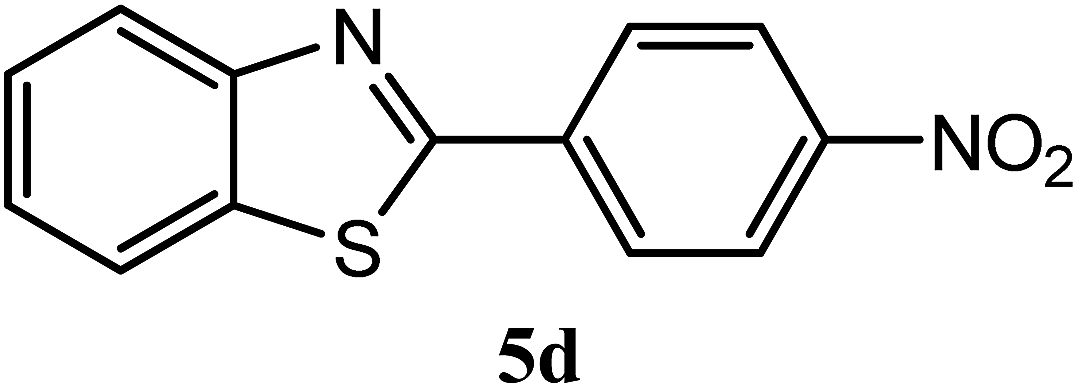 |
94 | 2.0 | 226–228 | 230–231 | 7m |
| 4-OH | 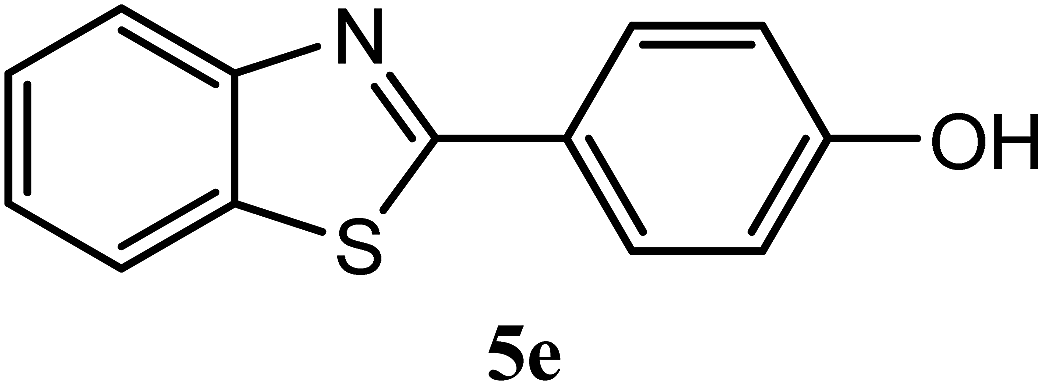 |
91 | 3.0 | 220–222 | 227 | 7m |
| 4-CH3 | 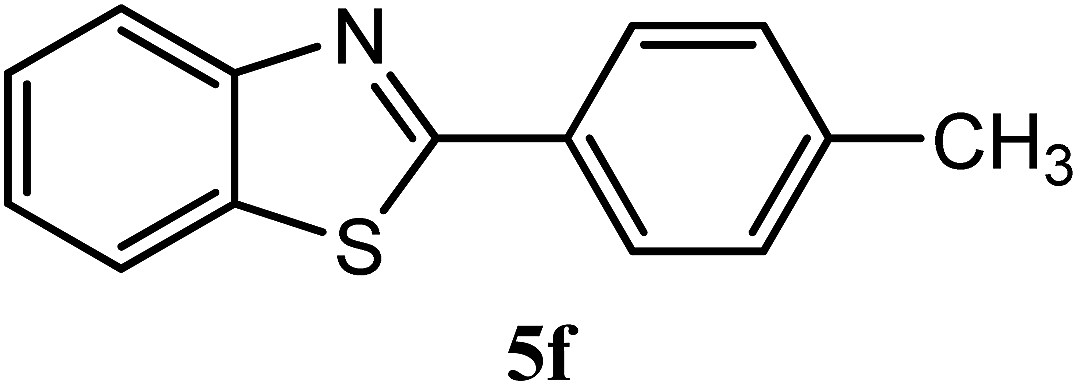 |
92 | 2.0 | 84–86 | 85 | 7m |
| 4-OCH3 | 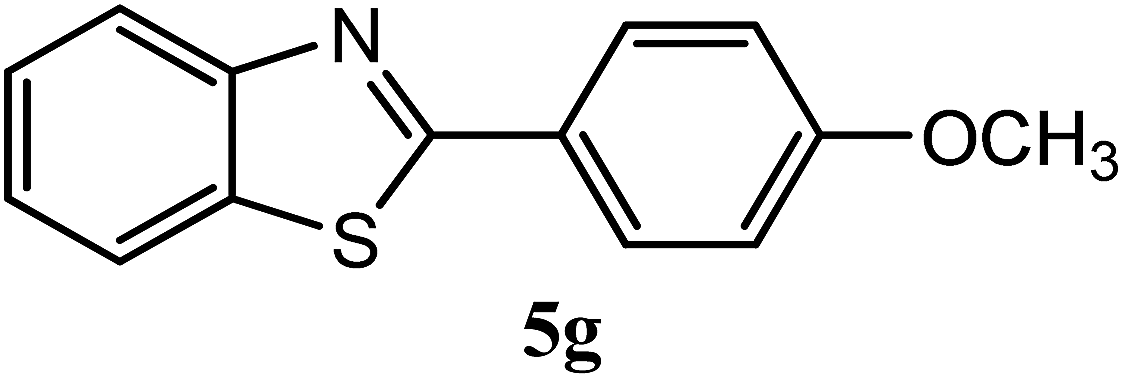 |
93 | 0.5 | 120–122 | 120–121 | 7k |
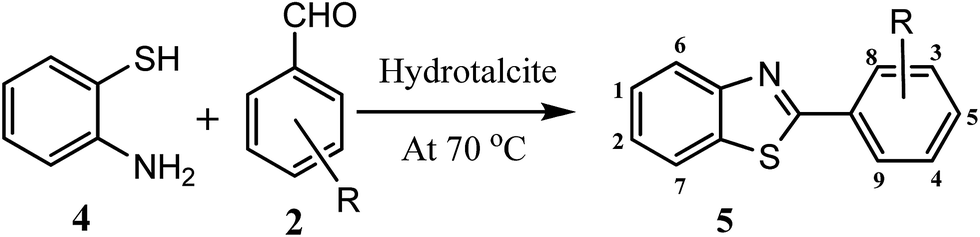
|
||||||
|---|---|---|---|---|---|---|
| R | Product | Yield (%) | Time (h) | MP (°C) | ||
| Found | Reported | Ref. | ||||
| a Reaction conditions: aldehyde (1 mmol), 2-aminophenol (1 mmol), hydrotalcite (20 mg), at 70 °C temp. | ||||||
| H | 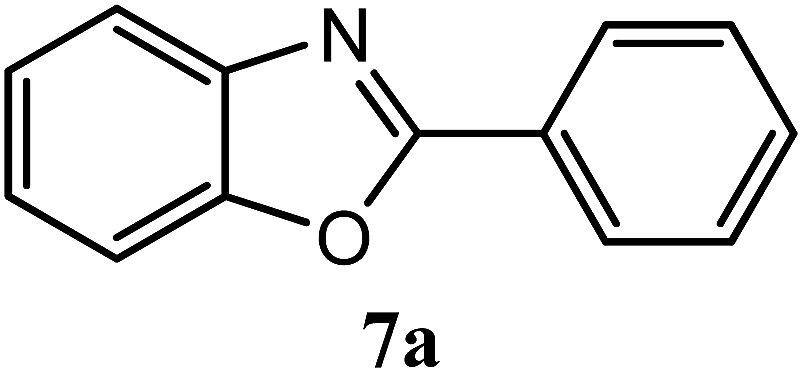 |
93 | 3.0 | 98–100 | 102–103 | 7f |
| 4-Cl | 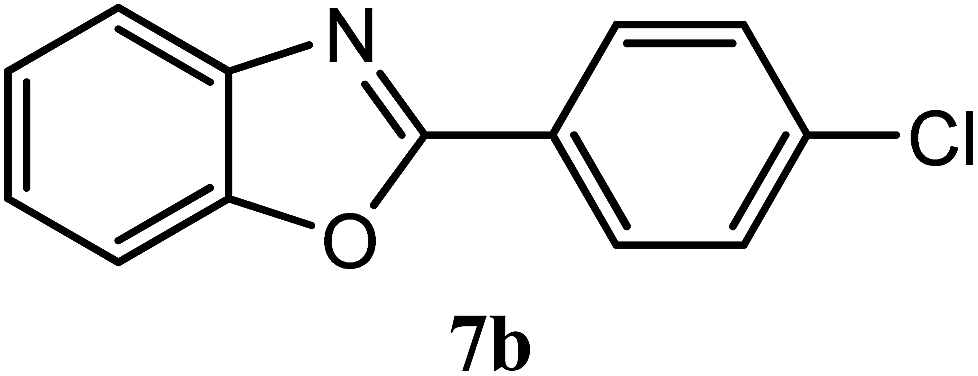 |
95 | 2.0 | 142–144 | 149–150 | 7f |
| 3,4-OCH3 | 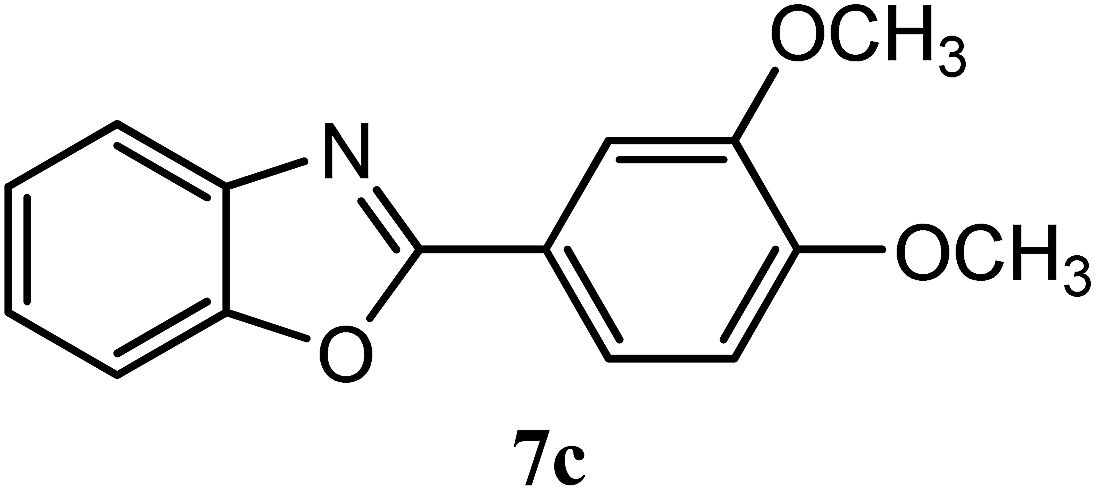 |
92 | 2.5 | 109–112 | ||
| 4-NO2 | 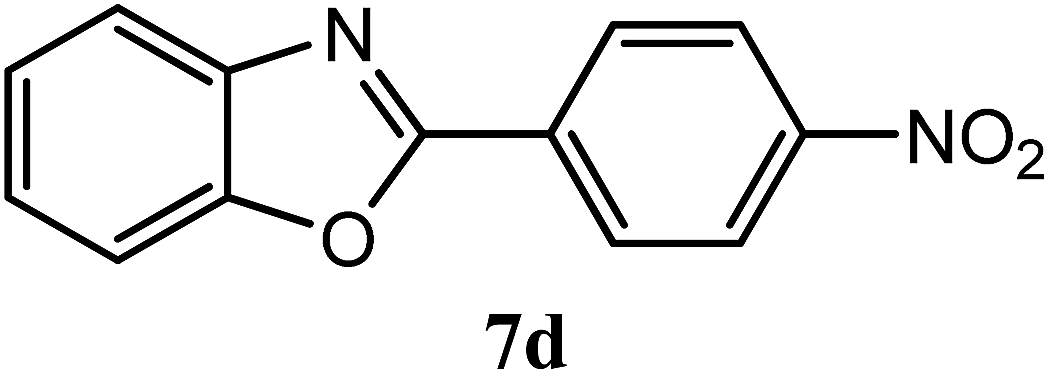 |
90 | 3.0 | 262–264 | 266–267 | 7f |
| 4-OH | 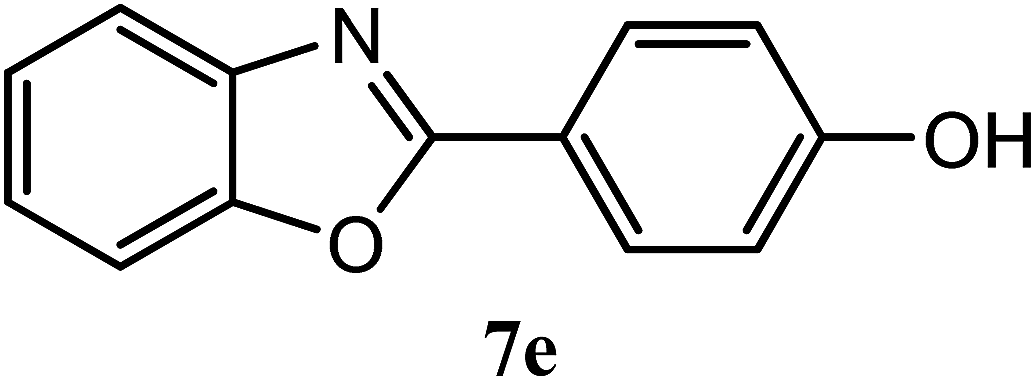 |
94 | 2.0 | 284–286 | 287–298 | 7j |
| 4-CH3 | 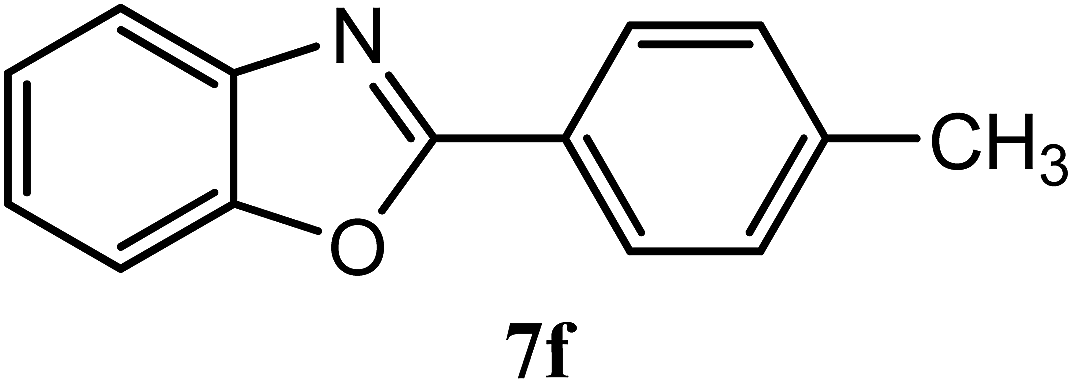 |
95 | 2.5 | 122–124 | 115–116 | 7o |
| 4-OCH3 | 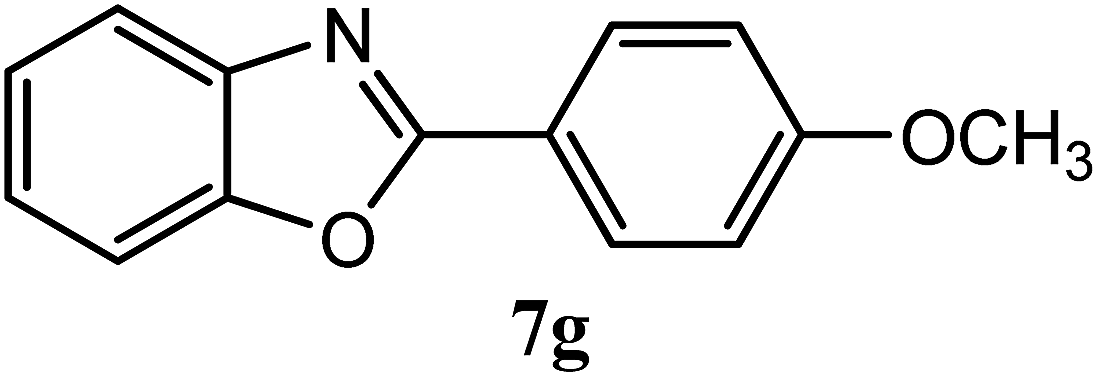 |
94 | 2.0 | 98–100 | 100–101 | 7f |
Leaching test
Filtrate was analyzed for leached metal content by ICP emission spectroscopy. No metal was detected. These results confirmed that the metal leaching did not occur. However, Mg–Al mixed oxide was found to be thermally and mechanically stable and no significant difference was observed in particle size and morphology of the used catalyst as evidenced by SEM.27 To ensue the sustainability of structure of recovered hydrotalcite, XRD has been carried out which showed the similar profile as fresh catalyst which confirmed that layered structure of hydrotalcite was maintained after the reaction.Effect of aging time and SEM analysis
To observe the effect of aging time on the morphology of the material, SEM images of hydrotalcite samples at Mg/Al molar ratio of 3.0 were recorded. The micrographs at different aging times are shown in Fig. 8. Micrographs of hydrotalcite show a well-developed layered and platelet structure of the hydrotalcite. However, a spongy type structure is exhibited due to overlapping of such platelets. The SEM images of the hydrotalcite showed a gradual crystallization during the hydrothermal treatment conditions. At the temperature 110 °C (hydrothermal treatment temperature) and 0 h aging time, crystallinity of the hydrotalcite was observed to be very poor; however, as aging time increases from 0 to 10 h, the crystallinity of the hydrotalcite also increased. These results confirmed an increase in the crystallinity of the hydrotalcite samples at Mg/Al under hydrothermal treatment conditions. | ||
| Fig. 8 SEM image of hydrotalcite at a Mg/Al (3:1) at different aging time at 110 °C. | ||
Basicity of hydrotalcite
Basicity of the catalyst was determined using Hammett indicator and benzoic acid titration method.28 All samples prepared in our study had the basic strength in the range of 9.3–15.0. The basicity of the catalysts with different Mg/Al molar ratios was shown in Fig. 9. The main basic sites with H− in the range of 7.2–9.8 and the other sites with H− in the range of 9.8–15.0 were observed, thus suggesting the calcined hydrotalcites contain different types of surface basic sites. Di Cosimo et al.29 suggested that pure MgO possesses strong basic sites consisting predominantly of O2− and calcined hydrotalcites contain surface basic sites. Our experimental results on the Hammett titration method conform to this viewpoint. It indicates a wide basic site distribution as far as the basicity is concerned.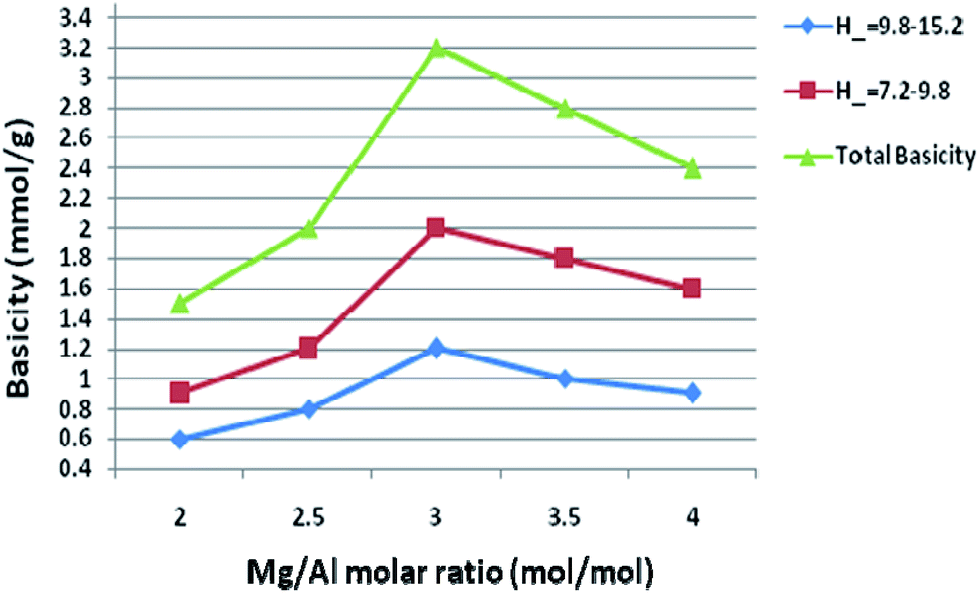 | ||
| Fig. 9 Basicity of hydrotalcite with different metal ratios. | ||
As molar ratio of Mg/Al reach up to the maximum value 3.0, the total basicity of the hydrotalcite (catalyst) was increased gradually. But basicity was decreased when further increase the molar ratio of Mg/Al, which resulted in a loss of the catalytic activity. Qualitatively similar trends were also reported by other researchers.30 Nakatsuka et al.30a found that the basicity measured by titration with benzoic acid reached a maximum for Mg/Al ratio of about 2.6. Also, Fishel and Davis30b measured the number of basic sites by TPD of CO2 and a maximum of the basic site density was observed at Mg/Al ratio of 3.0. Best results were obtained with 3.0 HT, therefore basicity of 3.0 HT calcined at different temperatures was measured with the same method; the results are illustrated in Fig. 10. From this figure, it can be seen that the maximum basicity (reaching 3.6 mmol g−1) is found at a calcinations temperature of 750 K and a low level of basicity is observed below 573 K and above 750 K. The increased basicity could be expected to correlate with an increase of the catalyst activity.
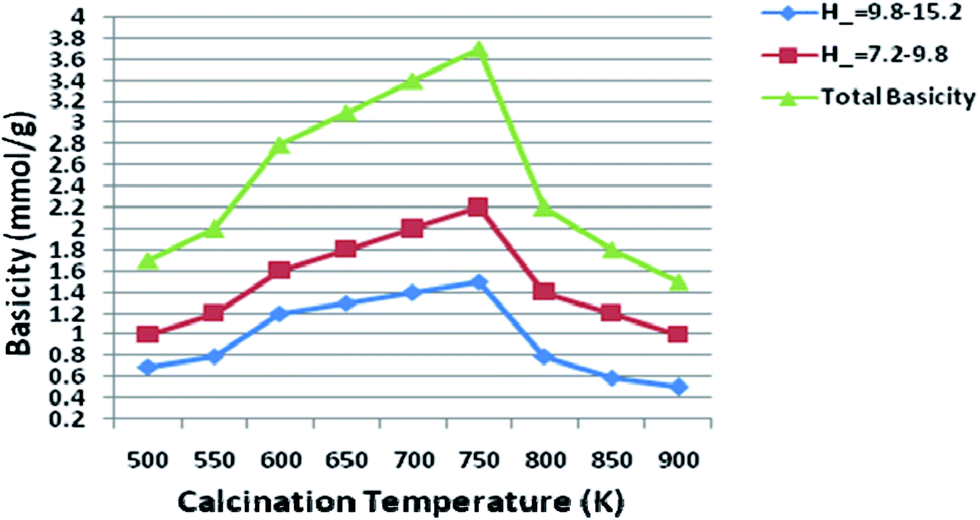 | ||
| Fig. 10 Basicity of 3.0 hydrotalcite at different temperature. | ||
Conclusion
In conclusion, we have developed green methodology for synthesis of hydrotalcite by grinding method. Prepared hydrotalcite could be used efficiently in selective, eco-friendly and green synthesis for variety of substituted benzimidazoles, benzothiazoles and benzoxazoles from 1,2-phenylenediamine, 2-aminothiophenol, and 2-aminophenol with substituted aromatic aldehydes under solvent free conditions. The catalytic activities of the calcined hydrotalcite show a striking correlation with their corresponding basic properties. The prepared catalyst were characterized with Hammett titration method, SEM, TG, IR and XRD showing the strong basic sites in double layers and coordinatively unsaturated O2− ion acting as basic sites in the calcined hydrotalcite may be responsible for their catalytic activity. The crystallization of hydrotalcite was significantly affected by hydrothermal treatment temperature and time.This is first attempt to synthesize substituted benzimidazoles, benzothiazoles and benzoxazoles using hydrotalcite as heterogeneous catalyst which is easily separable and recyclable up to eight runs. Compared to previously reported methods, moreover, the mild reaction conditions, easy work-up, clean reaction profiles, lower catalyst loading and cost efficiency render this approach as an interesting alternative to the existing methods.
Materials and methods
Experimental
The 1H NMR spectra were measured using BRUKER AVANCE II 400 NMR spectrometer with tetramethylsilane as an internal standard at 20–25 °C; data for 1H NMR are reported as follows: chemical shift (ppm), integration, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet, and br, broad), coupling constant (Hz). IR spectra were recorded by SHIMADZU; IR spectrometer of sample dispersed in KBr pellet or Nujol is reported in terms of frequency of absorption (cm−1). E-Merck pre-coated TLC plates and RANKEM silica gel G were used for preparative thin-layer chromatography. Melting points were determined in open capillaries and are uncorrected. AR grade of o-phenylenediamine, 2-aminothiophenol, 2-aminophenol, aldehydes and other catalysts were purchased from Himedia Laboratory Ltd., Mumbai, India and used without further purification.Preparation of hydrotalcite (HT)
Characterization
Conflicts of interest
Author has no conflict of interest.Acknowledgements
We are grateful thanks to Chandigarh and Punjab University, Chandigarh for spectral analytical data.References
- (a) C. Chen and Y. J. Chen, Tetrahedron Lett., 2004, 45, 113 CrossRef CAS; (b) N. Siddiqui, A. Rana, S. A. Khan, M. A. Bhat and S. E. Haque, Bioorg. Med. Chem. Lett., 2007, 17, 4178 CrossRef CAS PubMed; (c) C. J. Lion, C. S. Matthews, G. Wells, T. D. Bradshaw, M. F. G. Stevens and A. D. Westwell, Bioorg. Med. Chem. Lett., 2006, 16, 5005 CrossRef CAS PubMed; (d) S. T. Huang, I. J. Hsei and C. Chen, Bioorg. Med. Chem., 2006, 14, 6106 CrossRef CAS PubMed.
- (a) J. L. Girardet and L. B. J. Townsend, J. Org. Chem., 1999, 64, 4169 CrossRef CAS; (b) C.-M. Yeh, C.-L. Tung and C.-M. Sun, J. Comb. Chem., 2000, 2, 341 CrossRef CAS PubMed; (c) J. J. Chen, Y. Wei, J. C. Drach and L. B. Townsend, J. Med. Chem., 2000, 43, 2449 CrossRef CAS PubMed; (d) D. Tumelty, K. Cao and C. P. Holmes, Org. Lett., 2001, 3, 83 CrossRef CAS PubMed; (e) J. Mann, A. Baron, Y. Opoku-Boahen, E. Johansson, G. Parkinson, L. R. Kelland and S. Neidle, J. Med. Chem., 2001, 44, 138 CrossRef CAS PubMed; (f) B. Raju, N. Nguyen and G. W. Holland, J. Comb. Chem., 2002, 4, 320 CrossRef CAS PubMed; (g) H. Akamatsu, K. Fukase and S. Kusumoto, J. Comb. Chem., 2002, 4, 475 CrossRef CAS PubMed; (h) C. E. Hoesl, A. Nefzi and R. A. Houghten, J. Comb. Chem., 2003, 5, 155 CrossRef CAS PubMed; (i) D. Vourloumis, M. Takahashi, K. B. Simonsen, B. K. Ayida, S. Barluenga, G. C. Winters and T. Hermann, Tetrahedron Lett., 2003, 44, 2807 CrossRef CAS.
- (a) D.-F. Shi, T. D. Bradshaw, S. Wrigley, C. J. McCall, P. Lelieveld, I. Fichtner and M. F. G. Stevens, J. Med. Chem., 1996, 39, 3375 CrossRef CAS PubMed; (b) X. Beebe, D. Wodka and T. J. Sowin, J. Comb. Chem., 2001, 3, 360 CrossRef CAS PubMed; (c) A. Hari, C. Karan, W. C. Rodrigues and B. L. Miller, J. Org. Chem., 2001, 66, 991 CrossRef CAS PubMed; (d) R. S. Pottorf, N. K. Chadha, M. Katkevics, V. Ozola, E. Suna, H. Ghane, T. Regberg and M. R. Player, Tetrahedron Lett., 2003, 44, 175 CrossRef CAS; (e) F. Chen, C. Shen and D. Yang, Tetrahedron Lett., 2011, 52, 2128 CrossRef CAS.
- (a) M.-S. Chua, D.-F. Shi, S. Wrigley, T. D. Bradshaw, I. Hutchinson, P. N. Shaw, D. A. Barrett, L. A. Stanley and M. F. G. Stevens, J. Med. Chem., 1999, 42, 381 CrossRef CAS PubMed; (b) E. Kashiyama, I. Hutchinson, M.-S. Chua, S. F. Stinson, L. R. Phillips, G. Kaur, E. A. Sausville, T. D. Bradshaw, A. D. Westwell and M. F. G. Stevens, J. Med. Chem., 1999, 42, 4172 CrossRef CAS PubMed; (c) I. Hutchinson, M.-S. Chua, H. L. Browne, V. Trapani, T. D. Bradshaw, A. D. Westwell and M. F. G. Stevens, J. Med. Chem., 2001, 44, 1446 CrossRef CAS PubMed; (d) W. Leng, Y. Zhou, Q. Xu and J. Liu, Macromolecules, 2001, 34, 4774 CrossRef CAS; (e) I. Hutchinson, S. A. Jennings, B. R. Vishnuvajjala, A. D. Westwell and M. F. G. Stevens, J. Med. Chem., 2002, 45, 744 CrossRef CAS PubMed.
- (a) V. Kannan and K. Sreekumar, J. Mol. Catal. A: Chem., 2013, 376, 34 CrossRef CAS; (b) J. Yuan, Z. Zhao, W. Zhu, H. Li, X. Qian and Y. Xu, Tetrahedron, 2013, 69, 7026 CrossRef CAS; (c) Y. H. Cho, C. Y. Lee and C. H. Cheon, Tetrahedron, 2013, 69, 6565 CrossRef CAS; (d) G. Bramhachari, S. Laskar and P. Barik, RSC Adv., 2013, 3, 14245 RSC; (e) A. Teimouri, A. N. Chermahini, H. Salavati and L. Ghorbanian, J. Mol. Catal. A: Chem., 2013, 373, 38 CrossRef CAS; (f) S. Santra, A. Majee and A. Hajra, Tetrahedron Lett., 2012, 53, 1974 CrossRef CAS; (g) M. M. Guru, M. A. Ali and T. Punniyamurthy, J. Org. Chem., 2011, 76, 5295 CrossRef CAS PubMed; (h) J. P. Wan, S. F. Gan, J. M. Wu and Y. Pan, Green Chem., 2009, 11, 1633 RSC.
- (a) N. B. Valentin, J. B. Patrick, A. M. John, J. S. Colin and L. Stuart, J. Org. Chem., 2013, 78, 1471 CrossRef PubMed; (b) H. C. Yeon, Y. L. Chun, C. H. Deok and H. C. Cheol, Adv. Synth. Catal., 2012, 354, 29926 Search PubMed; (c) S. Hashem, A. Mahdi and M. D. Mohammad, J. Iran. Chem. Soc., 2012, 9, 189 CrossRef; (d) R. Yassin, M. Rachid, A. Rachid, E. H. Mohammadine, R. Sylvain, G. Geraid and L. Said, Tetrahedron Lett., 2011, 52, 3492 CrossRef; (e) W. Ying, S. Kathy, R. S. Daryl and W. D. Stevan, Tetrahedron Lett., 2006, 47, 4823 CrossRef.
- (a) R. J. Perry, B. D. Wilson and R. J. Miller, J. Org. Chem., 1992, 57, 2883 CrossRef CAS; (b) D. Alagille, R. M. Baldwin and G. D. Tamagnan, Tetrahedron Lett., 2005, 46, 1349 CrossRef CAS; (c) C. Benedi, F. Bravo, P. Uriz, E. Fernandez, C. Claver and S. Castillon, Tetrahedron Lett., 2003, 44, 6073 CrossRef CAS; (d) R. S. Varma, R. K. Saini and O. Prakash, Tetrahedron Lett., 1997, 38, 2621 CrossRef CAS; (e) R. S. Varma and D. Kumar, J. Heterocycl. Chem., 1998, 35, 1533 CrossRef CAS; (f) K. Bougrin, A. Loupy and M. Souuflaoui, Tetrahedron, 1998, 54, 8055 CrossRef CAS; (g) T. Yoshiyuki, Y. Kazuaki and K. H. Hokkaido Daigaku Koagakubu, Chem. Abstr., 1980, 93, 45–49204537k Search PubMed; (h) L. S. Gadekar, B. R. Arbad and M. K. Lande, Chin. Chem. Lett., 2010, 21, 1053 CrossRef CAS; (i) M. Lei, L. Ma and L. Hu, Synth. Commun., 2012, 42, 2981 CrossRef CAS; (j) R. Fazaeli and H. Aliyan, Appl. Catal., A, 2009, 353, 74 CrossRef CAS; (k) C. Mukhopadhyay and A. Datta, Heterocycles, 2007, 71, 1837 CrossRef CAS; (l) D. L. Boger, J. Org. Chem., 1978, 43, 2296 CrossRef CAS; (m) M. Kodomari, Y. Tamaru and T. Aoyama, Synth. Commun., 2004, 34, 3029 CrossRef CAS; (n) S. E. López, J. Restrepo, B. Pérez, S. Ortiz and J. Salazar, Bull. Korean Chem. Soc., 2009, 30, 1628 CrossRef; (o) V. R. Devalla and K. Ethirajulu, J. Chem. Soc., 1995, 1497 Search PubMed; (p) A. Teimouri, A. N. Chermahini, H. Salavati and L. Ghorbanian, J. Mol. Catal. A: Chem., 2013, 373, 38 CrossRef CAS; (q) D. Yang, X. Zhu, W. Wei, N. Sun, L. Yuan, M. Jiang, J. You and H. Wang, RSC Adv., 2014, 4, 17832 RSC; (r) D. Yang, X. Zhu, W. Wei, M. Jiang, N. Zhang, D. Ren, J. You and H. Wang, Synlett, 2014, 25, 0729 CrossRef CAS; (s) D. Yang, K. Yan, W. Wei, L. Tian, Y. Shuai, R. Li, J. You and H. Wang, Asian J. Org. Chem., 2014, 3, 969 CrossRef CAS; (t) D. Yang, P. Liu, N. Zhang, W. Wei, M. Yue, J. You and H. Wang, ChemCatChem, 2014, 6, 3434 CrossRef CAS.
- N. Ahmet, Z. K. Birgul and M. Z. Luis, Z. Anorg. Allg. Chem., 2009, 635, 1470 CrossRef.
- (a) M. S. Newton, J. D. Morrison, R. D. Pennell, N. P. Phodes and A. J. Toft, US Pat., Appl., 203152, 2010; (b) V. Rives, Layered double hydroxides: present and future, Nova Science Publishers, New York, 2001 Search PubMed; (c) S. K. Sharma, P. A. Parikh and R. V. Jarra, J. Mol. Catal. A: Chem., 2007, 278, 135 CrossRef CAS; (d) C. O. Veloso, C. N. Perez, B. M. de Souza, E. C. Lima, A. G. Dias, J. L. F. Monteiro and C. A. Henriques, Microporous Mesoporous Mater., 2008, 107, 23 CrossRef CAS.
- (a) W. Kagunya, Z. Hassan and W. Janes, Inorg. Chem., 1994, 35, 5970 CrossRef; (b) M. Ulibarri, F. M. Labajos, V. Rives, R. Trujillano, W. Kagunya and W. Janes, Inorg. Chem., 1994, 33, 2592 CrossRef CAS.
- C. M. Jinesh, C. A. Antonyraj and S. Kannan, Catal. Today, 2009, 141, 176 CrossRef CAS.
- (a) M. A. Ulibarri, I. Pavlovic, C. Barriga, M. C. Hermosin and J. Cornejo, Appl. Clay Sci., 2001, 18, 17 CrossRef CAS; (b) G. Centi and S. Perathoner, Microporous Mesoporous Mater., 2008, 107, 3 CrossRef CAS.
- K. Motocura, N. Fujita, K. Mori, T. Mizugaki, K. Ebitani, K. Htsukawa and K. Kanedar, Chem.–Eur. J., 2006, 12, 8228 CrossRef PubMed.
- M. Turco, G. Bagnasco, U. Costantino, F. Marmottini, T. Montanari, G. Ramis and G. Busca, J. Catal., 2004, 228, 56 CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, Catal. Rev.: Sci. Eng., 2001, 43, 443 CAS.
- Y. Xi and R. J. Davis, J. Catal., 2008, 254, 190 CrossRef CAS.
- S. K. Yun and T. Pinnavaia, J. Mater. Chem., 1995, 7, 348 CrossRef CAS.
- (a) P. K. Sahu, P. K. Sahu, S. K. Gupta, D. Thavaselvam and D. D. Agarwal, Eur. J. Med. Chem., 2012, 54, 366 CrossRef CAS PubMed; (b) P. K. Sahu, P. K. Sahu, D. Thavaselvam, A. M. Alafeefy and D. D. Agarwal, Med. Chem. Res., 2015, 24, 725 CrossRef CAS; (c) P. K. Sahu, P. K. Sahu and D. D. Agarwal, RSC Adv., 2013, 3, 9854 RSC; (d) P. K. Sahu, P. K. Sahu, S. K. Gupta and D. D. Agarwal, Ind. Eng. Chem. Res., 2014, 53, 2085 CrossRef CAS; (e) P. K. Sahu, P. K. Sahu, S. K. Gupta and D. D. Agarwal, Catal. Sci. Technol., 2013, 3, 1520 RSC; (f) P. K. Sahu, P. K. Sahu and D. D. Agarwal, RSC Adv., 2014, 4, 40414 RSC; (g) P. K. Sahu, P. K. Sahu, Y. Sharma and D. D. Agarwal, J. Heterocycl. Chem., 2014, 51, 1193 CrossRef CAS; (h) P. K. Sahu, P. K. Sahu and D. D. Agarwal, J. Indian Chem. Soc., 2015, 92, 169 CAS.
- S. J. Palmer, T. Nauyen and R. L. Frost, Coord. Chem. Rev., 2009, 253, 250 CrossRef CAS.
- S. J. Palmer and R. L. Frost, Ind. Eng. Chem. Res., 2011, 50, 5346 CrossRef CAS.
- M. Valcheva-Traykova, N. Davidova and A. Weiss, J. Mater. Sci., 1993, 28, 2157 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- F. M. Labojos, V. Rives and M. A. Ulibarri, J. Mater. Sci., 1992, 27, 1546 CrossRef.
- A. S. Bookin and A. Dritis, Clays Clay Miner., 1993, 41, 551 CAS.
- W. T. Reichle, CHEMTECH, 1986, 16, 58 CAS.
- K. R. Kottapalli, G. Monique, S. V. Jaime and F. Francois, J. Catal., 1998, 173, 115 CrossRef.
- (a) Y. Liu, E. Lotero, J. J. Goodwin and X. Mo, Appl. Catal., A, 2007, 331, 138 CrossRef CAS; (b) P. Chauyplod and W. Trakarnpruk, Ind. Eng. Chem. Res., 2009, 48, 4177 CrossRef.
- (a) D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183 CrossRef CAS; (b) J. M. Fraile, N. Garcia, J. A. Mayoral, E. Pires and L. Rodan, Appl. Catal., A, 2009, 364, 87 CrossRef CAS; (c) H. J. Hattori, J. Jpn. Pet. Inst., 2004, 47, 67 CrossRef CAS.
- J. J. Di Cosimo, V. K. Di'ez, M. Xu, E. Iglesia and C. R. Apesteguia, J. Catal., 1998, 178, 499 CrossRef CAS.
- (a) T. Nakatsuka, H. Kawasaki, S. Yamashita and S. Kohjiya, Bull. Chem. Soc. Jpn., 1979, 52, 2449 CrossRef CAS; (b) C. T. Fishel and R. Davis, Langmuir, 1994, 10, 159 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |