
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient access to triarylmethanes through decarboxylation†
Tiash Sahaa,
M. Srinivas Lavanya Kumara,
Saurav Beraa,
Bidhu Bhusan Karkaraa and
Gautam Panda *ab
*ab
aMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, BS 10/1, Jankipuram Extension, Sitapur Road, Lucknow 226031, India. E-mail: gautam.panda@gmail.com; gautam_panda@cdri.res.in; Fax: +91-522-2771941; Tel: +91-522-2772450 ext. 4661 Tel: +91-522-2772550 ext. 4662
bAcademy of Scientific and Innovative Research, New Delhi 110001, India
First published on 20th January 2017
Abstract
A new synthetic approach has been developed for efficient access to triarylmethanes by palladium catalyzed decarboxylative cross coupling reactions. The reaction features sp2–sp3 coupling where benzoic acids upon decarboxylation reacted with diaryl methyl iodides having both electron donating and withdrawing functionalities, leading to the synthesis of diverse triarylmethanes.
Introduction
The triarylmethane class of molecules contains several biologically significant motifs displaying a wide range of pharmaceutical profiles.1 It forms the basic skeleton of several antitubercular,2a anticancer,2b,c antidiabetic2d and other medicinally important molecules, Fig. 1. These privileged architectures can be diversely functionalized in the quest for new therapeutic agents and unexplored biologically relevant chemical spaces.3 Several triarylmethane based dyes were also developed as sensitizers, exhibiting selective phototoxicity towards cancerous cell lines and thus forms the basic principle of photodynamic therapy.4 These triarylmethanes exhibit interesting properties like photo lability, photo ionization which were successfully exploited for the development of many organic based dyes, photochromatic devices and fluorescent probes.4b,5 Apart from this, these class of compounds often found their explosive growth in the construction of supramolecular architectures.6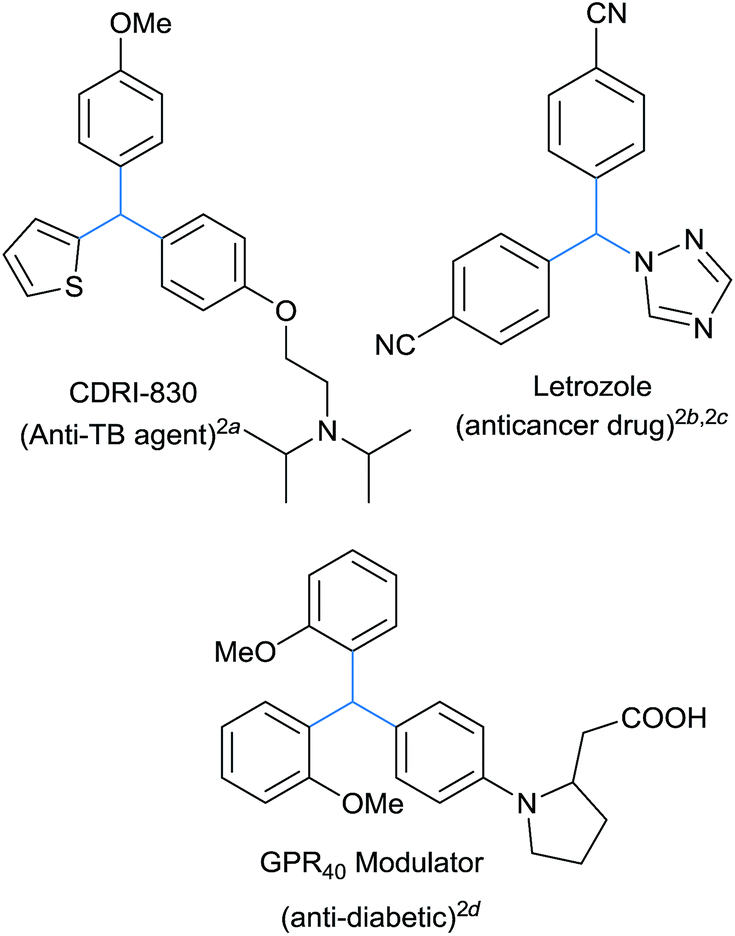 | ||
| Fig. 1 Representative triarylmethane containing architectures. | ||
Conventionally, triarylmethanes are synthesized through typical Friedel–Crafts alkylation of diarylcarbinols with aromatic ring as nucleophile7 or hydroarylation of arenes with aromatic aldehydes or imines in the presence of either suitable Bronsted or Lewis acid systems.1a Recently, Crudden et al. reported the synthesis of triarylmethanes through arylative desulfonation8 whereas Chakravarty et al. reported Friedel–Crafts reaction on benzylic phosphates for the synthesis of triarylmethanes.9a These procedures often suffer from lack of selectivity, involves multistep synthesis and moreover is limited to only electron rich arene systems.9b,c
Due to this, cross coupling based strategies emerged as promising avenue for the short and efficient synthesis of triarylmethanes.10 Oshima et al. reported palladium catalyzed direct C–H bond activation11 of aryl(azaaryl)methanes for synthesis of triarylmethanes. L. A. López et al. reported zinc catalyzed cascade reaction between enynones and azoles resulting in the diversely substituted triarylmethanes synthesis.12 A reductive cross coupling was developed by Zhang et al. using benzophenone tosylhydrazones using palladium catalyst.13 Jarvo et al. synthesized enantioenriched triarylmethanes by stereospecific cross coupling in presence of nickel catalyst.14 Similarly, palladium catalysed C–H/C–O coupling of oxazoles with diarylmethanol derivatives have also been reported for synthesis of triarylmethanes.15
Decarboxylative cross coupling reaction has been emerged as a powerful strategy for C–C bond formation16 in the recent past and had been largely explored. Organometallic species formed via expulsion of CO2 from commercially available arene carboxylic acids were ventured in this cross coupling strategy. Goossen et al. synthesized arylalkenes via decarboxylative cross coupling.17 Palladium catalyzed decarboxylative cross coupling, as demonstrated by Becht and coworkers18 were used in the synthesis of biaryl motifs. Even industrial scale production of commercially important biaryls like intermediates of agrochemicals boscalid, bixafen has been demonstrated by this strategy.19 Decarboxylation reactions were well explored in sp–sp2, sp2–sp2 cross couplings whereas sp2–sp3 coupling was hardly explored.20 In our quest for highly desirable triarylmethanes,1b,2b,7 we here reported an efficient palladium catalyzed decarboxylative sp2–sp3 cross coupling for the access to triarylmethanes with both electron demanding and donating substitutions using commercially available benzoic acids with tailor made diarylmethyl iodides.
Results and discussion
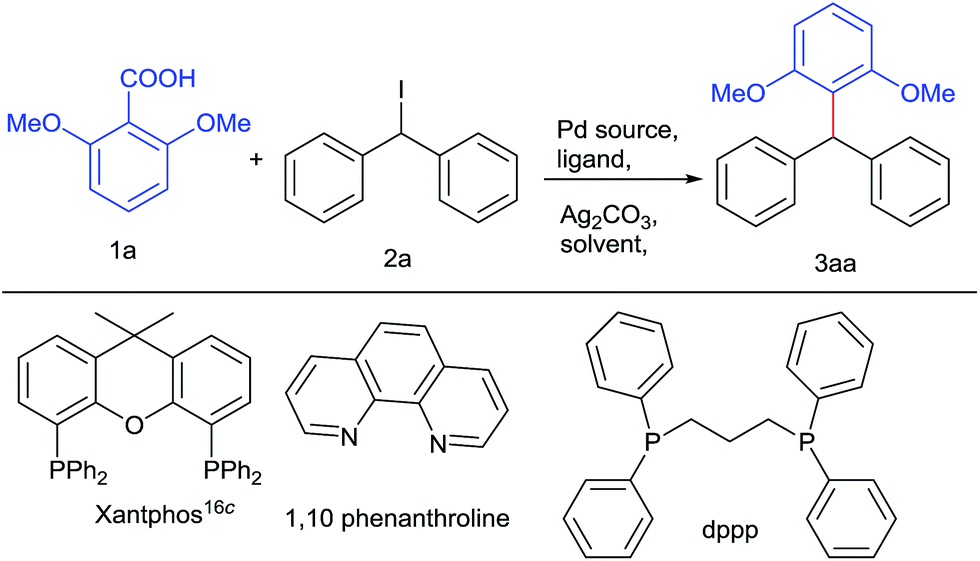
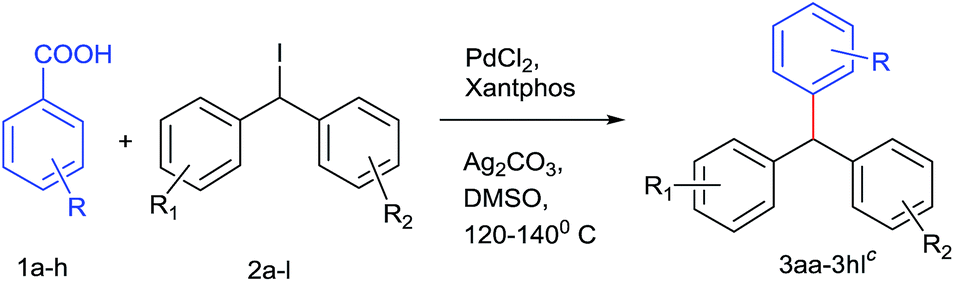
In principle, we presumed that aromatic carboxylic acids16a can be used as boronic acid surrogate and palladium catalyst could synchronize coupling16b between decarboxylated benzoic species with diarylmethyl iodides leading to these valuable architectures. For the test reaction, we have chosen 2,6 dimethoxy benzoic acid and diphenylmethyl iodide21 as model substrates. Preliminary screening using Pd(OAc)2 and dppp as a ligand in the presence of Ag2CO3 (Table 1, entries 1 and 2) was unsuccessful. Then changing the palladium catalyst to Pd2(dba)3 (Table 1, entry 3) resulted in 22% yield. Altering the bases to Cs2CO3, Li2CO3, Cs2CO3, AgOAc, DBU and K2CO3 were unable to increase the yield of the desired product to significant level. Employing additive such as CuI, resulted in trace amount of product. Keeping palladium source intact, varying the ligands to triphenyl phosphine and 1,10-phenanthroline resulted in more or less comparable yields but xantphos gave promising yields (Table 1, entries 4–6). Some by-products due to protodecarboxylation of carboxylic acids and deiodination of diphenyl methyl iodides were also isolated.22,23 Then surveying various palladium catalysts like Pd(CH3CN)2Cl2 and Pd(Cl)2(PPh3)2, Pd(acac)2, PdCl2 and careful solvent screening revealed that PdCl2 along with xantphos ligand, Ag2CO3 as base in presence of DMSO offered the ideal conditions for the reaction (Table 1, entry 11).
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | Ligand | Solvent | Yieldf (%) |
| a Reaction conditions: Pd source (0.1 equiv.), ligand (0.2 equiv.), Ag2CO3 (1.5 equiv.), arene carboxylic acid (1.2 equiv.), methyl iodide (1.0 equiv.), 100 °C.b Reaction was performed at 65 °C.c CuI additive (0.2 eq.) is used.d Pd catalyst (0.2 eq.) was used.e Reaction was performed at 120 °C.f Overall isolated yield.g Nd means not determined. | ||||
| 1b | Pd(OAc)2 | Dppp | THF | 0 |
| 2 | Pd(OAc)2 | Dppp | DMF | Ndg |
| 3 | Pd2(dba)3 | Dppp | DMF | 22 (trace)c |
| 4 | Pd2(dba)3 | 1,10-Phenanthroline | DMF | Ndg |
| 5 | Pd2(dba)3 | Xantphos | DMF | 42 |
| 6 | Pd2(dba)3 | PPh3 | DMF | 34 |
| 7 | PdCl2 | Xantphos | DMF | 59 (47)d |
| 8 | Pd(acac)2 | Xantphos | DMF | 31 |
| 9 | Pd(Cl)2(PPh3)2 | Xantphos | DMF | 40 |
| 10 | Pd(OAc)2 | Xantphos | DMF | 42 |
| 11 | PdCl2 | Xantphos | DMSO | 62 (68)e |
| 12 | PdCl2 | Xantphos | NMP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mesitylene (1:1) :mesitylene (1:1) |
Trace |
| 13 | PdCl2 | Xantphos | Diglyme | 15 |
After optimizing the catalyst system for decarboxylative coupling reactions, the scopes of substrates were evaluated with reference to both the coupling partners (Table 2). Poor yields were obtained in the case of diphenylmethyl bromide (38%) and reaction failed when performed with diphenylmethyl chloride. The electron rich 2,6 dimethoxy benzoic acid 1a was successfully coupled with various diphenylmethyl iodides which provided sterically hindered triarylmethanes 3aa–3ae, 3ah in good yields. Similarly, 2-methoxy benzoic acid 1b was successfully cross coupled with diphenylmethyl iodide derivatives 2a, 2f, 2g and even with the substrate 2i resulting in triarylmethane derivatives 3ba, 3bf, 3bg and 3bi. Thus, the reaction was also compatible with unsubstituted benzene carboxylic acids affording the products 3ca, 3cj in 67% and 56% yield, respectively. Even sterically demanding benzoic acid 1e also worked well in this strategy leading to cross coupled product 3ef. In order to explore the feasibility of this reaction further, cross coupling reaction was performed between electron poor counterparts. Slight modification in the reaction conditions needed for the elaboration of this cross coupling reaction to electron poor triaryl methanes. Doubling the amount of catalyst and ligand and changing the solvent system to DMSO:DMF (90:10) resulted the coupling of 2-fluoro 5-nitro benzoic acid 1d and 2-nitrobenzoic acid 1f with the methyl iodide 2k successfully in moderate yields leading to 3dk and 3fk respectively.
|
|---|
| a Reaction conditions: substituted diphenylmethyl iodide (1 equiv.), aryl carboxylic acid (1 equiv.), PdCl2 (0.1 equiv.), xantphos (0.2 equiv.), Ag2CO3 (1.5 equiv.), DMSO.b Substituted diphenylmethyl iodide (1 equiv.), aryl carboxylic acid (1 equiv.), PdCl2 (0.2 equiv.), xantphos (0.4 equiv.), Ag2CO3 (1.5 equiv), DMSO:DMF (90:10).c Isolated yields after column chromatography in silica gel. |
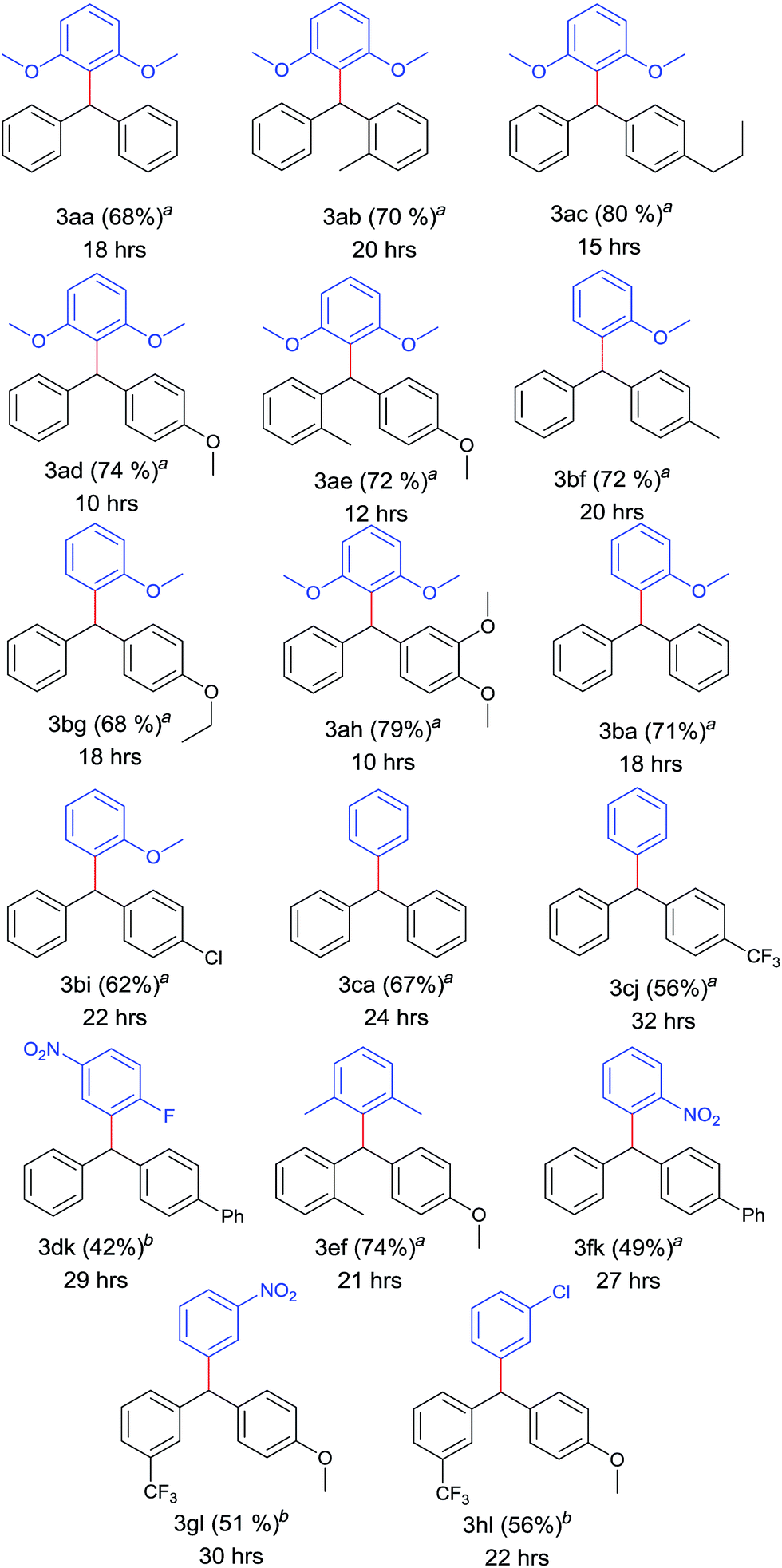 |
Electron density on aromatic backbone showed strong influence on reactivity and yields. Thus, the catalyst system showed its suitability for wide variety of diphenylmethyl iodides and both electron rich and electron poor benzoic acids successfully underwent decarboxylation to yield diverse triarylmethanes.
After establishing the decarboxylative cross coupling reaction with arenes, we verified the methodology towards synthesizing heteroaryl containing triarylmethanes. Attempted decarboxylative cross coupling (Fig. 2) between phenyl thiophenyl methyl iodide and benzoic acid was also successful using 0.2 equiv. PdCl2, 0.4 equiv. xantphos, 2.0 equiv. Ag2CO3 at 140 °C in DMSO:DMF (90:10) for 24 h. Agreeably, the cross coupling product 5 was observed in good yield and it provided a new horizon in the synthesis of antitubercular analogue of CDRI-830 using sp2–sp3 decarboxylative cross coupling.2a
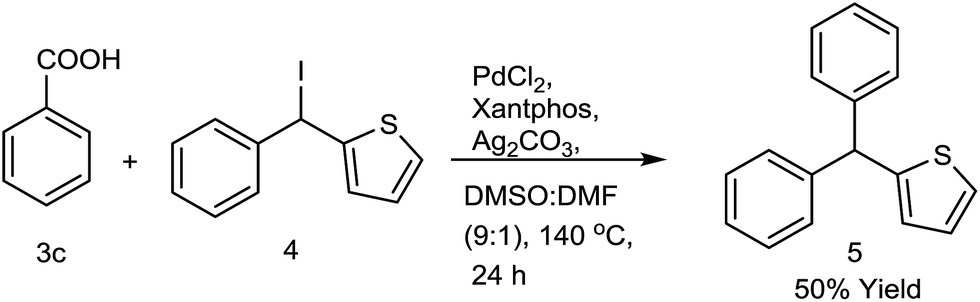 | ||
| Fig. 2 Synthesis of heteroaryl substituted triarylmethanes. | ||
Experimental section
General
All the reactions were carried out in oven-dried glassware under argon atmosphere. Carboxylic acids were purchased from Sigma Aldrich. Palladium catalysts were purchased from Sigma Aldrich. Phosphine ligands were bought from Sigma-Aldrich or Alfa Aesar and used as such. DMSO and DMF was dried according to standard procedure and stored on molecular sieves 4 Å. All the other reagents and solvents mentioned in this text were bought from Sigma Aldrich or Alfa Aesar or spectrochem and purified if necessary. NMR spectra were recorded on 300, 400 or 500 MHz spectrometer for 1H NMR, 75 or 100 or 125 MHz for 13C NMR spectroscopy. Chemical shifts are reported in parts per million (δ) downfield relative to the residual signals of tetramethylsilane in CDCl3. Spin multiplicities are indicated by the following symbol: s (singlet), d (doublet), t (triplet) and m (multiplet).General procedure for synthesis of diphenyl carbinols
A 100 ml double-necked round bottom flask was charged with magnesium (2 equiv.), pinch of iodine and after flushing with nitrogen, dried THF was added and stirred. Then corresponding bromoarene (1.5 equiv.) was added, the colour of iodine disappeared slowly on time, indicating the generation of Grignard reagent. It was stirred for one hour followed by addition of corresponding carbaldehye (1 equiv.). Completion of reaction was determined by TLC. Reaction was quenched by saturated NH4Cl solution. Organic layer was extracted thrice with ethyl acetate and concentrated in vacuo. Compound was purified by column chromatography.General procedure for synthesis of diphenyl methyl iodide
To oven dried round bottom flask under nitrogen, the alcohol (1 equiv.) and KI (1 equiv.) were added and dissolved in dry 1,4-dioxane and stirred for 5 min. Then BF3·Et2O (1 equiv.) was added and stirred at room temperature. Completion of reaction was monitored by TLC. The reaction mixture was poured in cold water and extracted with dichloromethane. The organic layer was washed with water and dried by sodium sulphate; solvent was evaporated under reduced pressure. The crude was purified by quickly passing through short pad of silica using hexane/ethyl acetate (8:2) as eluent to obtain diphenylmethyl iodides and should be used immediately.
General procedure A for synthesis of various substituted triarylmethanes by decarboxylative cross coupling
To the oven dried round bottom flask was charged with aryl carboxylic acid (1 equiv.), Ag2CO3 (1.5 equiv.), PdCl2 (0.1 equiv.), xantphos (0.2 equiv.), diphenyl methyl iodides (1 equiv.) were dissolved in DMSO. The reaction mixture was degassed with argon thrice and stirred in preheated oil bath. Reaction as monitored by TLC after completion, the reaction mixture was cooled to room temperature and filtered through celite bed. The organic layer was washed with saturated NH4Cl solution and dried on sodium sulphate and concentrated under reduced pressure. It was later purified by column chromatography on silica gel to obtain corresponding triarylmethanes.General procedure B for synthesis of substituted triarylmethanes by decarboxylative cross coupling
To the oven dried round bottom flask was charged with aryl carboxylic acid (1 equiv.), Ag2CO3 (1.5 equiv.), PdCl2 (0.2 equiv.), xantphos (0.4 equiv.), diphenyl methyl iodides (1 equiv.) were dissolved in DMSO:DMF 90:10. The reaction mixture was degassed with argon thrice and stirred in preheated oil bath at requisite temperature and was monitored by TLC. After the completion of reaction, mixture was cooled to room temperature and filtered through a bed celite with ethyl acetate. The organic layer was washed with saturated NH4Cl solution and dried on sodium sulphate and concentrated in vacuo. It was later purified by column chromatography on silica gel to obtain corresponding triarylmethanes.
Spectroscopic data of synthesized compounds
Conclusion
In conclusion, we reported an efficient pathway for synthesizing various triarylmethanes using sp2–sp3 decarboxylative cross coupling reactions. These reactions showed functional group tolerance, providing moderate to good yields of diverse triarylmethanes in less complicated steps. Decarboxylative cross coupling on heteroaryl methyl iodide provides an opportunity for accessing diverse biologically important triarylmethanes and work in this direction is currently underway.Acknowledgements
This research project was partly supported by DST project (SB/S1/OC-93/2013). SLKM and SB thank Council of Scientific and Industrial Research (CSIR), India for research fellowship. Instrumental facilities from SAIF, CDRI is acknowledged (CDRI Communication No. 9408).Notes and references
- (a) S. Mondal and G. Panda, RSC Adv., 2014, 4, 28317–28358 RSC; (b) S. K. Das Shagufta and G. Panda, Tetrahedron Lett., 2005, 46, 3097 CrossRef.
- (a) P. Singh, S. K. Manna, A. K. Jana, T. Saha, P. Mishra, S. Bera, M. K. Parai, M. S. L. Kumar, S. Mondal, P. Trivedi, V. Chaturvedi, S. Singh, S. Sinha and G. Panda, Eur. J. Med. Chem., 2015, 95, 357 CrossRef CAS PubMed; (b) R. Palchaudhuri, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2008, 130, 10274 CrossRef CAS PubMed; (c) R. S. Dothager, K. S. Putt, B. J. Allen, B. J. Leslie, V. Nesterenko and P. J. Hergenrother, J. Am. Chem. Soc., 2005, 127, 8686–8696 CrossRef CAS PubMed; (d) B. A. Ellsworth, W. R. Ewing and E. Jurica, US Pat., 2011/0082165A1, 2011.
- J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629 CrossRef CAS PubMed.
- (a) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897 CrossRef CAS PubMed; (b) S. K. Das, Shagufta and G. Panda, Tetrahedron, 2006, 62, 6731 CrossRef.
- (a) C. D. Mason and F. F. Nord, J. Org. Chem., 1951, 16, 722 CrossRef CAS; (b) V. V. Ghaisas, B. J. Kane and F. F. Nord, J. Org. Chem., 1958, 23, 560 CrossRef CAS; (c) M. Irie, J. Am. Chem. Soc., 1983, 105, 2078 CrossRef CAS; (d) R. Muthyala, A. R. Katritzky and X. F. Lan, Dyes Pigm., 1994, 25, 303 CrossRef CAS.
- (a) D. F. Duxbury, Chem. Rev., 1993, 93, 381 CrossRef CAS; (b) M. S. Shchepinov and V. A. Korshun, Chem. Soc. Rev., 2003, 32, 170 RSC.
- G. Panda, M. K. Parai, S. K. Das Shagufta, M. Sinha, V. Chaturvedi, A. K. Srivastava, Y. S. Manju, A. N. Gaikwad and S. Sinha, Eur. J. Med. Chem., 2007, 42, 410 CrossRef CAS PubMed.
- M. Nambo, Z. T. Ariki, D. Canseco-Gonzalez, D. D. Beattie and C. M. Crudden, Org. Lett., 2016, 18, 2339 CrossRef CAS PubMed.
- (a) G. Pallikonda and M. Chakravarty, J. Org. Chem., 2016, 81, 2135 CrossRef CAS PubMed; (b) Y. Z. Li, B. J. Li, X. Lu, S. Lin and Z. J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS PubMed; (c) X. Ji, T. Huang, W. Wu, F. Liang and S. Cao, Org. Lett., 2015, 17, 5096 CrossRef CAS PubMed.
- M. Nambo and C. M. Crudden, ACS Catal., 2015, 5, 4734 CrossRef CAS.
- T. Niwa, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9(12), 2373 CrossRef CAS PubMed.
- J. González, C. P. Calleja, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2013, 52, 5853 CrossRef PubMed.
- Y. Xia, F. Hu, Z. Liu, P. Qu, R. Ge, C. Ma, Y. Zhang and J. Wang, Org. Lett., 2013, 15, 1784 CrossRef CAS PubMed.
- B. L. Taylor, M. R. Harris and E. R. Jarvo, Angew. Chem., Int. Ed. Engl., 2012, 51, 7790 CrossRef CAS PubMed.
- S. Tabuchi, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2014, 79, 5401 CrossRef CAS PubMed.
- (a) Y. Luo and J. Wu, Chem. Commun., 2010, 46, 3785 RSC; (b) N. Rodriquez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC; (c) J. Zhang, A. Bellomo, N. Trongsiriwat, T. Jia, P. J. Carroll, S. D. Dreher, M. T. Tudge, H. Yin, J. R. Robinson, E. J. Schelter and P. J. Walsh, J. Am. Chem. Soc., 2014, 136, 6276 CrossRef CAS PubMed.
- J. Tang and L. J. Goossen, Org. Lett., 2014, 16, 2664 CrossRef CAS PubMed.
- J. M. Becht, C. Catala, C. L. Drian and A. Wagner, Org. Lett., 2007, 9, 1781 CrossRef CAS PubMed.
- K. Park, G. Bae, J. Moon, J. Choe, K. H. Song and S. Lee, J. Org. Chem., 2010, 75, 6244 CrossRef CAS PubMed.
- (a) W. W. Zhang, X. G. Zhang and J. H. Li, J. Org. Chem., 2010, 75, 5259 CrossRef CAS PubMed; (b) F. Bilodeau, M. C. Brochu, N. Guimond, K. H. Thesen and P. Forgione, J. Org. Chem., 2010, 75, 1550 CrossRef CAS PubMed.
- L. J. Goossen, C. Linder and A. Cotte, Saltigo GmbH, DE 102007016971, 2007.
- B. P. Bandgar, V. S. Sadavarte and L. S. Uppalla, Tetrahedron Lett., 2001, 42, 951 CrossRef CAS.
- L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25429b |
| This journal is © The Royal Society of Chemistry 2017 |