
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic impact of alloyed Al on the corrosion behavior of Co50Ni23Ga26Al1.0 magnetic shape memory alloy and catalysis applications for efficient electrochemical H2 generation†
Mohammed. A. Amin*ab,
Nader El-Bagouryac,
M. H. H. Mahmoudac,
M. M. Hessienac,
Sayed S. Abd El-Rehimb,
Joanna Wysockad and
Jacek Ryld
aMaterials Science and Engineering Group, Department of Chemistry, Faculty of Science, Taif University, 888 Hawiya, Saudi Arabia. E-mail: maaismail@yahoo.com
bDepartment of Chemistry, Faculty of Science, Ain Shams University, 11566 Abbassia, Cairo, Egypt
cCentral Metallurgical Research and Development Institute (CMRDI), P.O. Box: 87 Helwan, Cairo, Egypt
dDepartment of Electrochemistry, Corrosion and Materials Engineering, Chemical Faculty, Gdańsk University of Technology, Narutowicza 11/12, 80-233 Gdańsk, Poland
First published on 13th January 2017
Abstract
The electrochemical and corrosion behaviour of Co50Ni23Ga27−xAlx (x = 0 and 1.0 wt%) magnetic shape memory alloys (MSMAs) was studied in 0.5 M NaCl solutions using various electrochemical techniques. Results showed remarkable activation of the tested MSMA toward pitting corrosion upon alloying it with Al. XPS examination confirmed the activation influence of alloyed Al. It proved that the presence of Al in the alloy's matrix weakens its passivity, as manifested by a lower amount of gallium oxides and Cl− adsorption in the aluminium containing MSMA sample. Alloyed Al also activated significantly the tested MSMA for the hydrogen evolution reaction (HER), as indicated by cathodic polarization, electrochemical impedance spectroscopy (EIS), and faradaic efficiency (FE) measurements. Such measurements were performed in 0.1 M KOH solutions and showed that the Co50Ni23Ga26Al1.0 alloy is much more active for the HER than Co, Ni, Co50Ni50, and Co50Ni23Ga27 electrodes. The catalytic impact of pitting corrosion, proved to be catalyzed by Al, on the HER activity of the CoNiGaAl alloy was also studied. The pitted Co50Ni23Ga26Al1.0 MSMA, the best catalyst here, exhibited high HER catalytic performance with an exchange current density (jo) of 0.2 mA cm−2 and FE 96%, and thus approached Pt/C (jo = 0.6 mA cm−2 and FE ∼ 100%). Our best catalyst also showed good stability and durability after 3000 cycles of cathodic polarization between the corrosion potential (Ecorr) and −1.0 V vs. RHE, and 24 h of electrolysis at a high cathodic current density of 100 mA cm−2. Microstructure changes made by Al, together with findings obtained from SEM/EDX mapping and XPS studies, were used to interpret its activation influence towards the HER.
1. Introduction
Shape memory alloys (SMAs) are an important class of smart materials that have the ability to remember a shape. These alloys are unique materials having a different philosophy than the commercial ones, such as steel and titanium alloys. Large recoverable shape change is observed as a result of cooperative motion of atoms, which is the result of diffusion-less solid–solid phase transformation. The time-independent solid–solid phase transformation can be triggered by change in temperature, stress or magnetic field.1–3 Current practical uses of SMAs are limited to below 100 °C which is the limit for the transformation temperatures of most commercially successful SMAs such as NiTi and Cu-based alloys.A new class of these materials, termed as magnetic shape memory alloys (MSMAs), has recently discovered. Such materials have received much attention as they undergo both magnetic field induced strain and magnetic shape memory effect. This interesting effect has been first discovered in the Ni–Mn–Ga system.4,5 So far, the highest field-induced strain was only achieved for off-stoichiometric single crystals,6,7 where application of an external magnetic field has resulted in a strain of up to 10%.7 These changes provided this class of materials with additional properties, such as the elastocaloric and magnetocaloric effects.8,9 Such properties are of interest for applications in solid state cooling near to the room temperature.10 But, bulk polycrystalline materials are of technological interest, as they are easier to produce and less expensive than the single crystal ones. In addition, the fragility of single crystal MSMAs limits their practical applications. For these reasons, numerous studies have been performed on several candidates for MSMAs to find out new alternative materials with lower price and brittleness. These alternatives include: Fe–Pd,11 Fe–Pt,12 Ni–Mn–Al,13 and Co–Ni–Al.14,15 Recently, Ni–Fe–Al16 and Co–Ni–Ga17 have been emerged as new MSMAs, which are good alternatives to NiMnGa alloys, as they contain a γ phase capable of improving their ductility.
Since under application conditions MSMAs may undergo some corrosion problems,18 corrosion research studies on MSMAs have become essential. However, literature revealed very limited number of publications concerning corrosion behaviour of these materials.18–20 In addition, and to the best of our knowledge, there are no reports in the literature concerning the effect of Al addition as an alloying element on the corrosion behaviour of CoNiGa MSMA.
Successful electrode materials for the HER have to be stable, showing no signs of corrosion or passivation while having low overpotential. Paunovic et al.21 investigated numerous modifications of CoNi SMA concluding the necessity of further modification to increase the catalytic activity of HER.
Morphology, microstructure, internal stress and catalytic ability of the layers are strongly influenced by the structure of the substrate. In particular, morphology of alloy has a decisive influence on the kinetics of the surface redox reaction. The most practical approach to overcome the kinetic and diffusional limitations is to increase the real surface area of the electrode. Several studies on CoNi alloys report performance of submicron size powder electrodes,22 porous foams,23 or even encapsulating CoNi nanoalloys in ultrathin layers of graphene.24 Unlike other attempts to replace precious-metal-based electrocatalysts of HER, lowering efficiency and stability of such compounds, CoNi@C fabricated by Deng et al.24 are claimed to possess high performance. The optimized catalyst exhibits high stability and activity with an onset overpotential of almost zero versus the reversible hydrogen electrode (RHE) and an overpotential of only 142 mV at 10 mA cm−2, which is quite close to that of commercial 40% Pt/C catalysts.
On the other hand, electrochemical properties of thermally treated CoNiGa MSMA were investigated by Sanchez-Carrillo et al.25 It was revealed, that thermal treatments improve corrosion properties of alloy in neutral and acidic solutions while chlorides prevent spontaneous passivation which is otherwise observable at anodic oxidation.
Based on the literature presented above, the objective of the present work is two-fold; first, to study the effect of Al addition on the anodic behavior (including passivity and its breakdown and subsequent formation of pits and their repassivation) of the CoNiGa MSMA. It was also the purpose of this part to use chronoamperometry measurements to study the role of Al in the growth kinetics of the passive layer and pits. Such corrosion studies were performed in 0.5 M NaCl solutions. SEM/EDX and XPS examinations were used to study morphologies and compositions of the corroded surfaces devoid of and containing the alloyed Al.
The second objective of the present work is to evaluate, for the first time, the impact of alloyed Al on the electrocatalytic activity of CoNiGaAl SMA for the HER. HER activity measurements were conducted in deaerated aqueous KOH solutions (0.1 M) using linear sweep voltammetry and electrochemical impedance spectroscopy techniques. The HER activity of the best catalyst (the pitted CoNiGaAl MSMA) approached that of Pt/C. Such corrosion pretreatment, together with the activation influence of alloyed Al, led to the formation of deep pits with edges and walls proved to be active for the HER. Thus, pitting corrosion (a highly undesirable process) can be used to fabricate catalysts for wide applications. This may turn the attention of a number of researchers for future studies covering the title “pitting corrosion for useful materials fabrication”.
2. Experimental
2.1. Materials and solutions
Both of Co50Ni23Ga27Al0 and Co50Ni23Ga26Al1 alloys were produced by using arc-melting for the pure elements (99.99 wt% Co, 99.99 wt% Ni, 99.99 wt% Ga and 99.99 wt% Al) under controlled atmosphere (argon gas), in a water-cooled copper crucible. To get high level of homogenization, these alloys were melted for four times. After casting, both alloys were sliced into small-sized parts to be used in the various experimental studies. The same procedure was used for the manufacture of the Co50Ni50 alloy, which was used in HER catalytic activity measurements for comparison. These alloys were cast as cylindrical rods for the electrochemical tests. These rods machined carefully and mounted in polyester resin after the electric contact, with special care taken to prevent the presence of crevices. The exposed area was ∼1.0 cm2. Before each run, the samples were wet ground with 600-grit silicon carbide (SiC) paper and finally washed in distilled water, followed by immediate rinsing with absolute ethanol. The microstructural changes were investigated by a scanning electron microscope (JEOL JSM5410). The equilibrium phase composition was detected by an energy X-ray dispersive spectroscopy (EDS) attached to SEM. For the metallography study, agent of Nital's (15% nitric acid in ethanol) was used as chemical etching.All solutions used in this study were analytical grade chemical reagents purchased from Sigma-Aldrich. These solutions were freshly prepared using water purified by a Millipore Milli-Q system (resistivity: 18.2 MΩ cm).
2.2. Electrochemical study
A standard jacketed three-electrode cell was used for electrochemical measurements. A Pt electrode and a saturated calomel electrode (SCE) were used as auxiliary electrode and reference electrode, respectively. All potentials were measured against SCE. Electrochemical experiments were performed via connecting the cell to a potentiostat/galvanostat AUTOLAB (PGSTAT30) coupled to an Autolab frequency response analyzer (FRA) with FRA2 module connected to PC. Measurements were conducted in a 200 ml test solution (large enough to resist significant drifts in composition during the run). Prior to any electrochemical run, oxygen was removed from the test solutions by bubbling Ar through the solution for at least 30 min. The temperature was set at (25 °C ± 0.2 °C) using a temperature-controlled bath with water circulating through the outer cell jacket.For each electrochemical run, at least three separate experiments were performed to ensure results' reproducibility. Data obtained were found to be statistically significant. We calculated and reported their arithmetic mean and standard deviation accordingly.
ICP-AES (inductively coupled plasma atomic emission spectrometry) was also employed as an independent method of chemical analysis to confirm results obtained from polarizations and impedance measurements. In this method, the concentrations of both Ni2+ and Co2+ ions were determined for each alloy in 0.5 M NaCl solutions at 25 °C as a function of the time of immersion. In this respect, full immersion tests (1–15 days) were performed following the ASTM-G31 standard.26 The obtained results are collected in Table S2 (ESI†), where the amounts of Ni2+ and Co2+ ions released from the alloy into the corrosive medium, as a result of the aggressive attack of Cl−, were taken as a measure of the corrosion rate. ICP measurements were performed using Perkin-Elmer Optima 2100 Dual View inductively coupled plasma atomic emission spectrometry (ICP-AES) instrument connected with AS 93 Plus autosampler.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v water/Nafion by sonication. Typically, 5 μl of this catalyst ink were covered on a glassy carbon electrode (3 mm in diameter) and then dried in an ambient environment for measurements. The Pt/C catalyst was prepared with a catalyst loading of ∼0.14 mg cm−2. Measurements were conducted in deaerated 0.1 M KOH solutions. To run a LSV experiment, the working electrode is cathodically scanned starting from the corrosion potential (Ecorr) at a scan rate of 5.0 mV s−1 up to a cathodic potential of −1.0 V vs. SCE. All potentials in the HER study were converted from the SCE scale to that of the reversible hydrogen electrode (RHE), as described in the ESI file,† Section 1. Impedance measurements were performed at selected cathodic overpotential values, employing AC signals of amplitude 5 mV peak to peak covering the frequency range 100 kHz to 10 mHz.
:1 v/v water/Nafion by sonication. Typically, 5 μl of this catalyst ink were covered on a glassy carbon electrode (3 mm in diameter) and then dried in an ambient environment for measurements. The Pt/C catalyst was prepared with a catalyst loading of ∼0.14 mg cm−2. Measurements were conducted in deaerated 0.1 M KOH solutions. To run a LSV experiment, the working electrode is cathodically scanned starting from the corrosion potential (Ecorr) at a scan rate of 5.0 mV s−1 up to a cathodic potential of −1.0 V vs. SCE. All potentials in the HER study were converted from the SCE scale to that of the reversible hydrogen electrode (RHE), as described in the ESI file,† Section 1. Impedance measurements were performed at selected cathodic overpotential values, employing AC signals of amplitude 5 mV peak to peak covering the frequency range 100 kHz to 10 mHz.The durability and stability of the best catalyst was evaluated by 24 h of galvanostatic measurements at a fixed cathodic current density of 100 mA cm−2 and continuous cathodic potential cycling. For potential cycling, the potential was swept linearly from the starting potential (Ecorr) towards the cathodic direction at a scan rate of 50 mV s−1 till a catholic potential of −1.0 V vs. RHE, and then reversed with the same scan rate till the starting potential to form one complete cycle. The process of cycling was repeated 3000 times without withdrawing the electrode from the solution and without time delay. Measurements were carried out in deaerated 0.1 M KOH solutions at 25 °C.
2.2.3.1. Faradaic efficiency for the HER. Faradaic efficiencies of the studied catalysts towards the HER were evaluated via quantifying the amount of H2 liberated during a controlled potential electrolysis by a gas chromatography. Then, this measured amount of H2 is divided by the amount of H2 calculated from the charge passed (assuming 100% faradaic efficiency) through the working electrode (WE) during that electrolysis. See more details in the ESI file,† Section 2.
2.3. Characterization techniques
Morphology, elemental composition, and mapping studies of the pitted surfaces were performed using an Analytical Scanning Electron Microscope JEOL JSM 6390 LA with an EDS attachment (JEOL EDS EX-54175JMU) on the JEOL SEM. X-Ray photoelectron spectroscopy (XPS) investigation was carried out using Escalab 250Xi (ThermoFisher Scientific, United Kingdom) to determine chemical binding properties of the surface, utilizing monochromatic Al Kα source gun and a spot diameter of 650 μm with charge neutralization implemented by means of a flood gun. High-resolution spectra used during deconvolution were recorded at a pass energy 10 eV and energy step size of 0.1 eV. In order to normalize spectroscopic measurements, binding energy (BE) was calibrated for peak characteristics of neutral carbon 1s (BE = 284.6 eV). Data analysis was performed using Avantage v.5 software provided by the manufacturer.3. Results and discussion
3.1. Cyclic polarization measurements (passivity, pitting corrosion, and repassivation processes)
Fig. 1 shows cyclic polarization curves (linear and logarithmic scales) measured for CoNiGa (alloy I) and CoNiGaAl (alloy II) SMAs in 0.5 M NaCl solution at 25 °C. The two cyclic voltammograms were recorded between −2.0 V(SCE) and +1.0 V(SCE) at a scan rate of 5.0 mV s−1.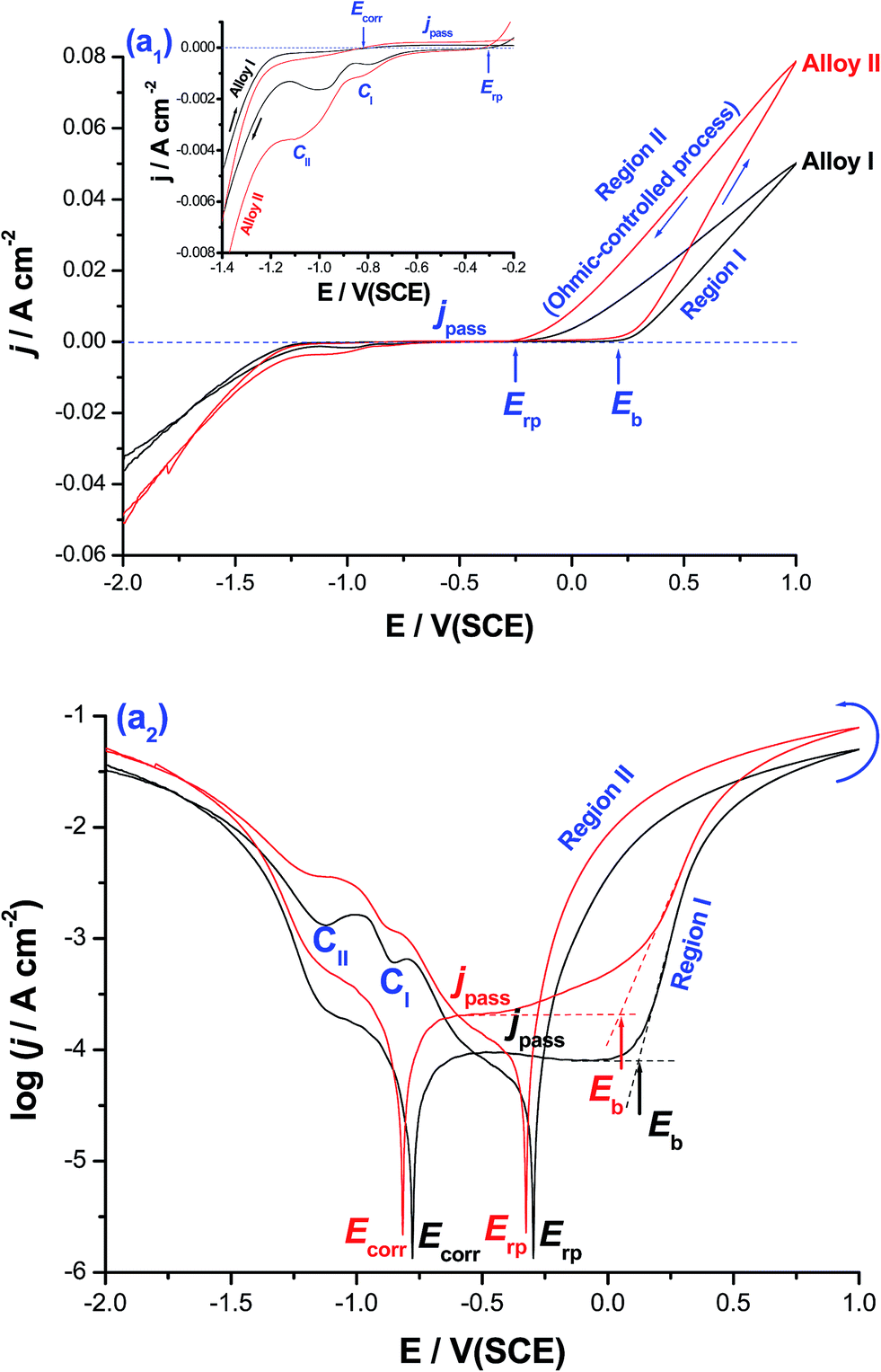 | ||
| Fig. 1 Cyclic polarization curves (upper) linear and (lower) logarithmic scales recorded for CoNiGa (alloy I) and CiNiGaAl (alloy II) in 0.5 M NaCl solution at 25 °C. Measurements were conducted between −2.0 V(SCE) and +1.0 V(SCE) at a scan rate of 5.0 mV s−1. | ||
It follows from Fig. 1(a1) that for the two tested alloys on positive going scan, the cathodic current density decreases gradually reaching a zero value at the corrosion potential (Ecorr). The uniform corrosion accelerating influence of alloyed Al is obvious from the cathodic and anodic polarization curves around Ecorr (Ecorr ± 300 mV), Fig. 1(a2), and deserves further investigations. In this respect, the effect of alloyed Al on the uniform corrosion behavior of the two tested MSMAs is systemically studied employing various electrochemical techniques, and complemented with ICP-AES method of chemical analysis and SEM examinations (ESI, Fig. S1–S4 and Tables S1 and S2†). The objective is to gain insights into the uniform corrosion activation influence of alloyed Al. This will help interpret for the first time, as will be shown later, the catalytic impact of the free corrosion process, which is catalyzed by Al and led to the formation of pits on the surface of alloy II (ESI, Fig. S4†), for the HER.
Referring again to Fig. 1, it is obvious that the polarization curves lack active dissolution near Ecorr due to passivation (as evidenced from XPS measurements, see later). Passivity of such alloys extends with a very low passive current (jpass) up to a certain critical potential (designated here as the breakdown potential, Eb), where passivity breakdown and initiation of pitting attack occur. Breakdown of passivity and subsequent formation of pits initiate as a result of the adsorption of Cl− anions on the oxide/solution interface assisted by applied electric field.27 This adsorption process is favored at the active sites (defects and flawed regions27) of the passive layer and occurs in competition with the passivating (passive layer forming) species, namely dissolved O2, H2O molecules and OH−.28–30 Following adsorption, a chemical reaction occurs between the adsorbed Cl− anions and metal cations within the passive oxide film,31 forming M–Cl soluble species.29–32 Once formed, the soluble species leave the oxide lattice and goes in solution, causing thinning and localized dissolution of the oxide film.31 Once the passive film is locally dissolved, pit nucleates at Eb and dissolution of the base metal commences. When the potential exceeds Eb, pit initiates. This in turn makes the medium locally acidic (where the solution chemistry inside the pit is different from that outside it28,29), hence allowing for efficient oxide dissolution and pit growth.28–31 This makes jpass increase drastically (region I).
Upon reversing the potential, the high-current regime (where the backward scan passes over the forward one) is maintained down to a potential known as the repassivation potential (Erp), region II, and a current hysteresis loop (characteristic of the pitting corrosion phenomena32) results. This loop indicates a delay in repassivation of an existing pit, the one that is formed during the forward scan (region I), when the potential is swept back negative. The location of Erp, defined as the potential on the reverse scan at which the anodic current becomes zero (i.e., the current changes polarity),32–34 with respect to Ecorr is well-defined in the logj vs. E plot, Fig. 1(a2). In pitting corrosion studies, Erp is the relevant potential instead of Eb, as the former helps establish the passive and pitting potential regions of the system. Further inspection of Fig. 1(a2) reveals that Erp is cathodic with respect to the potential necessary to activate the surface (i.e., towards passivity breakdown and initiation of pitting attack) on the forward potential scan, namely Eb. That means the anode is quite immune at potentials (E) more negative than Erp, where the electrode is protected by an oxide film and pitting will only take place at more positive potentials,32–34 and also safe from pitting attack within the region Erp < E < Eb. This safe location of Erp denotes that the electrode is able to repassivate after the breakdown of the passive film has occurred. On the contrary, other systems such as such as Zn/NO3− (ref. 33) and Al/gluconate,34 found it difficult to repassivate since their Erp values locate outside the passive region, thus suffering from severe pitting in these media during the reverse scan.
It is worth referring here to the linear j–E relationship observed in region II, Fig. 1(a1), which suggests an ohmic-controlled process. This process can be explained on the basis of pit growth and subsequent salt film formation.35–37 During pit growth, the concentration of metallic cations increases gradually due to the active dissolution within the pit. This active dissolution of the base metal continues till the saturated concentration is reached, and a salt film is subsequently formed at the bottom of the pit. The electric field applied across the salt film drives the dissolved metallic cations to move outward through the film. The stronger the field is, the faster the metallic cations move through the film. Hence, in this stage the growth of the pit is controlled by the ohmic potential drop across the salt film.35–37 At more cathodic potentials, two distinct cathodic current peaks (CI and CII) are formed. These two peaks are most probably assigned to the reduction of the free metal cations formed through the pits and the salt film (corrosion products) formed during pit growth.
Charge passage measurements showed that the total charge passed between Eb and Erp (regions I and II) for alloy II, that is alloyed with Al, was 38.9 C cm−2, which is ∼2.0 times greater than that recorded for alloy I (19.92 C cm−2), the Al-free alloy. The same trend was observed for the two cathodic peaks. In addition, jpass values at any given anodic potential (Ea) within the passive region are always greater, particularly when Ea is approaching Eb, for alloy II than those measured for alloy I. This increase in jpass of alloy II with Ea is quite clear in Fig. 1(a2). The increase in jpass with Ea refers to general weakness and thinning of the passive film as a result of Cl− adsorption, which promotes as Ea is made more positive.28 It seems therefore that the presence of alloyed Al in the matrix of alloy II weakens its passivity promoting Cl− adsorption, as supported by XPS (see later). On the other hand, the low values of jpass recorded for alloy I reflect the increased resistance of that alloy towards pitting corrosion. Also, Eb of alloy II is achieved at potentials more negative (active) than that of alloy I, as clearly seen in Fig. 1(a2). These findings are sufficient to conclude that alloy II is more susceptible to pitting corrosion in these solutions than alloy I.
3.2. Chronoamperometry measurements
Chronoamperometry (current vs. time) measurements were also performed to confirm the above findings and gain more information about the influence of alloyed Al on the kinetics of the passive layer growth and its breakdown. Fig. 2(a) and (b) show the j/t curves recorded for alloys I and II, respectively. Measurements were carried out in 0.5 M NaCl solution as a function of applied anodic potential (Ea: −0.2 to 0.5 V vs. SCE) at 25 °C.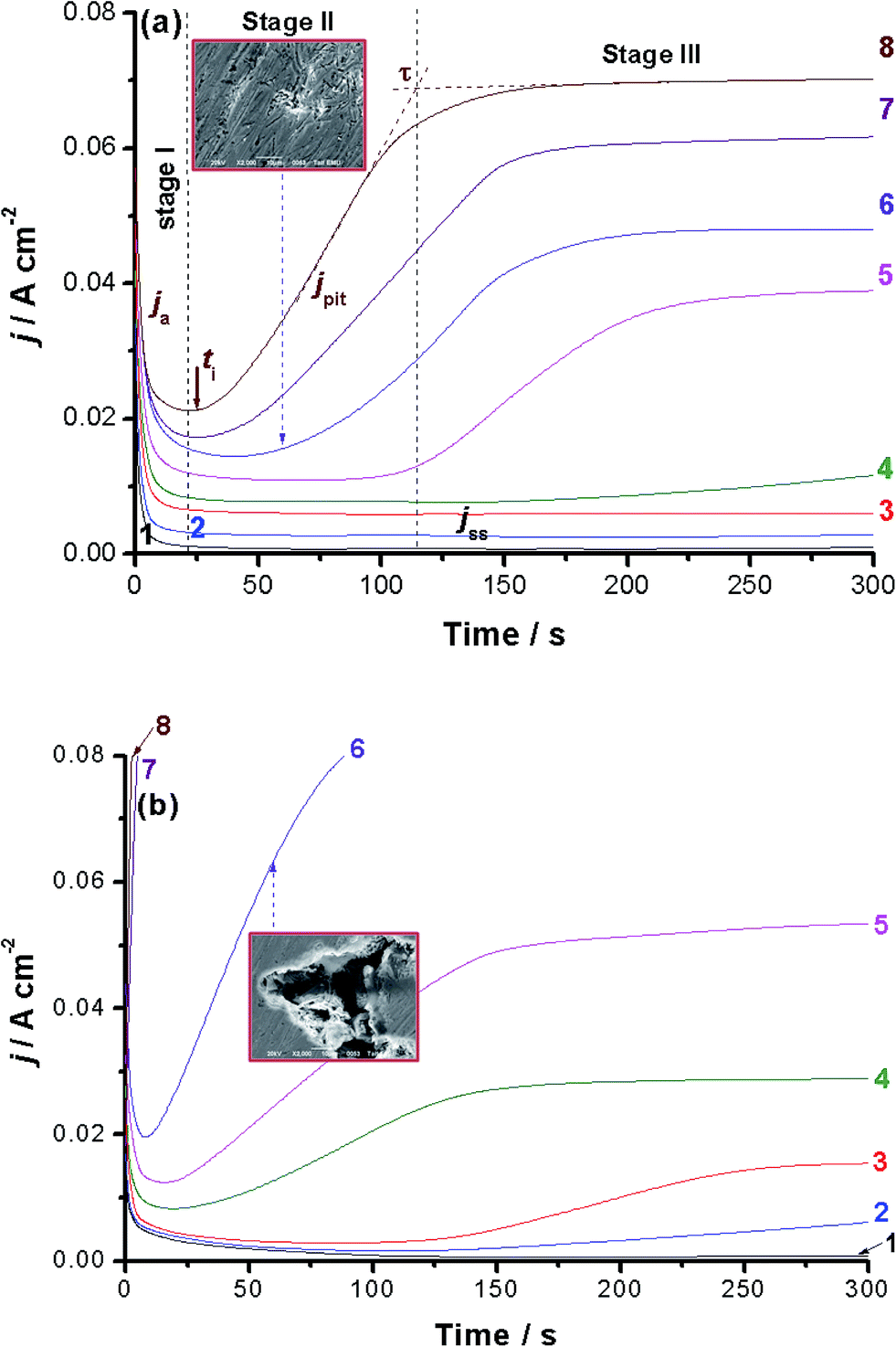 | ||
| Fig. 2 Chronoamperometry measurements recorded for (a) alloy I (CoNiGa SMA) and (b) alloy II (CoNiGaAl SMA) in 0.5 M NaCl solutions at 25 °C as a function of the applied anodic potential (Ea). (1) −0.2 V(SCE); (2) −0.1 V(SCE); (3) 0.0 V(SCE); (4) +0.1 V(SCE); (5) +0.2 V(SCE); (6) +0.3 V(SCE); (7) +0.4 V(SCE); (8) +0.5 V(SCE). Insets: SEM images captured for the two alloys in 0.5 M NaCl solutions at 25 °C after holding the alloy at Ea = 0.3 V(SCE) for 60 s. | ||
Different j/t profiles that deserve comments were obtained depending upon the location of Ea versus Eb and the chemical composition of the tested alloy. When Ea is cathodic to Eb, the transient current profile can be divided into two stages, as shown for alloy I (Fig. 2(a), curves 1–3) and alloy II (Fig. 2(b), curve 1). In the first stage, the anodic current (ja) first decreases as a result of the electroformation and growth of a passive oxide film on the anode surface. This decay in ja varies, as will be shown later, according to the chemical composition of the tested alloy. Following this decay, ja reaches a steady-state value, designated here as jss (related to jpass), the current of the second stage. Once jss is attained and well-established, the rates of the passive layer growth and its dissolution are balanced, so that the oxide film hardly grows and a constant passive current results. Close inspection of Fig. 2 (also inspect ESI Fig. S5†) reveals that the rate of ja decay, and hence the passivation rate, of alloy II, that is alloyed with Al, is higher than that of alloy I (the Al-free alloy). This indicates that alloy I tends to passivate in these solutions more effectively than alloy II. This in turn adds another evidence that Al when added to the CoNiGa SMA (alloy I) weakens its passivity making it less resistant to passivity breakdown and subsequent pitting attack.
Alloy I maintained its passive behavior even when Ea is approaching Eb. Only jss enhanced with Ea (Fig. 2(a), curves 1–4), denoting destabilization (oxide thinning/dissolution) of the passive oxide film as a result of the corrosive attack of Cl− anions, but still passive within this period of time reflecting its high corrosion resistance. On the contrary, and within the same potential range, alloy II exhibited new curve features, as shown in Fig. 2(b) (curves 3 and 4). This curve feature can be classified into the three stages, namely stages I–III for passivation stage, pit formation and growth stage, and the final steady-state stage, respectively. The stage I's current falls with time, denoting the electroformation and growth of the passive layer. Then it reaches its minimum value, corresponding to the maximum thickness and protectiveness of the passive layer, at a certain time known as the incubation time (ti); the time required for local removal of the passive film via the sequence of Cl− adsorption, penetration and formation of soluble complexes.29–32 It indicates the beginning of the pit nucleation period, where its magnitude reflects the susceptibility of the oxide film to breakdown.30–33
Stage II starts from ti and ends at another time designated here as τ. The current of this stage, termed here as pit current jpit, rises from the moment just after ti till τ. The current growth within the 2nd stage suggests that pit formation and growth are the dominant processes. New pits may also develop following this active period, i.e., ti. Following the current rise between ti and τ (i.e., stage II), jpit attains a steady-state, denoting stage III. This steady-state stage can be explained on the basis that pitting corrosion products formed during pit initiation and growth (stage II) precipitate inside the pits, blocking them up and therefore hinder the current flow (jpit) through the pits. It is most probable that a steady-state is established between the metal dissolution and oxide film formation including a blockade by pitting corrosion products in stage III of current transient after τ. Similar findings were previously obtained in our lab during pitting corrosion studies of some metals and alloys.38–41
Alloy I showed the same trend but with much longer ti and lower jpit values than those of alloy II at the same Ea values, compare for example curves 5 and 6. Beyond Eb, alloy I exhibited no change in its passivation and pit formation and growth behavior (Fig. 2(a) curves 7 and 8), even the final steady-state current persists. However, alloy II (Fig. 2(b) curves 7 and 8 and ESI, Fig. S6†) showed that stage I (the first passivation stage) is disappeared and the current grows very rapidly and almost linearly immediately after switching the applied anodic potential, then it reaches steady-state. Further inspection of Fig. 2(a) and (b) reveals that jpit enhances and ti gets shorter, and hence increased pitting attack results, as Ea is made more anodic. At the same Ea, the values of jpit are always smaller and those of ti are longer for alloy I than alloy II. These findings again confirm the higher susceptibility of alloy II towards pitting as compared with alloy I. SEM examinations, images in the insets of Fig. 2(a) and (b), recorded for the surfaces of alloys I and II after holding the alloy at Ea = 0.3 V(SCE) for 60 s in the test solution came to the same conclusion. See how the surface of alloy II suffers from intense pitting attack much more than alloy I.
3.3. Catalysis applications for the HER
Cyclic polarization and chronoamperometry measurements confirmed the obvious activation of CoNiGa MSMA towards pitting corrosion, even at the rest potential (revisit the surface morphologies of Fig. S4, ESI†), by adding Al as an alloying element. This activation influence of alloyed Al encouraged us to test these materials as active cathode materials for the HER. We propose here that pitting corrosion may increase the specific surface area of the alloy surface and create on it new O2-deficient (most probably due to thinning of the passive layer, depassivation, induced by adsorbed Cl−) active sites capable of catalyzing the HER. This proposal is supported here from SEM/EDX mapping and XPS studies (see Section 3.4) and may require additional future investigations. In addition, and to the best of our knowledge, there are no reports in the literature regarding hydrogen generation over CoNiGa and CoNiGaAl MSMAs. For the above reasons, the HER performance of the two tested MSMAs, namely alloys I and II was studied, as shown in Fig. 3(a). The results performed on pure Co, pure Ni, Co50Ni50, and Pt/C cathodes were also included for comparison. Curves 5 and 7 represent the cathodic polarization curves recorded for alloys I and II, respectively, but after 24 h of immersion in 0.5 M NaCl solutions at room temperature. The aim, as will be shown later, is to access the role of surface roughness, resulted due to corrosion induced by the aggressive Cl− anions, in improving the HER activity of the two tested SMAs.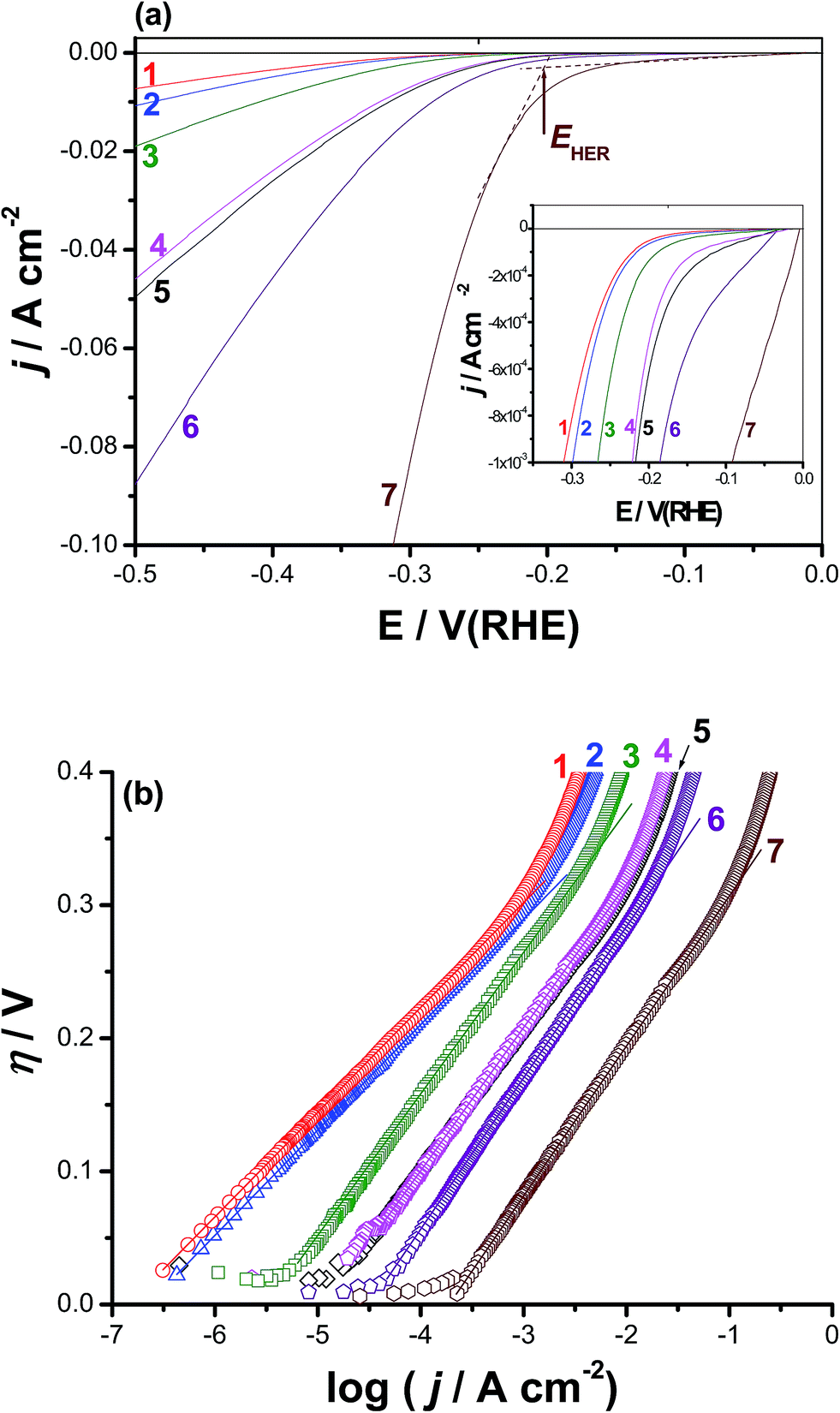 | ||
| Fig. 3 Electrocatalytic hydrogen evolution of catalysts 1–7: (a) polarization curves for the HER on the surfaces of such catalysts. (b) Tafel plots for the tested catalysts derived from (a). Measurements were conducted in deaerated 0.1 M KOH aqueous solutions at a scan rate of 5 mV s−1 at 25 °C. (1) pure Co; (2) pure Ni; (3) Co50Ni50 alloy; (4) Co50Ni23Ga27 (alloy I), (5) alloy I after 24 h of free corrosion in 0.5 M NaCl solution at 25 °C; (6) Co50Ni23Ga26Al1.0 (alloy II); (7) alloy II after 24 h of free corrosion in 0.5 M NaCl solution at 25 °C. | ||
The HER kinetics of the studied catalysts was probed by the corresponding η vs. logj plots, Fig. 3(b). The linear portions of these plots were fitted to the Tafel equation, expressed as shown in eqn (1):42
|
η = (2.3RT/nαF)logjo − (2.3RT/nαF)logj
| (1) |
| Tested cathode | EHER/mV(RHE) | −βc/mV dec−1 | jo/mA cm−2 | η10/mV |
|---|---|---|---|---|
| Pure Co | −385(8) | 82(2.05) | 1.5(0.1) × 10−4 | 382(9) |
| Pure Ni | −370(7) | 80(1.6) | 2.3(0.05) × 10−4 | 371(7) |
| Co50Ni50 alloy | −340(7) | 105(2.3) | 3.4(0.07) × 10−3 | 364(6) |
| Alloy I (NiCoGa) | −295(6) | 109(2.5) | 0.011(0.002) | 320(5) |
| Alloy I after 24 h of immersion in 0.5 M NaCl at 25 °C | −284(6) | 107(1.5) | 0.0124(0.002) | 313(4.5) |
| Alloy II (NiCoGaAl) | −250(5) | 115(1.9) | 0.04(0.003) | 285(4.8) |
| Alloy II after 24 h of immersion in 0.5 M NaCl at 25 °C | −220(5) | 113(1.7) | 0.2(0.025) | 194(4.3) |
Fig. 3(a) reveals that pure Co and Ni cathodes (curves 1 and 2) exhibit the worst catalytic activity towards the HER among the tested cathodes, as the proton reduction on their surfaces started at highly negative onset potentials, EHER. Moreover, small cathodic currents were obtained beyond EHER. The value of EHER (−340 mV vs. RHE) recorded for the Co50Ni50 alloy (curve 3) is smaller, accompanied with higher cathodic currents, than those measured for Co (−385 mV vs. RHE) and Ni (−370 mV vs. RHE). Also, the value of jo recorded for the Co50Ni50 alloy (3.4 × 10−3 mA cm−2) is ∼23 times greater than that measured for Co alone (1.5 × 10−4 mA cm−2) and ∼15 times that of Ni alone (2.3 × 10−4 mA cm−2). These results demonstrate that the Co50Ni50 alloy exhibits higher catalytic activity for the HER than Co and Ni alone, most probably as a consequence of the improvement in the intrinsic catalytic activity due to the synergetic effect of the two alloying metals.43–47 Further improvement in the HER catalytic performance of the Co50Ni50 alloy (curve 3) has occurred upon alloying it with Ga at the expense of Ni to yield the Co50Ni23Ga27 MSMA (alloy I), curve 4, reflecting the catalytic impact of alloyed Ga. This is clear from Table 1 where alloy I recorded a jo value of 0.011 mA cm−2, which is 3.2 times grater than that calculated for Co50Ni50 alloy (3.4 × 10−3 mA cm−2).
The addition of a small amount of alloyed Al (1.0%) to alloy I at the expense of Ga to yield alloy II (Co50Ni23Ga26Al1.0) has markedly enhanced the HER activity, denoting the important role the alloyed Al play in catalyzing the HER. In this respect, the value of EHER has decreased, as shown in Table 1, from −295 mV vs. RHE for alloy I to −250 mV vs. RHE for alloy II, and the cathodic polarization curves get much more steeper (i.e., higher cathodic currents and increased kinetics for the HER) in presence of alloyed Al (compare between curves 3 and 4). In addition, the value of jo measured for alloy II (0.04 mA cm−2) is found to be ∼4 times higher than that of alloy I (0.011 mA cm−2).
The overpotential (η10) requires to achieve a current density of 10 mA cm−2 is also considered an important controlling parameter for the apparent electrode activity for the HER.48–50 Data of Table 1 infer that the values of η10 decrease, and hence accelerated kinetics of the HER is achieved, following the sequence: Co < Ni < Co50Ni50 < alloy I < alloy II.
It is worth mention here that the jo value of alloy I is doubled and that of alloy II has increased 5 times, approaching that of Pt/C under the same experimental conditions (∼0.7 mA cm2 (ref. 51)) when the two alloys are subjected to a corrosion process (24 h of free corrosion in 0.5 M NaCl solution at 25 °C) prior to performing the cathodic polarization measurements. These results refer, for the first time, to the catalytic impact of corrosion, which is a well-known destructive process. This undesirable process can therefore be used to produce useful materials for wide applications, such as electrocatalysts here.
The HER proceeds in the alkaline solutions following either Volmer–Heyrovsky or Volmer–Tafel mechanisms:51,52
Volmer step:
| H2O + e− + S* ↔ Hads + OH− | (2) |
Heyrovsky step:
| Hads + H2O + e− ↔ H2 + OH− + S* | (3) |
Tafel step:
| 2Hads ↔ H2 + S* + S* | (4) |
Impedance measurements were also carried out at selected cathodic overpotentials, namely −0.3, −0.5, and −0.7 V vs. RHE to further clarify the activation influence of alloyed Al on the kinetics of the HER. Fig. 4 represents the electrochemical impedance spectra in the complex plane recorded at −0.5 V vs. RHE. The impedance behaviour of pure Co, pure Ni, CoNi, and Pt/C electrodes were also included in Fig. 4 for comparison. The obtained complex plane impedance spectra of all studied catalysts (except for Pt/C, which displayed a single capacitive loop) exhibit two well-defined depressed semicircles (two time constants). Similar results were obtained at −0.3 and −0.7 V vs. RHE (their plots are not included, but their fitting results are reported and discussed here, see Table 2). Such impedance responses are commonly obtained for electrocatalytic evolution of hydrogen on Ni,53 Co,54 and some of their alloys43–45,55 in alkaline solutions. Also, similar impedance profiles were previously obtained in our lab during catalytic HER studies in H2SO4 solutions on reduced graphene oxide nanosheets and titanium substrates decorated by some metallic nanoparticles.56–58
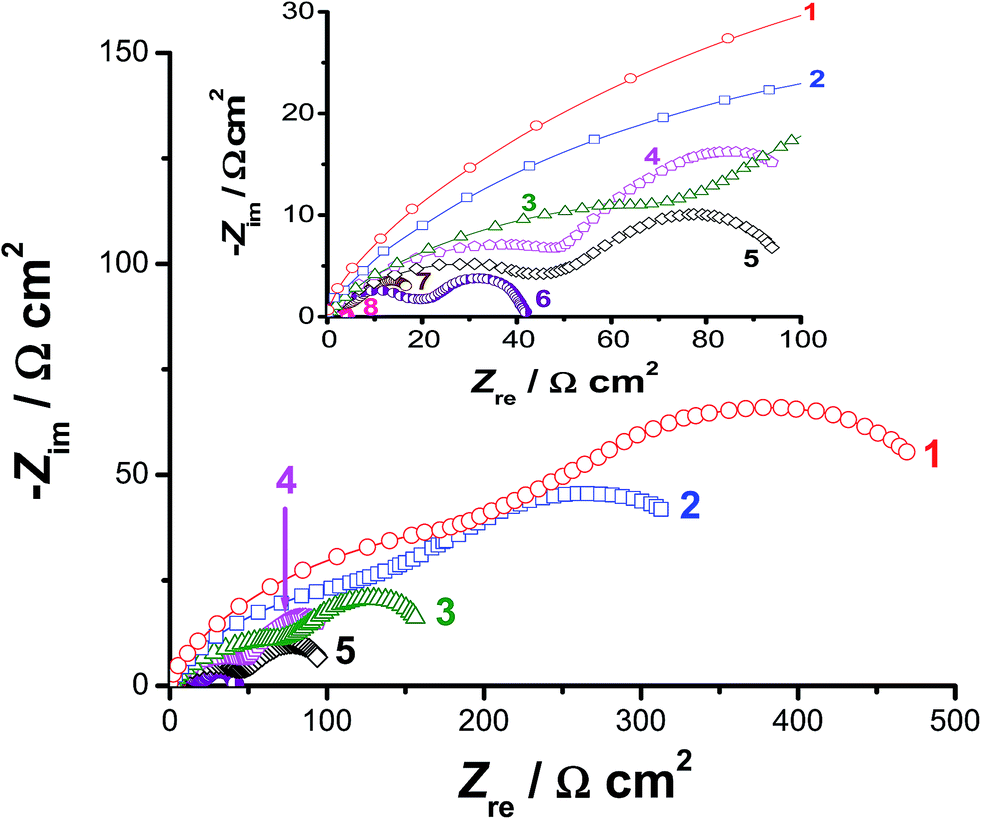 | ||
| Fig. 4 Complex-plane impedance plots recorded for the studied catalysts in deaerated 0.1 M KOH aqueous solutions at a cathodic overpotential of −0.5 V vs. RHE at 25 °C. (1) Pure Co; (2) pure Ni; (3) Co50Ni50 alloy; (4) Co50Ni23Ga27 (alloy I), (5) alloy I after 24 h of free corrosion in 0.5 M NaCl solution at 25 °C; (6) Co50Ni23Ga26Al1.0 (alloy II); (7) alloy II after 24 h of free corrosion in 0.5 M NaCl solution; (8) Pt/C catalyst. | ||
| Catalyst | E (V(RHE)) | Q1 (Sn (ω−1 cm−2)) | R1 (Ω cm2) | n1 | C1 (μF cm−2) | Q2 (Sn (ω−1 cm−2)) | R2 (Ω cm2) | n2 | C2 (μF cm−2) | Rct (Ω cm2) |
|---|---|---|---|---|---|---|---|---|---|---|
| Co | −0.3 | 22.1(0.4) | 426(7) | 0.94 | 16.4(0.24) | 19(0.4) | 457(7.8) | 0.95 | 14.8(0.22) | 883(14.8) |
| −0.5 | 28.2(0.5) | 308(5) | 0.93 | 19.7(0.29) | 28.1(0.5) | 386(6.2) | 0.92 | 19(0.28) | 694(11.2) | |
| −0.7 | 36.6(0.7) | 223(4) | 0.91 | 22.8(0.34) | 34.6(0.6) | 291(5.4) | 0.90 | 20.8(0.31) | 514(9.4) | |
| Ni | −0.3 | 26.1(0.5) | 388(5.4) | 0.93 | 18.5(0.27) | 24.4(0.4) | 405(5.8) | 0.94 | 18.2(0.27) | 793(11.2) |
| −0.5 | 35.6(0.7) | 206(3.1) | 0.93 | 24.6(0.36) | 35.5(0.7) | 266(4.2) | 0.92 | 23.7(0.34) | 472(7.3) | |
| −0.7 | 47.3(0.9) | 102(2) | 0.91 | 27.9(0.41) | 44.7(0.8) | 128(2.3) | 0.91 | 26.8(0.4) | 230(4.3) | |
| Co50Ni50 | −0.3 | 43.6(0.8) | 247(3.6) | 0.89 | 24.9(0.37) | 37.9(0.7) | 279(4.2) | 0.91 | 24.2(0.33) | 526(7.8) |
| −0.5 | 55.6(1.1) | 108(2.2) | 0.88 | 27.7(0.42) | 52.3(1.05) | 110(2.4) | 0.89 | 27.6(0.41) | 218(4.6) | |
| −0.7 | 76.1(1.5) | 63(1.2) | 0.86 | 31.9(0.47) | 69.5(1.39) | 69(1.4) | 0.87 | 31.3(0.45) | 132(2.6) | |
| Alloy I (without corrosion pretreatment) | −0.3 | 52(1.05) | 133(2.3) | 0.88 | 26.4(0.39) | 45.1(0.9) | 145(2.6) | 0.90 | 25.8(0.38) | 278(4.9) |
| −0.5 | 79.4(1.6) | 68(1.2) | 0.85 | 31.6(0.47) | 68.6(1.37) | 62(1.3) | 0.88 | 32.6(0.49) | 130(2.5) | |
| −0.7 | 109.8(2.2) | 44(0.8) | 0.84 | 39.8(0.6) | 89.6(1.8) | 47(0.9) | 0.87 | 39.6(0.58) | 91(1.7) | |
| Alloy I (after corrosion pretreatment) | −0.3 | 60.5(1.2) | 122(2.2) | 0.86 | 27.2(0.41) | 58.4(1.17) | 133(2.6) | 0.88 | 26.5(0.39) | 255(4.8) |
| −0.5 | 86.8(1.7) | 52(1.02) | 0.85 | 33.4(0.5) | 80.4(1.61) | 55(1.1) | 0.86 | 33.2(0.49) | 107(2.12) | |
| −0.7 | 127.1(2.5) | 38(0.7) | 0.83 | 42.7(0.64) | 109.5(2.2) | 41(0.8) | 0.85 | 42.2(0.63) | 79(1.5) | |
| Alloy II (without corrosion pretreatment) | −0.3 | 85.3(1.7) | 56(1.1) | 0.85 | 33.2(0.5) | 75(1.5) | 68(1.3) | 0.86 | 31.7(0.47) | 124(2.4) |
| −0.5 | 148.9(2.9) | 19(0.33) | 0.84 | 48.7(0.73) | 133.5(2.7) | 22(0.4) | 0.85 | 47.9(0.71) | 41(0.73) | |
| −0.7 | 208.1(4.1) | 11(0.2) | 0.82 | 54.8(0.82) | 178.9(3.6) | 15(0.32) | 0.83 | 53.2(0.79) | 26(0.52) | |
| Alloy II (after corrosion pretreatment) | −0.3 | 177.1(3.5) | 15.6(0.3) | 0.83 | 53(0.8) | 134.9(2.7) | 28.8(0.6) | 0.84 | 46.8(0.7) | 44.4(0.9) |
| −0.5 | 382.8(7.7) | 5.4(0.11) | 0.81 | 89.8(1.3) | 175.9(3.5) | 16.1(0.3) | 0.83 | 52.9(0.79) | 21.5(0.41) | |
| −0.7 | 832.9(14.6) | 2.6(0.05) | 0.78 | 147.6(2.2) | 347.8(7.2) | 7.2(0.14) | 0.80 | 77.8(1.17) | 9.8(0.19) |
The first semicircle (with diameter R1) was observed at high frequencies, while the second one (the diameter of which is R2) was formed at the low frequency values. Each of them can be assigned to the resistance–capacitance (RC) network. Each RC network consists of the charge-transfer resistance (R1 for the first semicircle and R2 for the second one) of the H+ reduction process and the corresponding double-layer capacitance (C1 for the first semicircle and C2 for the second one) at the catalyst/electrolyte interface. The total charge-transfer resistance (Rct) equals the summation of R1 and R2. To calculate the elements of such RC networks, and hence gain more insight into the HER kinetics associated with EIS measurements, the experimental impedance data were fitted using the equivalent circuit presented and fully described elsewhere.58 The obtained fitting parameters are presented in Table 2 as a function of the applied cathodic overpotentials. It follows from Table 2 that, for any tested catalyst, the values of R1 and R2 decrease as the applied cathodic overpotential made more negative, referring to enhanced HER kinetics. These results infer that the total impedance of the studied interfaces is associated to the HER kinetics.
Data of Table 2 reveal that the value of Rct, at any given applied potential, is smaller (i.e., accelerated HER kinetics) for alloy II (that is alloyed with Al) than those measured for the other studied catalysts. For instance, alloy II recorded an Rct value of 41 Ω cm2@−0.5 V(RHE), which is the smallest among the others (Table 2: Rct = 694, 472, 218, and 130 Ω cm2 for Co, Ni, CoNi, and alloy I, respectively) at the same overpotential. These findings support the catalytic influence of the alloyed Al. The Rct value of alloy II is diminished (i.e., faster and faster HER kinetics is achieved) from 41 Ω cm2@−0.5 V(RHE) to 21.5 Ω cm2 at the same overpotential after modifying its surface via the corrosion pretreatment process, approaching that of the commercial Pt/C catalyst (4.3 Ω cm2). These results confirm the high HER catalytic performance of the corroded surface of alloy II, highlighting again the catalytic impact of corrosion.
Further inspection of Table 2 reveals that, at any given potential, the largest capacitance (Q and C) value was recorded for alloy II after being activated by pitting corrosion, referring to its large active surface area. This increased active surface area of the pitted surface of alloy II promotes HER catalytic performance of the alloy. To confirm these findings, the electrochemical active surface area (EASA) of the studied catalysts was estimated. There are a number of different techniques described in literature for EASA measurements. One estimation procedure involves calculation of the roughness factor (Rf) from EIS data, more precisely from estimated double layer capacitance values.59,60 We considered here the values of Q rather than those of C for the calculation of Rf, as the former (Q, where Q = Rct−1(C × Rct)n) provides more information about surface inhomogeneity and roughness,61,62 and also includes the parameter n, which is also related to surface roughness.63,64 For a typical smooth surface, n = 1 and C = 20 μF cm−2.59,60 This gives a value of 20 Sn (ω−1 cm−2) for Qcalculated. The obtained Rf values (Rf = Qmeasured/Qcalculated; Qmeasured = Q1 + Q2) are shown in Table S3 (ESI†) for each studied catalyst as a function of the applied cathodic potential. Table S3† revealed that, for any studied catalyst, Rf increases as the applied potential is made more cathodic, where H2 evolves progressively and can incorporate in the crystal lattice of the investigated cathode. This in turn may create voids and flaws, resulting in increased surface roughness.65–67 This effect is quite clear in case of alloy II, particularly after the corrosion pretreatment process, where Rf varied markedly with potential (Rf = 15.6, 27.94, and 59.04 at −0.3, −0.5, and −0.7 V vs. RHE, respectively). In the absence of the corrosion pre-treatment process, the value of Rf at any given potential is doubled when we go from alloy I (for instance, Rf = 9.97@−0.7 V vs. SCE) to alloy II (Rf = 19.35 at the same potential) thus, confirming the catalytic impact of alloyed Al. The accelerating influence of pitting corrosion is also clear from Table S3;† the pitted surface of alloy II (the best catalyst here) recorded an Rf value of 59.04 at −0.7 V vs. SCE, which is 3 times greater than that measured for the non-pitted surface of the same alloy at the same potential (Rf = 19.35). Therefore, the values of Rf clearly indicate (in accordance with SEM images) development of surface area.
The faradaic efficiency (FE) of the HER was also determined for alloys I and II without and with the corrosion pretreatment process, see Table 3, to further evaluate their electrocatalytic performance and support electrochemical findings. This was achieved via dividing the experimentally measured volume of H2 by its theoretical volume (the procedure is reported in more details in the ESI,† Section 1). Fig. 5 shows the comparison of the theoretical volume of H2 and its experimentally measured volume for the best catalyst, namely the pitted surface of alloy II which produced via dipping alloy II (as-polished) in 0.5 M NaCl for 24 h at 25 °C. Similar results were obtained for Pt/C and the other tested catalysts (ESI, Fig. S8†). The charge vs. time plots obtained for the studied catalysts during 1 h of a controlled potential electrolysis process (a potentiostatic experiment where the tested catalyst was held at −0.8 V vs. RHE for 1 h in a 0.1 M KOH solution) are depicted in Fig. 6 in a comparison with Pt/C. Here again, as shown in Table 3, the experimentally measured volume of H2 produced by alloy II (11.8 μmol h−1, with a corresponding FE of 88%) is higher than that yielded by alloy I (7.3 μmol h−1, FE 75.6%), confirming the catalytic impact of alloyed Al. The catalytic influence of corrosion is also evident from Table 3. Alloy II, after being modified by the corrosion pretreatment process, produced 18 μmol h−1 of H2 (FE 96%, approaching that of Pt/C, ∼100%), which is 1.5 times greater than that recorded by the same alloy but without corrosion pretreatment (11.8 μmol h−1, FE 88%). That means 96% of the electrical charge that passes through the system was consumed for the HER. The rest of charge may be wasted in some parasitic electrochemical processes simultaneously occurring during the HER. These may be reduction of dissolved O2 and, although less common, metal ion reduction and metal deposition. O2 reduction is most probably excluded as the test solution was deaerated and the cell sealed, unless traces of O2 still exist in solution. Pt is used here as the counter electrode, and dissolution of minute amounts of Pt and its re-deposition at the cathode may occur, consuming part of electricity. This in turn may help improve the catalytic properties of the tested cathode. To exclude or ensure this possibility, a carbon electrode was used as the anode instead of Pt, and the same catalytic activity was obtained. Cathodic corrosion may also occur as a result of H2 incorporation in the cathode's crystal lattice, particular Ni during electrolysis,65–67 but ICP-AES did not detect any cations in solution after electrolysis. Perhaps once formed in minute amounts, metal ions (released from the cathode) in solution re-deposited during electrolysis and this may also consume part of electricity.
| Catalyst | H2 measured by gas chromatography (H2/μmol h−1) | Calculated H2 based on the charge passed during electrolysis | FE (%) | |
|---|---|---|---|---|
| Charge passed/C | H2/μmol h−1 | |||
| a A chronoamperometry run where the catalyst is held at −0.8 V vs. RHE for 1 h in 0.1 M KOH solution at 25 °C.b Prior to performing the chronoamperometry experiment, the electrode is first subjected to 24 h of free corrosion in 0.5 M NaCl solution at 25 °C. | ||||
| Alloy I (without corrosion pretreatment) | 6.5(0.1) | 1.66(0.023) | 8.6(0.12) | 75.6(0.11) |
| Alloy I (with corrosion pretreatment) | 7.3(0.11) | 1.8(0.03) | 9.3(0.16) | 78.5(0.17) |
| Alloy II (without corrosion pretreatment) | 11.8(0.18) | 2.58(0.038) | 13.4(0.2) | 88.06(0.03) |
| Alloy II (with corrosion pretreatment) | 18(0.26) | 3.6(0.05) | 18.7(0.26) | 96.3(0.07) |
| Pt/C | 20.5(0.28) | 3.98(0.06) | 20.6(0.32) | 99.5(0.18) |
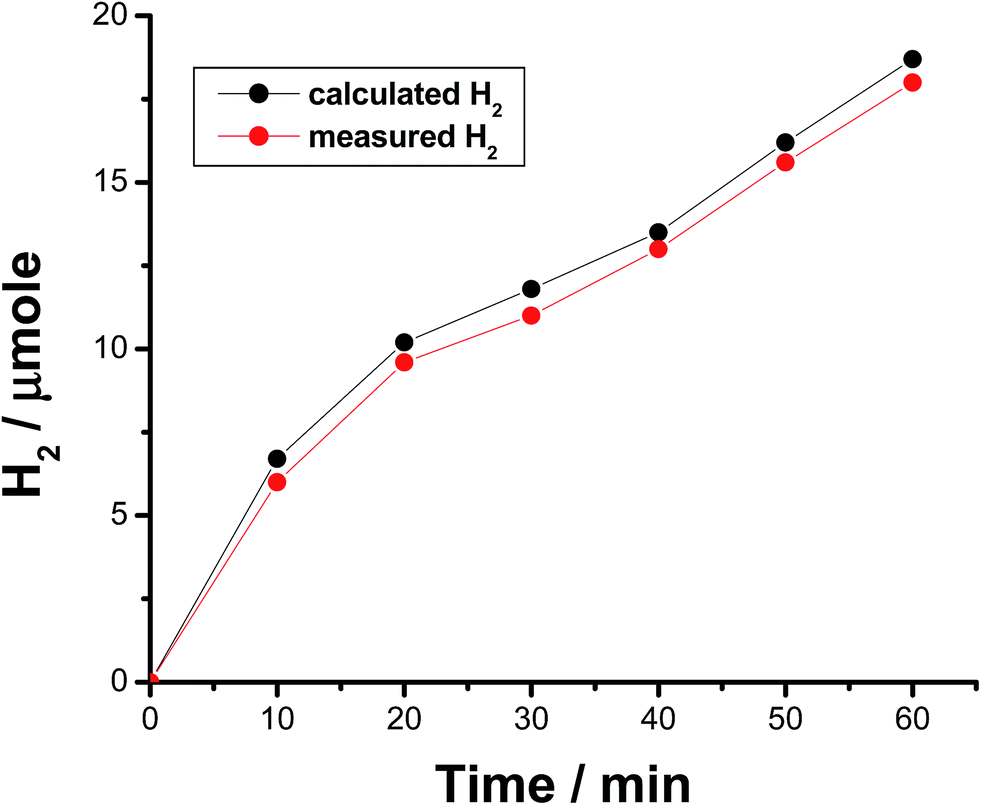 | ||
| Fig. 5 Volume of H2 calculated from the amount of charge passed (assuming 100% faradaic efficiency) and that of H2 measured from gas chromatography during 1 h of a controlled potential electrolysis run of the best catalyst (the pitted surface of alloy II) at −0.8 V vs. RHE in 0.1 M KOH aqueous solution. | ||
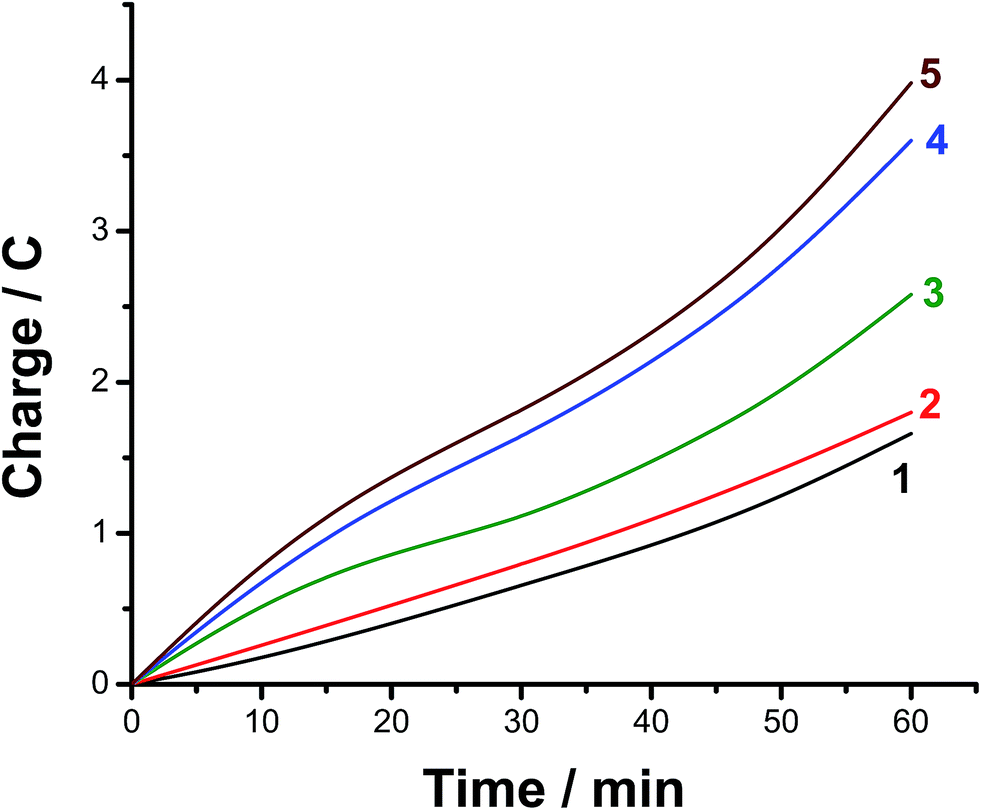 | ||
| Fig. 6 Charge vs. time plots of alloys I and II without and with corrosion pre-treatment, in a comparison with Pt/C, during 1 h of a controlled potential electrolysis process at −0.8 V vs. RHE in 0.1 M KOH aqueous solution. (1) Alloy I (without corrosion pre-treatment); (2) alloy I (with corrosion pre-treatment); (3) alloy II (without corrosion pre-treatment); (4) alloy II (with corrosion pre-treatment); (5) Pt/C. | ||
The above findings indicated that the investigated CoNiGaAl after pitting corrosion process is catalytically active for the HER. This high HER catalytic activity of the pitted surface of alloy II is found to be comparable, and even higher than that of other recently reported active Co- and Ni-based electrocatalysts.43–47,68–73
The activation influence of alloyed Al towards pitting attack and its catalytic impact on the kinetics of the HER on alloy II are evident from the above findings and deserves interpretation, vide infra.
3.4. Origin of catalytic activity
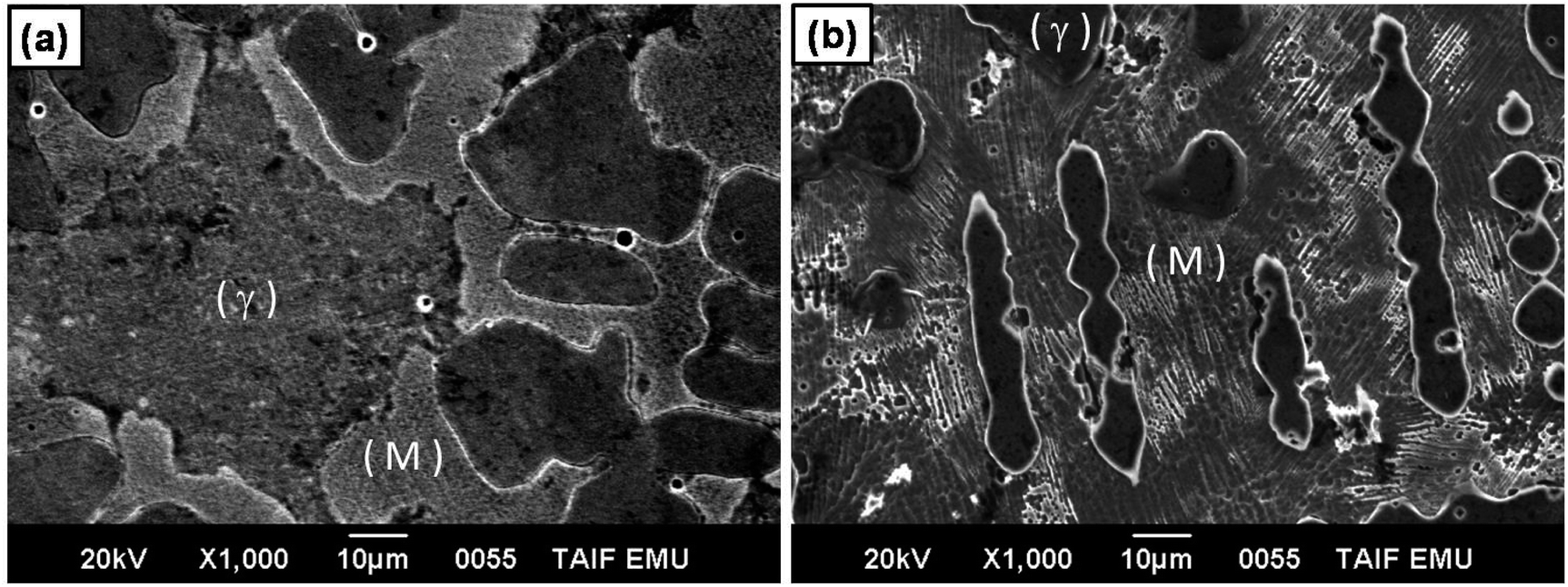 | ||
| Fig. 7 Microstructure of CoNiGa (a) and CoNiGaAl (b) alloys. | ||
It is well known that the γ phase has higher corrosion resistance than martensite phase. In this respect, the effect of martensite phase on decreasing corrosion resistance of stainless steel has been reported.74–76 Based on these considerations, the low corrosion resistance and high HER catalytic activity of alloy II can be attributed to the high Vf of the martensite phase in its microstructure, image (b), as compared with alloy I (that with low Vf of the martensite phase, image (a)). Moreover, the particle size of the γ phase in CoNiGaAl alloy is much smaller than that of the same phase in CoNiGa alloy. Hence, the grain boundary area in CoNiGaAl alloy is higher than in CoNiGa alloy. The area of the grain boundaries represent the imperfection in the polycrystalline alloys, therefore the candidate areas to be corroded are higher in CoNiGaAl than in CoNiGa alloy leading to higher corrosion rate in the former alloy than in the latter one. Such regions of imperfections may also catalyze the HER.
In an attempt to further assess the pitting corrosion activation influence of alloyed Al and its catalytic impact towards the HER, high resolution XPS study was performed for both investigated alloys before and after exposition in naturally aerated 0.5 M NaCl solution for period of 24 h (_ref and _24 h suffix in Table 4, respectively). Examination was carried out in the binding energy (BE) range of Ga3d5, Ni2p3, Co2p3, Al2p3, Cl2p3 and O1s peaks and corresponding XPS spectra are presented on Fig. 8. Before XPS examination ion gun etching was performed for each investigated sample (1000 V, 100 s). Its purpose was to remove carbon and oxygen contamination due to exposure in atmospheric air.
| BE [eV] | Co(1) | Co(2) | Ni(1) | Ni(2) | Ga(1) | Ga(2) | O(1) | O(2) | Cl |
|---|---|---|---|---|---|---|---|---|---|
| 778.4 | 782.0 | 853.0 | 854.2 | 18.6 | 20.3 | 531.0 | 532.4 | 200.3 | |
| CoNiGa_ref | 50.0 | 0.9 | 23.5 | 2.4 | 14.3 | 1.6 | 4.3 | 3.0 | — |
| CoNiGa_24 h | 37.4 | 1.0 | 19.2 | 2.9 | 6.6 | 7.0 | 14.0 | 11.9 | — |
| CoNiGaAl_ref | 44.7 | 1.2 | 22.5 | 2.5 | 13.3 | 3.1 | 7.1 | 5.6 | — |
| CoNiGaAl_24 h | 17.7 | 2.0 | 8.9 | 2.9 | 6.1 | 3.9 | 12.7 | 23.6 | 22.2 |
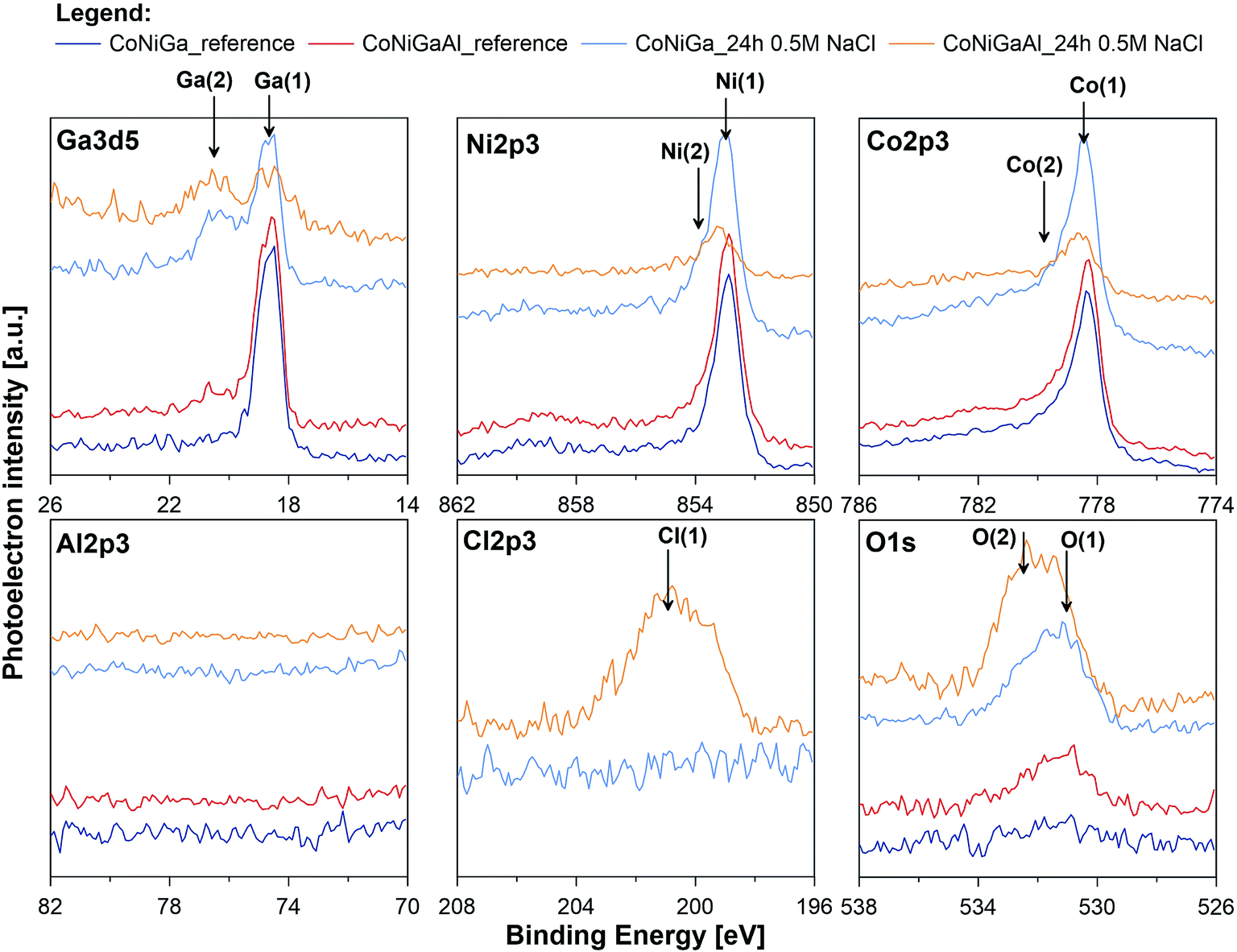 | ||
| Fig. 8 High resolution XPS spectra recorded at gallium, nickel, cobalt, aluminium, chlorine and oxygen for CoNiGa and CoNiGaAl samples before and after 24 h immersion in naturally aerated 0.5 M NaCl. | ||
The most dominant component was observed for Ga, Ni and Co in the energy range corresponding to chemical state of metal.77 A very small signal from non-metallic compounds was also present in the form of metal oxides (Ga(2), Ni(2), Co(2)) that originates from thin passive layer formed spontaneously when samples were exposed to atmospheric air.17 A well resolved Co2p and Ni2p spectra for metal shows complex structure. Asymmetric peaks were used during fitting procedure due to the presence of loss features and satellites.78 No visible peak was detected in the energy range of Al2p peak doublet for neither sample (reference or after the exposure). Authors suggest that the amount of aluminium in alloy is below the threshold of its identification, EDX studies indicate share of aluminium below 0.8 mass% (Fig. 9). Characteristic values of peak BE were presented in Table 4.
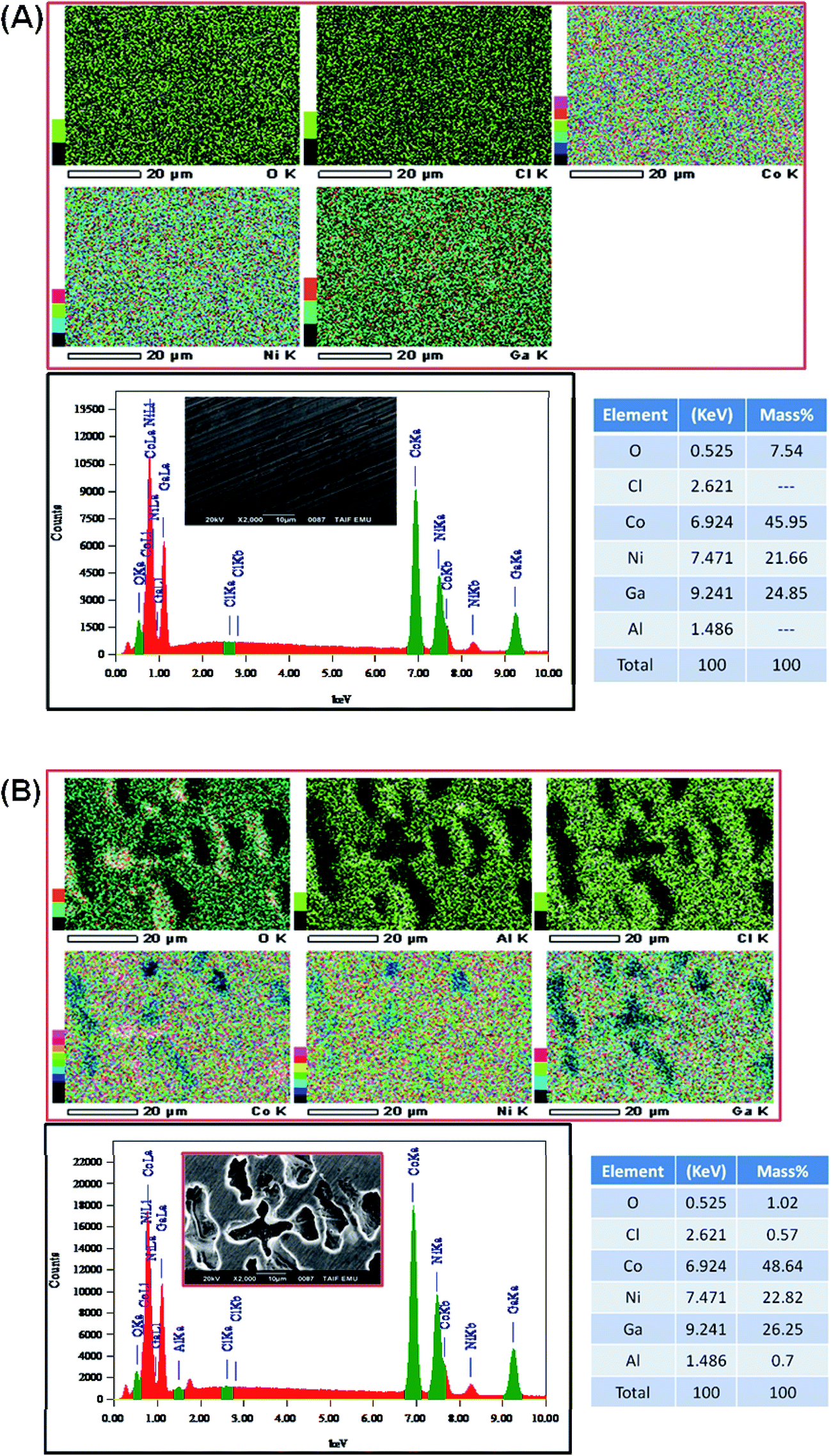 | ||
| Fig. 9 SEM micrographs and corresponding EDX chemical maps acquired for the alloying elements (Co, Ni, Ga, and Al), in addition to the EDX chemical analysis for Cl and O, recorded for alloys I and II after 24 h of free corrosion in 0.5 M NaCl solutions at 25 °C. | ||
Surface chemistry of investigated alloys change significantly as a result of 24 h exposition in naturally aerated 0.5 M NaCl. Gallium, which is next to Al, is the most active metal among the alloying components (−0.56 V compared to −0.28 V for Co and −0.24 V for Ni), oxidizes the most as a result of exposition. It can be seen, that surface coverage by gallium oxides is particularly significant for CoNiGa samples where Ga(1)/Ga(2) ratio drops from 8.9 merely to 0.9. In comparison, CoNiGaAl sample show decrease from 4.3 to 1.6. Second evident effect of sample exposure to 0.5 M NaCl is the appearance of Cl2p3 peak, manifesting itself in the energy range for metal chlorides, but only on CoNiGaAl alloy. Its share is above 20 at%. This high value may only indicate breakdown of the passive state combined with promotion of local corrosion of investigated alloy. The appearance of chlorides hinders Co(1), Ni(1) and Ga(1) peaks, which is to be expected. Considering the O1s spectra, the peak O(2) at 532.4 eV corresponding to hydroxide bonds is the main chemical state of oxygen for CoNiGaAl sample after exposure in NaCl. But for electrolyte-exposed CoNiGa samples O(1) peak at 531.0 eV is more pronounced, which indicates a higher fraction of oxidic bonding states and possibly a more stable passive state.77 This chemical state is also apparent in the residual peaks registered for pre-exposed samples.
The XPS examinations confirm the conclusions drawn on the base of electrochemical and microstructure studies. The passive state is significantly weaker due to Cl adsorption on aluminium-containing alloy (alloy II) and lower presence of gallium oxide in the passive layer. This weak passive state of alloy II may create active adsorption sites for the reduction of H+ on alloy II's surface, thus catalyzing the HER. This adds another reason behind the increased HER activity of alloy II as compared with alloy I; the later is characterized by stable passivity.
3.5. Catalyst stability and durability
Catalyst's stability and durability are highly important criteria to evaluate its catalytic performance. Stability and long-term durability of the best catalyst was evaluated here by means of repetitive cycling (3000 cycles) of the cathodic polarization curves starting from Ecorr up to a cathodic potential of −1.0 V(RHE), Fig. 10, and electrolysis at a high constant cathodic current density of 100 mA cm−2 for 24 h, see the inset of Fig. 10. It follows from Fig. 10 that the cathode exhibits excellent stability after 3000 times of continuous cycling up to a cathodic potential of −0.3 V vs. RHE. Beyond −0.3 V, it showed a slight degradation (due to small loss in the cathodic current density), which slightly enhances as the potential made more negative, most probably due to accumulation of the H2 bubbles at high cathodic potentials. This accumulated H2 bubbles may cause some sort of catalyst deactivation.79 In addition, a stable time-dependent overpotential curve was obtained around −0.31 V vs. RHE, as shown in the inset of Fig. 10. These findings reflect, besides its superior HER catalytic activity, the high stability and durability of our best catalyst (alloy II pre-treated by pitting corrosion).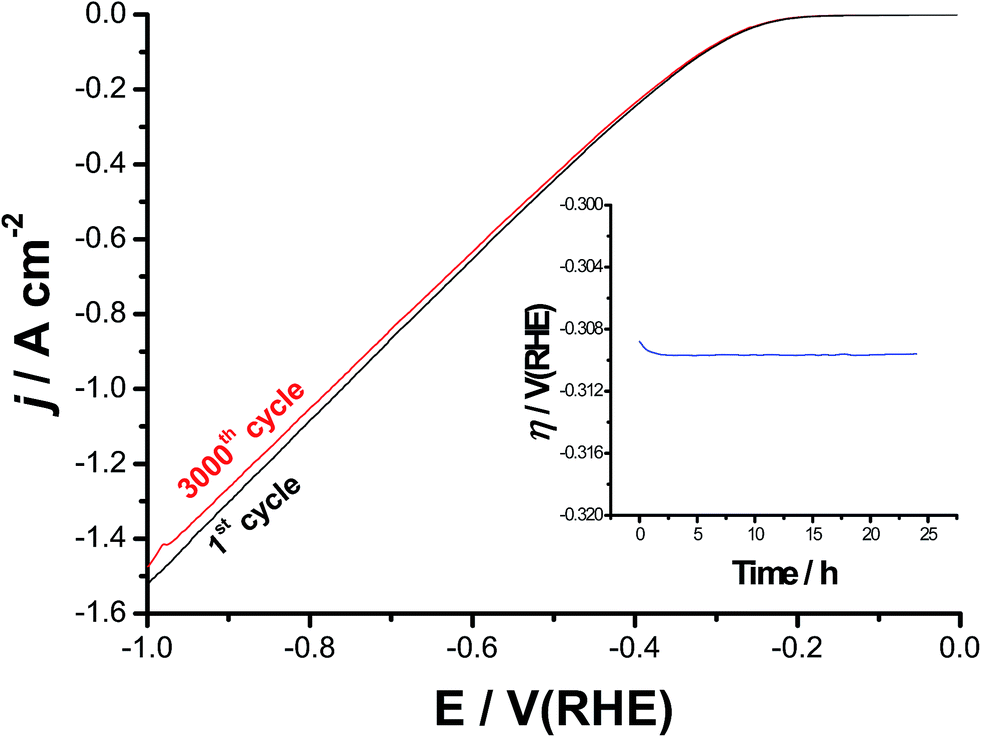 | ||
| Fig. 10 Long-term stability and durability tests for the best catalyst (alloy II after 24 h of free corrosion in 0.5 M NaCl solution at 25 °C). Measurements were carried out in 0.1 M KOH solutions at 25 °C. The main figure represents the cathodic polarization curves initially and after 3000 of continuous cycles between −1.0 V(RHE) and Ecorr at a scan rate of 50 mV s−1. Inset: time-dependent overpotential curve at a constant cathodic current density of 100 mA cm−2 for 24 h. | ||
4. Conclusion
The effect of alloyed Al addition (1.0 wt%) on the corrosion and electrochemical behavior of CoNiGa magnetic shape memory alloy was studied in 0.5 M NaCl solutions, employing various electrochemical techniques. Alloyed Al was found to enhance uniform corrosion of the tested MSMA. In this respect, Tafel extrapolation and linear polarization resistance methods showed an obvious increase in the uniform corrosion rate in presence of alloyed Al. Impedance measurements performed at the corrosion potential (Ecorr) came to the same conclusion and showed that the addition of Al to the tested MSMA decreased the total impedance (Z). Alloyed Al has also weakened passivity, and hence accelerated pitting attack induced by aggressive Cl− anions, of the studied MSMA, as evidenced from cyclic polarization and chronoamperometry measurements. XPS studies confirmed weakness of passivity in presence of Al, which was translated to enhanced adsorption of Cl−. Uniform and pitting corrosion processes promoted by alloyed Al were also supported from morphological studies based on SEM. Alloyed Al also increased the cathodic hydrogen evolution kinetics on the tested MSMA, as revealed by cathodic polarization and impedance measurements in 0.1 M KOH solutions. The electrocatalytic activity of CoNiGaAl MSMA towards the hydrogen evolution reaction in the KOH solutions was further improved approaching that of Pt/C via 24 h of free corrosion pre-treatment in 0.5 M NaCl solution at room temperature. Such corrosion pre-treatment has resulted in intense pitting attack. These results reveal that the combination of surface roughness (due to pitting corrosion catalyzed by alloyed Al) and high crystallinity (due to the martensite phase formation induced by alloyed Al) are highly beneficial for the greatly enhanced electrocatalytic activity of the pitted CoNiGaAl MSMA, which also exhibited good stability and durability.References
- H. E. Karaca, I. Karaman, Y. I. Chumlyakov, D. C. Lagoudas and X. Zhang, Scr. Mater., 2004, 51, 261–266 CrossRef CAS.
- H. E. Karaca, I. Karaman, D. C. Lagoudas, H. J. Maier and Y. I. Chumlyakov, Scr. Mater., 2003, 49, 831–836 CrossRef CAS.
- D. Meyer, H. J. Maier, J. Dadda, I. Karaman and H. E. Karaca, Mater. Sci. Eng., A, 2006, 438–440, 875–878 CrossRef.
- V. A. Chernenko, E. Cesari, V. V. Kokorin and I. N. Vitenko, Scr. Mater., 1995, 33, 1239–1244 CrossRef CAS.
- K. Ullako, J. K. Huang, C. Kantner, R. C. O'Handley and V. V. Kokorin, Appl. Phys. Lett., 1996, 69, 1966–1968 CrossRef.
- S. J. Murray, M. Marioni, S. M. Allen and R. C. O'Handley, Appl. Phys. Lett., 2000, 77, 886–888 CrossRef CAS.
- A. Sozinov, A. A. Likhachev, N. Lanska and K. Ullakko, Appl. Phys. Lett., 2002, 80, 1746–1748 CrossRef CAS.
- E. Bonnot, R. Romero, L. Mañosa, E. Vives and A. Planes, Phys. Rev. Lett., 2008, 100, 125901 CrossRef PubMed.
- A. Planes, L. Manosa and M. Acet, J. Phys.: Condens. Matter, 2009, 21, 233201 CrossRef PubMed.
- K. A. Gschneidner Jr, V. Pecharsky and A. Tsokol, Rep. Prog. Phys., 2005, 68, 1479 CrossRef.
- R. D. James and M. Wuttig, Philos. Mag. A, 1998, 77, 1273–1299 CAS.
- T. Kakeshita, T. Takeuchi, T. Fukuda, T. Saburi, R. Oshima, S. Muto and K. Kishio, Mater. Trans., JIM, 2000, 41, 882–887 CrossRef CAS.
- F. Gejima, Y. Sutou, R. Kainuma and K. Ishida, Metall. Mater. Trans. A, 1999, 30, 2721–2723 CrossRef.
- K. Oikawa, L. Wulff, T. Iijima, F. Gejima, T. Ohmori, A. Fujita, K. Fukamichi, R. Kainuma and K. Ishida, Appl. Phys. Lett., 2001, 79, 3290–3292 CrossRef CAS.
- H. Morito, A. Fujita, K. Fukamichi, R. Kainuma, K. Ishida and K. Oikawa, Appl. Phys. Lett., 2002, 81, 1657–1659 CrossRef CAS.
- S. Kaul, B. A. D'Santhoshini, A. Abhyankar, L. F. Barquin and P. Henry, Appl. Phys. Lett., 2006, 89, 093119 CrossRef.
- K. Oikawa, T. Ota, F. Gejima, T. Ohmori, R. Kainuma and K. Ishida, Mater. Trans., JIM, 2001, 42, 2472–2475 CrossRef CAS.
- A. Gebert, S. Roth, S. Oswald and L. Schultz, Corros. Sci., 2009, 51, 1163–1171 CrossRef CAS.
- M. A. Amin, N. El-Bagoury and H. Shokry, Int. J. Electrochem. Sci., 2013, 8, 2791–2805 CAS.
- L. L. Stepan, D. S. Levi, E. Gans, K. P. Mohanchandra, M. Ujihara and G. P. Carman, J. Biomed. Mater. Res., Part A, 2007, 82, 768–776 CrossRef CAS PubMed.
- P. Paunovic, O. Popovski, S. Hadzi Jordanov, A. Dimitrov and D. Slavkov, J. Serb. Chem. Soc., 2006, 71, 149–165 CrossRef CAS.
- P. Elumalai, H. N. Vasan, N. Munichandraiah and S. A. Shivashankar, J. Appl. Electrochem., 2002, 32, 1005–1010 CrossRef CAS.
- L. D. Rafailovic, C. Gammer, C. Rentenberger, C. Kleber, A. H. Whitehead, B. Gollas and H. P. Karnthaler, Phys. Chem. Chem. Phys., 2012, 14, 972–980 RSC.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- M. Sanchez-Carrillo, J. P. Flores de los Rios, E. Huape-Padilla, R. G. Bautista Margulis, H. Flores-Zuniga, M. I. Ferrer-Sanchez, G. Lopez-Ocana, L. G. Chacon-Nava and A. Martinez-Villafane, Int. J. Electrochem. Sci., 2014, 9, 7725–7735 CAS.
- ASTM G 31, Standard Practice for Laboratory Immersion Corrosion Testing of Metals, Licensed for BASF, p. 5 Search PubMed.
- J. A. Richardson and G. C. Wood, Corros. Sci., 1970, 10, 313–323 CrossRef CAS.
- A. Kolics, J. C. Polkinghorne and A. Wieckowski, Electrochim. Acta, 1998, 43, 2605–2618 CrossRef CAS.
- Z. A. Foroulis and M. J. Thubrikar, J. Electrochem. Soc., 1975, 122, 1296–1301 CrossRef CAS.
- R. T. Foley and T. H. Nguyen, J. Electrochem. Soc., 1982, 129, 464–467 CrossRef CAS.
- T. P. Hoar, Corros. Sci., 1967, 7, 341–355 CrossRef CAS.
- Z. Szklarska-Smialowska, Pitting Corrosion of Metals, NACE, Houston, TX, 1986 Search PubMed.
- M. A. Amin, H. H. Hassan and S. S. Abd El Rehim, Electrochim. Acta, 2008, 53, 2600–2609 CrossRef CAS.
- M. A. Amin, S. S. Abd El Rehim and A. S. El-Lithy, Corros. Sci., 2010, 52, 3099–3108 CrossRef CAS.
- A. G. Munoz and J. B. Bessone, Corros. Sci., 1999, 41, 1447–1463 CrossRef CAS.
- V. Moutarlier, M. P. Gigandet and J. Pagetti, Appl. Surf. Sci., 2003, 206, 237–249 CrossRef CAS.
- Z. Szklarska-Smialowska, Corros. Sci., 1999, 41, 1743–1767 CrossRef CAS.
- M. A. Amin, Electrochim. Acta, 2011, 56, 2518–2531 CrossRef CAS.
- M. A. Amin, N. El-Bagoury, M. Saracoglu and M. Ramadan, Int. J. Electrochem. Sci., 2014, 9, 5352–5374 CAS.
- M. A. Amin, S. S. Abd El-Rehim, E. E. F. El-Sherbini, S. R. Mahmoud and M. N. Abbas, Electrochim. Acta, 2009, 54, 4288–4296 CrossRef CAS.
- N. El-Bagoury, M. A. Amin and M. Saracoglu, Int. J. Electrochem. Sci., 2015, 10, 5291–5308 CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamental and Applications, John Wiley and Sons, Inc, New York, 2001 Search PubMed.
- C. Lupi, A. Dell'Era and M. Pasquali, Int. J. Hydrogen Energy, 2009, 34, 2101–2106 CrossRef CAS.
- F. Safizadeh, E. Ghali and G. Houlachi, Int. J. Hydrogen Energy, 2015, 40, 256–274 CrossRef CAS.
- F. Rosalbino, S. Delsante, G. Borzone and E. Angelini, Int. J. Hydrogen Energy, 2008, 33, 6696–6703 CrossRef CAS.
- M. M. Jaksic, Int. J. Hydrogen Energy, 2001, 26, 559–578 CrossRef CAS.
- M. Xia, T. Lei, N. Lv and N. Li, Int. J. Hydrogen Energy, 2014, 39, 4794–4802 CrossRef CAS.
- R. Solmaz and G. Kardas, Int. J. Hydrogen Energy, 2011, 34, 12079–12087 CrossRef.
- L. M. Rodrıguez-Valdez, I. Estrada-Guel, F. Almeraya-Calderon, M. A. Neri-Flores, A. Martınez-Villafane and R. Martınez-Sanchez, Int. J. Hydrogen Energy, 2004, 29, 1141–1145 Search PubMed.
- A. Barbucci, G. Cerisola and P. L. Cabot, J. Electrochem. Soc., 2002, 149, B534–B542 CrossRef CAS.
- Q. Lu, G. Hutchings, W. Yu, Y. Zhou, R. Forest, R. Tao, J. Rosen, B. Yonemoto, Z. Cao, H. Zheng, J. Xiao, F. Jiao and J. Chen, Nat. Commun., 2015, 6, 6567–6574 CrossRef CAS PubMed.
- J. Durst, A. Siebel, C. Simon, F. Hasché, J. Herranz and H. A. Gasteiger, Energy Environ. Sci., 2014, 7, 2255–2260 CAS.
- E. A. Franceschini, G. I. Lacconi and H. R. Corti, Electrochim. Acta, 2015, 159, 210–218 CrossRef CAS.
- D. Delgado, G. Hefter and M. Minakshi, Hydrogen Generation, in Alternative Energies, ed. G. Ferreira, Springer, Berlin, 2013, pp. 141–161 Search PubMed.
- P. Elumalai, H. N. Vasan, N. Munichandraiah and S. A. Shivashankar, J. Appl. Electrochem., 2002, 32, 1005–1010 CrossRef CAS.
- G. Darabdhara, M. A. Amin, G. A. M. Mersal, E. M. Ahmed, M. R. Das, M. B. Zakaria, V. Malgras, S. M. Alshehri, Y. Yamauchi, S. Szunerits and R. Boukherroub, J. Mater. Chem. A, 2015, 3, 20254–20266 CAS.
- M. A. Amin, S. A. Fadlallah and G. S. Alosaimi, Int. J. Hydrogen Energy, 2014, 39, 19519–19540 CrossRef CAS.
- M. A. Amin, S. A. Fadlallah, G. S. Alosaimi, F. Kandemirli, M. Saracoglu, S. Szunerits and R. Boukherroub, Int. J. Hydrogen Energy, 2016, 41, 6326–6341 CrossRef CAS.
- B. Losiewicz, A. Budniok, E. Rowinski, E. Lagiewka and A. Lasia, Int. J. Hydrogen Energy, 2004, 29, 145–157 CrossRef CAS.
- C. González-Buch, I. Herraiz-Cardona, E. M. Ortega, J. García-Antón and V. Pérez-Herranz, Chemical Engineering Transactions, 2013, 32, 865–870 Search PubMed.
- X. Wu, H. Ma, S. Chen, Z. Xu and A. Sui, J. Electrochem. Soc., 1999, 146, 1847–1853 CrossRef CAS.
- H. Ma, S. Chen, B. Yin, S. Zhao and X. Liu, Corros. Sci., 2003, 45, 867–882 CrossRef CAS.
- Z. B. Stoynov, B. M. Grafov, B. Savova-Stoynova and V. V. Elkin, Electrochemical Impedance, Nanka, Moscow, 1991 Search PubMed.
- F. B. Growcock and R. J. Jasinski, J. Electrochem. Soc., 1989, 136, 2310–2314 CrossRef CAS.
- M. Monev, Bulg. Chem. Commun., 2016, 48, 73–77 Search PubMed.
- D. M. Soares, O. Teschke and I. Torriani, J. Electrochem. Soc., 1992, 139, 98–105 CrossRef CAS.
- D. S. Hall, C. Bock and B. R. MacDougalla, J. Electrochem. Soc., 2013, 160, F235–F243 CrossRef CAS.
- C. Lupi, A. Dell'Era and M. Pasquali, Int. J. Hydrogen Energy, 2014, 39, 1932–1940 CrossRef CAS.
- F. Rosalbino, D. Macciò, A. Saccone and G. Scavino, Int. J. Hydrogen Energy, 2014, 39, 12448–12456 CrossRef CAS.
- W. Ben Aziza, J. F. Petit, U. B. Demirci, Q. Xu and P. Miele, Int. J. Hydrogen Energy, 2014, 39, 16919–16926 CrossRef CAS.
- M. I. Jamesh, J. Power Sources, 2016, 333, 213–236 CrossRef CAS.
- S. H. Hong, S. H. Ahn, I. Choi, S. G. Pyo, H.-J. Kim, J. H. Jang and S.-K. Kim, Appl. Surf. Sci., 2014, 307, 146–152 CrossRef CAS.
- V. Bachvarov, E. Lefterova and R. Rashkov, Int. J. Hydrogen Energy, 2016, 41, 12762–12771 CrossRef CAS.
- H. S. Shatak, Corrosion of Austenitic Stainless Steel, Narosa Publishing House, London, 2002, p. 41 Search PubMed.
- B. M. Gonzalez, C. S. B. Castro, V. T. L. Buono, J. M. C. Vilela, M. S. Andrade, J. M. D. Moraes and M. J. Mantel, Mater. Sci. Eng., A, 2003, 343, 51–56 CrossRef.
- F. S. Shieu, M. J. Deng and S. H. Lin, Corros. Sci., 1998, 40, 1267–1279 CrossRef CAS.
- M. C. Biesinger, B. P. Paynec, A. P. Grosvenor, L. W. M. Laua, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- D. Briggs, XPS: Basic Principles, Spectral Features and Qualitative Analysis, in Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, ed. D. Briggs and J. T. Grant, IM Publications, Chichester, 2003, pp. 31–56 Search PubMed.
- J. F. Xie, H. Zhang, S. Li, R. X. Wang, X. Sun, M. Zhou, J. F. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25384a |
| This journal is © The Royal Society of Chemistry 2017 |