
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New pentose dimers with bicyclic moieties from pretreated biomass†
H. Rasmussena,
H. R. Sørensena,
D. Tannerb and
A. S. Meyer *c
*c
aDONG Energy, Kraftværksvej 53, DK-7000 Fredericia, Denmark
bDept. of Chemistry, Technical University of Denmark, DK-2800 Lyngby, Denmark
cCenter for BioProcess Engineering, Dept. of Chemical and Biochemical Engineering, Technical University of Denmark, DK-2800 Lyngby, Denmark. E-mail: am@kt.dtu.dk
First published on 17th January 2017
Abstract
In lignocellulosic biorefinery processes involving enzyme catalysed reactions it is a challenge that enzyme inhibiting compounds are generated and liberated during pretreatment of the biomass. In this study the contribution to cellulase inhibition from xylooligosaccharides and newly discovered oligophenolic compounds from pilot scale pretreated wheat straw was assessed at two different pretreatment severities. An increase in severity of the pretreatment led to more oligophenol compounds and in turn the total overall cellulase inhibition increased. When the xylooligosaccharides were enzymatically degraded prior to cellulose hydrolysis, a relief in cellulase inhibition was observed, but some inhibition remained, suggesting that other components also played a role in inhibition. We propose that these components include dipentoses with bicyclic moieties and feruloylated tripentoses, because LC-MS/MS analysis revealed the presence of these components in the liquid from hydrothermal pretreated wheat straw after enzymatic treatment. The reaction mechanisms for synthesis of the new dipentoses having hydroxylated oxane bicyclic residues are considered and they are proposed to be formed as reaction products from either xylose or glucose reacting with glyceraldehyde during pretreatment. The data show that the main cellulase inhibition from hydrothermally pretreated wheat straw liquors is due to xylooligosaccharides followed by oligophenolic compounds and the newly discovered dipentose with bicyclic moieties and feruloylated tripentoses. The relative amounts and hence contribution to inhibition from each class of compounds changes with severity of the pretreatment.
Introduction
Plant biomass biorefinery processes can be tuned to produce environmentally friendly energy and important chemicals.1–6 In lignocellulose-based biorefinery processes applying enzyme catalysed reactions, it remains a challenge that enzyme inhibiting compounds are generated and liberated during pretreatment of biomass.7,8 Degradation compounds such as phenolic compounds, but also xylooligosaccharides have previously been identified as cellulase inhibitors in lignocellulosic biomass reactions.9–13It complicates the picture that both amount and type of degradation compounds as well as the biomass structural elements that are liberated during pretreatment, can vary with pretreatment process parameters.8 Hence, both the inhibitor potency and the actual amount of an enzyme inhibitor compound determine its contribution to the overall inhibition from the pretreatment liquors and the relative contribution of different types of inhibitor compounds may in turn change when process parameters during pretreatment are changed. To shed light on these relations an “inhibitor mass balance” method was applied to account for inhibition contributions from different compounds in liquors from hydrothermally pretreated wheat straw and in turn account for the inhibition exerted by the liquid fraction after hydrothermal pretreatment (LfHP) (“C5 liquid”, Fig. 1). As illustrated in Fig. 1, in the DONG Inbicon technology, this liquid from hydrothermal pretreatment is returned to the enzymatic hydrolysis, since it is possible to co-ferment C6 and C5 monosaccharides using newly developed microorganisms that can convert both C6 and C5 to ethanol. However, the LfHP xylooligosaccharide-rich “C5 liquid” fraction contains a wide array of inhibitors that negatively affect the enzyme catalyzed cellulose conversion.14 This inhibition is undesirable regardless of whether the liquid fraction is led back for hydrolysis or is not sufficiently separated from the cellulose fibre fraction (“C6 fibre, Fig. 1) in the first round. Our previous work has revealed that even though soluble oligophenols produced during hydrothermal pretreatment of wheat straw are potent enzyme inhibitors, the simultaneously released xylooligosaccharides are more abundant and thus contribute relatively more to the overall cellulase inhibition exerted by the LfHP.14
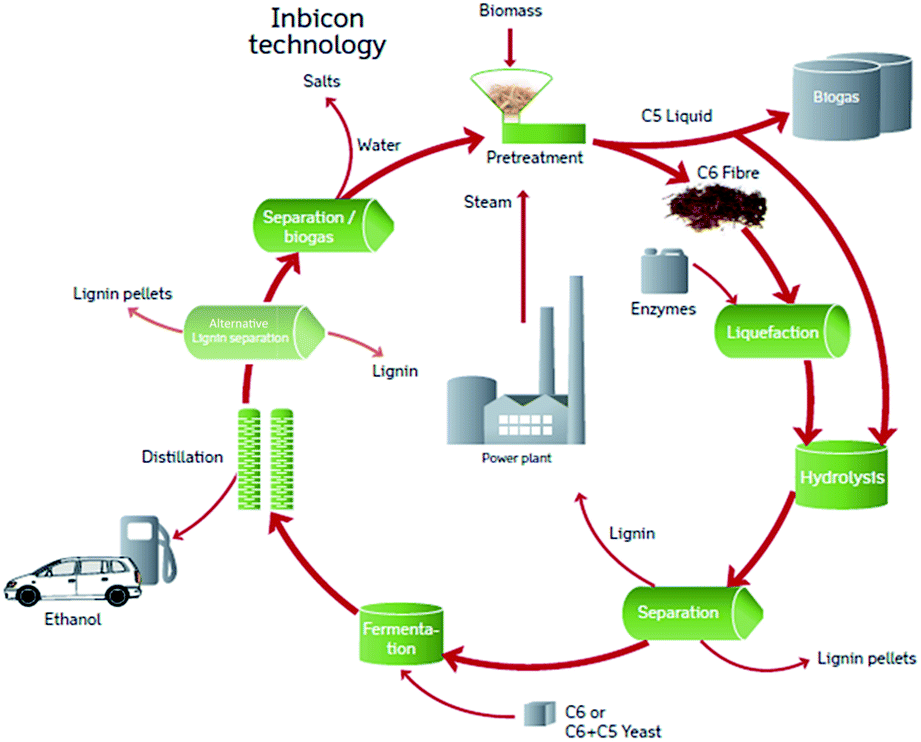 | ||
| Fig. 1 The overall DONG Inbicon technology process from where the liquid from hydrothermal pretreatment (“C5 liquid”) was investigated. | ||
The aim of the present work was to investigate (1) if pretreatment severity would change the relative level of oligophenolic compounds and xylooligosaccharides in liquids from hydrothermally pretreated wheat straw and in turn affect the contribution to total inhibition from that LfHP. (2) If other compounds in the LfHP beyond oligophenols and enzymatically degradable xylooligosaccharides may exert inhibitory action on cellulose degrading enzymes.
Experimental
Chemicals and enzymes
Buffer chemicals, solvents, acids and bases as well as Avicel (a tradename for microcrystalline acid treated cellulose powder derived from wood pulp) were purchased from Sigma Aldrich (Steinheim, Germany). Cellic CTec3 (1920 BHU(2)/g) was obtained from Novozymes A/S (Bagsværd, Denmark). This is a commercially available cellulase preparation based on the Trichoderma reesei cellulolytic enzyme complex. Apart from the cellulolytic enzyme base from T. reesei containing at least the two main cellobiohydrolases EC 3.2.1.91 (Cel6A and Cel7A), several different endo-1,4-β-glucanases EC 3.2.1.4 (Cel7B, Cel5A, Cel12A, and Cel45A), β-glucosidase EC 3.2.1.21, and a β-xylosidase,15 the preparation Cellic CTec3 also contains other proprietary hydrolysis-boosting proteins.Fractionation of pilot plant liquid from hydrothermal pretreatment (LfHP) of wheat straw
The wheat straw composition was determined according to ref. 16 to the following content (w/w%): cellulose 38% ± 0.4%, hemicellulose 27% ± 0.4%, lignin 21 ± 0.5%.Fractionation of the liquid from pilot plant scale hydrothermally pretreated wheat straw was done as previously described,14 in short:
Liquid from pilot scale hydrothermally pretreated wheat straw (12 minutes, 40% (w/w) dry matter, LfHP 1 = 183 °C, LfHP 2 = 191 °C) was extracted with 2-butanone. 170 g liquid from each pretreatment liquor was extracted with 2 × 160 g solvent. The organic phase was evaporated and freeze dried and the residual water phase was freeze dried.
Determination of inhibition from the fractions: enzyme inhibition assay
Enzymatic hydrolysis was carried out as previously described,14 in short:The freeze dried fractions were dissolved in 0.1 M acetate buffer (pH 5.1) to dry matter 13.6% (w/w) and Avicel was added to 12% (w/w). The flasks were incubated at 50 °C, 160 rpm and the enzyme preparation Cellic CTec3 was added to 0.6% (w/w). Upon sampling (200 μL) the enzyme was inactivated at 99 °C for 5 minutes.
Glucose release was measured with High Pressure Anion Exchange Chromatography (HPAEC) as described below.
Enzyme pre-hydrolysis of the water phase from 2-butanone extraction and LfHP (prior to enzyme assay)
The freeze dried compounds from the water phase from 2-butanone extraction was dissolved in 0.1 M acetate buffer (pH 5.1) to dry matter 16.6% (w/w): 15 g scale flasks were incubated at 50 °C, 160 rpm and the cellulase enzyme preparation (Cellic CTec3) was added to 2.4% (w/w). An additional enzyme dose (to a total of 4.8% (w/w)) was added after 3 days and the mixture was allowed to react for another 5 days until the enzyme was inactivated at 99 °C for 15 minutes.At the end of the prehydrolysis the glucose and xylose release were 1 and 0.7% (w/w) respectively. Thus, glucose and xylose were added to these concentrations to control experiments with 12% (w/w) Avicel in the enzyme inhibition assay above, to account for any product inhibition from glucose and xylose in the inhibition mass balance from the prehydrolysed experiments.
Determination of xylooligosaccharides in LfHP
Weak acid hydrolysis was carried out according to Sluiter et al.16 and the hydrolysates were analysed by HPLC as described below.Analytical methods
The column was a pentafluorophenyl (pfp) discovery HS F5, L × I.D. 15 cm × 4.6 mm, 5 μm particle size (Supelco).
Results and discussion
Consequences of increased severity in pretreatment
Hydrothermal pretreatments were performed at 183 °C for 12 minutes and 191 °C for 12 minutes and inhibitory action of the liquid fractions LfHP 1 (183 °C for 12 minutes) and LfHP 2 (191 °C in 12 minutes) and their fractions from extraction were compared. It was found that LfHP 2 was slightly more inhibiting toward cellulases than LfHP 1 (Fig. 2) when compared at the same dry matter level, but both LfHPs retarded the glucose release with ∼75% after 24 hours.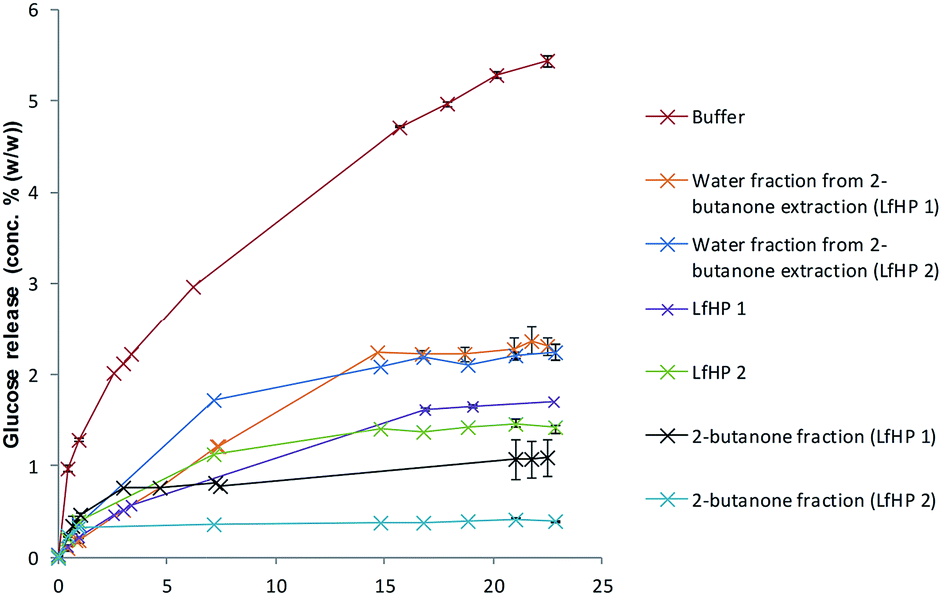 | ||
| Fig. 2 Enzymatic cellulose (Avicel) hydrolysis assay with LfHP from pretreatment for 12 minutes at 183 °C (LfHP 1) and 191 °C (LfHP 2), respectively and corresponding fractions from extractions. | ||
With the 2-butanone extraction method previously described14 it was established that highly potent inhibitory oligophenols were extracted into the 2-butanone organic phase, whereas xylooligosaccharides were left in the water phase (xylooligosaccharides include acetyl and feruloyl substituted xylooligosaccharides).
In the present study, the amount by weight of compounds in the 2-butanone fraction increased by 14% with the more severe pretreatment (LfHP 2), whereas the amount in the water fraction decreased correspondingly.
This indicates that more oligophenolic inhibitors were formed with increased pretreatment severity and more xylooligosaccharides were degraded to xylose and in turn resulted in oligophenolic compounds. The latter presumed to originate mainly from xylose degradation reactions and xylose reactions with lignin solubilisation/degradation compounds, although partial degradation of lignin could also contribute.14 Also the 2-butanone fraction from LfHP 2 was profoundly more inhibiting than the corresponding LfHP 1 2-butanone fraction when compared at the same dry matter basis (Fig. 2).
The LfHP 2 2-butanone fraction thus retarded glucose release by more than 90% compared to the control whereas the LfHP 1 2-butanone fraction retarded glucose release by ∼80% (Fig. 2). These data suggest that the more severe pretreatment had induced the formation of either new more potent inhibitory compounds or changed the profile of oligophenolic compounds extracted into the 2-butanone fraction. The inhibitory effects exerted by the water fractions from the 2-butanone extraction were similar for LfHP 1 and LfHP 2 and inhibited glucose release by ∼60% compared to the control after reaction for 24 h (Fig. 2).
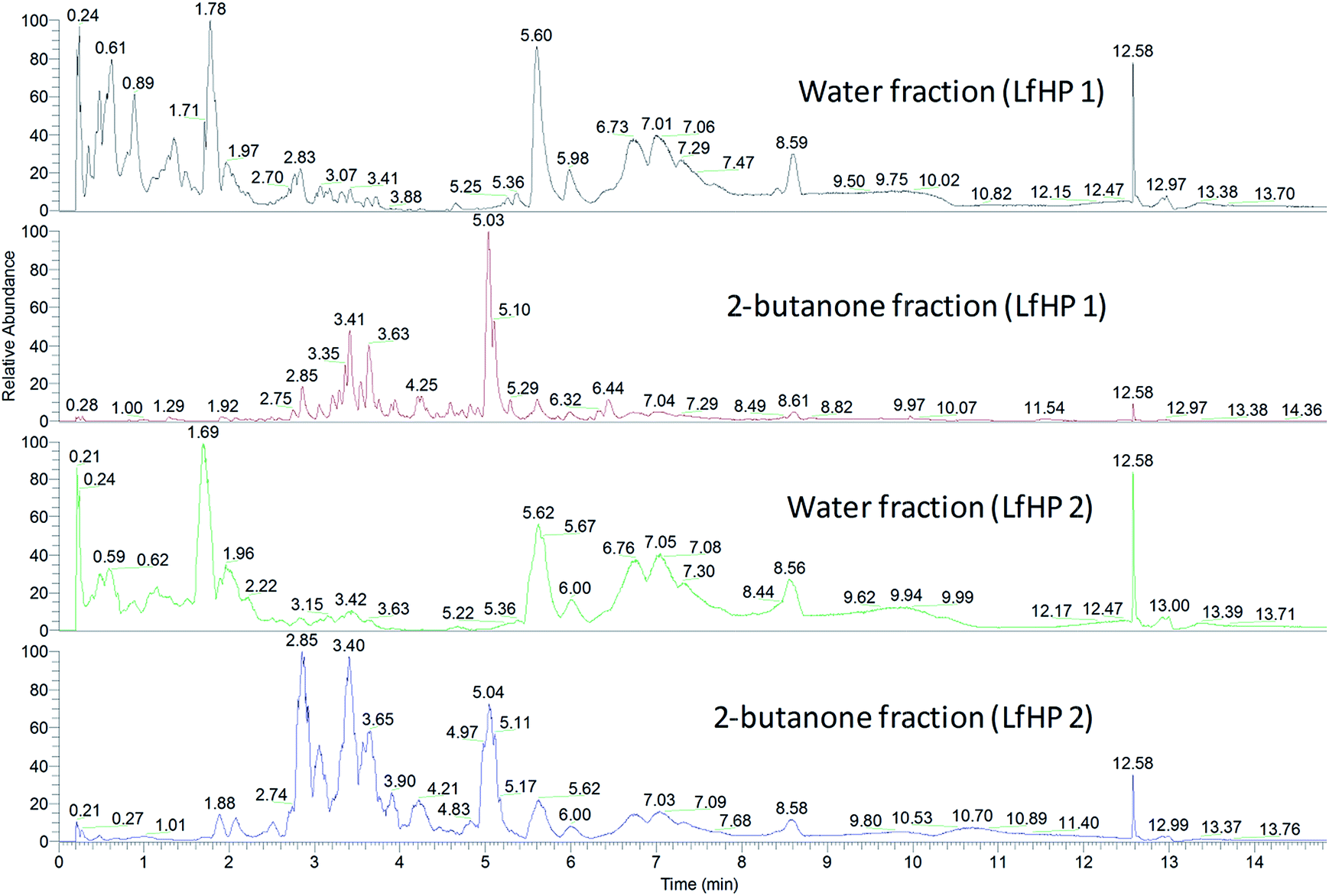 | ||
| Fig. 3 Base peak chromatograms (negative ionisation) of water fractions and 2-butanone fractions from LfHP 1 and LfHP 2. | ||
The compounds in the 2-butanone and water fractions were analysed in detail, regarding accurate mass, retention time and fragmentation pattern (ESI, Tables S1 and S2†). The compounds present were found to be similar in LfHP 1 and LfHP 2, even though the relative abundance had changed: in the water fractions the amount of xylooligosaccharides without ester substitution (rt 0.21–1.69, Fig. 3) were less abundant with more severe pretreatment. However, isomers of tetrapentoses and pentapentoses with 1 acetyl substituent (rt 1.78 Fig. 3 and rt 1.69 Fig. 3) were present at similar abundance in the water fractions from LfHP 1 and LfHP 2. In the 2-butanone fraction of LfHP 2, the shift in distribution of compounds as compared to in the LfHP 1 2-butanone fraction was towards earlier retention times and especially the compound at rt 2.85 had an increased relative abundance compared to its abundance in the 2-butanone fraction from LfHP 1 (Fig. 3, pane 2 and 4). This ion, rt 2.85, m/z 163.03938 ([M − H+]−) was subjected to fragmentation with HCD energies 25% and 50%, but did not result in any fragmentation, suggesting a condensed compound with no functionalities readily available for fragmentation.
The accurate mass corresponds to the molecular formula C9H8O3 with 6 double bond equivalents. On this basis compound 1 (Fig. 4) is suggested.
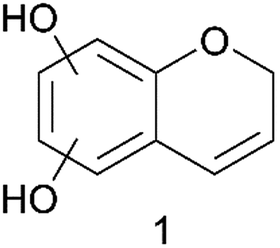 | ||
| Fig. 4 Proposed structure of the compound (m/z 163.03938) that is present at elevated relative amounts when the pretreatment temperature of wheat straw was increased from 183 °C to 191 °C. | ||
Compound 1 and other compounds eluting at the earlier retention time in the 2-butanone fraction from LfHP 2 may be responsible for the increased inhibition towards cellulases of this fraction (both in relation to the corresponding 2-butanone fraction of LfHP 1 and the water fraction of LfHP 2) (Fig. 3).
Inhibition mass balance
To compare the significance of the pretreatment severity in relation to overall inhibition from LfHP 1 compared to LfHP 2 a compound mass balance was performed. To evaluate how large a part of the total LfHP inhibition in the two cases each of the fractions 2-butanone and the residual water fraction comprised, the actual amounts of dry matter extracted into the fractions were considered.Despite the high inhibitor potency of the oligophenolic compounds per mass unit, the total mass of compounds in the organic fraction was much less than in the water fraction regardless of the severity of the pretreatment. Hence the contribution to the total LfHP inhibition from the compounds in the organic fraction (containing oligophenols) was smaller in total than the contribution from the compounds in the water fraction (containing xylooligosaccharides both without substitution and with acetyl, feruloyl and arabinose substitutions) in both LfHP 1 and LfHP 2 cases (Fig. 5).
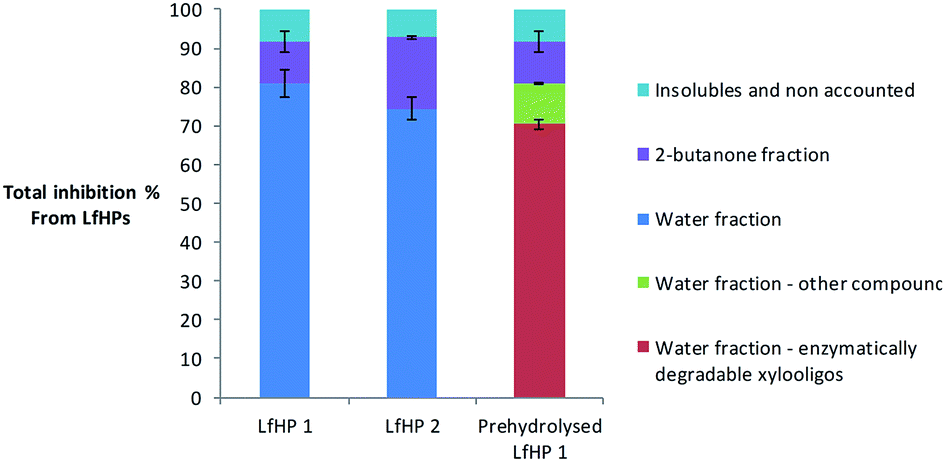 | ||
| Fig. 5 Inhibition of cellulases exerted by the compounds in LfHP 1, LfHP 2 and LfHP 1 with enzymatic removal of xylooligosaccharides (prehydrolysed LfHP 1). | ||
The water fraction containing the xylooligosaccharides (with and without substitutions) thus accounted for approximately 80% of the total inhibition exerted by the LfHP 1 (Fig. 5).
The content of xylooligosaccharides in LfHP 2 decreased to 22% (w/w of dry matter) from 37% in LfHP 1 and the inhibition from the 2-butanone fraction increased from 11% in LfHP 1 to 18% in LfHP 2.
Even though the more severe pretreatment LfHP 2 resulted in formation of more oligophenolic compounds and their potency increased (Fig. 2), the majority (75%) of the overall inhibition from LfHP 2 when compared on an equal mass base to LfHP 1 was still from the water fraction containing the xylooligosaccharides (with/without substitutions) (Fig. 5).
The above results and the recent finding that xylose is heavily involved in inhibitor formation by several synthesis routes14 stress, that pretreatment of biomass is a balance between degradation of xylooligosaccharides and reactions of xylose to form oligophenols. One solution, to have xylooligosaccharides hydrolysed to xylose and avoid xylose degradation at the same time, is to protect the anomeric center in xylose as earlier reported.14
Enzymatic removal of xylooligosaccharides and subsequent evaluation of cellulase inhibition
To investigate if all inhibition from the water phase was exerted solely by enzymatically degradable xylo-oligosaccharides (with and without substitution), they were enzymatically removed prior to testing in the Avicel assay. To remove xylooligosaccharides, the water fraction from the 2-butanone extraction was prehydrolysed with the same enzyme preparation (Cellic CTec3) as used in the enzyme inhibition assay, but at a higher dose and prolonged time: after 8 days 90% of the xylooligosaccharides were degraded to xylose (data not shown). However, even though the majority of xylooligosaccharides were removed, the prehydrolysates were still inhibiting the enzymatic glucose release, which suggests that some other components in the prehydrolysed water fraction were inhibitory to the cellulose degradation (Fig. 5).The compounds in the water fraction still contributed with 81% of the total inhibition from the LfHP 1, whereof 70% were contributions from xylooligosaccharides, that were hydrolysable in the prehydrolysis, and 11% were contributions from other components (Fig. 5). Thus, the vast majority of inhibition from the water fraction was confirmed to be due to hydrolysable xylooligosaccharides. However, other components also played a role. 10% of the xylooligosaccharides (including various substituted xylooligomers) were not degraded by the enzymatic treatment and are accounted for in the category of other components in the water fraction. Hence, these compounds most likely contributed to inhibition in this category.
Residual compounds after enzymatic hydrolysis
In order to investigate the structures of the residual compounds after enzymatic hydrolysis, the intensively enzymatically prehydrolysed water fraction was analysed with LC-MS/MS.The major compound in the TIC from the enzymatically prehydrolysed water fraction corresponded to a feruloylated tripentose, which was present as different isomers with retention times around 3.15 minutes (Fig. 6).
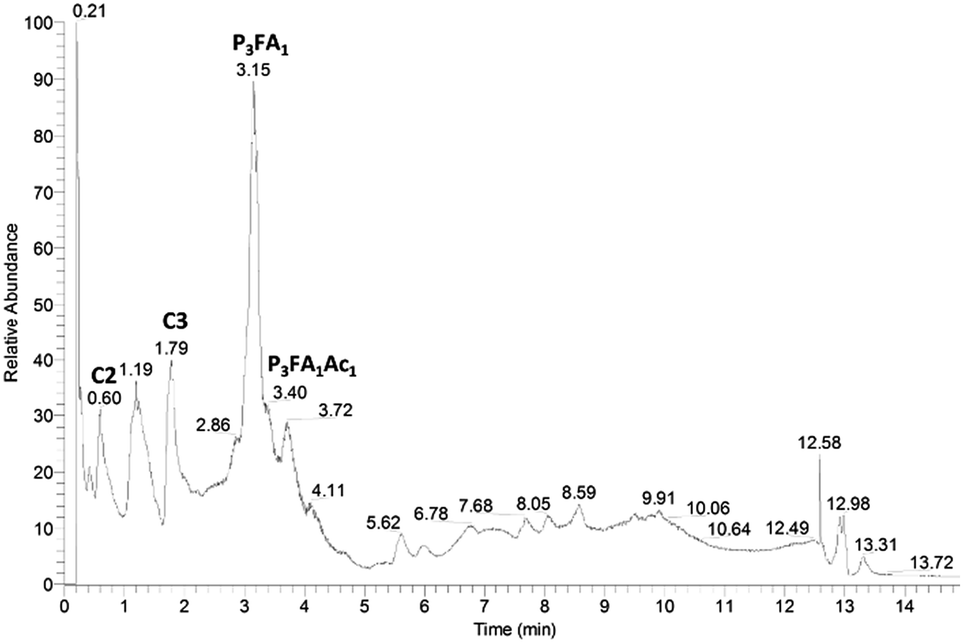 | ||
| Fig. 6 Total ion chromatogram (negative ionisation) of pre-hydrolysed water fraction from 2-butanone extraction of LfHP 1. C2: compound 2 (Fig. 7), C3: compound 3 (Fig. 7), P3FA1: feruloylated tripentose, P3FA1Ac1: feruloylated acetylated tripentose. | ||
Upon fragmentation (Table 1) it formed fragments, corresponding to feruloylated dipentose (m/z 457), feruloylated pentose (m/z 325) and ferulic acid (m/z 193) together with neutral mass losses of 90 and 60, which are characteristic for glycosides.17 Water loss was also evident and especially profound for the feruloylated dipentose fragment (m/z 439) and for the [M − H+ − 60]− fragment.
| Retention time (min) | Mass (m/z) [M − H+]− | Fragments MS/MS (relative intensity) | Compound |
|---|---|---|---|
| 0.60 | 499.13094 | 499.13094 (100), 439.10976 (27), 421.09933 (8), 367.08855 (6), 307.06721 (3), 235.04579 (0.1), 175.02422 (2) | C2 (Fig. 6 and 7) |
| 1.19 | 513.14941 | Could not be found in MS/MS. Possibly due to double charged ion (1027 ion observed) | Not determined |
| 1.79 | 513.14649 | 513.14649 (100), 453.12512 (25), 435.11457 (10), 381.10398 (8), 363.09342 (4), 321.08258 (2), 317.08769 (4), 249.06123 (9), 189.03979 (4) | C3 (Fig. 6 and 7) |
| 3.15 | 589.17976 | 589.17976 (0.5), 529.15801 (1), 511.14736 (18), 499.14763 (4), 457.13649 (17), 439.12602 (58), 325.09332 (100), 265.07214 (52), 235.06125 (10), 193.05033 (14), 175.03972 (4) | Feruloylated tripentose |
| 3.40 | 631.18663 | 631.18663 (3), 571.16618 (38), 553.15579 (100), 541.15589 (44), 511.14512 (26), 499.14525 (16), 493.13464 (12), 481.13468 (90), 457.13479 (14), 439.12414 (88), 421.11369 (9), 367.10300 (14), 325.09243 (86), 307.08182 (8), 265.07131 (51), 235.06051 (11), 193.04979 (35), 175.03918 (18) | Feruloylated acetylated tripentose |
Feruloylated pentoses up to DP 9 were observed in the mass spectra of the water phase (from 2-butanone extraction) after pretreatment i.e. before enzymatic treatment.14 Thus the enzymes seem able to degrade pentose oligomers with a feruloyl substituent down to tripentose level.
Feruloylated pentose oligomers from mildly acid pretreated corn fiber, have previously been reported to accumulate after treatment with enzyme mixtures not containing feruloyl esterase activities.18
In the present study, feruloylated acetylated tripentoses were also present (Table 1). They exhibited the same type of fragmentation pattern as feruloylated tripentoses i.e. breakdown to its substructures and neutral mass losses of 18, 60 and 90. One important fragment from the precursor ion 631.18663 is m/z 367.10300 (Table 1), which corresponds to a pentose substituted with both an acetyl group and a feruloyl group, thus suggesting that tripentoses with acetyl and feruloyl at the same pentose were present.
Two of the other major compounds in the total ion chromatogram (Fig. 6) were found to be a new type of residual compounds after enzymatic hydrolysis (Table 1 and Fig. 7). They are dipentoses, which have a highly hydroxylated bicyclic residue consisting of 2 oxanes with a double bond, attached at the unprotected anomeric center of the dipentose (Fig. 7). Both compounds displayed the same type of fragmentation pattern with fragments corresponding to the bicyclic residue with either none, one or two pentoses (Table 1). Within these substructures neutral mass losses of 60 and 18 were also observed (Table 1).
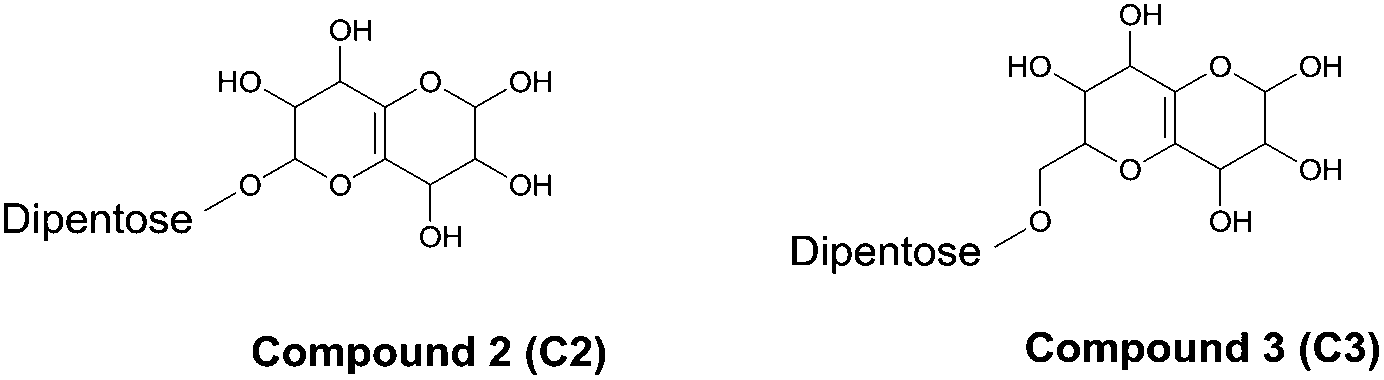 | ||
| Fig. 7 Proposed structures for two new residual compounds after enzymatic hydrolysis. Note that substitution position is given for clarity, but regioisomerism was not determined. | ||
Proposed reaction mechanism for formation of bicyclic residues
The bicyclic residues are proposed to be reaction products from the hydrothermal pretreatment, where xylose and glucose have undergone isomerisation to xylulose and fructose respectively,19,20 followed by aldol condensation with glyceraldehyde 4 (Fig. 8), which is present due to degradation of hexoses.21 Two ring closures lead to the final bicyclic residues 5 and 6 (Fig. 8).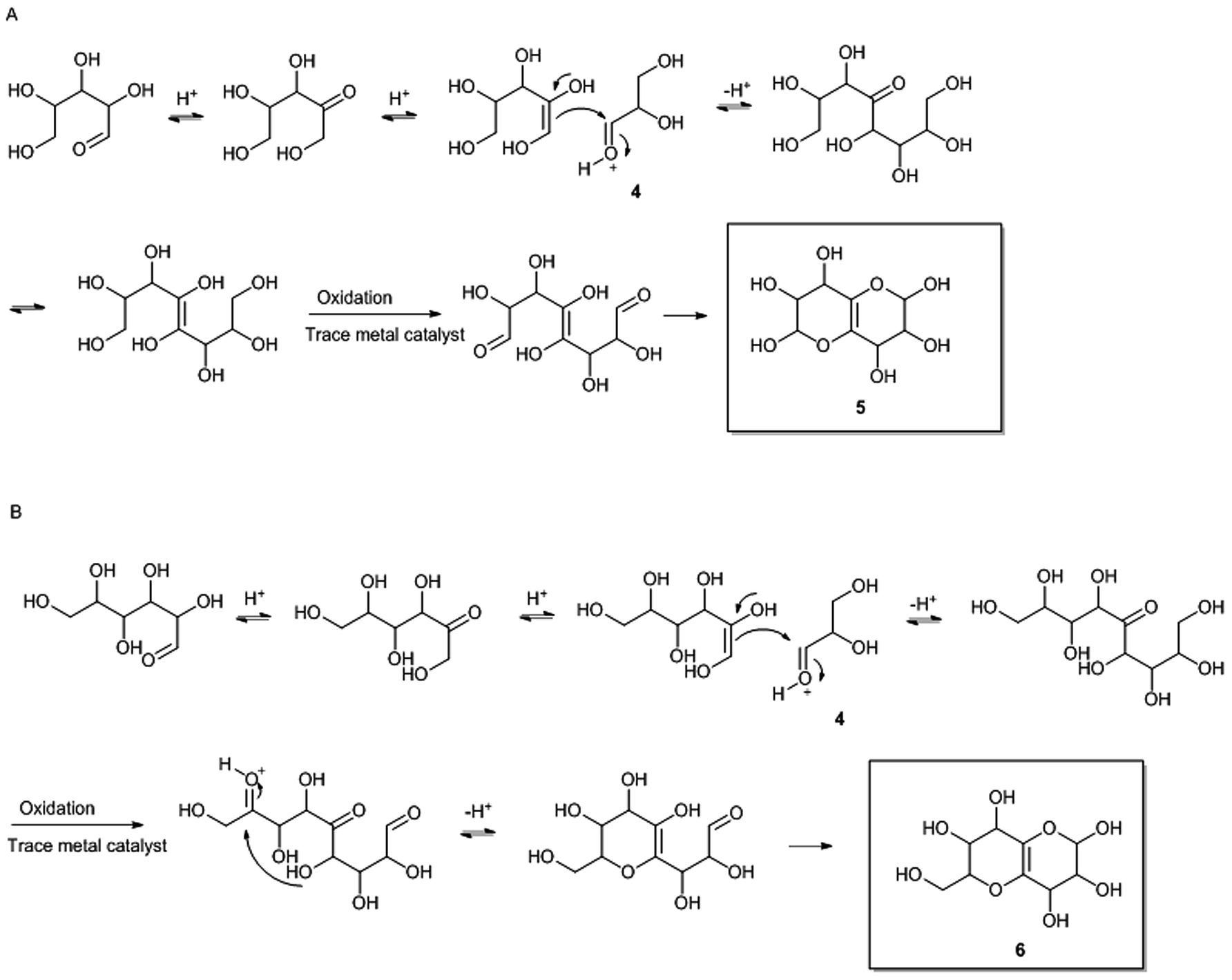 | ||
| Fig. 8 Proposed mechanisms for formation of the bicyclic residues 5 (A) and 6 (B) in compound 2 and compound 3 respectively. Compound 2 and 3 are shown in Fig. 7. | ||
To form the pentose dimers with bicyclic moieties (compounds 2 and 3 Fig. 7), compounds 5 and 6 (Fig. 8) are suggested to have reacted at the reducing end of an oligosaccharide, at any length, which is capable of forming an oxocarbenium ion. The oxocarbenium ion is readily available for nucleophile attack from the hydroxylated bicyclic compounds. Since pentapentose oligomers with bicyclic moieties are present in the water phase (from 2-butanone extraction) after pretreatment i.e. before enzymatic treatment (data not shown), it seems that the enzymes can degrade pentose oligomers with the bicyclic residue from pentapentose level down to dipentose level. It is unknown at the present point in time, if enzymes activities that catalyse cleavage of pentoses from the bicyclic residue in compound 2 and 3 exist.
In addition to their accumulation after enzymatic hydrolysis, it is also possible that both compounds 2 and 3 as well as acetylated and/or feruloylated tripentoses are inhibitors towards the cellulase enzymes. The bicyclic residues in compound 2 and 3 have structural similarities with compounds found in the 2-butanone fraction,14 which contained the most inhibitory compounds and shorter feruloyl substituted pentose oligomers were found in the very inhibiting 2-butanone fraction, whereas the longer feruloyl substituted pentose oligosaccharides were in the water fraction.14 Protection of the anomeric center in xylose,14 as well as the anomeric center at the reducing end of a xylooligosaccharide, will hinder formation of the enzyme resistant compounds 2 and 3.
Conclusions
It is a challenge in lignocellulosic biorefinery processes involving enzyme catalysed reactions that enzyme inhibiting compounds are generated and liberated during the hydrothermal pretreatment of the biomass.A solvent extraction approach was applied to separate xylooligosaccharides and oligophenolic compounds in liquors from two different severities of hydrothermal pretreatment of wheat straw to determine the contribution from oligophenolic compounds and enzyme hydrolysable xylooligosaccharides to total cellulase inhibition of the liquors. It was found that oligophenolic compounds are more potent cellulase inhibitors than xylooligosaccharides, when compared on a dry matter level and an increase in severity of the pretreatment lead to more oligophenol compounds and thus the overall inhibition increased. But due to the much higher abundance of xylooligosaccharides than oligophenolic compounds, compared on dry matter base, the xylooligosaccharides contributed most to the overall inhibition at both the investigated pretreatment severities. Although the relative abundance of different compounds changed with pretreatment severity, the compounds present in the pretreatment liquors were very similar.
After enzymatic removal of xylooligosaccharides from the water fraction from 2-butanone extraction some inhibition remained compared to the control, and two major types of residual compounds after enzymatic hydrolysis were found; feruloylated tripentoses and pentose dimers with bicyclic moieties. The latter are novel dipentose compounds with a hydroxylated oxane bicyclic residue and reaction mechanisms for their synthesis are proposed.
Acknowledgements
This work was supported by the Danish National Advanced Technology Foundation via the Technology Platform “Biomass for the 21st century – B21st”.Notes and references
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- X. Wang and R. Rinaldi, Angew. Chem., Int. Ed., 2013, 52, 11499–11503 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8559–8560 CrossRef CAS PubMed.
- L. J. Jönsson and C. Martín, Bioresour. Technol., 2016, 199, 103–112 CrossRef PubMed.
- H. Rasmussen, H. R. Sørensen and A. S. Meyer, Carbohydr. Res., 2014, 385, 45–57 CrossRef CAS PubMed.
- A. Tejirian and F. Xu, Enzyme Microb. Technol., 2011, 48, 239–247 CrossRef CAS PubMed.
- R. Kont, M. Kurasin, H. Teugjas and P. Väljemäe, Biotechnol. Biofuels, 2013, 6, 135 CrossRef CAS PubMed.
- Q. Qing, B. Yang and C. E. Wyman, Bioresour. Technol., 2010, 101, 9624–9630 CrossRef CAS PubMed.
- S. I. Mhlongo, R. d. Haan, M. Viljoen-Blooma and W. H. van Zyla, Enzyme Microb. Technol., 2015, 81, 16–22 CrossRef CAS PubMed.
- Y. Kim, E. Ximenes, N. S. Mosier and M. R. Ladisch, Enzyme Microb. Technol., 2011, 48, 408–415 CrossRef CAS PubMed.
- H. Rasmussen, D. Tanner, H. R. Sørensen and A. S. Meyer, Green Chem., 2017 10.1039/c6gc01809b.
- L. Rosgaard, S. Pedersen, J. Langston, D. Akerhielm, J. R. Cherry and A. S. Meyer, Biotechnol. Prog., 2007, 23, 1270–1276 CrossRef CAS PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of Sugars, Byproducts, and Degradation Products in Liquid Fraction Process Samples, NREL – Biomass Program, 2006 Search PubMed.
- A. G. A. W. Alakolanga, A. M. D. A. Siriwardane, N. S. Kumar, L. Jayasinghe, R. Jaiswal and N. Kuhnert, Food Res. Int., 2014, 62, 388–396 CrossRef CAS.
- M. M. Appeldorn, P. de Waard, M. A. Kabel, H. Gruppen and H. A. Schols, Carbohydr. Res., 2013, 381, 33–42 CrossRef PubMed.
- O. Ershova, J. Kanervo, S. Hellsten and H. Sixta, RSC Adv., 2015, 5, 66727 RSC.
- H. Kimura, M. Nakahara and N. Matubayasi, J. Phys. Chem. A, 2011, 115, 14013–14021 CrossRef CAS PubMed.
- T. Flannelly, M. Lopes, L. Kupiainen, S. Dooley and J. J. Leahy, RSC Adv., 2016, 6, 5797 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25432b |
| This journal is © The Royal Society of Chemistry 2017 |