
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResponsive copolymer–graphene oxide hybrid microspheres with enhanced drug release properties†
Fuping Dongab,
Izabela Firkowska-Bodena,
Matthias M. L. Arras ac and
Klaus. D. Jandt*ad
ac and
Klaus. D. Jandt*ad
aChair of Materials Science, Otto Schott Institute of Materials Research, Faculty of Physics and Astronomy, Friedrich Schiller University Jena, Löbdergraben 32, 07743 Jena, Germany. E-mail: K.Jandt@uni-jena.de
bDepartment of Polymer Materials and Engineering, College of Materials and Metallurgy, Guizhou University, Guiyang, 550025, China
cOak Ridge National Lab, Biology and Soft Matter Division, Oak Ridge, TN 37831, USA
dJena Center for Soft Matter (ICSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
First published on 13th January 2017
Abstract
The ability to integrate both high encapsulation efficiency and controlled release in a drug delivery system (DDS) is a highly sought solution to cure major diseases. However, creation of such a system is challenging. This study was aimed at constructing a new delivery system based on thermoresponsive poly(N-isopropylacrylamide-co-styrene) (PNIPAAm-co-PS) hollow microspheres prepared via two-step precipitation polymerization. To control the diffusion-driven drug release, the PNIPAAm-co-PS spheres were electrostatically coated with graphene oxide (GO) nanosheets. As a result of the coating the permeability of the copolymer–GO hybrid microspheres was reduced to an extent that suppressed the initial burst release and enabled sustained drug release in in vitro testing. The hybrid microspheres showed improved drug encapsulation by 46.4% which was attributed to the diffusion barrier properties and π-conjugated structure of GO. The system presented here is promising to advance, e.g., anticancer drug delivery technologies by enabling sustained drug release and thus minimizing local and systemic side effects.
Introduction
Known as a smart material, poly(N-isopropylacrylamide) (PNIPAAm) features a volume phase transition between a swollen state and a collapsed state under external stimuli such as temperature, pH or pressure.1 When the environmental temperature falls below the lower critical solution temperature (LCST ≈ 32 °C), PNIPAAm adsorbs much water and is in a swollen state, while above the LCST, it expels most of the free water inside the polymer and demonstrates abrupt volume shrinkage. Based on this excellent stimuli-responsive property, this smart material has been applied to the field of controlled drug delivery, tissue engineering and artificial muscles.2–4 PNIPAAm amide side groups and their variation with temperature were used to form materials which can release active molecules in a controlled fashion.5 For additional responsiveness, such as pH or photo sensitivity, different copolymers of PNIPAAm have been fabricated.6–8 However, an obvious limitation of PNIPAAm-based polymers is their poor mechanical properties in the highly swollen state, which leads to a highly undesired burst release of the carried drug molecules.9 A possible way to overcome this uncontrolled release is the use of a protective barrier layer.10 Ideally, the protective layer is thin, yet offering low permeability and good mechanical stability.Graphene, a single layer of a two-dimensional carbon network, is the ultimate thin membrane, which revolutionized chemistry and condensed matter physics owing to its outstanding chemical and physical properties. The excellent properties of graphene have opened a remarkable pathway for its potential application in consumer electronics, energy storage or selective water-gas separation.11–13
Notably, graphene oxide (GO), the derivative of graphene, has emerged as a diversified biocompatible material for biological applications, including bioimaging and biosensing.14–16 In particular, the huge specific surface area and aromatic structure render GO an excellent platform for the immobilization and delivery of biomolecular drugs.17 Other physical features of graphene derivatives, such as high optical absorbance and thermal conductivity, can be used to endow a thermoresponsive DDS with new properties, e.g., photo-response, as shown recently by Lu et al.18
On account of the unique properties of both, PNIPAAm and GO, their rational combination shows a promising route towards the development of a unique thermoresponsive hybrid composite material for drug delivery. Despite the exciting potential of GO nanosheets and PNIPAAm, a rationally designed system with improved drug encapsulation efficiency and sustained release has yet to be developed. Compared to solid particles, hollow spheres have drawn more interests for DDS because of their low density, high surface area, and large cavity volume suitable for material loading.19 However, the remaining problem associated with the application of hollow spheres is the difficulty of controlling either the loading or the release of drug molecules, since the loading and release are only driven by diffusion. The most practical way to overcome this problem is to develop thermoresponsive hollow spheres with an exterior ultrathin diffusion barrier layer. Such a strategy may represent an important innovation to create a long-acting DDS.
Herein we report the rational design and synthesis of PNIPAAm-co-PS hollow microspheres electrostatically coated with GO nanosheets, denoted as GO@PNIPAAm-co-PS. To the best of our knowledge, this is the first report on the creation of hollow, monodisperse thermoresponsive polymer microspheres with GO for DDS. By virtue of the microspheres' hollow structure and polymer composition the resulting hybrid system has a well-defined morphology and improved mechanical stability. Of particular interest is that GO layers could effectively hinder the out-diffusion of the enclosed drug molecules which results in (i) increased encapsulation efficiency, (ii) suppressed the initial burst release and (iii) endowed the copolymer microspheres with a more controlled drug release performance.
Results and discussion
Synthesis and characterization of GO@PNIPAAm-co-PS hollow microspheres
One of the physical attributes which influences the performance of DDS is the size distribution of the particles. Because the distribution of the particles within the body and the interaction with biological cells are greatly affected by the particle size,20 spheres with a defined size, i.e., narrow size distribution, are required. The drug release kinetics of such monodisperse spherical DDS can be engineered reproducibly, which makes it easier to formulate more sophisticated and intelligent DDS. In this work, a copolymer system, composed of PNIPAAm and PS was synthesized, in which PS was included to ensure monodispersity21 and to improve the poor mechanical stability of PNIPAM under load conditions.22 PNIPAAm provides the thermoresponse of the system. PNIPAAm-co-PS hollow spheres were prepared by a two-step precipitation polymerization method as shown in Fig. 1a. In the first step, the self-polymerization of NIPAAm at 60 °C (above the LCST) leads to oligomers' coagulation and nuclei formation. After further addition of the co-monomers (NIPAAm and styrene) and the cross-linker [N,N′-methylenebisacrylamide (MBAA)], copolymerization and formation of cross-linked shell take place. The final step involves a self-removal process of the oligomer core at a temperature below the LCST, i.e., in the swollen state. The interstices of the swollen PNIPAAm-co-PS shell provide channels for the non-cross-linked PNIPAAm units to be dissolved from the interior, yielding the final PNIPAAm-co-Ps hollow spheres. Scanning and transmission electron microscopy (SEM and TEM, respectively) images show the morphology of well-dispersed spherical particles with uniform diameter. The particles have a narrow poly(styrene) size distribution with an average hydrodynamic diameter of 520 ± 69 nm, as measured by dynamic light scattering technique at 25 °C. TEM images further confirm the hollow-shell structure with an estimated cavity size of 90 nm (see Fig. 1c inset).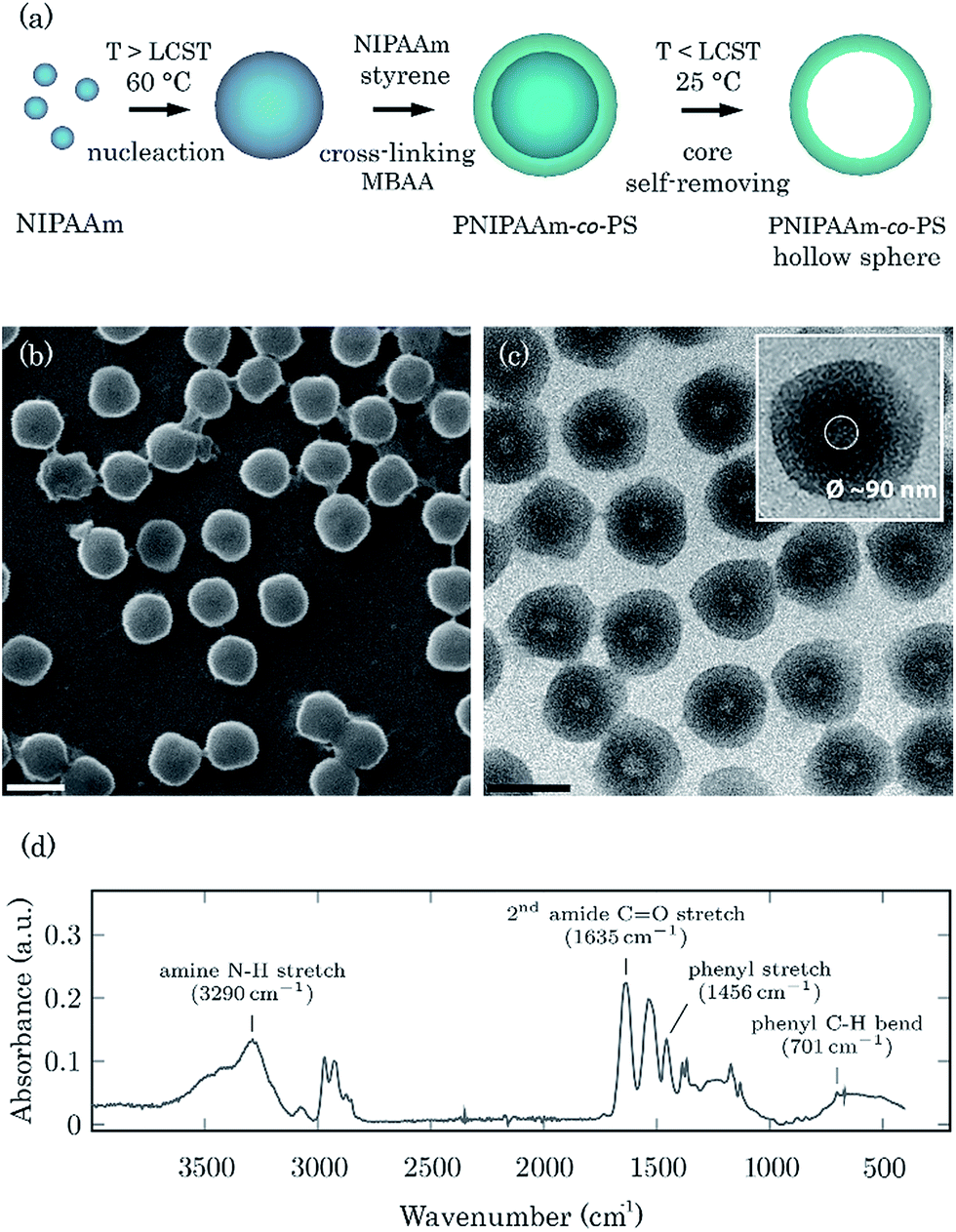 | ||
| Fig. 1 Schematic illustration of the synthesis process of PNIPAAm-co-PS hollow microspheres (a). SEM (b) and TEM (c) images of copolymer hollow microspheres, scale bar: 500 nm. FTIR spectrum of PNIPAAm-co-PS at room temperature (d). | ||
The chemical composition of the PNIPAAm-co-PS was analysed by Fourier transform infrared spectroscopy (FTIR). The amine N–H stretching (3290 cm−1), second amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching (1635 cm−1), second amide N–H stretching (1537 cm−1) and deformation of two methyl groups (1385 and 1367 cm−1), attributed to PNIPAAm, are clearly present in the spectrum (Fig. 1c) whereas, the phenyl stretching (1456 cm−1) and stretching of five adjacent H atoms in the benzene ring (753 and 701 cm−1) corresponds to PS. Thus, the FTIR results provide solid evidence that the hollow spheres contain both PNIPAAm and PS.
O stretching (1635 cm−1), second amide N–H stretching (1537 cm−1) and deformation of two methyl groups (1385 and 1367 cm−1), attributed to PNIPAAm, are clearly present in the spectrum (Fig. 1c) whereas, the phenyl stretching (1456 cm−1) and stretching of five adjacent H atoms in the benzene ring (753 and 701 cm−1) corresponds to PS. Thus, the FTIR results provide solid evidence that the hollow spheres contain both PNIPAAm and PS.
GO sheets were synthesized from crystalline graphite flakes via the improved Hummers method.23 The sheets were broken into small pieces via an ultrasonic probe treatment in an ice bath,24 resulting in GO nanosheets. As can be seen from the AFM images (Fig. 2a) the nanosheets, with lateral size in the range of 100–500 nm, are evenly distributed on the silicon wafer. The corresponding height profile demonstrates that the synthesized sheets are approximately 1.6 nm in thickness, suggesting that GO nanosheets contain 1–2 layers of graphene (considering the thickness of a single layer graphene (0.34 nm)25 and the interlayer spacing of graphene oxide (1.01 nm) measured by XRD (Fig. S1†)). To explore the chemical changes in the graphite structure after chemical exfoliation, FTIR and X-ray photoelectron spectroscopy (XPS) analyses were performed. As depicted in Fig. 2b, the most characteristic features in the FTIR spectrum are the broad band peak between 3400 and 3200 cm−1 assigned to an O–H stretching and absorption bands corresponding to CO carbonyl stretching at 1732 cm−1 and the COO− symmetric stretching at 1372 cm−1. The presence of local chemical bonding in the carbon structure confirms the XPS analysis as well. As seen in Fig. 2c, GO shows a typical C 1s peak at 284.2 eV attributed to carbon in the non-oxygenated ring C–C bonds, along with three other peaks assignable to C–O (hydroxyl and epoxy), CO (carbonyl) and OC–OH (carboxyl) bond at 286.2 eV, 287.2 eV and 288.5 eV, respectively. This comprehensive characterization showed that the GO nanosheets comprised of graphene modified with various oxygen functional groups.
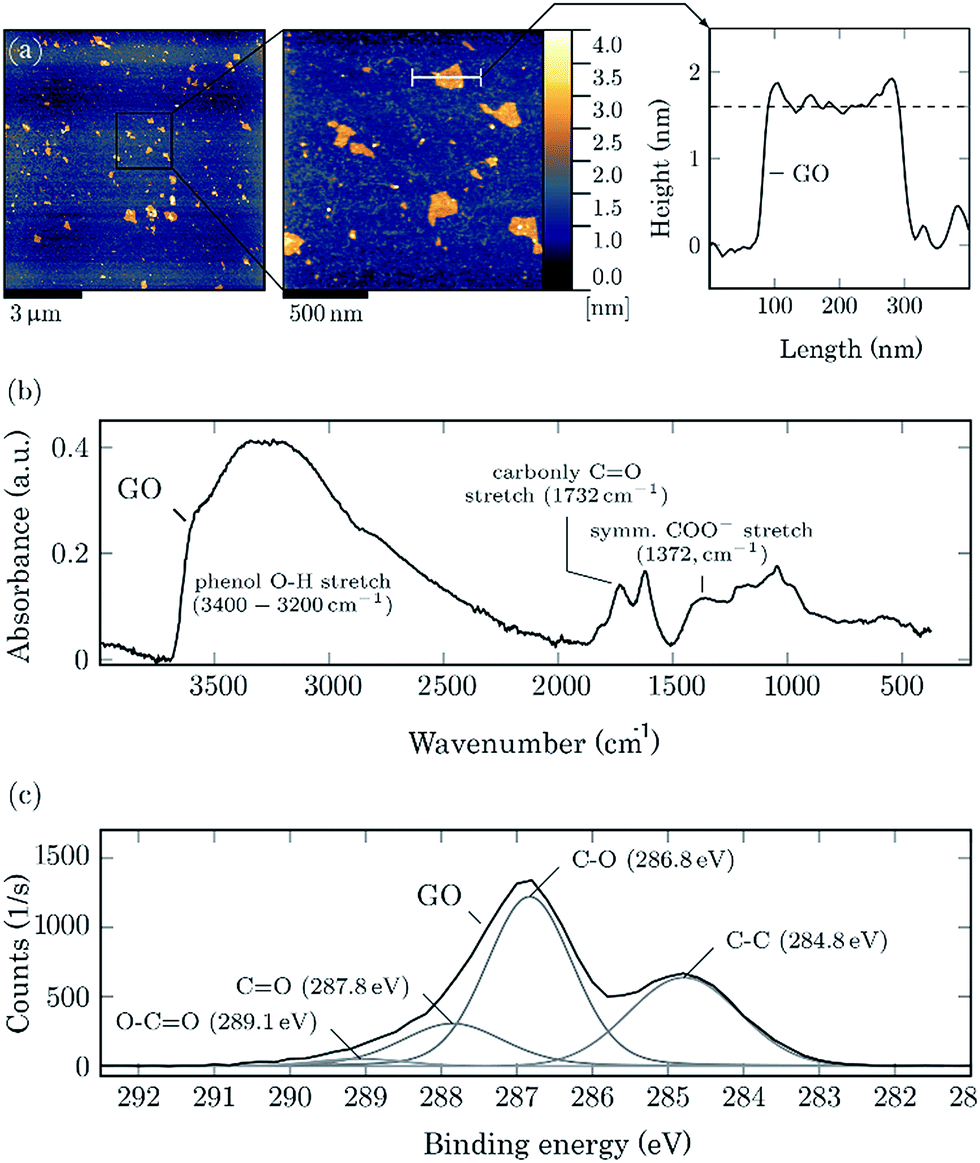 | ||
| Fig. 2 AFM topography images of graphene oxide sheets with height profile along the white line (a). FTIR (b) and XPS (c) spectra of graphene oxide. | ||
The self-assembly of GO on the PNIPAAm-co-PS microspheres was driven by the electrostatic interactions between positively charged copolymer (zeta potential of 23.0 ± 5 mV) and negatively charged GO (zeta potential of −50.3 ± 12.1 mV). The corresponding SEM and TEM images of GO@PNIPAM-co-PS composite are shown in Fig. 3. Clearly, the spheres are wrapped by GO nanosheets (confirmed by Raman measurements, cf. ESI, Fig. S2†). Remarkably, there is no sign of agglomeration, neither of the copolymer hollow spheres nor the GO nanosheets. Moreover, the TEM image reveals that the PNIPAAm-co-PS hollow spheres are entirely and tightly covered by GO nanosheets and the surface appears rough and wrinkled, on the contrary to the smooth surface of bare hollow spheres (see Fig. 1b and c). Based on the thickness of the individual GO sheet (∼1.6 nm) and taking into account the observed sheets overlap, one can give a lower bound of the shell thickness of approximately 4 nm. These observations imply that the flexibility and small lateral size of GO allow for the formation of an ultrathin layer.
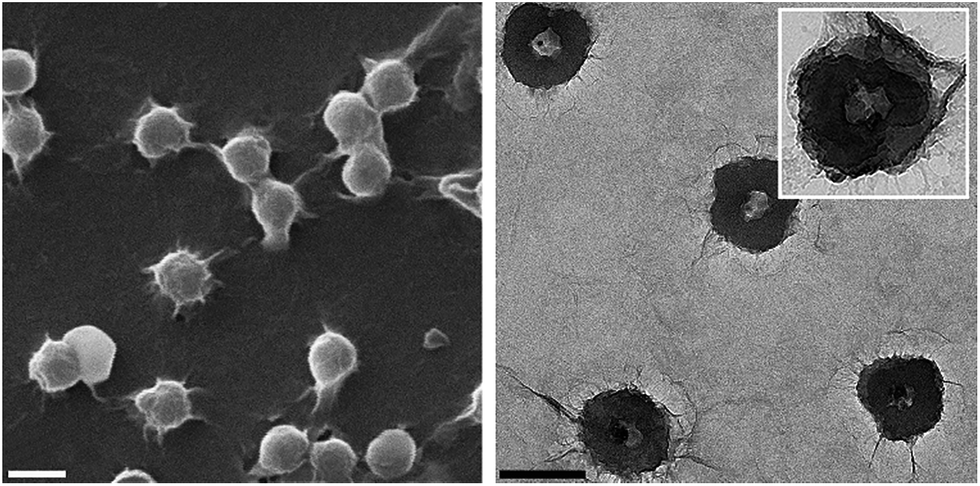 | ||
| Fig. 3 SEM (left) and TEM (right) images of GO@PNIPAAm-co-PS hollow microspheres, scale bar: 500 nm. | ||
Drug encapsulation and release
Vancomycin (VAN) is an antibiotic used to treat serious bacterial infections and widely applied in the clinical routine. We chose it as the model drug to evaluate the drug encapsulation and release ability of the GO@PNIPAAm-co-PS system in comparison to the bare copolymer hollow spheres. Fig. 4a shows the schematic representation of the copolymer loaded with vancomycin and wrapped with an ultrathin layer of GO. Note that the scheme illustrates the order of the creation process (more details in the Experimental section).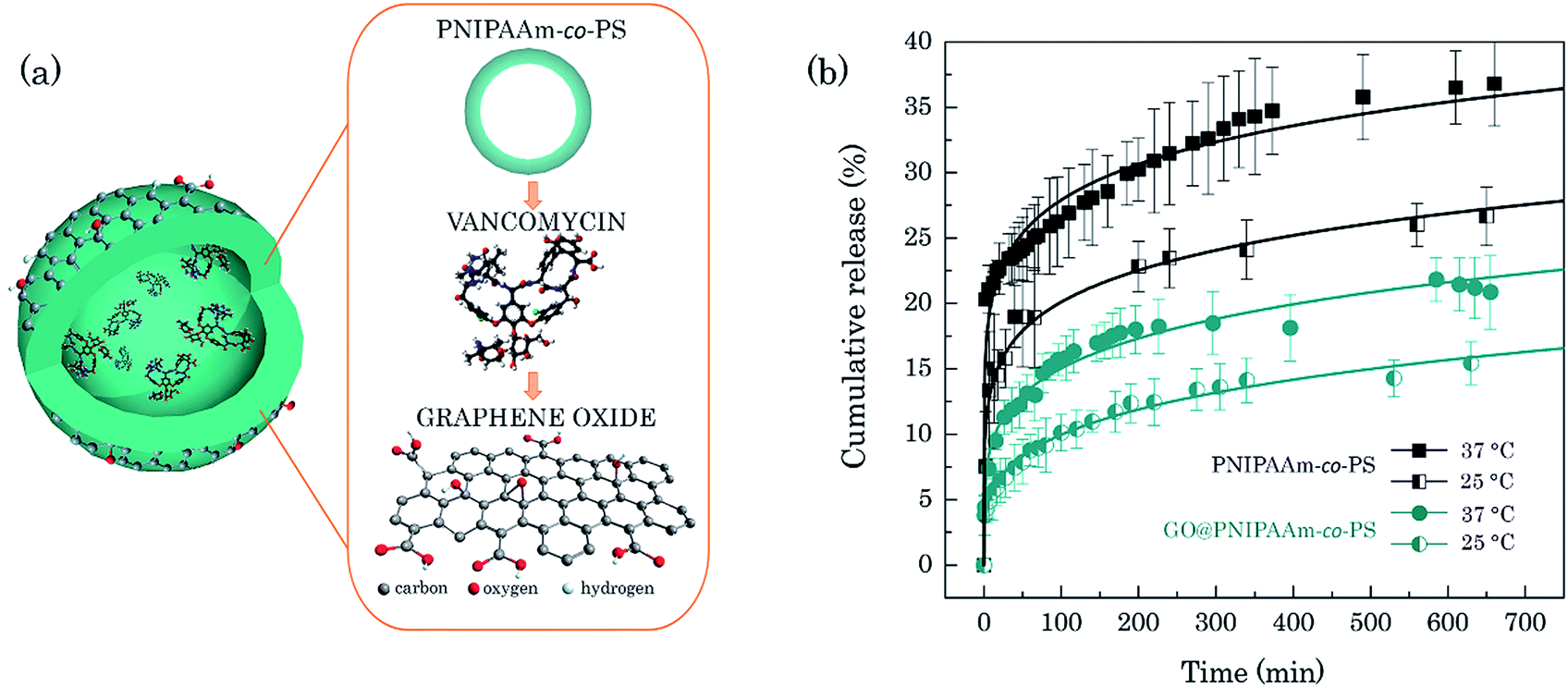 | ||
| Fig. 4 Illustration of the DDS based on PNIPAAm-co-PS hollow microsphere loaded with VAN molecules and wrapped with GO nanosheets (not in scale) (a). In vitro VAN release profile (b). The solid curves represent the fitted release from the Weibull model. | ||
The encapsulation efficiency (EE) of the GO@PNIPAAm-co-PS system was quantified by evaluating the concentration of the free VAN in the supernatant of the copolymer–VAN system by using the characteristic light absorption of VAN at 280 nm. The GO-wrapped copolymer hollow microspheres show an increased encapsulation efficiency when compared to unwrapped carriers, 26.5% and 18.1%, respectively. The improved encapsulation efficiency might be related to the enhanced mechanical stability of the copolymer in the swollen state gained by the ultrathin GO layer. Additionally, since the VAN loading and GO layer formation took place in the same reaction vessel, VAN molecules loaded directly onto the GO nanosheets are also thought to influence the overall EE. The large π conjugated structure of GO can form non-covalent π–π stacking interaction with the VAN molecules. In addition, the –OH and –COOH groups on the graphene sheets can form strong hydrogen bonding interaction with –OH and –NH2 groups in VAN. The presence as well as the nature of such interactions might be verified from the changes in the UV-vis absorption spectra of GO–VAN aqueous solution. Namely, the adhesion of VAN on graphene oxide via π–π stacking or hydrogen bonding should be reflected by a red-shift of the vancomycin characteristic absorption peak, due to the ground-state electron donor–acceptor interaction between GO and VAN.17,26 For control experiments, GO–VAN suspension, aqueous GO suspension and pure VAN solution were prepared and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min, respectively. On the one hand, in these three control systems, no precipitate was observed, indicating that GO sheets with the drug were not depleted from the solution when not attached to the copolymer hollow spheres. This means the encapsulation efficiency and drug release of GO@PNIPAAm-co-PS system can be directly compared with the results for the bare copolymer hollow spheres. On the other hand, however, it prevents the quantitative evaluation of the GO-loading capacity based on absorption intensity changes of free vancomycin in the supernatant. In addition, the control showed no distinct changes in the UV-vis absorption spectrum of VAN when binding to GO (details ESI, Fig. S3†). The recorded spectrum was a superposition of individual GO and VAN spectra, which excludes VAN binding to GO via non-covalent π stacking or covalent hydrogen bond interactions. The adhesion of VAN via electrostatic interaction was confirmed indirectly from AFM measurements of the GO surface roughness (Fig. S4†). Nevertheless, the quantitative contribution of GO–VAN loading efficiency to the entire encapsulation efficiency could not be determined.
000 rpm for 30 min, respectively. On the one hand, in these three control systems, no precipitate was observed, indicating that GO sheets with the drug were not depleted from the solution when not attached to the copolymer hollow spheres. This means the encapsulation efficiency and drug release of GO@PNIPAAm-co-PS system can be directly compared with the results for the bare copolymer hollow spheres. On the other hand, however, it prevents the quantitative evaluation of the GO-loading capacity based on absorption intensity changes of free vancomycin in the supernatant. In addition, the control showed no distinct changes in the UV-vis absorption spectrum of VAN when binding to GO (details ESI, Fig. S3†). The recorded spectrum was a superposition of individual GO and VAN spectra, which excludes VAN binding to GO via non-covalent π stacking or covalent hydrogen bond interactions. The adhesion of VAN via electrostatic interaction was confirmed indirectly from AFM measurements of the GO surface roughness (Fig. S4†). Nevertheless, the quantitative contribution of GO–VAN loading efficiency to the entire encapsulation efficiency could not be determined.
The in vitro release of VAN was studied by the dialysis bag diffusion technique performed in physiological conditions (PBS medium, pH 7.4, 37 °C). To show the thermoresponse of the system it was repeated at 25 °C. Fig. 4b shows the release profiles of VAN from bare and GO-wrapped PNIPAAm-co-PS hollow microspheres. A thermoresponsive drug release was observed for both systems which confirms that the thermo-responsive property of the copolymer hollow microspheres is preserved after modification with GO. As anticipated, the cumulative drug release rate is faster at higher temperatures for both systems which is caused by the collapse of the hydrogel at 37 °C.27 From the drug release profile one finds that 20.3% VAN molecules diffuse from the bare copolymer hollow microspheres into pristine PBS solution within two minutes. This indicates an extremely high initial burst release from the PNIPAAm-co-PS which comes from the rapid collapse of the copolymers. This drug release profile is similar to the one of the semi-hollow PNIPAAM spheres.28 It is worth noting that even in the absence of thermal stimulation, i.e., below the LCST, one can observe a relatively high burst from the bare copolymer hollow microspheres. On the contrary, the GO-wrapped PNIPAAm-co-PS drug carriers show a more controlled drug release, indicated by a five times lower concentration of released drug molecules (4.4% VAN at 25 °C) within the first two minutes, and thus, low burst release. Furthermore, 36.8% of the drug molecules were released from the bare copolymer hollow microspheres and only 20.8% of VAN from GO-wrapped copolymer after 600 min. The accumulated release amount of the latter is nearly half the value of the bare PNIPAAm-co-PS hollow microspheres. Such a slow drug-release characteristic is desired in clinical applications of anticancer drugs, where controlled release of drugs for the chosen period of time may help to overcome the adverse side effects on other healthy organs. To predict how the VAN release develops over time, the experimental data have been fitted with the Weibull and Korsemeyer-Peppas models.29,30 Both models can describe our data. For the sake of clarity, Fig. 4b shows only the regression line fit from the Weibull model. Using the obtained fit we can predict the cumulative VAN release from GO@PNIPAAm-co-PS of 40% within two weeks.
It is safe to say that GO nanosheets work as a diffusion barrier layer to prevent the early-stage and too fast release of the drug from the polymer carriers which is supported by a previous report31 where bilayers of GO incorporated in a protein-release platform were found to tune the protein release in a time-controlled and sequential fashion. The slower drug release in our DDS may be attributed to the reduced water diffusion coefficient through GO interlayers as a result of strong hydrogen bonded interaction with the water molecules.32 As the loaded VAN molecules are expelled together with the water from the shrunk PNIPAAm-co-PS network, the drastic water release and thus the gradual release of the VAN can be well controlled through the ultrathin GO shell. Our hybrid system shows advantages over reported drug-loaded PNIPAAm spheres where the drug release influenced by temperature cannot be controlled or requires either a sophisticated approach of drug loading or complex methods for the preparation of the polymer capsules.33 Based on presented results, we believe that GO@PNIPAAm-co-PS could be further optimized by tuning the thickness of the graphene layer, and chemical modification of functional groups on GO nanosheets in order to improve the release behaviour for different drugs and increase the encapsulation efficiency, respectively. Taking the advantage of conducting properties of graphene nanosheets, the GO-wrapped copolymer hollow microspheres could be enhanced with the on-demand drug release properties in response to electrical stimulation, as emphasized in recent research reports.34,35
Conclusions
A DDS based on GO and hollow copolymer microspheres was successfully created in a rational strategy. The GO@PNIPAAm-co-PS hybrid system integrates the advantages of well-defined morphology, excellent hydrophilicity, high encapsulation efficiency, and sustained release. Apart from the diffusion barrier properties, GO layers have the potential to enable several degrees of customizability to the thermoresponsive PNIPAAm-co-PS hollow microspheres, indicating that the GO@PNIPAAm-co-PS could be a valuable system for therapeutics within the field of biomedicine. Cytotoxicity of the GO@PNIPAAm-co-PS system still needs to be investigated, even though GO nanosheets have been reported as having good biocompatibility and low cytotoxicity.36Experimental section
Materials
All chemicals were purchased from Sigma-Aldrich GmbH (Schnelldof, Germany). NIPAAm was purified by recrystallization in hexane. MBAA, ammonium persulfate (APS, (NH4)2S2O8) styrene, vancomycin hydrochloride, expandable graphite powder, hydrogen peroxide, potassium permanganate (KMnO4), concentrated H2SO4 and H3PO4 were used as received.Analytical and physical methods
The FTIR spectrum was recorded using a Bruker-FTIR Alpha instrument (Bruker, Ettlingen, Germany) at room temperature. AFM images were obtained using the Dimension 3100 (Digital Instruments, CA, USA) equipped with a Nanoscope Controller IV controller (ibidem) in tapping mode. For AFM observations, the samples were dispersed in water with the aid of ultrasonication, dropped cast on a silicon wafer and dried in a desiccator at room temperature. Raman spectra were recorded in a confocal Raman microscope spectrometer (Bruker Senterra, Ettlingen, Germany) with a laser wavelength of 514 nm. XPS measurements were conducted on a PHI Quantum 2000 spectrometer (PHI Co., Chanhassen, MN, USA). The SEM images were obtained using the Leica LEO 440i (Bensheim, Germany). One drop of sample suspension was applied onto a silica wafer, dried under ambient conditions overnight and a gold (Au) layer was coated before the SEM analysis. The TEM images were obtained using a JEOL 3010 (JEOL Ltd., Tokyo, Japan) at an acceleration voltage of 300 kV. UV-vis spectra were recorded using a Perkin Elmer Lambda 25 (Cambridge, UK) spectrometer with 1 cm quartz cuvettes. Particle size and zeta potential were measured using a Zetasizer Nano-ZS instrument (Malvern Instruments, Worcestershire, UK).Synthesis of GO nanosheets
GO sheets were synthesized by the improved Hummers method with slight modifications. Expandable graphite powder (1.0 g), concentrated H2SO4 (45 mL) and H3PO4 (5 mL) were mixed together in a one neck flask. After keeping it in the ice bath for 30 min, KMnO4 (7 g) was added slowly to the system while keeping the system temperature below 20 °C. Subsequently, the reaction mixture was heated to 50 °C and stirred continuously for 10 h. 200 mL 5% H2O2 aqueous solution was slowly added to the reaction vessel placed in an ice bath, stirred for another 15 min and diluted further with 700 mL warm distilled water (40 °C). The resulting suspension was centrifuged, washed with 5% HCl and then dispersed in water. As-synthesized GO was subject to repeated washing with distilled water to completely remove residual salts and acids. The GO was exfoliated by ultrasonication and was broken into small pieces utilizing an ultrasonic probe (UP200S-Hielscher Ultrasonics GmbH, Teltow, Germany) in an ice bath resulting in GO nanosheets.Preparation of PNIPAAm-co-PS hollow microspheres
The crosslinked PNIPAAm-co-PS hollow microspheres were synthesized by a precipitation polymerization method as described previously.37 0.85 g of NIPAAm, was dissolved in 100 mL of deionized water. After the solution was purged with nitrogen for 35 min, 0.11 g ammonium persulfate was added (the APS to NIPAAm ratio of 0.12 was necessary to initiate rapid oligomerization without much chain propagation and thus formation of numerous oligomers seeds). Then the reaction was allowed to proceed at 60 °C under nitrogen atmosphere. When the solution showed a light blue colour, 5 mL of MBAA (0.02 g mL−1) including 0.270 g NIPAAm and 0.210 g styrene was injected into the solution. The polymerization continued to proceed at 60 °C for 8 h. Subsequently, the temperature of the solution was reduced to room temperature and the solution was stirred for 36 h to carry out the “self-removal” process. Finally, the hollow spheres were separated by centrifugation at 10000 rpm and cleaned by repetitive redispersion and centrifugation in deionized water.
Preparation of VAN-loaded GO@PNIPAAm-co-PS DDS
The VAN loaded GO@polymer hollow microspheres for drug delivery were prepared by mixing VAN (8.8 mg) and the polymer hollow microsphere suspension (22 mL, 0.40 wt% in aqueous solution). The mixture was stirred at room temperature for 24 h to diffuse the drug into the polymer spheres. A GO aqueous suspension (2 mL, 0.16 wt%) was added to the polymer suspension, and the mixture was stirred for 12 h to complete the electrostatic wrapping process. The suspensions were centrifuged at 10000 rpm for 30 min and washed with water to remove the unloaded GO and drugs. For comparison, VAN loaded bare polymer spheres were also fabricated with the same procedure without adding GO. The supernatants were collected to calculate the unloaded drug amount by using UV-vis absorption spectroscopy (from the intensity difference between the VAN stock solution and the supernatant solution after loading, the loaded VAN concentration in the polymer spheres was 2.33 mg). For further control experiments, a suspension of VAN and GO without adding any polymers was also prepared with the same procedure and centrifuged at 10000 rpm for 30 min. Pure VAN solution and pure GO suspension were also prepared and centrifuged respectively at the same condition.
Drug delivery assay
The encapsulation efficiency was calculated according to the equation: EE = (mi − ms)/mi, where mi is the initial mass of drug and ms is the mass of drug detected only in the supernatant after centrifugation (from the intensity difference between the VAN stock solution and the supernatant solution after loading, the loaded VAN concentration in the polymer spheres was 2.33 g). The release of the drug from GO@PNIPAAm-co-PS hollow microspheres was evaluated by the dialysis bag diffusion technique.38 3 mL of VAN loaded bare polymer or GO@polymer hollow spheres suspension was injected into a small dialysis bag. Then the dialysis bag with the drug-loaded sample was immersed into 27 mL PBS (pH = 7.4) in a glass vessel. Subsequently, the solution was kept in the dark while stirring it gently for a certain time and its UV-vis absorbance was measured by transferring the 3 mL solution into a standard UV-vis cuvette. After each spectral measurement, the solution in the cuvette was returned into the vessel. The amount of the released VAN was calculated according to the absorbance of the solution at 280 nm. The cumulative release curves were plotted for the average data of three measurements.Acknowledgements
We thank Mr R. Wagner and Mr M. Mühlstädt for XPS measurement/analysis and Ms H. Garlipp for her help with the SEM measurements. The research at Oak Ridge National Laboratory's Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Science, U.S. Department of Energy.References
- M. Motornov, Y. Roiter, I. Tokarev and S. Minko, Prog. Polym. Sci., 2010, 35, 174 CrossRef CAS.
- J. T. Zhang, T. F. Keller, R. Bhat, B. Garipcan and K. D. Jandt, Acta Biomater., 2010, 6, 3890 CrossRef CAS PubMed.
- Y.-M. Liu, W. Wu, X.-J. Ju, W. Wang, R. Xie, C.-L. Mou, W.-C. Zheng, Z. Liua and L.-Y. Chu, RSC Adv., 2014, 4, 46568 RSC.
- S.-H. Lee, T. H. Kim, M. D. Lima, R. H. Baughmann and S. J. Kim, Nanoscale, 2016, 8, 3248 RSC.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655 CrossRef CAS PubMed.
- Z. Zhu and S. A. Sukhishvili, ACS Nano, 2009, 3, 3595 CrossRef CAS PubMed.
- H. Liu, C. Li, D. Tang, X. An, Y. Guo and Y. Zhao, J. Mater. Chem. B, 2015, 3, 3959 RSC.
- F. Zhang and C.-C. Wang, Colloid Polym. Sci., 2008, 286, 889 CAS.
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3 CrossRef CAS.
- A. S. Hasan, M. Socha, A. Lamprecht, F. E. Ghazouani, A. Sapin, M. Hoffman, P. Maincent and N. Ubrich, Int. J. Pharm., 2007, 344, 53 CrossRef CAS PubMed.
- H. Bi, J. Chen, W. Zhao, S. Sun, Y. Tang, T. Lin, F. Huang, X. Zhou, X. Xie and M. Jiang, RSC Adv., 2013, 3, 8454 RSC.
- H. Gwon, H. S. Klim, K. U. Lee, D.-H. Seo, Y. C. Park, Y. S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277 CAS.
- Q. Xu, H. Xu, J. Chen, Y. Lv, C. Dong and T. S. Sreeprasad, Inorg. Chem. Font., 2015, 2, 417 RSC.
- M. Nurunnabi, K. Parvez, M. Nafiujjaman, V. Revuri, H. A. Khan, X. Feng and Y.-K. Lee, RSC Adv., 2015, 5, 42141 RSC.
- J. A. Tyson, V. Mirabello, D. G. Calatayuda, H. Ge, G. Kociok-Köhn, S. W. Botchway, G. D. Pantos and S. I. Pascu, Adv. Funct. Mater., 2016, 26, 5641 CrossRef CAS.
- Y. Liu, D. Yu, C. Zeng, Z. Miao and L. Dai, Langmuir, 2010, 9, 6158 CrossRef PubMed.
- X. Yang, X. Zhang, Z. Liu, Y. Ma, Y. Huang and Y. Chen, J. Phys. Chem., 2008, 112, 17554 CAS.
- N. Lu, J. Liu, J. Li, Z. Zhang, Y. Weng, B. Yuan, K. Yanga and Y. Ma, J. Mater. Chem. B, 2014, 2, 3791 RSC.
- Z. Teng, X. Su, Y. Zheng, J. Zhang, Y. Liu, S. Wang, J. Wu, G. Chen, J. Wang, D. Zhao and G. Lu, J. Am. Chem. Soc., 2015, 137, 7935 CrossRef CAS PubMed.
- K. Shiga, N. Muramatsu and T. Kondo, J. Pharm. Pharmacol., 1996, 48, 891 CrossRef CAS PubMed.
- T. Hellweg, C. D. Dewhurst, W. Eimer and K. Kratz, Langmuir, 2004, 20, 4330 CrossRef CAS PubMed.
- T. R. Matzelle, G. Geuskense and N. Kurse, Macromolecules, 2003, 38, 2926 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806 CrossRef CAS PubMed.
- H. Shen, L. Zhang, M. Liu and Z. Zhang, Theranostics, 2012, 2, 283 CrossRef CAS PubMed.
- S. R. Wang, Y. Zhang, N. Abidi and L. Cabrales, Langmuir, 2009, 25, 11078 CrossRef CAS PubMed.
- Z. Liu, J. Liu, D. Li, P. S. Francis, N. W. Barnett, C. J. Barrow and W. Yang, Chem. Commun., 2015, 51, 10969 RSC.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301 CrossRef CAS.
- J. Qian and F. Wu, J. Mater. Chem. B, 2013, 1, 3464 RSC.
- A. Mendyk, R. Jachowicz, K. Fijorek, P. Dorożyński, P. Kulinowski and S. Polak, Dissolution Technol., 2012, 19, 6 CrossRef.
- K. H. Ramteke, P. A. Dighe, A. R. Kharat and S. V. Patil, Scholars Acad. J. Pharm., 2014, 3, 388 Search PubMed.
- J. Hong, N. J. Shah, A. C. Drake, P. C. DeMuth, J. B. Lee, J. Chen and P. T. Hammond, ACS Nano, 2012, 6, 81 CrossRef CAS PubMed.
- R. Devanathan, D. Chase-Woods, Y. Shin and D. W. Gotthold, Sci. Rep., 2016, 6, 29484 CrossRef PubMed.
- B. Miksa, RSC Adv., 2015, 5, 87781 RSC.
- C. L. Weaver, J. M. LaRosa, X. Luo and X. T. Cui, ACS Nano, 2014, 8, 1834 CrossRef CAS PubMed.
- M. Choi, K.-G. Kim, J. Heo, H. Jeong, S. Y. Kim and J. Hong, Sci. Rep., 2015, 5, 17631 CrossRef CAS PubMed.
- H. Hu, X. Wang, K. I. Lee, K. Ma, H. Hu and J. H. Xin, Sci. Rep., 2016, 6, 31815 CrossRef CAS PubMed.
- J. Qian and F. P. Wu, Chem. Mater., 2007, 19, 5839–5841 CrossRef CAS.
- S. D'Souza, Adv. Pharmacol., 2014, 2014, 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25353a |
| This journal is © The Royal Society of Chemistry 2017 |