
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile synthesis of MOFs with uncoordinated carboxyl groups for selective CO2 capture via postsynthetic covalent modification
Feng Zhoua,
Jingjing Zhouab,
Xuechao Gaoa,
Chunlong Kong*a and
Liang Chen*a
aInstitute of New Energy Technology, Ningbo Institute of Material Technology and Engineering, Ningbo, Zhejiang 315201, China. E-mail: kongchl@nimte.ac.cn; chenliang@nimte.ac.cn
bSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China
First published on 17th January 2017
Abstract
A postsynthetic covalent strategy involving dual-acyl chloride has been developed to introduce uncoordinated carboxyl groups into amine containing metal–organic frameworks (MOFs). The carboxyl group functionalized MOFs have been characterized by various techniques, including X-ray diffraction patterning, scanning electron microscopy, Fourier transform infrared spectroscopy, nuclear magnetic resonance, thermal gravimetric analysis, and gas adsorption. Results clearly indicated uncoordinated carboxyl groups were successfully grafted to the MIL-101(Cr)–NH2 framework. In addition, most of the amine groups (>80%) were grafted with carboxyl groups, which indicates this method is very effective. The thermal stability and adsorption selectivity of CO2/N2 were substantially enhanced, albeit the BET surface areas and total pore volumes were reduced. These observations could be explained by the effect of elimination of macropores in the framework due to the projecting of new functional groups in pore apertures. Here the successful fabrication of a MOF with uncoordinated carboxyl groups provides the possibility of efficiently modifying other MOFs.
Introduction
Metal–organic frameworks (MOFs), a new class of porous materials, are made by linking tunable inorganic and organic units.1 In the last two decades, a particularly large number of MOFs have been successfully synthesized,2–7 and the versatile characteristics of their chemical moieties have endowed MOFs with many special properties relevant to fuel cell,8 catalysis9 and biomedical applications.10 Because of their ultrahigh porosity and the possibility of functionality, they play an important role in gas adsorption and separation.11–15 For instance, MOFs are becoming one of the most promising candidates to reduce carbon dioxide (CO2) emissions, which is useful to resolve the increasingly severe global warming problem.13,16,17 However, very limited MOFs show satisfactory CO2 capture performance at low pressure and ambient temperature due to the weak interaction between CO2 and the frameworks. In order to improve the adsorption performance, tremendous efforts have been devoted to functionalization of MOFs. To design MOFs with desired properties, the introduction of various functional groups into MOFs has attracted considerable interests. Postsynthetic covalent modification provides a variety of means to chemically modify the structures of MOFs,18–21 among which the direct grafting of functional groups on the organic linkers is effective, and has been widely applied.22–24Interestingly, considerable reports on postsynthetic covalent modification involve the use of amine-contained MOFs, which is beneficial for enhancing the CO2 capture performance. This is partly because of their convenient property to incorporate the 2-amino-1,4-benzenedicarboxylate into a number of BDC-derived MOFs. Amine groups in MOFs not only exhibit affinities with acidic gases molecules such as CO2, but also offer binding sites to accept functional groups. Previous studies have shown that amine groups in framework could be covalently modified by reagents such as peptide coupling,25 acetic anhydride26 and aldehyde.27,28 Indeed, a wide range of chemical moieties has been successfully incorporated into MOFs via amine groups such as photo-switchable functional groups,29 halo- and azo dye-functional groups22 et al. Recently, Bai and co-workers have inserted –CONH– groups into rht-type MOFs through acylamide-bridging linkers by in situ solvothermal reaction.30,31 Especially, uncoordinated carboxyl group functionalized MOFs exhibit promise for application in separation of transition metal ions,32 catalysis33 and sensing of ions.34
In a previous study, we obtained a type of amine-functionalized MIL-101(Cr) nanoparticles via a direct synthetic method.35 The resulting MIL-101(Cr)–NH2 possesses high surface area, good thermal stability, and selective adsorption of CO2.13,36 In addition, it contains mesoporous cavities, which are likely to preserve sufficient remaining guest accessibility after functionalization with a bulky group. Inspired by the aforementioned advantages of MIL-101(Cr)–NH2 and carboxyl group functionalized MOFs, it was selected in our project to explore the possibility of grafting uncoordinated carboxyl group via postsynthetic covalent modification in the present study. In this article, MIL-101(Cr)–NH2 was covalently modified by two kinds of dual-acyl chlorides for the first time, under a mild condition and simple process. Results show that uncoordinated carboxyl groups are successfully introduced to MIL-101(Cr)–NH2 framework. In addition, functionalized MIL-101(Cr)–NH2 exhibited enhanced thermal stabilities, and higher CO2/N2 ideal selectivities than the pristine MIL-101(Cr)–NH2. We anticipate that the proposed strategy is also applicable for the functionalization of other MOFs.
Experimental
Materials
Chromium nitrate [Cr(NO3)3·9H2O, TCI], 2-aminoterephthalic acid (H2N-H2BDC, Sigma-Aldrich), sodium hydroxide (NaOH, Aladdin), N,N-dimethylformamide (DMF, TCI), tetrahydrofuran (THF, TCI), oxalyl chloride [(COCl)2, TCI], triethylamine (TEA, TCI), and terephthaloyl dichloride (C8H4Cl2O2, TCI) were used as received from vendors without further purification. Deionized water and THF were used as solvents.Synthesis of MIL-101(Cr)–NH2
MIL-101(Cr)–NH2 was synthesized mainly according to the procedure reported in our previous work,35 with an optimized amount of sodium hydroxide. Typically, 2-aminoterephthalic acid (H2N-H2BDC) (800 mg, 2 mmol), chromic nitrate (360 mg, 2 mmol) and sodium hydroxide (4 mmol) were dispersed in deionized water (15 mL) and ultrasonically mixed for 30 min. Subsequently, the solution was heated in a 50 mL Teflon-lined autoclave at 150 °C for 12 h under autogenous pressure, and then left to cool down to ambient temperature. The green products were collected by centrifugation and washed by DMF first to remove the most unreacted 2-aminoterephthalic acid on the surface, and then the obtained powder was cleaned by hot alcohol at 90 °C for 6 h twice in an autoclave to remove unreacted 2-aminoterephthalic acid in the pores. Finally, the products were dried at 90 °C under vacuum.Postsynthetic covalent modification
Instruments in the reactions were thoroughly dried prior to use. Firstly, MIL-101(Cr)–NH2 (150 mg) powder was placed in a quartz tube and activated at 160 °C and 10−2 kPa for 10 h to remove the remained guest molecules. Then the MIL-101(Cr)–NH2 powder was gradually dispersed into the solution, containing dry tetrahydrofuran (THF) (8 mL) and oxalyl chloride (156 mg, 1.23 mmol) or terephthaloyl dichloride (250 mg, 1.23 mmol), under continuous fierce stirring. A certain amount of triethylamine (TEA) was dropwisely added into the mixture after 2 h, and further stirred overnight at ambient temperature. The precipitates were collected by centrifugation and then thoroughly washed by THF and water before drying at 100 °C under vacuum. The obtained samples were denoted as MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 to represent the treatment by oxalyl chloride and terephthaloyl dichloride, respectively. All processes were carried out under anhydrous and argon atmosphere. The reaction process was illustrated as Scheme 1.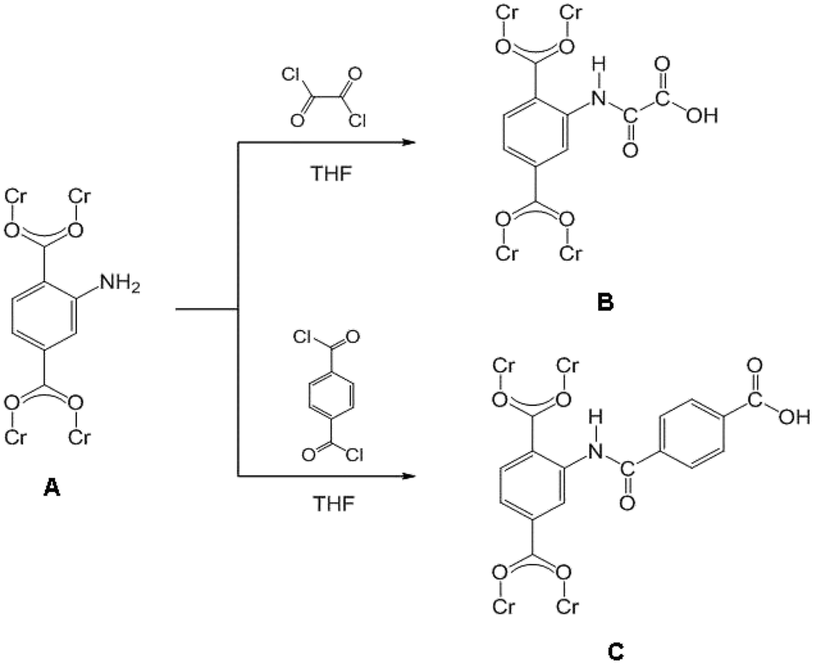 | ||
| Scheme 1 Schematic illustration of postsynthetic covalent modification of MIL-101(Cr)–NH2 (A) using dual-acyl chloride. The resulting products: MIL-101(Cr)–amide-1 (B) and MIL-101(Cr)–amide-2 (C). | ||
Characterization
Powder X-ray diffraction (PXRD) patterns were obtained on a Bruker AXS D8 Advance diffractometer using Cu Kα radiation at room temperature at a voltage of 40 kV and 40 mA. The morphologies of the as-prepared samples were examined using a field emission scanning electron microscope (FESEM, Hitachi, S-4800). Nuclear magnetic resonance (NMR) measurements were performed using a Bruker FT-NMR-spectrometer, Avance III (1H: 400 MHz). Thermal gravimetric analysis (TGA) was conducted in air from room temperature to 800 °C at a heating rate of 10 °C min−1 using a system provided by Mettler Toledo (model TGA/DSC1) apparatus. Fourier transform infrared spectroscopy (FTIR) of the samples was recorded on KBr/sample pellets in a TENSOR 27 apparatus to determine the functional groups. The BET surface area was calculated over the range of relative pressures between 0.05 and 0.10 bar with a N2 adsorption isotherm at 77.3 K (ASAP 2020, Micromeritics). Adsorption of CO2 and N2 at pressures up to 0.1 MPa were measured at 298 K by a standard volumetric analyzer (ASAP 2020, Micromeritics). Before each measurement, the samples were evacuated at 150 °C for 6 h to remove the guest molecules. All the gases used in the study had a minimum purity of 99.99%.Adsorption
The as-prepared functionalized MIL-101(Cr) samples were immediately measured after moving from the vacuum. Before each measurement, the sample was evacuated at 160 °C for 12 h. In the adsorption measurement, the apparatus first gave a desired pressure of the gas to the sample holder, and the pressure values were recorded in the sample holder to calculate the adsorption amount.The adsorption isotherms of CO2 and N2 were measured using a volumetric apparatus (ASAP 2020, Micromeritics). The relationship of adsorption amount versus gas bulk pressure was determined using the Langmuir–Freundlich37,38 fit for the isotherms:
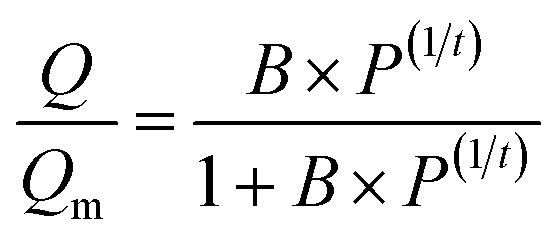
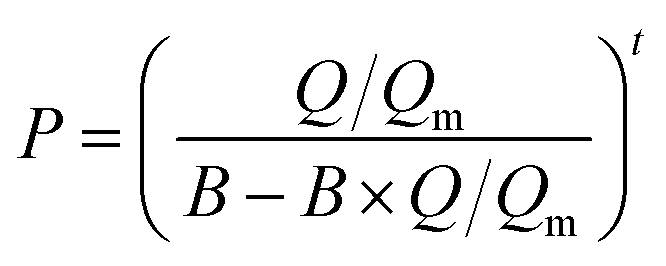
The adsorption selectivities of CO2/N2 (α) for all the samples were estimated by the following equation,39,40 where Qi is the
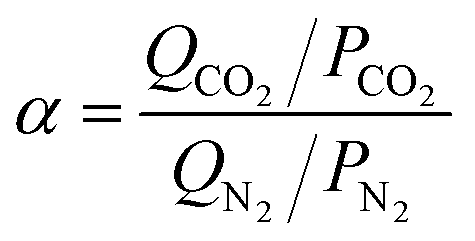
Results and discussion
Fig. 1A describes the PXRD results for the as-synthesized MIL-101(Cr), MIL-101(Cr)–NH2 and the two samples prepared by postsynthetic covalent modification. It is evident that the XRD patterns of the three samples are similar to the characteristic peaks for the MIL-101(Cr), indicating high purity of our products,31 thus the pore structures are preserved during the covalent modification process. Here the three modified samples also show broader and weaker Bragg reflections of the XRD patterns than the MIL-101(Cr), because the corresponding particle sizes are smaller than the latter.41 These observations are comparatively verified in the SEM images. As shown in Fig. 1B, the morphologies of MIL-101(Cr)–NH2 remain nearly intact after the functional incorporation, while the crystal size is very small, much smaller than that of MIL-101(Cr).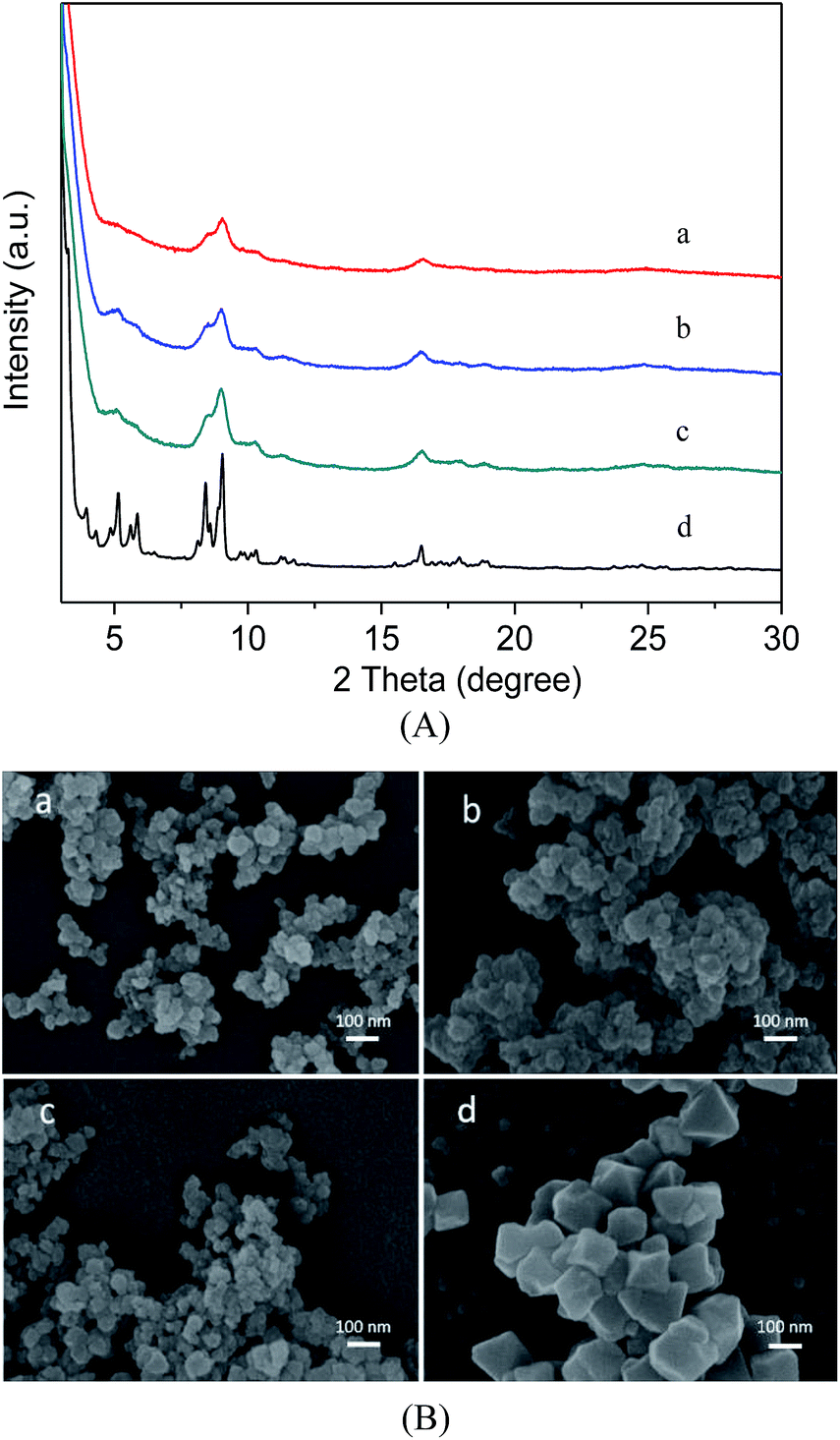 | ||
| Fig. 1 Powder XRD patterns (A) and SEM images (B) of MIL-101(Cr)–NH2 (a), MIL-101(Cr)–amide-1 (b), MIL-101(Cr)–amide-2 (c), MIL-101(Cr) (d). | ||
To further ensure the successful incorporation of the MIL-101(Cr)–NH2 framework, IR spectra was collected on the as-synthesized MIL-101(Cr)–NH2 and products by postsynthetic covalent modification. For MIL-101(Cr)–NH2, the double peaks at 3490 cm−1 and 3363 cm−1 are clearly observed (Fig. 2), which are assigned to the asymmetrical and symmetrical stretching vibration of the amine groups. In the lower frequency region, the two peaks around 1593 cm−1 correspond to the N–H bending vibration and the C–N stretching vibrations (1340 cm−1 and 1259 cm−1) of aromatic amines, respectively.29 Besides, the incorporation of amino-dicarboxylate within the structure is confirmed by the characteristic –(O–C–O)– stretching vibrations at 1500 cm−1 and 1430 cm−1.42 As for MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2, three additional signals including N–H stretching vibration (3310 cm−1 and 3313 cm−1) of amide groups, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (1700 cm−1 and 1710 cm−1) and C–N stretching vibration (1271 cm−1 and 1281 cm−1) are directly observed. Here the IR spectra of the samples have validated the presence of amide and carboxyl groups in the MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 samples, respectively.
O stretching vibration (1700 cm−1 and 1710 cm−1) and C–N stretching vibration (1271 cm−1 and 1281 cm−1) are directly observed. Here the IR spectra of the samples have validated the presence of amide and carboxyl groups in the MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 samples, respectively.
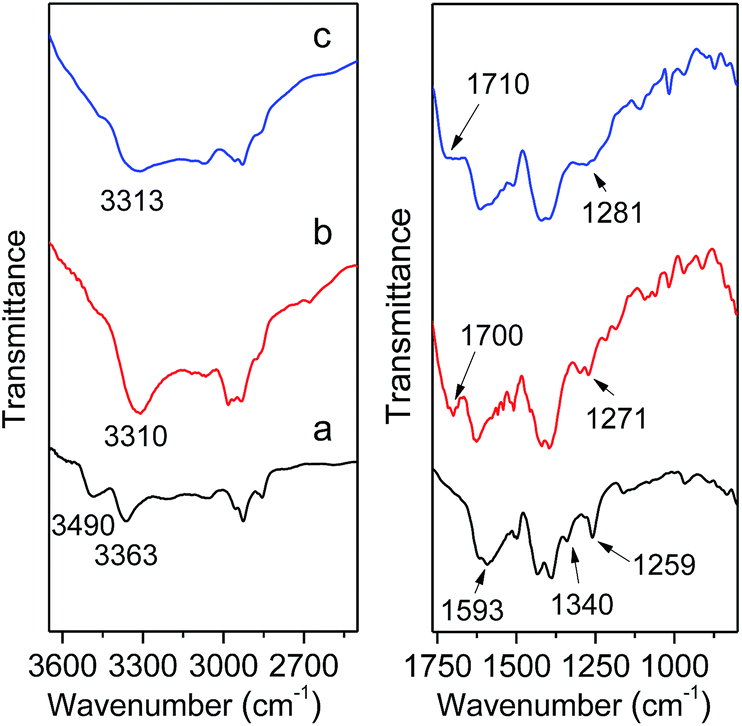 | ||
| Fig. 2 IR spectra of MIL-101(Cr)–NH2 (a), MIL-101(Cr)–amide-1 (b), MIL-101(Cr)–amide-2 (c). | ||
Meanwhile, NMR-measurements were also conducted to inspect the linker structures of the original and the modified samples. Fig. 3 illustrates the 1H-NMR spectra of 2-amino-terephthalic acid (H2N-H2BDC) and the digested products from the two samples. For H2N-H2BDC in Fig. 3a, signals at 7.75, 7.37 and 7.00 ppm are assigned to protons A, B and C on the benzene. For H2BDC–amide-1 in Fig. 3b, the signals are mainly lumped together, and the characteristic peaks for H2N-H2BDC is hardly seen. The peak at 12.57 ppm is assigned to the amide proton, with the signals at 9.23, 8.09, 7.70 ppm for the proton 3, 1 and 2 (numbering in the figure), respectively. Similarly, Fig. 3c and d describe the majority of characteristic information for H2BDC–amide-2, despite having few weak signals due to the presence of unreacted H2N-H2BDC. The signal at 12.12 ppm and 9.20 ppm are produced by the amide proton and the proton 3 (numbering in the figure), respectively, and the other protons are marked in the region of 8.2 ppm to 7.6 ppm. The conversion rates of amine groups calculate from the integral data are above 97% and nearly 87% for MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2, respectively, implying the high conversion rate of the amino groups in our work. On the contrary, the conversion rates of amine in IRMOF-3 and MIL-68(In)–NH2 are less than 13% and 10% according to the research, respectively.27,43 Cohen and co-workers have reported that the rate for the covalent modification of IRMOF-3, UMCM-1-NH2 and DMOF-1-NH2 are around 32–76%.44 Therefore, dual-acyl chloride is very suitable for the amine functionalization of MOFs modified by postsynthetic covalent. In conclusion, the IR and 1H-NMR data have successfully verified the accomplishment of covalent modification of MIL-101(Cr)–NH2 by reacting with dual-acyl chloride.
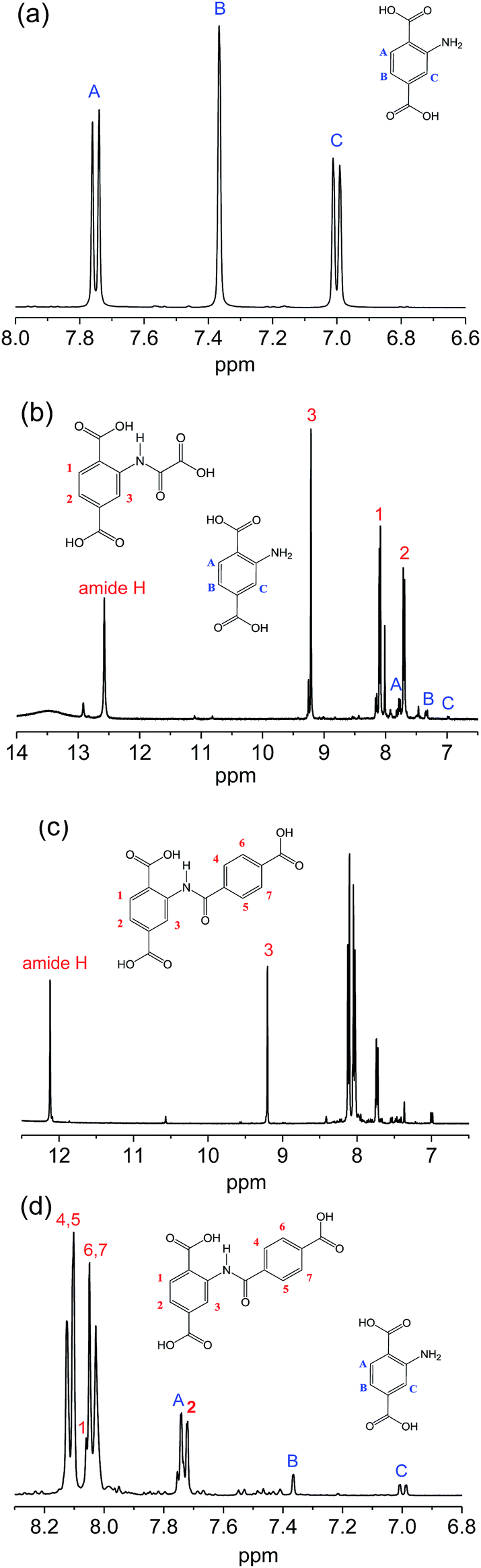 | ||
| Fig. 3 1H-NMR spectra of 2-aminoterephthalic acid (a), the digested products from MIL-101(Cr)–amide-1 (b), and MIL-101(Cr)–amide-2 (c and d). | ||
Moreover, the pore networks of the samples are explored to elucidate a better understanding of their adsorption mechanism.
Fig. 4 plot the adsorption–desorption loops of the three samples by N2, and a type I adsorption behavior occurred, with small hysteresises due to the interstitial voids formed by the small crystal particles. Comparing with MIL-101(Cr)–NH2, BET surface areas and total pore volumes are substantially reduced in both MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2, due to the presence of carboxyl groups projecting into the pores. The observed decrements of surface areas have been also found in many other studies for various MOFs modified by postsynthetic covalent.45 Fig. 5 provides the pore size distributions of the samples based on the interpretation of nonlinear density functional theory (NLDFT), and the results implies that the average pore sizes of MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 become smaller than that of MIL-101(Cr)–NH2. It is evident that such a result is caused by the elimination of the larger pore in MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2. On the other hand, despite two smallest pores are also eliminated in MIL-101(Cr)–amide-1, some pores are newly-created in MIL-101(Cr)–amide-2, locating at 7.7 Å, 12.4 Å and 15.2 Å, and this is due to the partial occupation of larger groups. The results of the pore parameters are outlined in Table 1. It have found that the surface area and pore volume of MIL-101(Cr) with functional groups decreased with the increases in group sizes. Further, the proportion of micropores agree with the size variation of the functional groups, and this is important to enhance the CO2/N2 selectivity for its smaller kinetic size than N2.
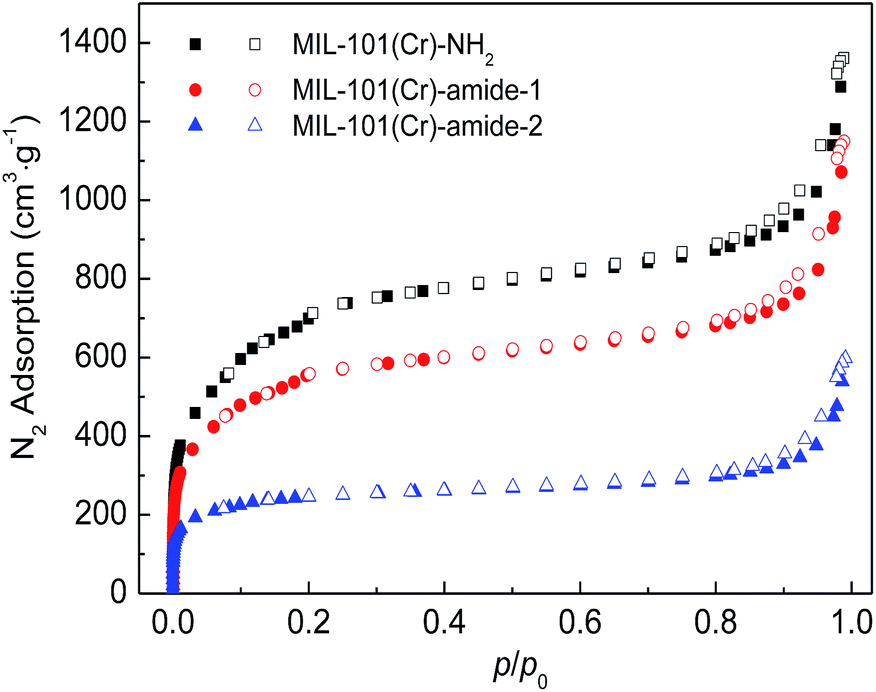 | ||
| Fig. 4 N2 adsorption–desorption isotherms on MIL-101(Cr)–NH2, MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 samples at 77.3 K. | ||
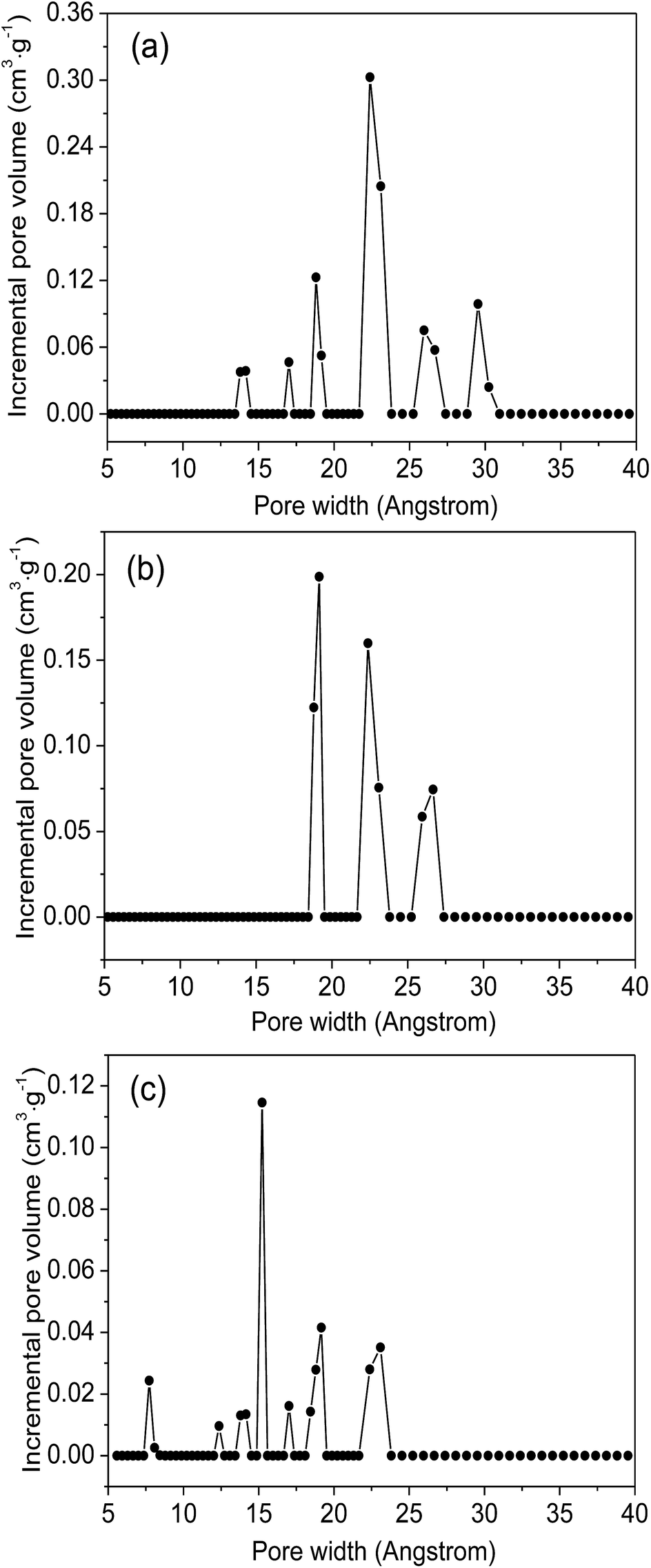 | ||
| Fig. 5 NLDFT pore size distribution of MIL-101(Cr)–NH2 (a), MIL-101(Cr)–amide-1 (b) and MIL-101(Cr)–amide-2 (c). | ||
| Sample | SBET (m2 g−1) | Vtotal (cm3 g−1) | Vmicro (cm3 g−1) | Vmicro/Vtotal |
|---|---|---|---|---|
| MIL-101(Cr)–NH2 | 2558 | 2.11 | 0.18 | 8.53% |
| MIL-101(Cr)–amide-1 | 2005 | 1.78 | 0.16 | 8.99% |
| MIL-101(Cr)–amide-2 | 861 | 0.93 | 0.20 | 21.51% |
To evaluate the stability of as-prepared MIL-101(Cr)–NH2 and the samples under postsynthetic covalent modification, the thermal gravimetric analysis was carried out. As shown in Fig. 6, MIL-101(Cr)–NH2 starts to decompose at 250 °C in the air, similar to the discovery reported in the literature.35 In contrast, the decomposing temperatures of MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 increase up to 280 °C and 380 °C, respectively, which is slightly lower than that of the value for ZIF-8 (ca. 500 °C), but higher than most of MOF materials.46 It have reported that the thermal stability of MOFs is improved by reducing the primary amine to a secondary one,28 thus the incorporation of larger groups in the framework favors the improvement of the thermal stability.
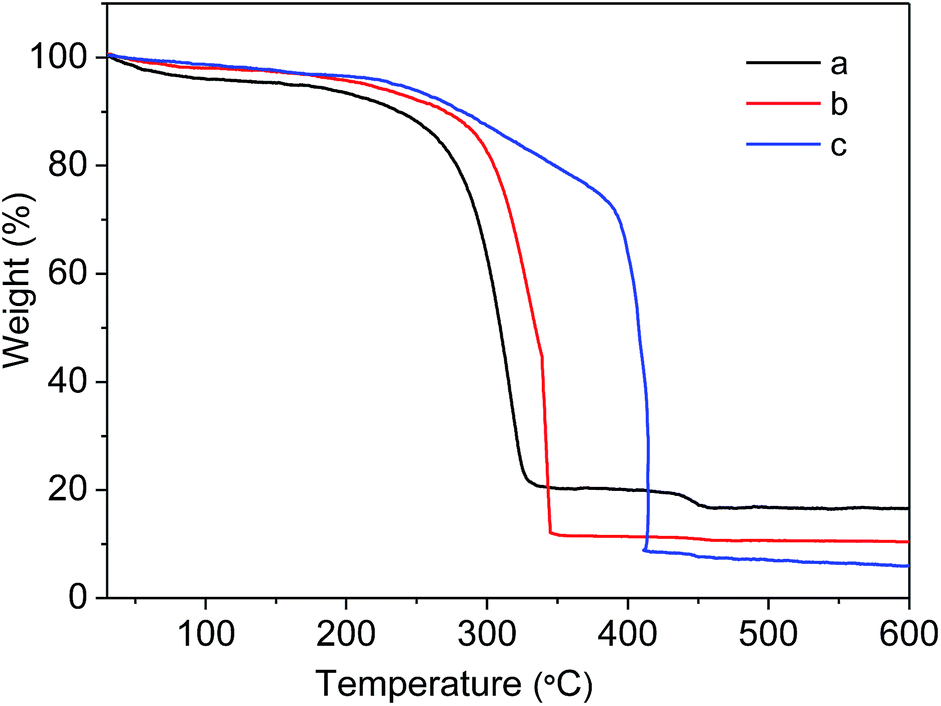 | ||
| Fig. 6 TGA curves: (a) MIL-101(Cr)–NH2, (b) MIL-101(Cr)–amide-1 and (c) MIL-101(Cr)–amide-2. | ||
Finally, the CO2 and N2 adsorption isotherms were conducted at a temperature of 298 K to evaluate the adsorption capability of the as-prepared functionalized MIL-101(Cr) materials. As shown in Fig. 7, although the CO2 adsorption amounts of the two modified samples are consistently smaller than the corresponding adsorption of MIL-101(Cr)–NH2 within the entire pressure range, relatively higher ideal selectivities of CO2 over N2 are produced by the incorporation of amide and carboxyl groups (Fig. 8). Specifically, the ideal selectivities at 0.1 MPa are up to 10.93, 12.49 and 16.44 for MIL-101(Cr)–NH2, MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2, respectively. Amide groups have been previously reported to enhance CO2 adsorption.31 Further, the incorporation of carboxyl groups into the MOFs is also beneficial to the CO2 capture, and the performance is even better than the results achieved by amine groups,47 due to a higher heat of adsorption for carboxyl groups, thus a higher efficiency of adsorption behavior at low pressure occurred. Herein the high selectivities in MIL-101(Cr)–amide-1 and MIL-101(Cr)–amide-2 are physically caused by the strong interactions between the modified groups and CO2 molecules. Moreover, since the quadrupole moment of N2 (−4.6 × 10−40 C m2) is much lower than the value of CO2 (−14 × 10−40 C m2),48 the enhanced polarizability (electrostatic interaction) of the framework caused by the incorporated groups is also an important factor for the selectivity enhancement.49 Thus, all of these factors contributes to a higher uptake of CO2 than of N2.
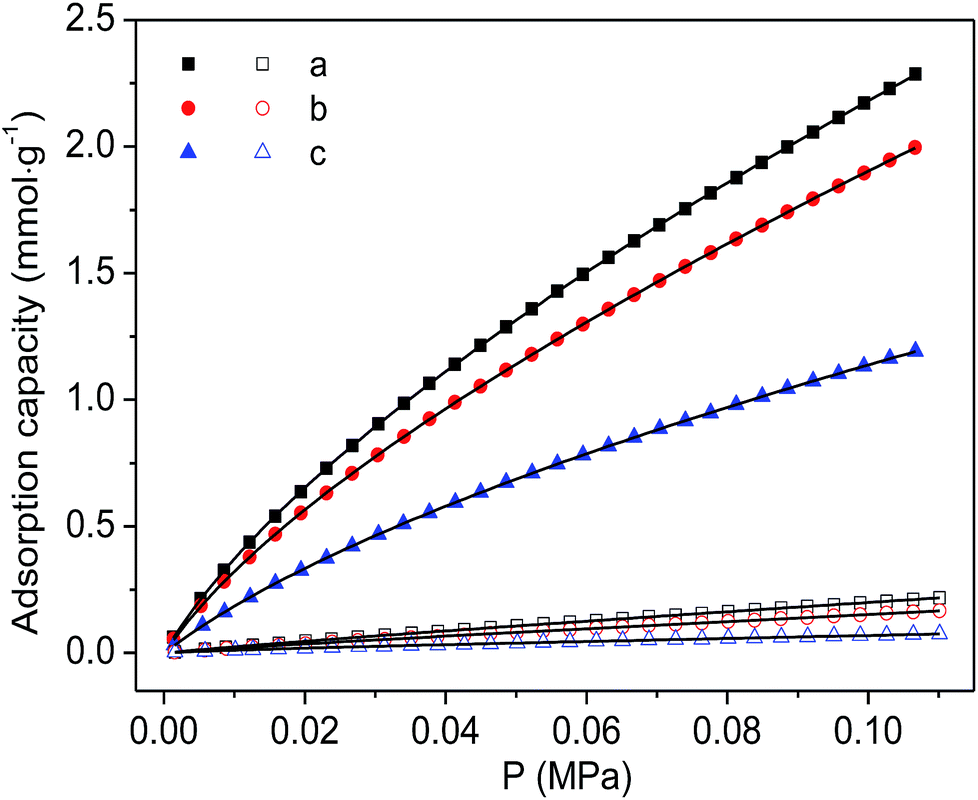 | ||
| Fig. 7 Adsorption isotherms of CO2 (solid) and N2 (hollow) on the samples at 298 K and the fitted isotherms by DSL model: (a) MIL-101(Cr)–NH2, (b) MIL-101(Cr)–amide-1 and (c) MIL-101(Cr)–amide-2. | ||
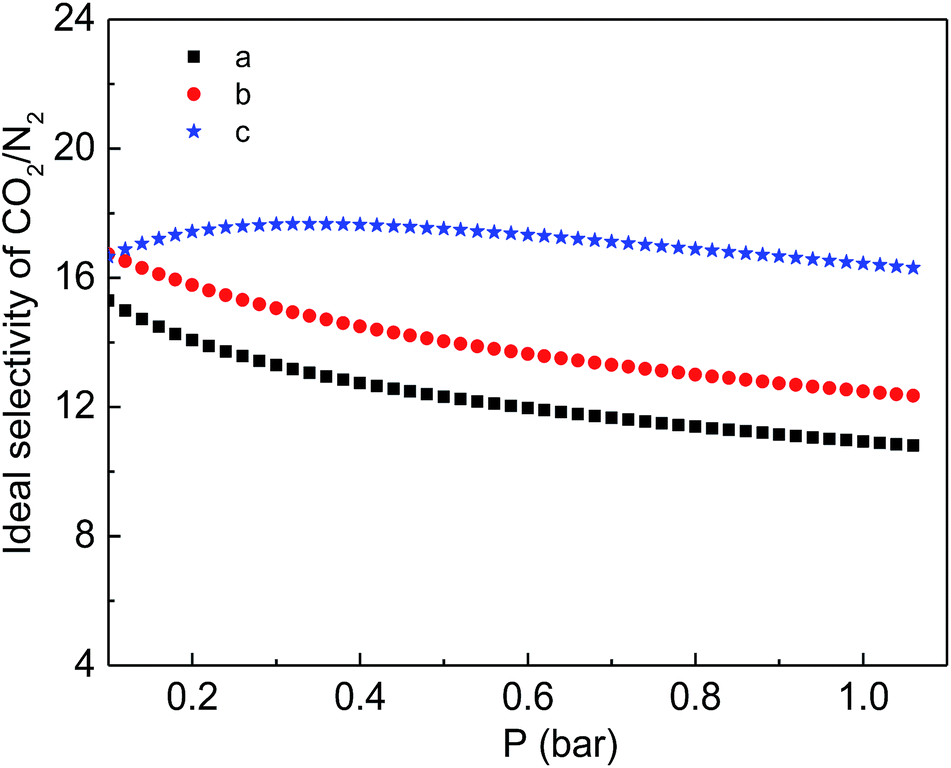 | ||
| Fig. 8 Ideal separation factor of CO2/N2 for the samples at 298 K: (a) MIL-101(Cr)–NH2, (b) MIL-101(Cr)–amide-1 and (c) MIL-101(Cr)–amide-2. | ||
Conclusions
In summary, uncoordinated carboxyl groups were successfully grafted on MIL-101(Cr)–NH2 framework, and a general and facile method was developed to modify MOFs by dual-acyl chloride at mild conditions. In addition, most of the amine groups of MIL-101(Cr)–NH2 were successfully grafted with carboxyl groups in the present work, indicating the proposed strategy is very efficient. The carboxyl groups functionalized samples exhibited enhanced selectivities of CO2/N2 in comparison with the original MIL-101(Cr)–NH2 at low pressure and ambient temperature. Besides, the thermal stabilities of the functionalized samples were also enhanced. This could be caused by the positive evolution of pore due to the introducing of carboxyl groups into the pores apertures. The uncoordinated carboxyl groups functionalized samples will be used for fabrication of MOF-based mixed matrix membrane and separation of transition metal ions, which are currently underway in our laboratory. Overall, the present work provides a high efficient strategy to functionalize amine-containing MOF with uncoordinated carboxyl groups.Acknowledgements
We gratefully acknowledge the financial support by the National Science Foundation of China (Grants No. 51272260, 51572272), the aided program for Science and Technology Innovative Research Team of Ningbo Municipality (Grants No. 2015B11002 and 2014B81004), Natural Science Foundation of Zhejiang Province (Grants No. LY15E020008), and the Natural Science Foundation of Ningbo (Grant No.: 2015A610087).Notes and references
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
.
- L.-B. Sun, J.-R. Li, J. Park and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 126 CrossRef CAS PubMed
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018 CrossRef CAS PubMed
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed
- H. S. Cho, H. Deng, K. Miyasaka, Z. Dong, M. Cho, A. V. Neimark, J. K. Kang, O. M. Yaghi and O. Terasaki, Nature, 2015, 527, 503 CrossRef PubMed
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gandara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365 CrossRef CAS PubMed
- M. Tafipolsky, S. Amirjalayer and R. Schmid, J. Comput. Chem., 2007, 28, 1169 CrossRef CAS PubMed
- Y. Ren, G. H. Chia and Z. Gao, Nano Today, 2013, 8, 577 CrossRef CAS
- D. Y. Shi, C. He, B. Qi, C. Chen, J. Y. Niu and C. Y. Duan, Chem. Sci., 2015, 6, 1035 RSC
- M. C. Das, S. Xiang, Z. Zhang and B. Chen, Angew. Chem., 2011, 50, 10510 CrossRef CAS PubMed
- Y. Lin, Q. Yan, C. Kong and L. Chen, Sci. Rep., 2013, 3, 1859 Search PubMed
- Z. R. Herm, E. D. Bloch and J. R. Long, Chem. Mater., 2014, 26, 323 CrossRef CAS
- X. Huang, J. Lu, W. Wang, X. Wei and J. Ding, Appl. Surf. Sci., 2016, 371, 307 CrossRef CAS
- Y. Lin, C. Kong, Q. Zhang and L. Chen, Adv. Energy Mater. DOI:10.1002/aenm.201601296
- Y. Lin, H. Lin, H. Wang, Y. Suo, B. Li, C. Kong and L. Chen, J. Mater. Chem. A, 2014, 2, 14658 CAS
- R. Zou, P.-Z. Li, Y.-F. Zeng, J. Liu, R. Zhao, H. Duan, Z. Luo, J.-G. Wang, R. Zou and Y. Zhao, Small, 2016, 12, 2386 CrossRef PubMed
- D. Danaci, R. Singh, P. Xiao and P. A. Webley, Chem. Eng. J., 2015, 280, 486 CrossRef CAS
- S. M. Cohen, Chem. Rev., 2012, 112, 970 CrossRef CAS PubMed
- B. Liu, S. Jie, Z. Bu and B.-G. Li, RSC Adv., 2014, 4, 62343 CAS
- F. Yang, C.-X. Yang and X.-P. Yan, Talanta, 2015, 137, 136 CrossRef CAS PubMed
- M. A. Gotthardt, S. Grosjean, T. S. Brunner, J. Kotzel, A. M. Gaenzler, S. Wolf, S. Braese and W. Kleist, Dalton Trans., 2015, 44, 16802 RSC
- D. Jiang, L. L. Keenan, A. D. Burrows and K. J. Edler, Chem. Commun., 2012, 48, 12053 RSC
- W. Ma, L. Xu, Z. Li, Y. Sun, Y. Bai and H. Liu, Nanoscale, 2016, 8, 10908 RSC
- L. Xu, Y. P. Luo, L. Sun, S. Pu, M. Fang, R. X. Yuan and H. B. Du, Dalton Trans., 2016, 45, 8614 RSC
- H. Hintz and S. Wuttke, Chem. Commun., 2014, 50, 11472 RSC
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368 CrossRef CAS PubMed
- M. J. Ingleson, J. P. Barrio, J.-B. Guilbaud, Y. Z. Khimyak and M. J. Rosseinsky, Chem. Commun., 2008, 23, 2680 RSC
- A. D. Burrows and L. L. Keenan, CrystEngComm, 2012, 14, 4112 RSC
- A. Modrow, D. Zargarani, R. Herges and N. Stock, Dalton Trans., 2012, 41, 8690 RSC
- B. Zheng, Z. Yang, J. Bai, Y. Li and S. Li, Chem. Commun., 2012, 48, 7025 RSC
- B. Zheng, J. Bai, J. Duan, L. Wojtas and M. J. Zaworotko, J. Am. Chem. Soc., 2011, 133, 748 CrossRef CAS PubMed
- X. Meng, R. L. Zhong, X. Z. Song, S. Y. Song, Z. M. Hao, M. Zhu, S. N. Zhao and H. J. Zhang, Chem. Commun., 2014, 50, 6406 RSC
- X. Jia, S. Wang and Y. Fan, J. Catal., 2015, 327, 54 CrossRef CAS
- W. Sun, J. Wang, G. Zhang and Z. Liu, RSC Adv., 2014, 4, 55252 RSC
- Y. Lin, C. Kong and L. Chen, RSC Adv., 2012, 2, 6417 RSC
- K. Zhang, Y. Chen, A. Nalaparaju and J. Jiang, CrystEngComm, 2013, 15, 10358 RSC
- E. J. Granite and H. W. Pennline, Ind. Eng. Chem. Res., 2002, 41, 5470 CrossRef CAS
- J. An and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 5578 CrossRef CAS PubMed
- Q. Yan, Y. Lin, C. Kong and L. Chen, Chem. Commun., 2013, 49, 6873 RSC
- T. M. McDonald, D. M. D'Alessandro, R. Krishna and J. R. Long, Chem. Sci., 2011, 2, 2022 RSC
- S. Wuttke, S. Braig, T. Preiss, A. Zimpel, J. Sicklinger, C. Bellomo, J. O. Radler, A. M. Vollmar and T. Bein, Chem. Commun., 2015, 51, 15752 RSC
- S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Ferey and N. Stock, Inorg. Chem., 2008, 47, 7568 CrossRef CAS PubMed
- J. Canivet, S. Aguado, G. Bergeret and D. Farrusseng, Chem. Commun., 2011, 47, 11650 RSC
- Z. Wang, K. K. Tanabe and S. M. Cohen, Chem.–Eur. J., 2010, 16, 212 CrossRef CAS PubMed
- H. Hintz and S. Wuttke, Chem. Mater., 2014, 26, 6722 CrossRef CAS
- J. Qian, F. Sun and L. Qin, Mater. Lett., 2012, 82, 220 CrossRef CAS
- Z. Xiang, S. Leng and D. Cao, J. Phys. Chem. C, 2012, 116, 10573 CAS
- C. Graham, J. Pierrus and R. E. Raab, Mol. Phys., 1989, 67, 939 CrossRef CAS
- X. Gao, J. C. Diniz da Costa and S. K. Bhatia, J. Membr. Sci., 2014, 460, 46 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |