
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ultraviolet upconversion emission of Pb2+ ions sensitized by Yb3+-trimers in CaF2
Tuerxun Aidilibikeab,
Junjie Guoa,
Lili Wanga,
Xiaohui Liua,
Yangyang Lia and
Weiping Qin *a
*a
aState Key Laboratory on Integrated Optoelectronics, College of Electronic Science and Engineering, Jilin University, Changchun, Jilin 130012, China. E-mail: wpqin@jlu.edu.cn
bYili Normal University, Electronic and Information Engineering, Laboratory on Micro-Nano Electro Biosensor and Bionic Devices, Yining, Xinjiang 835000, China
First published on 12th January 2017
Abstract
Under 978 nm near infrared (NIR) excitation, ultraviolet (UV) upconversion (UC) emissions from Pb2+ ions in CaF2:Pb2+,Yb3+ were first observed at room temperature. The UC emission centered at ∼383 nm was ascribed to the 3P0 → 1A1g (1S0) transition of Pb2+ ions. From transient measurements, the UC process was found to be dominated by the energy transfer process: three excited Yb3+ ions simultaneously transfer their energy to one Pb2+ ion. With the increase in Pb2+ concentration, the cooperative luminescence from 3-Yb3+ clusters decreased gradually until it was extinguished. The cooperative sensitization to one Pb2+ ion comes from three excited Yb3+ ions, which is a four-ion process and exhibits a third power dependence on the NIR pumping power.
Optical frequency UC is a luminescence technology developed since the 1960s. Thanks to the successful fabrications of powerful infrared (IR) diode lasers and heavy metal fluoride glasses, visible (VIS) upconversion luminescence (UCL) including red, green, and blue has been performed since the 1980s.1–7 With the development of manufacturing technology for nanometer materials, UCL labelling has gathered great attention.8–11 The study on UCL was derived from the detection of IR light; however, it has a great number of potential applications in the fields of solid-state lasers, 3D displays, IR quantum counters, optical probes in fluorescent imaging techniques, anti-counterfeiting, NIR photocatalysis, and temperature sensors.6,7,9,10,12–15 Most of UC materials are rare-earth doped solid compounds; in which, a lanthanide ion absorbs more than one photon with lower energy and converts them to one photon with higher energy by using its metastable energy levels. Yb3+ ion has been extensively adopted as a sensitizer in UC processes because it has a long excited state lifetime and a relatively large absorption cross-section in the NIR region.16
No UCL from Pb2+ ions have been observed until now, although the down-conversion (DC) luminescence of Pb2+ ions in the UV and VIS regions has been reported and applied.17–19 As we know, the absorption and emission bands of Pb2+ ions usually appear in the UV region, and their electronic configuration is 6s2 in the ground state (the 1S0 level) and (6s1)(6p1) in the excited states. The 1S0 ground state is expressed by 1A1g and has an even parity. The excited state expressed by (6s1)(6p1) has an odd parity and consists of triplet states (3P0, 3P1, and 3P2) and singlet excited state 1P1. After absorbing a UV photon (∼330 nm), Pb2+ ion make an optical transition from 3P0 to 1A1g.18,20 The energy difference between the ground state and the triplet state of Pb2+ is higher than the energy of two 980 nm photons or two excited Yb3+ ions. Such an energy-level characteristic determines that neither the energy of two 978 nm photons nor the energy of an excited Yb3+-dimer (two excited Yb3+ ions) can excite or sensitize a Pb2+ ion efficiently. Although, in the view of energy, three-photon excitation indeed can lead to the UV UCL of Pb2+ with a very low probability,16 however, until now, there has been no report on the UCL of Pb2+ ions under NIR excitation. Even so, the cooperative energy transfer from three or four excited Yb3+ ions (Yb3+-trimer or Yb3+-tetramer) can excite Pb2+ ion from the view of energy.
Cooperative transition usually means the physical processes in which identical ions or atoms simultaneously make transitions by absorbing (or emitting) one photon with the sum of all transition energies, or transferring their energies to another ion or atom. Cooperative transition includes cooperative luminescence (CL), cooperative absorption (CA), and cooperative energy transfer (CET) and is closely relative to clustered lanthanide ions in solid hosts. Clusters made of lanthanide ions can easily form in many materials, especially in alkaline-earth fluoride crystal AF2 (A = Ca, Sr, Ba). When a lanthanide ion with +3 oxidation state is doped in an alkaline-earth fluoride, non-equilibrium of charge arises in the lattice due to the lanthanide ion usually occupy the position of an alkaline-earth ion with +2 oxidation state. The non-equilibrium of charge produces crystal defects such as the interstitials of F− ions and cation vacancies in order to compensate excess plus charges, which gives non-uniform distribution of lanthanide ions. Different clusters, such as dimers, trimers, and even hexamers, can form in crystal lattices depending on the kind of lanthanide ions and their doping densities.21,22 The first observations of cooperative absorption and cooperative luminescence of Yb3+ dimers were reported in 1961 and 1970,23,24 respectively. Since then, cooperative transitions of Yb3+-dimers have been investigated in many Yb3+-doped materials.25–27 Recently, Qin et al. reported the triplet cooperative luminescence (TCL) at ∼343 nm from three excited Yb3+ ions (Yb3+-trimer) and the cooperative energy transfer from four excited Yb3+ ions (Yb3+-tetramer) to one Gd3+ ion, which open the way to investigate the cooperative transitions of many-body systems.28,29 Furthermore, by considering the electric multipole interactions between ions and using the coupled wave functions of a many-lanthanide-ion system, Qin et al. proposed main cooperative transition mechanisms in a multi-ion system and calculated their probabilities.30
In the present work, we report the first experimental observation of UV UCL peaked at 383 nm from Pb2+ ions sensitized by three excited Yb3+ ions cooperatively.
The XRD patterns of CaF2:1% Yb3+, CaF2:0.5% Pb2+ and CaF2:1% Yb3+,0.5% Pb2+ samples are shown in Fig. 1. The narrow and intense diffraction peaks indicate the well crystallization of the samples. The diffraction peaks of all the samples are consistent with the standard card (PDF #35-0816), suggesting that these materials have a cubic crystal structure.
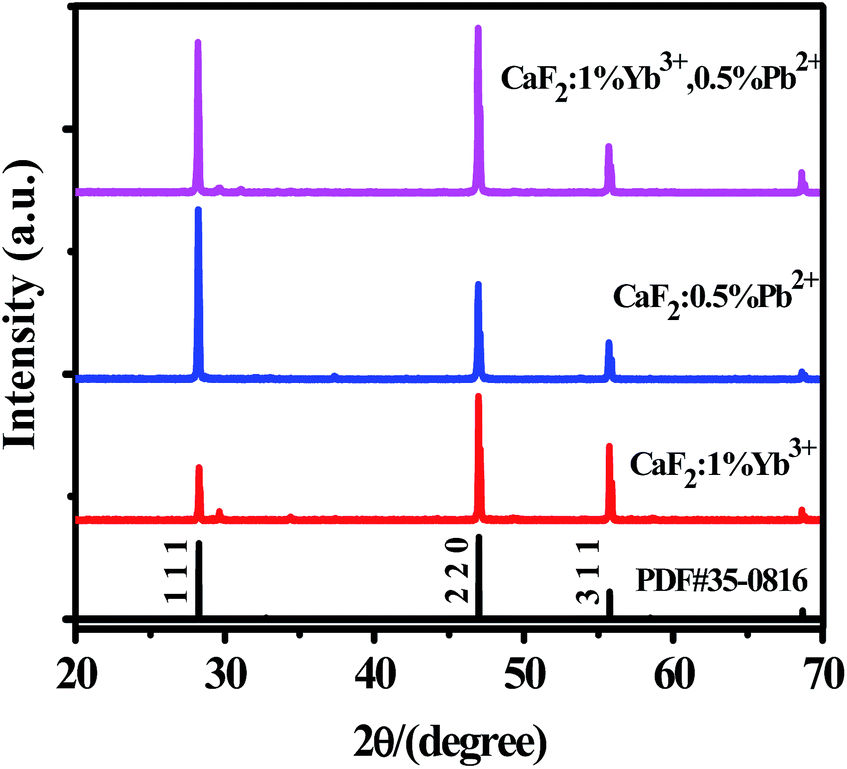 | ||
| Fig. 1 XRD patterns of CaF2:x% Yb3+,y% Pb2+. | ||
We have measured the absorption spectra of CaF2, CaF2:0.5% Pb2+, CaF2:1% Yb3+, and CaF2:1% Yb3+,0.5% Pb2+ powder samples in a wide spectral range from 200 to 1200 nm (Fig. 2). The spectra reveal the existence of Yb3+ ion (in NIR domain). The Yb3+ ions are characterized by a very broad band in the NIR spectral domain, corresponding to the 2F7/2–2F5/2 transition (f–f transition). The ground state 2F7/2 split into three or four components and the excited state 2F5/2 split into two or three sublevels depending on the crystal field symmetry. The strongest absorption band peak appears at 979 nm.31,32 Fig. 2 shows the strong “absorption” in UV region (<400 nm) should be attributed to the Mie scattering. The absorption in NIR region show that the samples with 1% Yb3+ dopant have significant absorption, and the sample of CaF2:0.5% Pb2+ has not. Pb2+ ions cannot absorb NIR light directly therefore cannot influence the NIR absorption spectra of Yb3+ ions.
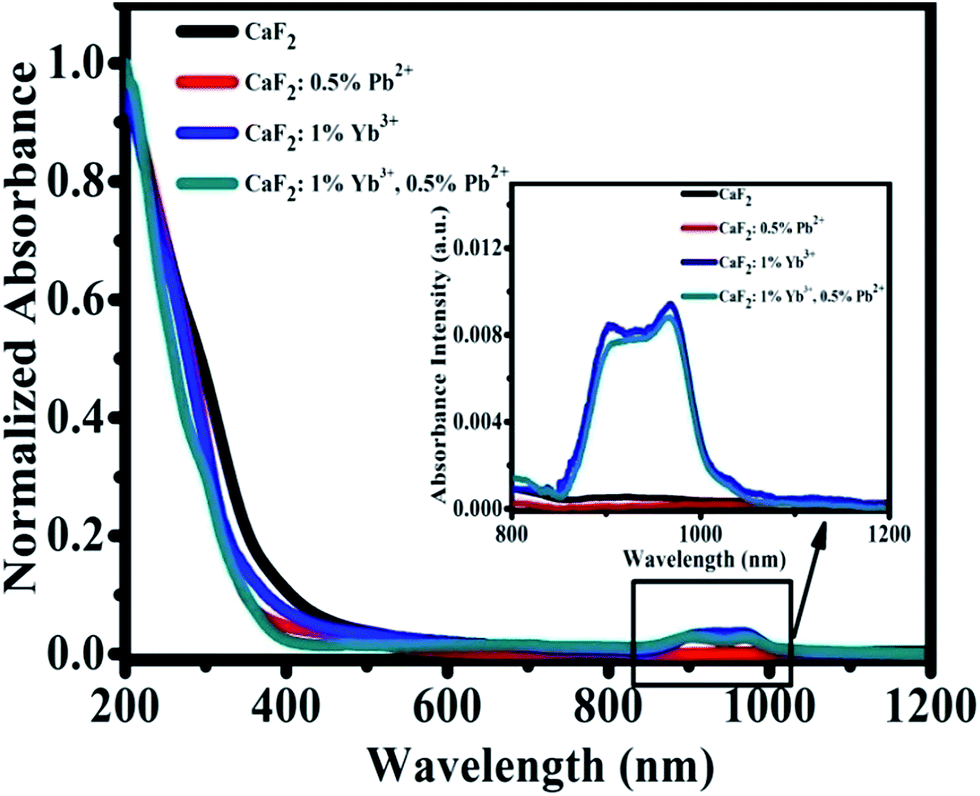 | ||
| Fig. 2 Optical absorption spectra of CaF2, CaF2:0.5% Pb2+, CaF2:1% Yb3+, and CaF2:1% Yb3+,0.5% Pb2+ samples. | ||
Under the excitation of a 978 nm laser, the CaF2:Yb3+ polycrystalline powders showed 343 nm and 500 nm UC emission spectra, which has been previously reported.28 Our previous works has demonstrated that the 343 nm UC fluorescence is the TCL of Yb3+-trimers, as shown in Fig. 3(a). With the increase of doped Yb3+ concentration, the UV emission peaked at 343 nm first increases and then decreases, Fig. 3(b). The maximum UC emission intensity of Yb3+-trimers is obtained at the concentration of Yb3+ reaches 1 mol%. Spectral analysis indicated that the 500 nm UC emission came from Yb3+-dimers in the CaF2:Yb3+ polycrystalline, as shown in Fig. 3(c). With the increase of doped Yb3+ concentration, the maximum UC emission intensity of Yb3+-dimers is obtained at the concentration of Yb3+ reaches 1 mol%, Fig. 3(d).
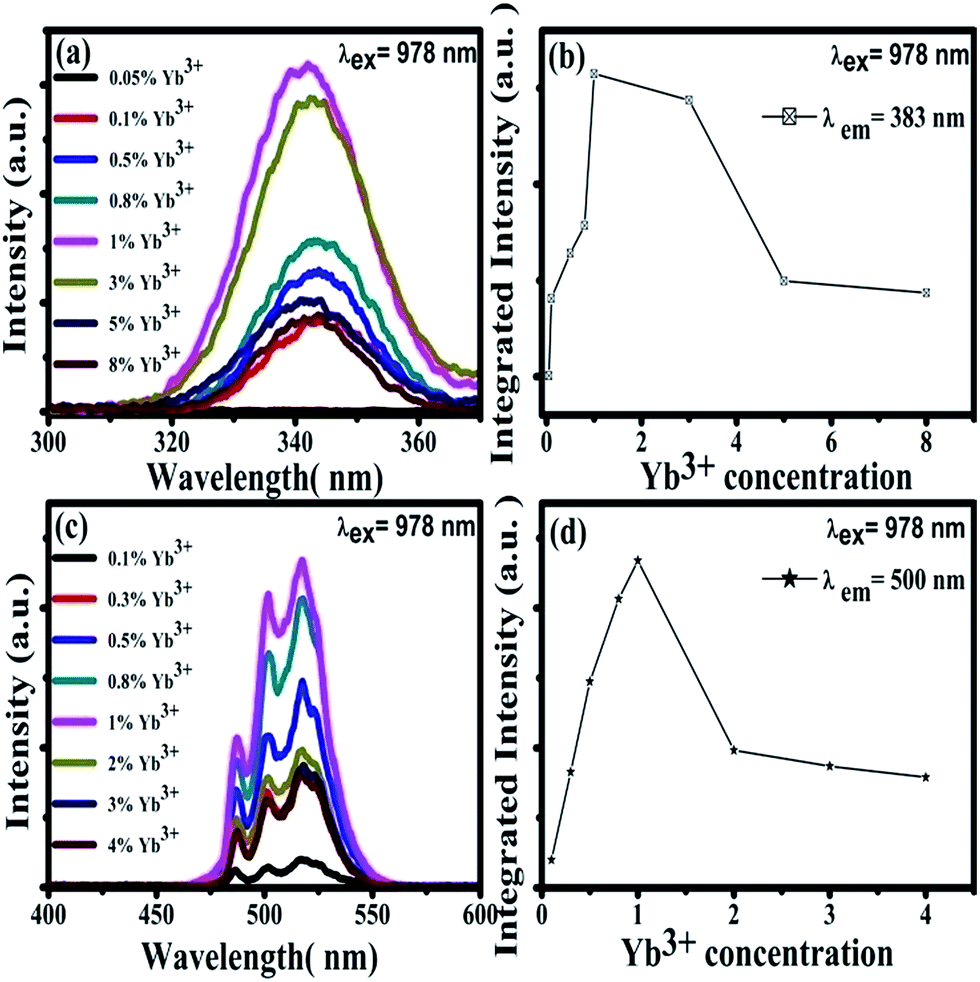 | ||
| Fig. 3 (a) Emission spectra (300–400 nm) of CaF2:x% Yb3+ (x = 0.05, 0.1, 0.5, 0.8, 1, 3, 5, 8) upon 978 nm excitation at room temperature. (b) Integrated intensity dependence of 343 nm UC emissions on the doping concentration of Yb3+. (c) Emission spectra (400–600 nm) of CaF2:x% Yb3+ (x = 0.1, 0.3, 0.5, 0.8, 1, 2, 3, 4) upon 978 nm excitation at room temperature. (d) Integrated intensity dependence of 500 nm UC emissions on the doping concentration of Yb3+. | ||
Under 978 nm excitation, the UV emission spectra of CaF2:x% Yb3+,y% Pb2+ (x = 0, 1; y = 0, 0.3, 0.5, 1, 2, 3, 4) were recorded at room temperature, as shown in Fig. 4(a). No luminescence can be found in the sample of CaF2:0.5% Pb2+ throughout the entire spectral region because the NIR excitation cannot be absorbed by Pb2+ ions.33 On the other hand, an obvious UV fluorescence peaked at 343 nm can be seen in the spectrum of CaF2:1% Yb3+. Our previous works has demonstrated that the 343 nm upconversion fluorescence is the TCL of Yb3+-trimers.28 After doping small amounts of Pb2+ ions (e.g. 0.3%) in CaF2:1% Yb3+, the TCL intensity became lower and a new UV luminescence appeared at 383 nm. Fig. 4(a) show the cooperative upconversion spectra against Pb2+ concentrations. It is quite clear that the TCL (343 nm) peak follow a similar trend, that is, a gradual decrease in the emission intensities until they disappear when Pb2+ concentration reaches 4 mol%. With the increase of doped Pb2+ concentration, the UV emission peaked at 383 nm first increases and then decreases. It reaches the maximum at 0.5 mol% doping concentration of Pb2+ ions and disappears at 4 mol% Pb2+ ions. Therefore, it is reasonable for us to deduce that the emission peaked at 383 nm comes from the excited Pb2+ ions, and further investigation (show later) exhibits that it comes from the 3P0 → 1A1g transition of Pb2+ ions. Now, the problem is, in this system, what induces the Pb2+ ions excited under the NIR irradiation? In ref. 28, we reported the UV UCL from Gd3+ ions upon 978 nm excitation and verified that the excitation of Gd3+ ions came from the cooperative energy transfer of four excited Yb3+ ions (Yb3+-tetramer). In the case of Pb2+ ions, the same mechanism is also existence; namely, the excitation of Pb2+ ions may originate from the cooperative sensitization of Yb3+-trimers or Yb3+-tetramers.
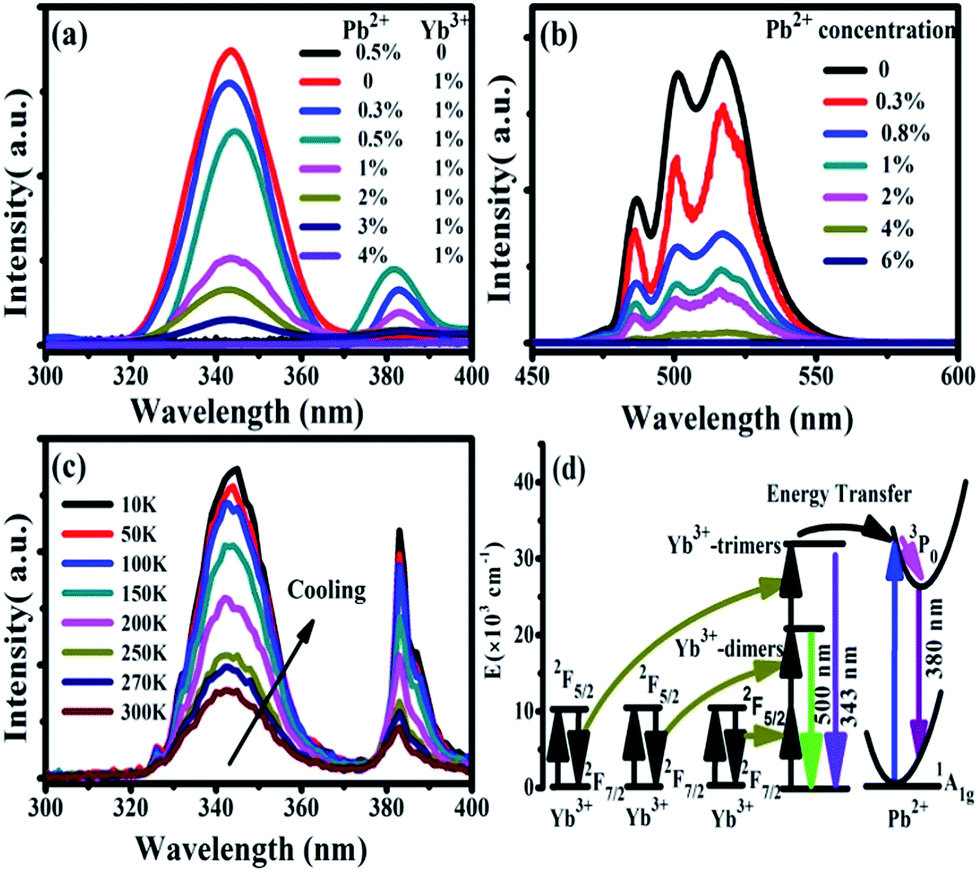 | ||
| Fig. 4 (a) Emission spectra (300–400 nm) of CaF2:x% Yb3+,y% Pb2+ (x = 0, 1; y = 0, 0.3, 0.5, 1, 2, 3, 4) upon 978 nm excitation at room temperature (b) emission spectra (450–600 nm) of CaF2:1% Yb3+,y% Pb2+ (y = 0, 0.3, 0.8, 1, 2, 4, 6) upon 978 nm excitation at room temperature (c) temperature dependent UC emission spectra of CaF2:1% Yb3+,0.5% Pb2+ during cooling (d) schematic energy level diagram of Yb3+ and Pb2+. The cooperative energy transfer from three excited Yb3+ ions to one Pb2+ ion is exhibited by using black arrows. The magenta arrow indicates the non-radiative relaxation process in Pb2+ ions. | ||
We have already shown that the cooperative transition probability of an Yb3+-trimer is much higher than that of an Yb3+-tetramer,30 indicating that it has a larger possibility for excited Yb3+-trimers to transfer their energy to the Pb2+ ions than that for excited Yb3+-tetramers. Moreover, it can be seen from Fig. 4(a) that the increase of Pb2+ concentration induces not only the fluorescence of Pb2+ ions but also the decrease of TCL from Yb3+-trimers. Combining the above experimental phenomena with the fact that Pb2+ ions cannot absorb 978 nm excitation light directly, it is not difficult to deduce that the excitation of Pb2+ ions originates from the cooperative sensitization of more than two excited Yb3+ ions, in other words, from the cooperative energy transfer of Yb3+-trimers. It can be seen from Fig. 4(a) that the increment of Pb2+ luminescence is much smaller than the decrement of TCL, indicating that not all excitation energy of Yb3+-trimers lost in the energy transfer to Pb2+ ions. The UV emissions of 343 nm and 383 nm completely disappear when the doping concentration of Pb2+ ions reaches 4%, which strongly suggests that doped Pb2+ ions lead to the occurrence of cooperative energy transfer from Yb3+-trimers. According to above analysis, we propose here that the excitation of Pb2+ ions comes from the energy transfer from excited Yb3+-trimers.
Furthermore, we studied the influence of Pb2+ concentration to the cooperative luminescence from Yb3+-dimers (DCL). As shown in Fig. 4(b), the intensity of green DCL from Yb3+-dimers becomes lower when the concentration of Pb2+ ions increases gradually. The DCL emission band in the green spectral region has unique spectral structure even at room temperature. When the doping concentration of Pb2+ ions increases gradually from 0% to 6% in CaF2:1% Yb3+ samples, the DCL decreases and until completely disappears at 6% Pb2+ doping concentration. The transition probabilities of DCL and TCL in CaF2 are calculated to be 2.6 × 10−2 s−1 and 1.8 × 10−6 s−1, respectively.30 Although the TCL from Yb3+-trimers disappears when the doped concentration of Pb2+ ions is 4%, the DCL from Yb3+-dimers exists until the doping concentration of Pb2+ ions reaches 6%. Fig. 4(c) shows the temperature-dependent UC emission spectra of CaF2:1% Yb3+,0.5% Pb2+ during cooling under the excitation of a 978 nm laser. As the temperature decreases, the peak positions of all emission bands (343 and 383 nm) exhibit no significant changes, whereas the UC emission intensities increase monotonously. It is well known that the luminescence efficiencies are strongly temperature dependent, and usually the higher temperature would result in lower luminescence efficiency due to the strong nonradiative loss as the temperature rises. Fig. 4(d) shows a schematic of energy diagram for the cooperative energy transfer of three Yb3+ ions. In the schematic diagram, Pb2+ ions are excited by the sensitization of Yb3+-trimers, and then relax to the bottom of the excited state through a non-radiative way. When the excited Pb2+ ions make a transition from the excited state 3P0 to the ground state 1A1g, the UC emission at 383 nm can be observed. The cooperative UC process includes four ions, one Pb2+ ion and three Yb3+ ions. Under the excitation of 978 nm NIR, the multi-ion system emits an UV photon of 383 nm. The 3P0 → 1A1g transition of Pb2+ ions is forbidden since ΔS = 1 and ΔJ = 0. However, selection rules are not absolute, especially for heavy ions (such as Pb2+). In a heavy ion, the coupling of spin–orbit is strong, which mixes spin triplet (S = 1) and singlet (S = 0) and lifts the spin forbidden partly. As a result, the spin forbidden transitions in some heavy ions can be observed. It can be seen from Fig. 4(d) that the purple line shows the radiative transition of Pb2+ ions. Fig. 5 shows the intensity dependence of 343 nm, 383 nm, and 500 nm UC emissions on the doping concentration of Pb2+. While the concentration of Yb3+ keeps at 1%, the best concentration of Pb2+ ions is 0.5% for the emission of Pb2+ ions at 383 nm.
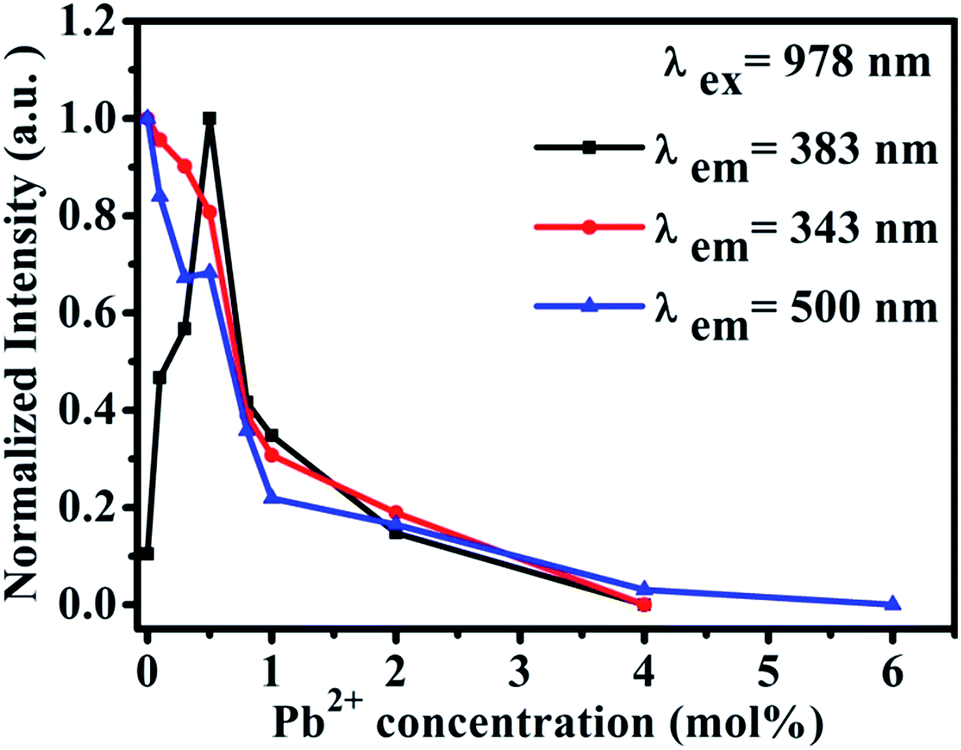 | ||
| Fig. 5 Intensity dependence of 343 nm, 383 nm and 500 nm UC emissions on the doping concentration of Pb2+ in CaF2:1% Yb3+,x% Pb2+ (x = 0, 0.3, 0.5, 0.8, 1, 2, 3, 4, 6). | ||
The UV excitation spectrum of 383 nm emission from CaF2:1% Yb3+,0.5% Pb2+ is peaked at 330 nm (due to Pb2+ ions), as shown in Fig. 6(a). The excitation band is highly overlapped with the TCL band peaked at 343 nm, which satisfies the condition of cooperative sensitization from excited Yb3+-trimers to Pb2+ ions and gives further the excitation mechanism of 383 nm UCL. The 383 nm luminescence of CaF2:0.5% Pb2+ upon 330 nm excitation shows that this luminescence comes from Pb2+ ions, as shown in Fig. 6(b). Under 330 nm excitation, we also recorded the UV emission spectrum of CaF2:1% Yb3+,0.5% Pb2+ (red line). This spectral result is similar to that of the sample without Yb3+ doping, indicating that the down-conversion emission of Pb2+ ions is not influenced by Yb3+ doping. The absorption bands of Pb2+ ions in CaF2 are peaked at 246 nm, 306 nm, and a broad band between 310 nm and 450 nm.33 On the other hand, upon 978 nm NIR excitation, a similar UV emission (Fig. 6(b), blue line) peaked at 383 nm was also observed in CaF2:1% Yb3+,0.5% Pb2+. Therefore, it is reasonable for us to attribute the UV emission peaked at 383 nm to the 3P0 → 1A1g transition of Pb2+ ions. The widths of black and red peaks are almost the same and wider than that of the blue one. There are various activator symmetry sites for the codoped Pb2+ ions in the powder samples but only a special part of them can be excited by the ET from the excited Yb3+-trimers, therefore the UCL band from Pb2+ ions is rather narrow.
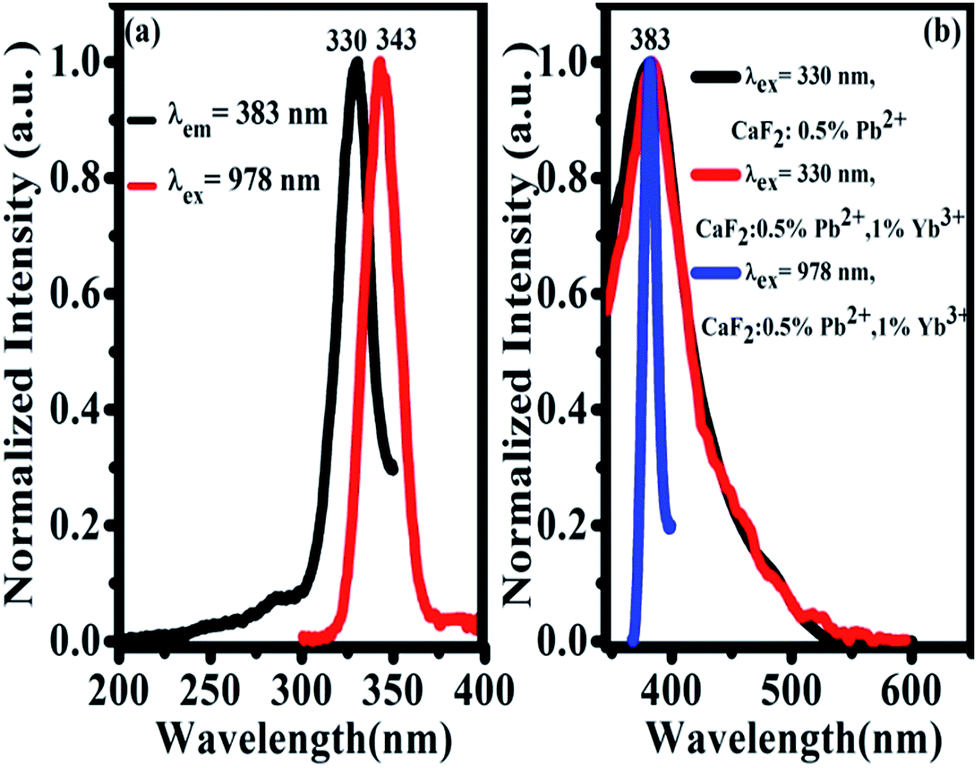 | ||
| Fig. 6 (a) UC emission spectrum upon 978 nm excitation of Yb3+-trimers (red line) and excitation spectrum for 383 nm Pb2+ emission in CaF2:1% Yb3+,0.5% Pb2+ (black line) at room temperature; (b) emission spectra of CaF2:0.5% Pb2+ (black) and CaF2:1% Yb3+,0.5% Pb2+ (red) upon 330 nm excitation, and the UCL spectrum of CaF2:1% Yb3+,0.5% Pb2+ (blue) upon 978 nm excitation at room temperature, respectively. | ||
In order to study the cooperative energy transfer from Yb3+-trimers to Pb2+ ions, we studied the excited state dynamics of CaF2:1% Yb3+,0.5% Pb2+ under the excitation of 978 nm. At 10 K and 300 K, we recorded the decay curve of TCL from CaF2:1% Yb3+,0.5% Pb2+ under the chopped 978 nm excitation by using an oscilloscope. The energy transfer processes are often dependent on temperature. It can be seen from Fig. 7(a) that these decay curves have no rising edge, which indicates that TCL (∼343 nm) come from the direct excitation of 978 nm and no energy transfer process exists. The lifetime of Yb3+-trimers a little decrease from 0.487 ms to 0.423 ms. In addition, the higher the temperature the shorter the life time due to dependence of nonradiative multiphonon relaxation processes on temperature. However, the 383 nm UCL of Pb2+ ions shows a rising edge, as shown in Fig. 7(b), which means that the excitation of Pb2+ comes from energy transfer. The lifetime of Pb2+ excited state is long (0.475 ms) due to the 3P0 → 1A1g transition is strongly forbidden. As can be seen in Fig. 7(c), the lifetime of TCL gradually decreases with the increase of Pb2+ doping concentration, which indicates the cooperative energy transfer from Yb3+-trimers to Pb2+ ions. Fig. 7(d) shows the typical double-logarithmic plots of luminescence intensity If versus the pump power density INIR. The n value can be obtained from the slope of the linear fit. The fitting results of square and circle are approximately 2.87 ± 0.07 and 2.71 ± 0.05 for 343 and 383 nm emissions, respectively. These n values reveal that both the emissions at 343 and 383 nm belong to three-photon UC processes.
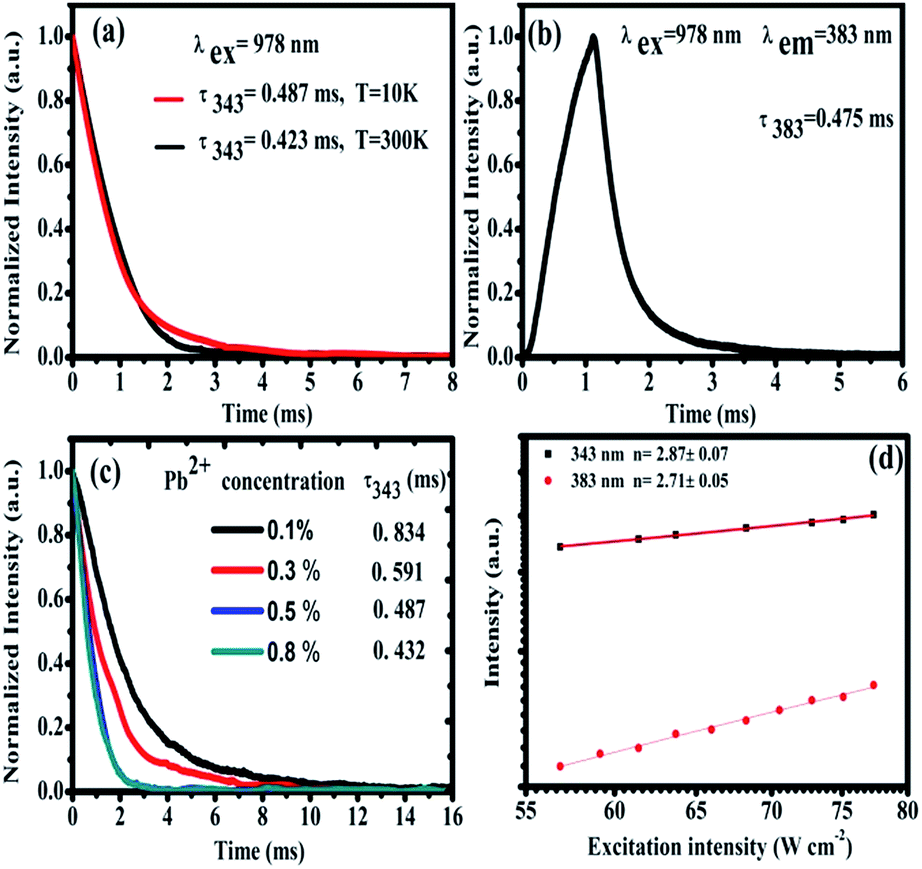 | ||
| Fig. 7 (a) Temperature dependent fluorescence decay curves of 343 nm TCLs. (b) Fluorescence decay curve of the UCL at 383 nm from Pb2+ ions upon 978 nm excitation at 10 K. (c) Fluorescence decay curves of 343 nm TCL from Yb3+-trimers with different Pb2+ concentrations under 978 nm excitation in CaF2:1% Yb3+,x% Pb2+ (x = 0.1%, 0.3%, 0.5%, and 0.8%) at 10 K, respectively. (d) Double-logarithmic plots of the pump-power dependent UC emission intensity recorded under 978 nm excitation. | ||
Conclusions
In summary, we synthesized CaF2:Pb2+,Yb3+ phosphors through a coprecipitation method. Under the excitation of 978 nm, not only the TCL at 343 nm from Yb3+-trimers was observed but also the UV UCL at 383 nm from Pb2+ ions was recorded for the first time. Experimental results shows that the population of Pb2+ excited states originates from the cooperative energy transfer from excited Yb3+-trimers. With the increase of Pb2+ concentration, both of the TCL and the UV UCL from Pb2+ ions decrease and disappear.Experimental section
Powder samples of CaF2 doped with Yb3+ (0, 1 mol%) and Pb2+ (0, 0.3, 0.5, 0.8, 1, 2, 3, 4 mol%) were prepared by a conventional co-precipitation method by using Ca(NO3)2·4H2O (99%, Aladdin), Yb(NO3)3·5H2O (99.99%, Aladdin), Pb(NO3)2 (99.999%, Aladdin), and NH4HF2 (99.99%, Aladdin) as the raw materials. Firstly, standard aqueous solutions of 0.5 mol l−1 Yb(NO3)3·5H2O, 0.01 mol l−1 Pb(NO3)2, and 0.5 mol l−1 Ca(NO3)2·4H2O were prepared. A stoichiometric amounts of Yb(NO3)3·5H2O, Pb(NO3)2, and Ca(NO3)2·4H2O aqueous solutions were put in a beaker with 30 min constant stirring, and then the mixture was added dropwise to NH4HF2 aqueous solution under constant stirring for 1 h to get uniform precipitations. The obtained precipitations were then washed several times with distilled water to remove the remained NO3− and F− ions. The products were dried at 95 °C for 12 h and then calcined at 1200 °C for 2 h in air. All the samples were synthesized under the identical conditions. The luminescence spectra were recorded with a one-meter monochromator (SPEX 1000M; HORIBA Jobin Yvon Inc., Edison, NJ, USA) equipped with an 1800 lines per mm grating. The excitation light source was a power-adjustable continuous wave laser diode (978 nm, 10 W; BWT Beijing Ltd, Beijing, China). The mean power of this excitation light source is 10 W, and the NIR laser was focused on the sample by a lens. A digital oscilloscope (DPO4104B, bandwidth 1 GHz, sampling rate 5 GS s−1; Tektronix, Shanghai, China), a power-adjustable continuous wave laser diode (CW 978 nm, 10 W), and a chopper were used to record decay curves.Acknowledgements
This work supported by the National Natural Science Foundation of China (NSFC) (11274139, 11474132) and the Natural Science Foundation of Xinjiang Province, China (2015211C295).Notes and references
- A. J. Silversmith, W. Lenth and R. M. Macfarlane, Green infrared-pumped erbium upconversion laser, Appl. Phys. Lett., 1987, 51, 1977 CrossRef CAS.
- R. M. Macfarlane, F. Tong, A. J. Silversmith and W. Lenth, Violet cw neodymium upconversion laser, Appl. Phys. Lett., 1988, 52, 1300 CrossRef CAS.
- T. Danger, J. Koetke, R. Brede, E. Heumann, G. Huber and B. H. T. Chai, Spectroscopy and green upconversion laser emission of Er3+-doped crystals at room temperature, J. Appl. Phys., 1994, 76, 1413 CrossRef CAS.
- L. F. Johnson and H. J. Guggenheim, Laser emission at 3 μ from Dy3+ in BaY2F8, Appl. Phys. Lett., 1973, 23, 96 CrossRef CAS.
- M. F. Joubert, Photon avalanche upconversion in rare earth laser materials, Opt. Mater., 1999, 11, 181 CrossRef CAS.
- J. F. Suyver, A. Aebischer, D. Biner, P. Gerner, J. Grimm, S. Heer, K. W. Kramer, C. Reinhard and H. U. Gudel, Novel materials doped with trivalent lanthanides and transition metal ions showing near-infrared to visible photon upconversion, Opt. Mater., 2005, 27, 1111 CrossRef CAS.
- M. Pollnau, D. R. Gamelin, S. R. Luthi and H. U. Gudel, Power dependence of upconversion luminescence in lanthanide and transition-metal-ion systems, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337 CrossRef CAS.
- E. H. Song, S. Ding, M. Wu, S. Ye, F. Xiao, S. F. Zhou and Q. Y. Zhang, Anomalous NIR Luminescence in Mn2+-Doped Fluoride Perovskite Nanocrystals, Adv. Opt. Mater., 2014, 2, 670 CrossRef CAS.
- L. L. Wang, X. J. Xue, F. Shi, D. Zhao, D. S. Zhang, K. Z. Zheng, G. F. Wang, C. F. He, R. Kim and W. P. Qin, Ultraviolet and violet upconversion fluorescence of europium(III) doped in YF3 nanocrystals, Opt. Lett., 2009, 34, 2781 CrossRef CAS PubMed.
- E. H. Song, S. Ding, M. Wu, S. Ye, F. Xiao, G. P. Dong and Q. Y. Zhang, Temperature-tunable upconversion luminescence of perovskite nanocrystals KZnF3:Yb3+,Mn2+, J. Mater. Chem. C, 2013, 1, 4209 RSC.
- N. Rakov, R. B. Guimaraes and G. S. Maciel, Photon up-conversion production in Tb3+–Yb3+ co-doped CaF2 phosphors prepared by combustion synthesis, Mater. Res. Bull., 2016, 74, 103 CrossRef CAS.
- F. F. Huang, X. Q. Liu, Y. Y. Ma, S. Kang, L. L. Hu and D. P. Chen, Origin of near to middle infrared luminescence and energy transfer process of Er3+/Yb3+ co-doped fluorotellurite glasses under different excitations, Sci. Rep., 2015, 5, 8233 CrossRef CAS PubMed.
- W. Strek, B. Cichy, L. Radosinski, P. Gluchowski, L. Marciniak, M. Lukaszewicz and D. Hreniak, Laser-induced white-light emission from graphene ceramics-opening a band gap in graphene, Light: Sci. Appl., 2015, 4, e237 CrossRef CAS.
- W. P. Qin, D. S. Zhang, D. Zhao, L. L. Wang and K. Z. Zheng, Near-infrared photocatalysis based on YF3:Yb3 Tm3+/TiO2 core/shell nanoparticles, Chem. Commun., 2010, 46, 2304 RSC.
- X. Y. Guo, W. H. Di, C. F. Chen, C. X. Liu, X. Wang and W. P. Qin, Enhanced near-infrared photocatalysis of NaYF4:Yb, Tm/CdS/TiO2 composites, Dalton Trans., 2014, 43, 1048 RSC.
- F. Auzel, Upconversion and anti-stokes processes with f and d ions in solids, Chem. Rev., 2004, 104, 139 CrossRef CAS PubMed.
- C. P. Joshi and S. V. Moharil, Luminescence of Pb2+ in some aluminates prepared by combustion synthesis, Phys. Status Solidi B, 2000, 220, 985 CrossRef CAS.
- H. F. Folkerts, J. Zuidema and G. Blasse, The luminescence of Pb2+ in lead compounds with one-dimensional chains, Solid State Commun., 1996, 99, 655 CrossRef CAS.
- H. F. Folkerts and G. Blasse, Two types of luminescence from Pb2+ in alkaline-earth carbonates with the aragonite structure, J. Phys. Chem. Solids, 1996, 57, 303 CrossRef CAS.
- D. F. Anderson, J. A. Kierstead, P. Lecoq, S. Stollb and C. L. Woody, A search for scintillation in doped and orthorhombic lead fluoride, Nucl. Instrum. Methods Phys. Res., Sect. A, 1994, 342, 473 CrossRef CAS.
- S. A. Kazanskii, A. I. Ryskin, A. E. Nikiforov, A. Y. Zaharov, M. Y. Ougrumov and G. S. Shakurov, EPR spectra and crystal field of hexamer rare-earth clusters in fluorites, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 014127 CrossRef.
- V. A. Chernyshev, A. E. Nikiforov and A. D. Nazemnikh, Hexamer clusters in MeF2: Yb3+ (Me = Ca, Sr, Ba), J. Phys.: Conf. Ser., 2011, 324, 012025 CrossRef.
- E. Nakazawa, Cooperative Luminescence in YbPO4, Phys. Rev. Lett., 1970, 25, 1710 CrossRef CAS.
- F. Varsanyi and G. H. Dieke, Ion-pair resonance mechanism of energy transfer in rare earth crystal fluorescence, Phys. Rev. Lett., 1961, 7, 442 CrossRef CAS.
- J. Wang, R. R. Deng, M. A. MacDonald, B. L. Chen, J. K. Yuan, F. Wang, D. Z. Chi, T. S. A. Hor, P. Zhang, G. K. Liu, Y. Han and X. Liu, Enhancing multiphoton upconversion through energy clustering at sublattice level, Nat. Mater., 2014, 13, 157 CrossRef CAS PubMed.
- X. T. Wei, J. B. Zhao, W. P. Zhang, Y. Li and M. Yin, Cooperative energy transfer in Eu3+, Yb3+ codoped Y2O3 phosphor, J. Rare Earths, 2010, 28, 166 CrossRef CAS.
- J. L. He, Z. G. Zhou, H. Zhan and A. X. Lin, Intense red fluorescence from Ho/Yb codoped tellurite glasses, J. Non-Cryst. Solids, 2014, 383, 157 CrossRef CAS.
- W. P. Qin, Z. Y. Liu, C. N. Sin, C. F. Wu, G. S. Qin, Z. Chen and K. Z. Zheng, Multi-ion cooperative processes in Yb3+ clusters, Light: Sci. Appl., 2014, 3, e193 CrossRef CAS.
- T. Aidilibike, Y. Y. Li, J. J. Guo, X. H. Liu and W. P. Qin, Blue upconversion emission of Cu2+ ions sensitized by Yb3+-trimers in CaF2, J. Mater. Chem. C, 2016, 4, 2123 RSC.
- W. P. Qin, S. Cholnam, Z. Y. Liu, G. S. Qin and C. F. Wu, Theory on cooperative quantum transitions of three identical lanthanide ions, J. Opt. Soc. Am. B, 2015, 32, 303 CrossRef CAS.
- W. Low, Paramagnetic and Optical Spectra of Ytterbium in the Cubic Field of Calcium Fluoride, Phys. Rev., 1960, 118, 1608 CrossRef CAS.
- V. Petit, P. Camy, J. L. Doualan, X. Portier and R. Moncorge, Spectroscopy of Yb3+: CaF2: From isolated centers to clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085131 CrossRef.
- I. Nicoara, M. Munteanu, N. Pecingina-Garjoaba, M. Stef, E. Preda and A. Lucaci, Dielectric spectra of doped and X-ray irradiated calcium fluoride crystals, Phys. Status Solidi C, 2007, 4, 1234 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |