DOI:
10.1039/C6RA25318K
(Paper)
RSC Adv., 2017,
7, 2186-2192
Composite fibers prepared from multi-walled carbon nanotubes/cellulose dispersed/dissolved in ammonium/dimethyl sulfoxide mixed solvent†
Received
16th October 2016
, Accepted 14th December 2016
First published on 12th January 2017
Abstract
A co-dispersion of cellulose and multi-walled carbon nanotubes (MWCNTs) was prepared in a tetrabutylammonium acetate/dimethyl sulfoxide solvent mixture, to yield hydroxide radical functionalized-MWCNTs. The rheological properties of the mixture were investigated, and its morphology was investigated by optical microscopy. The mixture was then used to prepare composite fibers by a dry-wet spinning process at room temperature. The regenerated composite fibers prepared by coagulation in water were characterized by scanning electron microscopy, X-ray diffraction, Fourier-transform infrared spectroscopy, thermogravimetric analysis, and electronic tensile testing and four-probe testing measurements. The results showed that the mixture was well dispersed, with good interaction between the cellulose and MWCNTs. Introducing an appropriate amount of MWCNTs could enhance the crystallinity, thermal stability, mechanical properties, and electrical conductivity of the composite fibers.
Introduction
Cellulose is the most abundantly created and widely degraded polymer on earth, and harbors half of all organic carbon.1 Cellulose consists of glucose units and has a long thread-like structure. The molecular structure of cellulose imparts important characteristics, such as excellent thermal and mechanical properties, biocompatibility, biodegradability, and cost-effectiveness. Glucose units in cellulose are connected by acetal functional groups, but this functionality is largely absent in commercial polymers. The main applications of cellulose are pulp and paper, textiles, and other traditional uses. Increasing concern about environmental and energy issues has led to increased attention on expanding the applications of cellulose to more advanced fields. Much research is required to better synthetically use this natural resource.
Many studies have shown that introducing a small amount of nanomaterials into polymers can change the materials' performance, and impart new functionality. This has been demonstrated using carbon-based materials such as carbon nanotubes (CNTs)2,3 and graphene,4,5 metallic nanomaterials such as Fe3O4 nanoparticles6 and silver nanoparticles,7 and inorganic nanomaterials such as SiO2 nanoparticles8 and CaCO3 nanoparticles.9,10 CNTs have been widely used as one-dimensional reinforcing nanomaterials for cellulose-based composite films and fibers.11,12 CNTs exhibit an exceptional carrier mobility of 1000–4000 cm2 V−1, tensile strength of 50–200 GPa, Young's modulus of 0.27–1.25 TPa, electrical conductivity of 104 to 105 S cm−1, thermal conductivity of 3000–6600 W mK−1, and thermal stability of up to 700 °C in air.
In 2007, Zhang et al. reported multi-walled carbon nanotube (MWCNT)-reinforced regenerated cellulose fibers, which were prepared from an ionic acid by dry-jet wet-spinning.2 The resulting fibers exhibited excellent mechanical properties and thermal stability, because of the strong interaction between the cellulose and MWCNT. The fibers containing 4 wt% MWCNTs exhibited a high electrical conductivity of 8.3 × 10−3 S cm−1. Fibers containing >4 wt% MWCNTs did not exhibit any further increase in electrical conductivity, because of alignment difficulties and severe aggregation of the MWCNTs. Lu et al. prepared cellulose/MWCNT fibers under varying draw ratios by dry-wet spinning, and investigated their structures and properties.13 A higher MWCNT content and lower draw ratio improved the electrical conductivity of the resulting fibers. Fibers containing 5 wt% MWCNTs exhibited a volume conductivity of 8.8 × 10−4 S cm−1.
The maximum theoretical performance has not been close to approached for CNT/cellulose composites, in contrast with other nanocomposites. The explanation has always been because of the non-uniform dispersion of CNTs in the matrix, and weak interaction between the cellulose and CNTs. CNT/polymer composites can generally be synthesized by mixing CNTs and polymers in solutions or dispersing CNTs in polymer melts, depending on whether the matrix polymer is soluble or meltable. The melting temperature of cellulose is lower than its decomposition temperature, so cellulose composites can only be processed by the solution method. In this process, CNTs are dispersed in a cellulose solvent, so the solvent plays a key role in preparing the cellulose/CNT composites.
We recently reported a tetrabutylammonium acetate (TBAA)/dimethyl sulfoxide (DMSO) mixture as a cellulose solvent.14 This solvent has advantages over traditional solvents, such as its higher efficiency and lower viscosity. Quaternary ammonium compounds such as TBAA are often used as cationic surfactants and phase transfer catalysts. The presence of TBAA allows CNTs to be well dispersed in this solvent mixture. Han et al. recently prepared dispersant-free conducting pastes based on organic solvents with MWCNTs and graphene nanoplatelets. The MWCNTs were chemically modified to have quadruple hydrogen bonding motifs.15 The MWCNTs containing quadruple hydrogen bonding sites were used as a reinforcing nanomaterial to prepare poly(vinyl alcohol)-based composite fibers.
In the current study, MWCNTs and cellulose were dispersed/dissolved in a TBAA/DMSO solvent mixture. This mixture was used to prepare composite fibers by a dry-wet spinning process at room temperature. The composite fibers were characterized by scanning electron microscopy (SEM), Fourier-transform infrared (FT-IR) spectroscopy, X-ray diffraction (XRD), and thermogravimetric analysis (TGA). The mechanical and electrical properties of the composite fibers are also discussed.
Experimental
Materials
Cellulose powder (W100, α-cellulose content of ≥95%) was supplied from Nippon Paper Industries Co., Ltd., Tokyo, Japan. MWCNTs with an average diameter of 9.5 nm and length and 1.5 μm, and hydroxide radical-functionalized MWCNTs (MWCNTs–hydroxyl) with an average diameter of 9.5 nm, length of 1.5 μm, and carboxylic acid content of >8%, were supplied by Shanghai Aladdin Bio-Chem Technology Co., Shanghai, P. R. China. TBAA was purchased from Tokyo Chemical Industry Co., Ltd., Shanghai, P. R. China. Analytically pure DMSO was purchased from Beijing Chemical Works, Beijing, P. R. China. All chemicals were used as received without further purification.
Preparation of dispersed/dissolved cellulose/MWCNT and cellulose/MWCNTs–hydroxyl solutions
Transparent cellulose solutions were prepared by dissolving cellulose powder (12.0 wt%) in a TBAA/DMSO solvent mixture (1/4 ratio by weight), and then stirring at 60 °C for 8 h. Predetermined amounts of MWCNTs (5–15 wt%) and MWCNTs–hydroxyl (5–15 wt%) were then added to TBAA/DMSO solvent mixtures (1/4 ratio by weight), and stirred in a bath-type ultrasonicator (200 W) to obtain dispersed liquids. Each of these dispersions was then mixed with the above-prepared cellulose solution, and stirred for 30 min at 50 °C. The resulting solutions were stirred for 4 h at room temperature, to obtain the final cellulose/MWCNTs and cellulose/MWCNTs–hydroxyl solutions.
Preparation of composite fibers
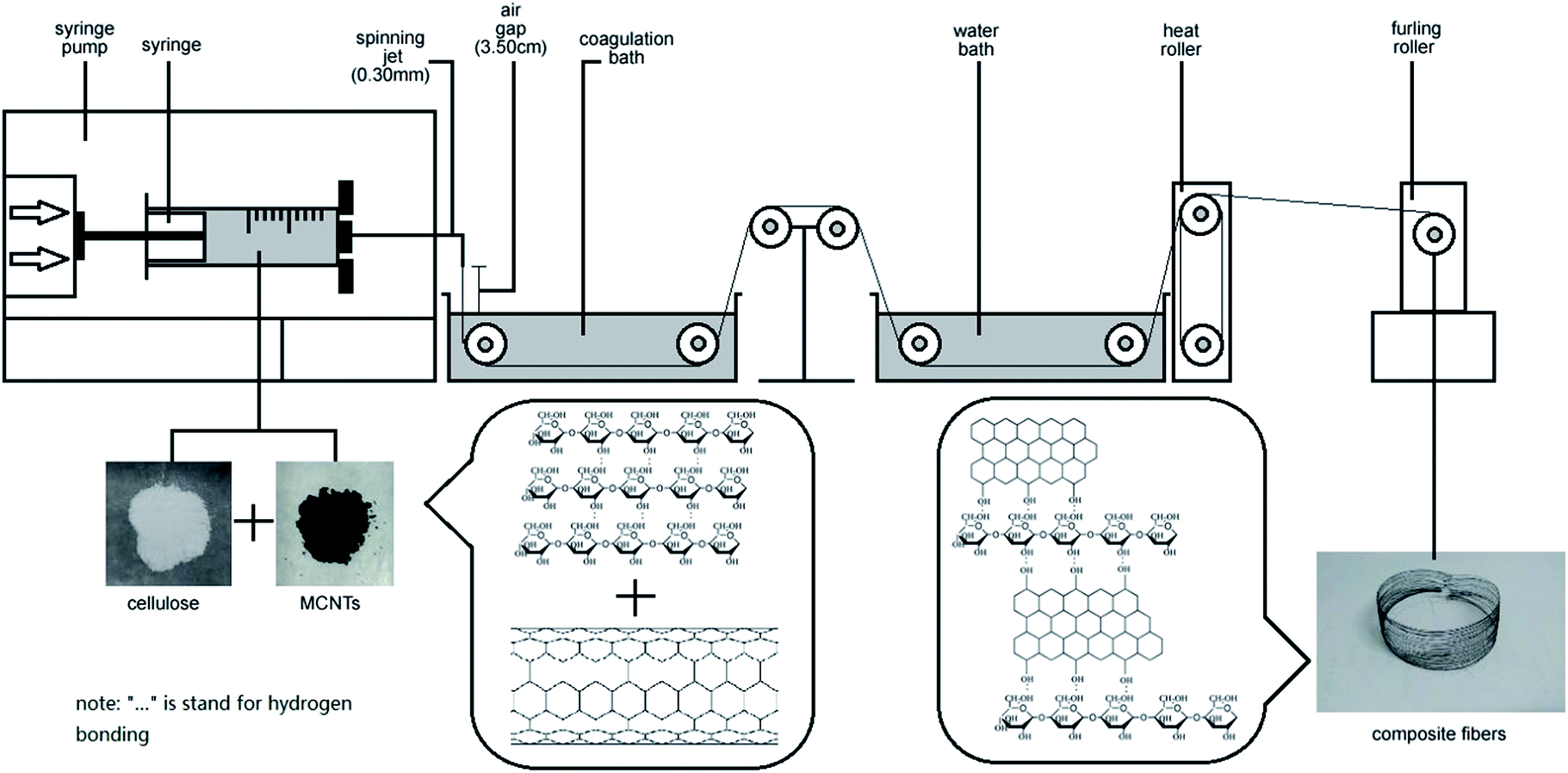
Regenerated cellulose/MWCNTs–hydroxyl fibers were prepared by dry-wet spinning. Specifically, the above-prepared cellulose/MWCNTs–hydroxyl solution was spun into a water bath at room temperature, through a spinneret needle with an inner diameter of 0.24 mm. The air gap was 3.5 cm, and the spinning speed was 0.3 mL min−1. The first take-up speed was controlled at 5.6 m min−1. After dry-wet spinning, the composite fibers were then solidified, washed, and dried with heat rollers at 120 °C. Finally, a furling roller was used as the take-up device. The MWCNT contents of the fibers were controlled at 2.5, 5.0, and 7.50 wt%. The MWCNT contents of the fibers were controlled at 2.5, 5.0, and 7.50 wt%. The final composite fibers were denoted xCC and xCCH, where x indicates the MWCNTs and MWCNTs–hydroxyl content in wt%, respectively. For comparison, a neat regenerated cellulose fiber (RC) was also prepared under the same conditions, but with the absence of MWCNTs or MWCNTs–hydroxyl.
Characterization
The observation of cellulose/MWCNTs–hydroxyl solutions was performed on a microscope with a digital camera. The sample was placed between a microscope glass and a cover slip. The rheological properties of the mixtures were determined from viscometry and oscillatory measurements, which were obtained using a rheometer (Bohlin CVO-100-901, Malvern, UK) equipped with a PP20 plate measuring system. To observe the surfaces and cross-sections of the composite fibers, fibers were frozen in liquid nitrogen, immediately snapped, and then vacuum-dried. Samples were sputter-coated with gold using a sputter coater (SBC-12, KYKY, P. R. China) prior to observation. SEM images were collected using a scanning electron microscope (S-3400N, Hitachi, Japan). XRD patterns were recorded on a XRD diffractometer (Shimadzu XRD-6000, Japan). Samples were vacuum-dried for 24 h before measurement, and then ground into fine powders to negate the influence of crystallographic orientation. XRD patterns were collected using Cu Kα radiation (λ = 0.154 nm), at an operating voltage and current of 40 kV and 30 mA, respectively. Powder samples were scanned from 5 to 40° 2θ, at a scanning rate of 2° min−1. Crystallinities were calculated using the MDI jade 5.0 software package (Materials Data Incorporated, USA). FT-IR spectra were recorded using an FT-IR spectrometer (VERTEX 70/70v FT-IR Spectrometer, Germany). Samples were ground into powders, and were then vacuum-dried for 24 h. Spectra were then collected from samples prepared as KBr disks. The thermostability of samples was studied by TGA (STA 449 F3 Jupiter, Netzsch, Germany) in the range of 50 to 400 °C at a rate of 10 °C min−1 under a nitrogen flow. The tensile strength and mechanical elongation of the fiber samples were measured at room temperature using an electronic tensile tester (Instron 5848, Germany), with an extension rate of 1 cm min−1. The sample length was 20 mm. Each specimen was tested 30 times, and the average value is reported. The electrical conductivities of the fibers were measured at room temperature, using a multifunction digital four-probe tester (ST-2258C Jingge, Suzhou, P. R. China).
Results and discussion
Characterization of cellulose/MWCNTs–hydroxyl solutions
Optical images of cellulose/MWCNTs–hydroxyl solutions. Digital photos of the cellulose/MWCNTs or MWCNTs–hydroxyl mixed liquor are shown in Fig. 2. Fig. 2a and b show that using DMSO/TBAA as a dispersion medium and solvent resulted in MWCNTs–hydroxyl being well dispersed in the cellulose solution. The diameters of the MWCNTs–hydroxyl fibers were approximately 1–5 μm. Increasing the MWCNTs–hydroxyl content to 7.5 wt% resulted in agglomeration of the MWCNTs–hydroxyl, as shown in Fig. 2c. Fig. 2d shows that MWCNTs were dispersed in cellulose solution, but that the diameters of the MWCNT were larger than those of the MWCNTs–hydroxyl. The diameters of the MWCNTs were approximately 15–20 μm. Increasing the MWCNTs content to 5.0 and 7.5 wt% resulted in serious agglomeration, as shown in Fig. 2e and f, respectively. Coagulations with diameters of approximately 30 μm were observed in the mixed liquor. CNTs tend to strongly aggregate, because of their van der Waals' forces. It was therefore important to prevent the dispersed CNTs from self-agglomerating, before preparing the cellulose fibers. This result showed that the presence of TBAA allowed the CNTs to be dispersed in the TBAA/DMSO mixed solvent. MWCNTs–hydroxyl were more readily dispersed in this mixed solvent than MWCNT, probably because the hydroxyl groups improved the interaction between the solvent and MWCNTs–hydroxyl. Surface-modified MWCNTs are reportedly more compatible with cellulose than unmodified MWCNTs.16 Thus, MWCNTs–hydroxyl were used to prepare composite fibers in the subsequent experiments (Fig. 1).
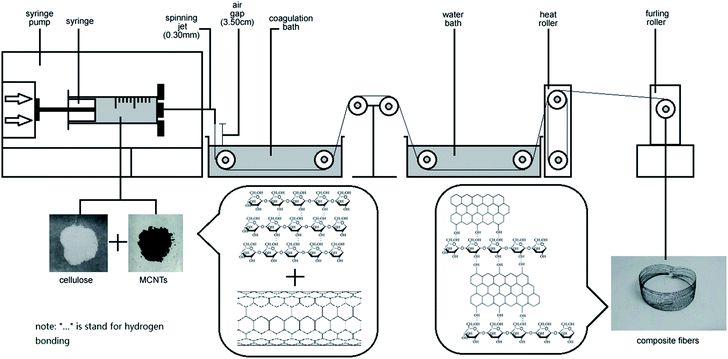 |
| | Fig. 1 Schematic of the preparation of MWCNT–hydroxyl/cellulose composite fibers via a dry-jet spinning process. | |
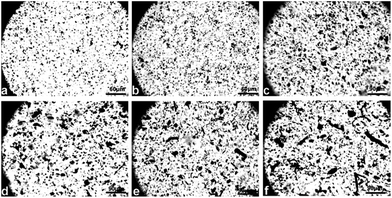 |
| | Fig. 2 Optical images of cellulose/MWCNTs or MWCNTs–hydroxyl samples in ammonium/DMSO mixed solvent: (a) 2.5CC; (b) 5.0CC; (c) 7.5CC; (d) 2.5CCH; (e) 5.0CCH; (f) 7.5CCH. | |
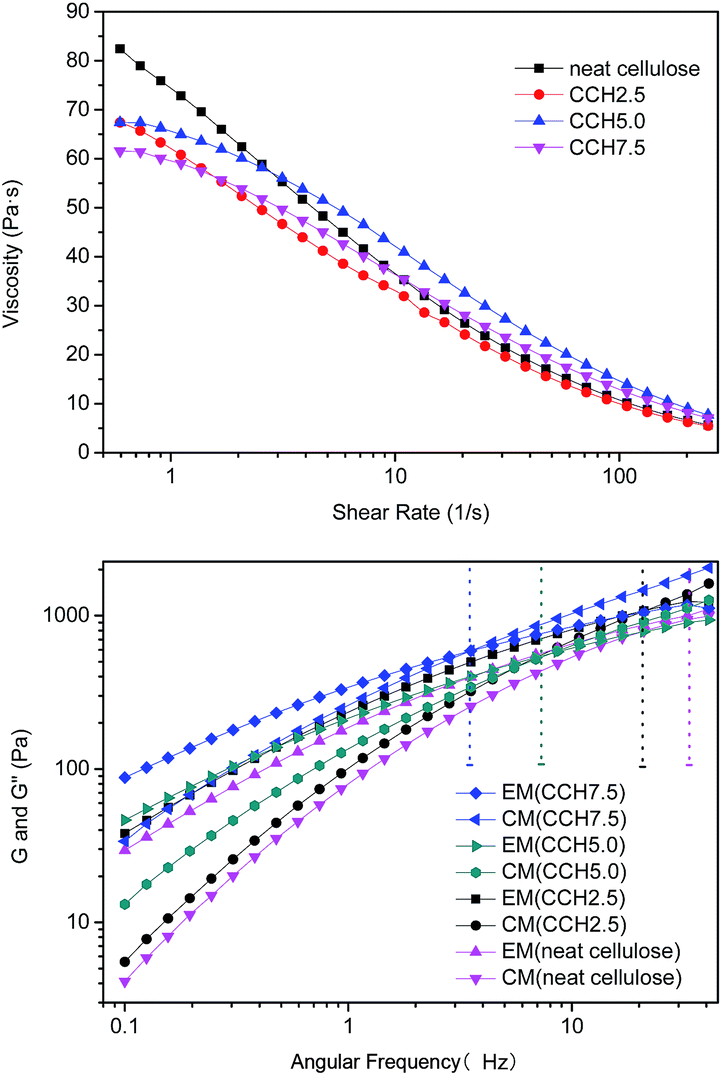
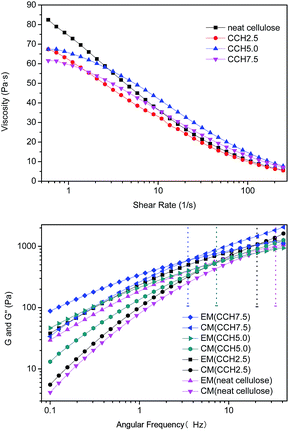
Rheological properties of cellulose/MWCNTs–hydroxyl solutions. The properties of composite fibers are generally considered to be closely related to the viscosities of their solutions. The viscosity properties of the current composite fibers are shown in Fig. 3, in which all mixtures exhibited typical shear thinning behavior with varying shear rate. The shear thinning behavior of the solutions resulted from the shear-induced reduction in the number of entanglements, and macromolecules dissolving under the high shear rate. The high concentration also led to a critical shear rate of non-Newtonian flow transition, and moved it toward lower shear rates. This result is similar to reports of neutral polymer solutions and studies of cellulose in ionic liquids.17 Fig. 3 also shows that the zero-shear viscosity decreased with increasing MWCNTs–hydroxyl concentration, which may have been caused by the existence of particles consist of agglomerating MWCNTs–hydroxyl.
 |
| | Fig. 3 Rheological properties of ammonium/DMSO mixtures containing different MWCNTs–hydroxyl contents, measured at 25 °C. | |
Rheology is a powerful tool for evaluating the structures of polymer solutions or suspensions.18 The dynamic viscoelastic properties of mixed liquors containing 2.5, 5.0, and 7.5 wt% MWCNTs were investigated at 50 °C, to characterize the degree of MWCNTs–hydroxyl dispersion in the cellulose solution. The storage modulus (G′) and loss modulus (G′′) of the cellulose solution and mixed dispersions are shown in Fig. 3b. G′ and G′′ tended to increase with increasing MWCNTs–hydroxyl concentration from 0 to 7.5%. G′ increased with frequency more sharply than G′′. This meant that all samples exhibited a crossover point, where G′(w) = G′′(w), so the curves could be divided into two parts. For neat cellulose, G′′ was larger than G′ at frequencies of <40 Hz, which is typical of polymer melts and solutions. Incorporating MWCNTs–hydroxyl caused the crossover point to move toward lower frequencies. Mixed dispersions with MWCNTs–hydroxyl concentrations of >5 wt% exhibited typical strong gel-like behavior. Similar observations have been reported for aqueous single-wall carbon nanotubes (SWCNTs) suspensions, where the SWCNTs concentration was below the critical “entangled” concentration.14 In a polymer solution or melt, liquidity is dominant when G′′ > G′, while the opposite is true for semisolids such as gels.19 Thus, the mixed dispersions behaved like viscous liquids at low deformation rate, where G′′ was larger than G′. Elastic properties developed at higher angular velocities. A higher MWCNTs–hydroxyl content resulted in an increase in the segment density and number of entanglement points. This could be explained by the chains in dispersions having room to counteract the externally imposed deformation at low MWCNTs–hydroxyl concentration. The deformation occurred slowly in the cellulose solution, and the majority of the energy was dissipated by the viscous flow. These results show that entanglement of the network increased with increasing MWCNTs–hydroxyl concentration, which was attributed to an increase in the segment density of the macromolecules. The MWCNTs–hydroxyl could be well dispersed in the TBAA/DMSO mixed solvent, similar to the situation for ammonium ion-based aqueous solution.20
Morphology and interactions
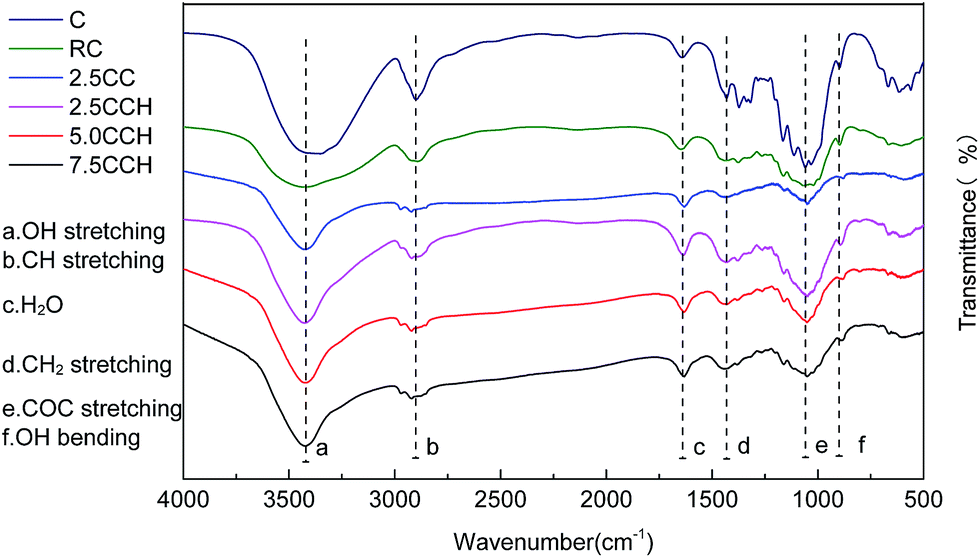
Interactions between cellulose and MWCNTs. FT-IR spectra of neat cellulose, RC fibers, and the composite fibers were recorded to investigate the interaction between the MWCNTs–hydroxyl and cellulose, as shown in Fig. 4. The spectrum of RC fibers contained peaks at around 3379, 2903, 1641, 1038, 1434 and 899 cm−1, which were assigned to the O–H stretching, C–H stretching, water in the amorphous region, C–O–C stretching, C–H2 bending and glycosidic C–H deformation/O–H bending vibrations, respectively.23 The recorded spectra exhibited similar profiles. The absence of new peaks in the spectra of the composite fibers indicated no chemical bonding between the cellulose and MWCNTs–hydroxyl. The O–H stretching bands shifted with increasing MWCNTs–hydroxyl content, from 3443 cm−1 for the RC fibers to 3423 cm−1 for the composite fibers containing 5.0 and 7.5 wt% MWCNTs–hydroxyl. The peak of the O–H stretching vibration broadened with increasing MWCNTs–hydroxyl content. This may have been due to the interaction between the –OH groups of the MWCNTs–hydroxyl and cellulose, such as the development of complex hydrogen bonding. The spectrum of cellulose contained weak peaks at 1641 and 891 cm−1, which are characteristic of crystalline and amorphous regions, respectively.21
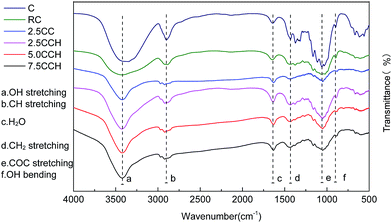 |
| | Fig. 4 FT-IR spectra of neat cellulose fiber and its composite fibers containing different MWCNTs–hydroxyl contents. | |
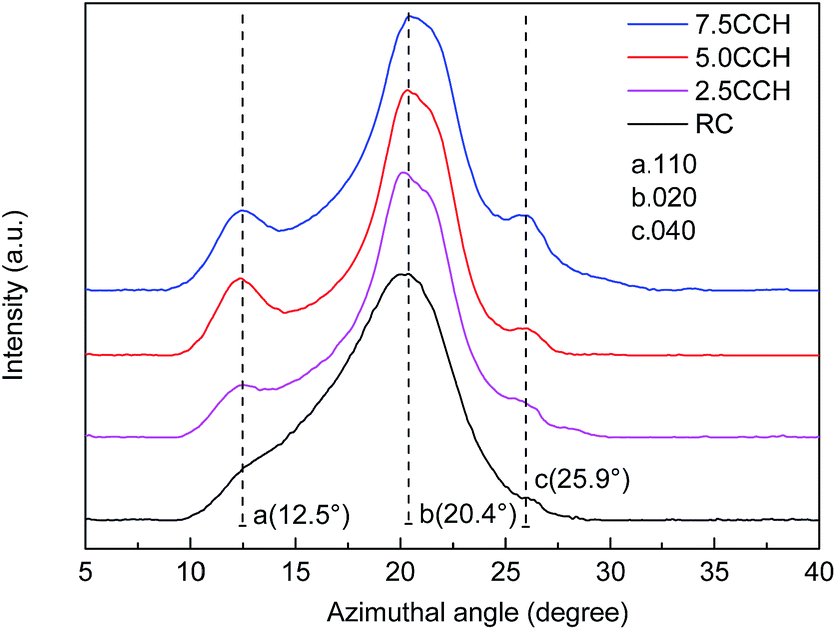
XRD patterns of cellulose fibers and cellulose/MWCNTs–hydroxyl fibers are shown in Fig. 5. The XRD pattern of the RC fibers contained two obvious peaks at 12.5° and 20.4° 2θ, as shown in Fig. 5. These could be indexed to the (110) and (020) planes of the known cellulose II crystal structure, respectively.22 The XRD patterns of the composite fibers 2.5CCH, 5.0CCH, and 7.5CCH contained an extra peak at 25.9° 2θ, which was assigned to the MWCNTs–hydroxyl. Its intensity increased with increasing MWCNTs–hydroxyl content. The degree of crystallinity was quantitatively calculated using the Jade 5.0 software package. A degree of crystallinity of 32.1% was obtained for neat cellulose fibers, and degrees of crystallinities of 41.5, 48.1, and 46.0% were obtained for 2.5CCH, 5.0CCH, and 7.5CCH, respectively. This indicated that the degree of crystallinity of the composite increased upon incorporating the MWCNTs–hydroxyl. This may have been due to the MWCNTs–hydroxyl possessing hydroxide groups. Cellulose is a hydrophilic biopolymer that possesses three hydroxyl groups in each hydroglucose unit. It was expected that some interactions such as hydrogen bonding would occur between the cellulose and MWCNTs–hydroxyl.23 The compatibility and strong interaction between the MWCNTs–hydroxyl and cellulose enhanced the interfacial adhesion, and significantly increased the degree of crystallinity of the composite fibers. The dry-wet spinning process could also increase the crystallographic orientation of the composite fibers.
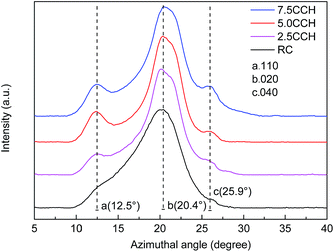 |
| | Fig. 5 XRD patterns of neat cellulose fiber and its composite fibers containing different MWCNTs–hydroxyl contents. | |
Morphologies of the composite fibers. SEM images of sample cross-sections and surfaces were used to characterize the morphologies of the composite fibers, and the dispersion of the MWCNTs–hydroxyl. SEM images of composite fibers with MWCNTs–hydroxyl contents of 2.5, 5.0, and 7.5 wt% are shown in Fig. 6 and 7. The SEM image of 2.5CCH showed a smooth surface and dense section without micropores. This indicated that the MWCNTs–hydroxyl were well dispersed in the cellulose matrix, and that introducing the MWCNTs–hydroxyl had no obvious effect on the fiber morphology. 5.0CCH had a rougher surface than 2.5CCH, but its cross-sectional SEM image still showed a dense structure, indicating good adhesion between the MWCNTs–hydroxyl and cellulose. 7.5CCH had an even rougher surface, with sunken regions observed at higher MWCNTs–hydroxyl content. The cross-sectional SEM image of 7.5CCH showed severe aggregation of the MWCNTs–hydroxyl, indicating poor interfacial adhesion between the MWCNTs–hydroxyl and cellulose. This result showed that an appropriate amount of MWCNTs–hydroxyl could be well dispersed in the TBAA/DMSO mixed solvent, and that homogeneous composite fibers could be obtained. Excess MWCNTs–hydroxyl caused agglomeration and decreased adhesion between the MWCNTs–hydroxyl and cellulose.
 |
| | Fig. 6 SEM images of composite fibers containing different MWCNTs–hydroxyl contents: (a)–(c) 2.5CCH; (d)–(f) 5.0CCH; (g)–(i) 7.5CCH. | |
 |
| | Fig. 7 Cross-sectional SEM images of composite fibers containing different MWCNTs–hydroxyl contents: (a)–(c) 2.5CCH; (d)–(f) 5.0CCH; (g)–(i) 7.5CCH. | |
Mechanical properties
Tensile strength, elongation at break, and Young modulus of the composite fibers. The mechanical properties of the composite fibers with different MWCNTs–hydroxyl contents were measured at 25 °C. The tensile strength, elongation at break, and Young modulus are summarized in Table 1. Neat cellulose fibers had a tensile strength of 2.12 cN dtex−1, elongation at break of 9.3–11.1%, and Young modulus of 0.801 GPa. 2.5CCH and 5.0CCH exhibited increasing tensile strengths with increasing MWCNTs–hydroxyl contents. An initial modulus except for the strain to break was attained, in contrast to the situation for neat cellulose fibers. This was a result of the reinforcing effect of the MWCNTs–hydroxyl. Compared with RC fibers, fibers containing 5.0 wt% MWCNTs–hydroxyl exhibited a relatively high tensile strength and Young modulus. This may have been due to hydroxyl groups in the MWCNTs–hydroxyl. Interactions such as hydrogen bonding between cellulose and the MWCNTs–hydroxyl could have enhanced the mechanical properties. Composite fibers containing 7.5 wt% MWCNTs–hydroxyl exhibited a slightly lower tensile strength and strain to break. This was attributed to the agglomeration of MWCNTs–hydroxyl influencing the structure of the composite fibers.
Table 1 Mechanical properties of cellulose fiber and composite fibers containing different MWCNTs–hydroxyl contents
| MWCNT concentration (wt%) |
Tensile strength (cN dtex−1) |
Elongation at break (%) |
Young modulus (Mpa) |
| 0 |
2.12–2.31 |
9.3–11.1 |
801 |
| 2.5 |
2.42–2.53 |
10.6–13.0 |
2105 |
| 5.0 |
2.68–2.74 |
11.5–13.6 |
3105 |
| 7.5 |
2.55–2.64 |
8.9–10.2 |
2646 |
Thermal properties
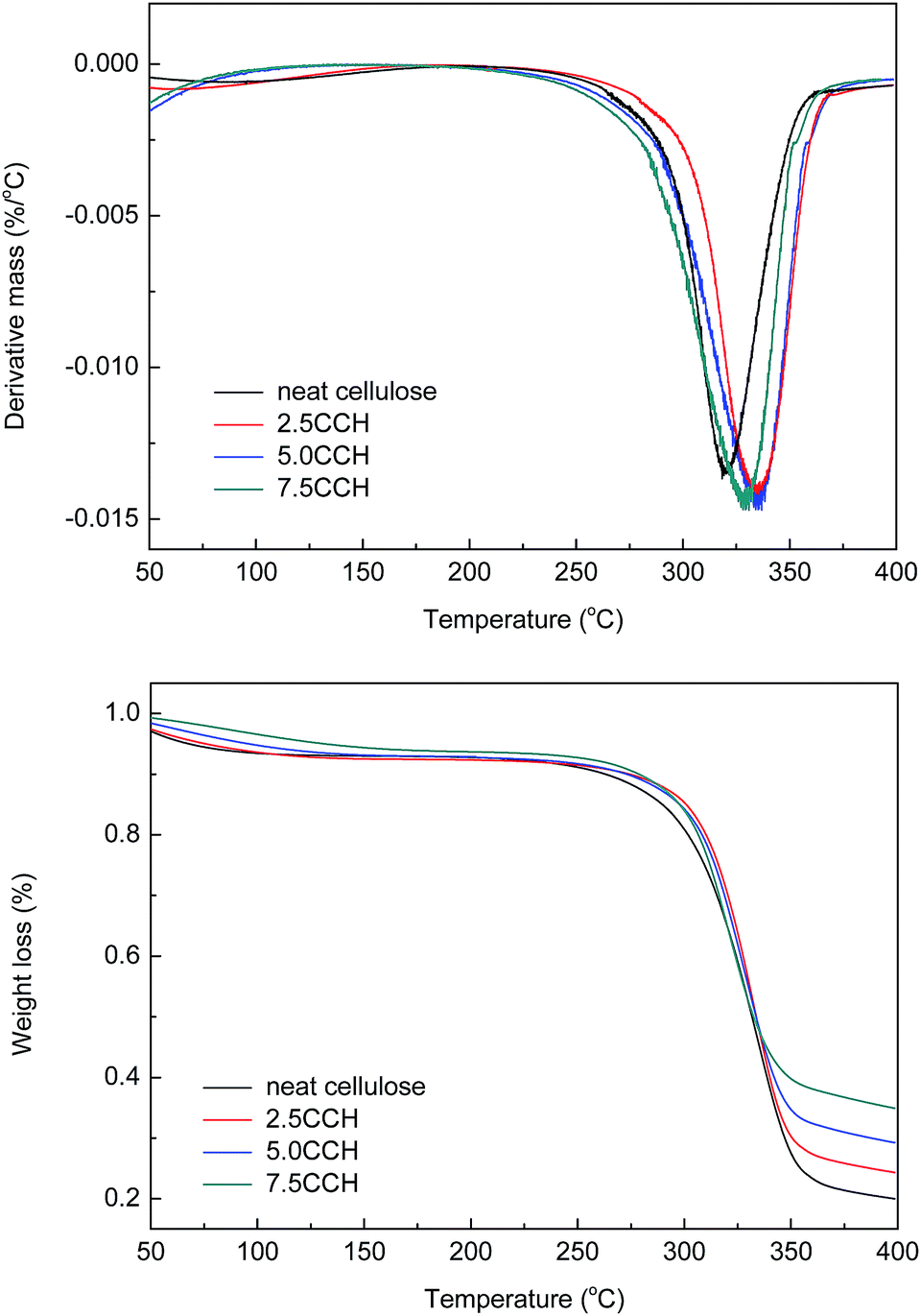
Differential thermogravimetric (DTG) and TGA curves of the composite fibers are shown in Fig. 8. The decomposition of neat cellulose, 2.5CCH, 5.0CCH, and 7.5CCH began at 253, 282, 283, and 272 °C, respectively, and a rapid weight loss occurred at 265–360, 262–367, 276–368, and 251–361 °C respectively. Thus, the initial decomposition temperature of cellulose increasing with increasing MWCNTs–hydroxyl content. The DTG curves of the composite fibers showed a characteristic change in thermal decomposition with increasing MWCNTs–hydroxyl content. The maximum decomposition temperature of neat cellulose, 2.5 CCH, 5.0 CCH, and 7.5 CCH was 319, 335, 336, and 319 °C, respectively. This demonstrated that incorporating MWCNTs–hydroxyl into the cellulose matrix could enhance the thermal stability of the resulting composite. The crystallite size, crystallinity index, and degree of polymerization can all affect the transfer of heat during the decomposition of cellulose. All samples were prepared from the same crude cellulose material, and thus had comparable degrees of polymerization. The improved thermal stability of cellulose was therefore attributed to the crystallite size and crystallinity index. The temperature of the initial thermal decomposition and peak temperature in the DTA and DTG curves generally increase with increasing crystallite size and degree of crystallinity.24 For composites with larger CNT loadings (≥3%), the intrinsic high thermal conductivity of the CNTs could also have improved the thermal properties of the composite.25 The enhanced thermal properties were therefore attributed to the intrinsic high thermal conductivity of MWCNTs–hydroxyl, and its effect on improving the crystallinity of the composite. The high MWCNTs–hydroxyl loading in the 7.5CCH composite resulted in a slightly lower thermal stability than that of the 5.0CCH composite. Thus, the enhanced thermal properties were predominantly attributed to the MWCNTs improving the crystallinity of the composite, as evidenced by the XRD results.
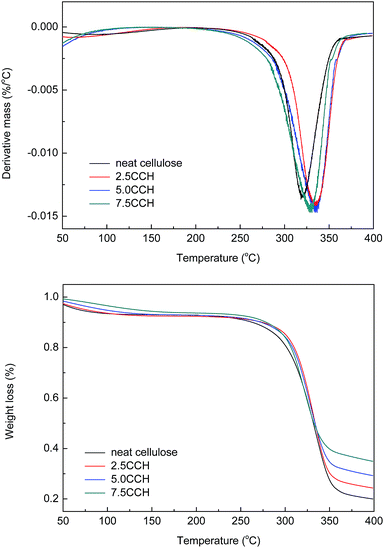 |
| | Fig. 8 DTG and TGA curves of cellulose fiber and composite fibers containing various MWCNTs–hydroxyl contents. | |
Electrical properties
The electrical conductivities of the cellulose/MWCNTs–hydroxyl fibers were measured, and are shown at Table 2. Cellulose fibers containing 7.5 wt% MWCNTs–hydroxyl exhibited the highest electrical conductivity of 6.70 × 10−2 S cm−1. The electrical conductivities of fibers containing >5 wt% MWCNTs–hydroxyl increased more slowly than for fibers containing <5 wt% MWCNTs–hydroxyl. The electrical conductivities of the cellulose/MWCNTs–hydroxyl fibers were measured, and are shown at Table 2. Cellulose fibers containing 7.5 wt% MWCNTs–hydroxyl exhibited the highest electrical conductivity of 6.70 × 10−2 S cm−1. The electrical conductivity of fibers containing >5 wt% MWCNTs–hydroxyl increased more slowly with increasing fiber content, than for fibers containing <5 wt% MWCNTs–hydroxyl. This may have reflected the degree of dispersion of fibers in the composite. Adding a small amount of CNTs (≤0.1%) into the matrix could cause percolation, leading to large changes in the properties of the composites.26,27 Table 2 shows that the conductivity curve date for the 5.0CCH and 2.5CCH composites was similar to data reported for functionalized CNT–polymer composites. Alignment problems and poor dispersion of the MWCNTs–hydroxyl at high concentration (≥5%) resulted in agglomeration of the MWCNTs. Thus, the conductivity increased more slowly with increasing MWCNTs–hydroxyl concentration. Similar phenomena have been reported by Maiti et al. for polycarbonate/MWCNT composites.28
Table 2 Electrical conductivities of composite fibers containing different MWCNTs–hydroxyl contents
| MWCNT concentration (wt%) |
0 |
2.5 |
5.0 |
7.5 |
| Electrical conductivity (S cm−1) |
∼10−9 |
2.25 × 10−4 |
6.21 × 10−2 |
6.70 × 10−2 |
Conclusions
MWCNTs–hydroxyl and cellulose were dispersed/dissolved in TBAA/DMSO mixed solvent. FT-IR spectroscopy and XRD confirmed the presence of MWCNTs–hydroxyl within the composite fibers. SEM images showed that the morphology of the composite fibers become rougher and more uniform, as their MWCNTs–hydroxyl content increased from 2.50 to 7.50 wt%. The presence of MWCNTs–hydroxyl enhanced the crystallinity of the composite fibers, which increased their tensile strength. Fibers containing 5.00 wt% of MWCNTs–hydroxyl had a Young's modulus (3105 MPa) three times higher than that of pure cellulose fiber (801 MPa). The tensile strength of cellulose fiber (2.12–2.31 MPa) increased to 2.68–2.74 MPa, and its elongation at break (9.3–11.1%) increased to 11.5–13.6%, upon incorporating 5.00 wt% of MWCNTs–hydroxyl. DTG and TGA measurements showed that the presence of MWCNTs–hydroxyl increased the thermal stability of the composite fibers. The conductivity of the composite fibers increased significantly upon incorporating MWCNTs–hydroxyl.
Acknowledgements
The authors are grateful for support from the Specialized Research Fund for the Forestry Public Welfare Industry (201504602-2).
Notes and references
- H. P. S. Abdul Khalil, A. H. Bhat and A. F. Ireana Yusra, Carbohydr. Polym., 2012, 87, 963–979 CrossRef CAS.
- H. Zhang, Z. G. Wang, Z. N. Zhang, J. Wu, J. Zhang and J. S. He, Adv. Mater., 2007, 19, 698–704 CrossRef CAS.
- H.-D. Huang, C.-Y. Liu, D. Zhou, X. Jiang, G.-J. Zhong, D.-X. Yan and Z.-M. Li, J. Mater. Chem. A, 2015, 3, 4983–4991 CAS.
- X. Zhang, X. Liu, W. Zheng and J. Zhu, Carbohydr. Polym., 2012, 88, 26–30 CrossRef CAS.
- J. Yang, E. Zhang, X. Li, Y. Zhang, J. Qu and Z.-Z. Yu, Carbon, 2016, 98, 50–57 CrossRef CAS.
- J. Zhou, R. Li, S. Liu, Q. Li, L. Zhang, L. Zhang and J. Guan, J. Appl. Polym. Sci., 2009, 111, 2477–2484 CrossRef CAS.
- M. Yadollahi, H. Namazi and M. Aghazadeh, Int. J. Biol. Macromol., 2015, 79, 269–277 CrossRef CAS PubMed.
- M. He, B. Duan, D. Xu and L. Zhang, Cellulose, 2015, 22, 553–563 CrossRef CAS.
- X.-F. Cheng, H. Qian, S.-W. Zhang, Z.-S. Zhang, Y. He and M.-G. Ma, BioResources, 2016, 11, 4392–4401 CAS.
- M.-G. Ma, Y.-Y. Dong, L.-H. Fu, S.-M. Li and R.-C. Sun, Carbohydr. Polym., 2013, 92, 1669–1676 CrossRef CAS PubMed.
- G. Y. Li, P. M. Wang and X. Zhao, Cem. Concr. Compos., 2007, 29, 377–382 CrossRef CAS.
- W.-T. Hong and N.-H. Tai, Diamond Relat. Mater., 2008, 17, 1577–1581 CrossRef CAS.
- J. Lu, H. Zhang, Y. Jian, H. Shao and X. Hu, J. Appl. Polym. Sci., 2012, 123, 956–961 CrossRef CAS.
- H. Sun, J. Miao, Y. Yu and L. Zhang, Appl. Phys. A: Mater. Sci. Process., 2015, 119, 539–546 CrossRef CAS.
- Y.-L. Liu, W.-H. Chen and Y.-H. Chang, Carbohydr. Polym., 2009, 76, 232–238 CrossRef CAS.
- B. Singh, D. Singh, R. Mathur and T. Dhami, Nanoscale Res. Lett., 2008, 3, 444 CrossRef CAS.
- Y. Lv, J. Wu, J. Zhang, Y. Niu, C.-Y. Liu, J. He and J. Zhang, Polymer, 2012, 53, 2524–2531 CrossRef CAS.
- J. D. Ferry, Viscoelastic properties of polymers, John Wiley & Sons, 1980 Search PubMed.
- C. Olsson and G. Westman, J. Appl. Polym. Sci., 2013, 127, 4542–4548 CrossRef CAS.
- F. Lu, S. Zhang and L. Zheng, J. Mol. Liq., 2012, 173, 42–46 CrossRef CAS.
- U. P. Agarwal, R. R. Reiner and S. A. Ralph, J. Agric. Food Chem., 2013, 61, 103–113 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 1 CrossRef PubMed.
- H. Qi, J. Liu, S. Gao and E. Mäder, J. Mater. Chem. A, 2013, 1, 2161–2168 CAS.
- G. Rodrigues Filho, R. M. de Assunção, J. G. Vieira, C. d. S. Meireles, D. A. Cerqueira, H. da Silva Barud, S. J. Ribeiro and Y. Messaddeq, Polym. Degrad. Stab., 2007, 92, 205–210 CrossRef CAS.
- R. Gulotty, M. Castellino, P. Jagdale, A. Tagliaferro and A. A. Balandin, ACS Nano, 2013, 7, 5114–5121 CrossRef CAS PubMed.
- G. Ambrosetti, C. Grimaldi, I. Balberg, T. Maeder, A. Danani and P. Ryser, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155434 CrossRef.
- M. Castellino, M. Rovere, M. I. Shahzad and A. Tagliaferro, Composites, Part A, 2016, 87, 237–242 CrossRef CAS.
- S. Maiti, N. Shrivastava, S. Suin and B. Khatua, eXPRESS Polym. Lett., 2013, 7(6), 505–518 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25318k |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Danni Chen,
Yongqi Yu,
Jiaojiao Miao,
Yang Liu and
Liping Zhang*
,
Danni Chen,
Yongqi Yu,
Jiaojiao Miao,
Yang Liu and
Liping Zhang*