
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of the electron donor groups on the optical and electrochemical properties of borondifluoride complexes of curcuminoid derivatives: a joint theoretical and experimental study†
Gabriel Canard *a,
Miguel Ponce-Vargasb,
Denis Jacquemincd,
Boris Le Guennic*b,
Abdellah Felouata,
Morgane Rivoala,
Elena Zaborovaa,
Anthony D'Aléoa and
Frédéric Fages*a
*a,
Miguel Ponce-Vargasb,
Denis Jacquemincd,
Boris Le Guennic*b,
Abdellah Felouata,
Morgane Rivoala,
Elena Zaborovaa,
Anthony D'Aléoa and
Frédéric Fages*a
aAix Marseille Univ, CNRS, CINaM, Campus de Luminy, Case 913, 13288 Marseille, France. E-mail: frederic.fages@univ-amu.fr
bInstitut des Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1, 263 Avenue du Général Leclerc, 35042 Rennes Cedex, France
cLaboratoire CEISAM, UMR CNRS 6230, Université de Nantes, 2 Rue de la Houssinière, BP 92208, 44322 Nantes Cedex 3, France
dInstitut Universitaire de France, 1 rue Descartes, 75231 Paris Cedex 05, France
First published on 6th February 2017
Abstract
Molecules displaying a π-conjugated D–A–D structure, in which D and A are electron donor and acceptor groups, respectively, represent an important class of dyes. In this work, we present a series of borondifluoride complexes of curcuminoid derivatives in which the central dioxaborine ring acts as a strong electron acceptor unit. Compounds 1–15 differ by the nature of the terminal D groups. We have also studied unsymmetrical compounds 16–23 that contain two different terminal donor groups, as well as compounds 24–32 in which an electron acceptor or donor unit has been introduced at the meso position of the dioxaborine ring. We describe the synthesis of the dyes that have not yet been reported in the literature, and report the electrochemical, absorption and fluorescence properties of all the compounds in dichloromethane. To gain insights into the electronic structures and optical properties, we performed DFT/TD-DFT calculations for a panel of representative compounds. We established correlations between the reduction potential and the Hammett σ and σ+ constants, as well as between the redox gap and the emission wavelength. The electron donor character of one terminal D unit strongly influences (88 mV per unit of σ) the reduction potential due to the strong resonance interaction along the curcuminoid backbone. The correlation points to the smaller effect of the meso substituent (50 mV per unit of σ), which we relate to the twisted ground-state geometry of the meso aryl substituent. The correlation models established in this study may be useful to anticipate the optical and electrochemical properties of borondifluoride complexes of curcuminoids with good reliability.
Introduction
Difluoroboron β-diketonate complexes represent an important family of photo- and electroactive molecules with applications in (bio)imaging, photovoltaics, organic electronics, and display technology. Their optical1–11 and electrochemical11–16 properties have been described in previous studies and it has been shown that they can be used as versatile fluorophores,1–5 electron acceptors17,18 in one-electron transfer processes19 or in charge-transport materials in both organic field effect transistors and organic light emitting diodes.20–25 One-electron reduction proceeds via the formation of a radical anion where the unpaired electron is delocalized on the π-system of the boron-containing six-membered chelate ring, as demonstrated by electron paramagnetic resonance studies.13Cucurminoids displaying the diarylheptanoid backbone represent a particular case of β-diketonate ligands.26 Owing to their extended π-conjugated structure, their borondifluoride complexes exhibit strong visible absorption and fluorescence,27–33 and unique near-infrared (NIR) emission in both solution and in the solid state using one- and two-photon excitation.34,35 As a result of these features, they are attractive molecules for bioimaging and virtually any application requiring highly absorbing and emitting materials such as organic photovoltaics and optoelectronics.
In our recent studies,32–35 we showed that the unique linear and nonlinear optical properties of borondifluoride complexes of curcuminoids also stem from the combination of electron donor (D) and acceptor (A) units in a D–A–D quadrupolar-like architecture in which A is the central dioxaborine ring and D is a terminal aromatic moiety acting as an electron donor substituent. As such, these compounds are highly solvatochromic dyes and their emission wavelengths can be tuned over the entire visible spectrum up to NIR region by controlling the strength of the terminal donor group. Moreover, we also showed that the introduction of an electron donor subunit D′ connected at the meso position of the curcuminoid skeleton resulted in T-shaped molecules displaying a complex interplay of charge transfer processes in the excited state depending on the nature of the D′ group.36
The color of borondifluoride complexes of curcuminoids depends on the gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) which itself depends on the substituent attached to the terminal aromatic units, and to a lesser extent on that at the meso position.32 A variety of symmetrical and unsymmetrical D–A–D molecules have been investigated, which provided valuable insights into the photophysics of those systems.37–40 In order to provide comprehensive structure–property relationships, we sought to establish correlations between Hammett parameters41 and optical and electrochemical properties in series of borondifluoride complexes of curcuminoids. To this end, we focused on compounds 1–15 (Chart 1) and 16–23 (Chart 2) that feature (i) a H atom at the meso position, and (ii) two identical or different terminal aromatic D groups. Electron donor groups of different strengths were considered but the study was also extended to compounds that contain electron acceptor groups, such as cyano and ester moieties. Furthermore, in order to get some insights into the influence of lateral D′ groups on the photophysical and electrochemical properties of the borondifluoride-containing curcuminoid core, we also investigated the series of compounds 24–32 (Chart 3) composed of a meso-substituted symmetrical curcuminoid backbone. We describe herein the optical and electrochemical study of compounds 1–32 in dichloromethane (DCM) solution, the synthesis of the new molecules and the results of first principles calculations performed with Density Functional Theory (DFT) and its time-dependent counterpart (TD-DFT). To our knowledge, there is only one report in literature describing the cyclic voltammetry (CV) of the BF2 complex of the naturally occurring curcumin.13
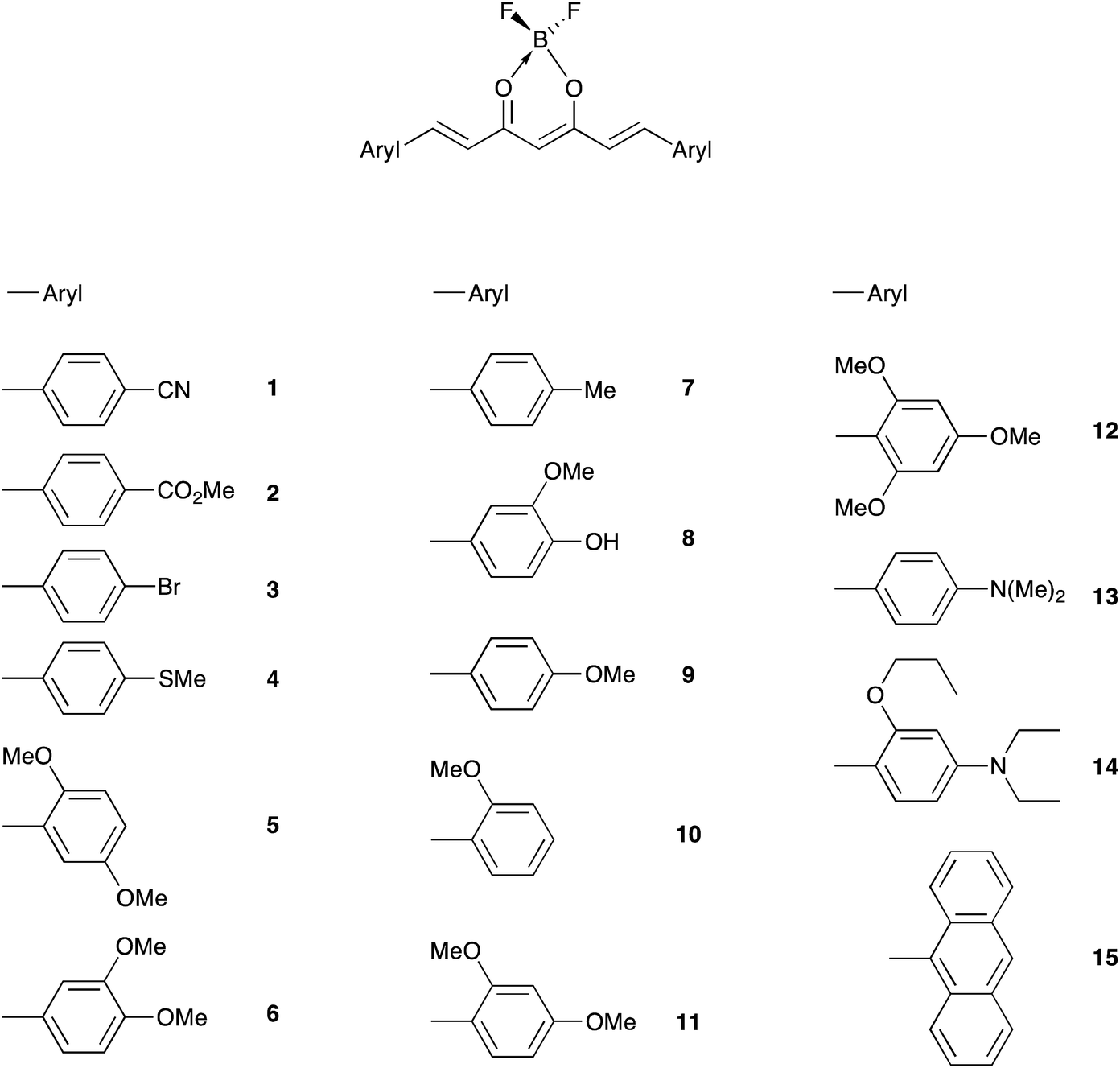 | ||
| Chart 1 | ||
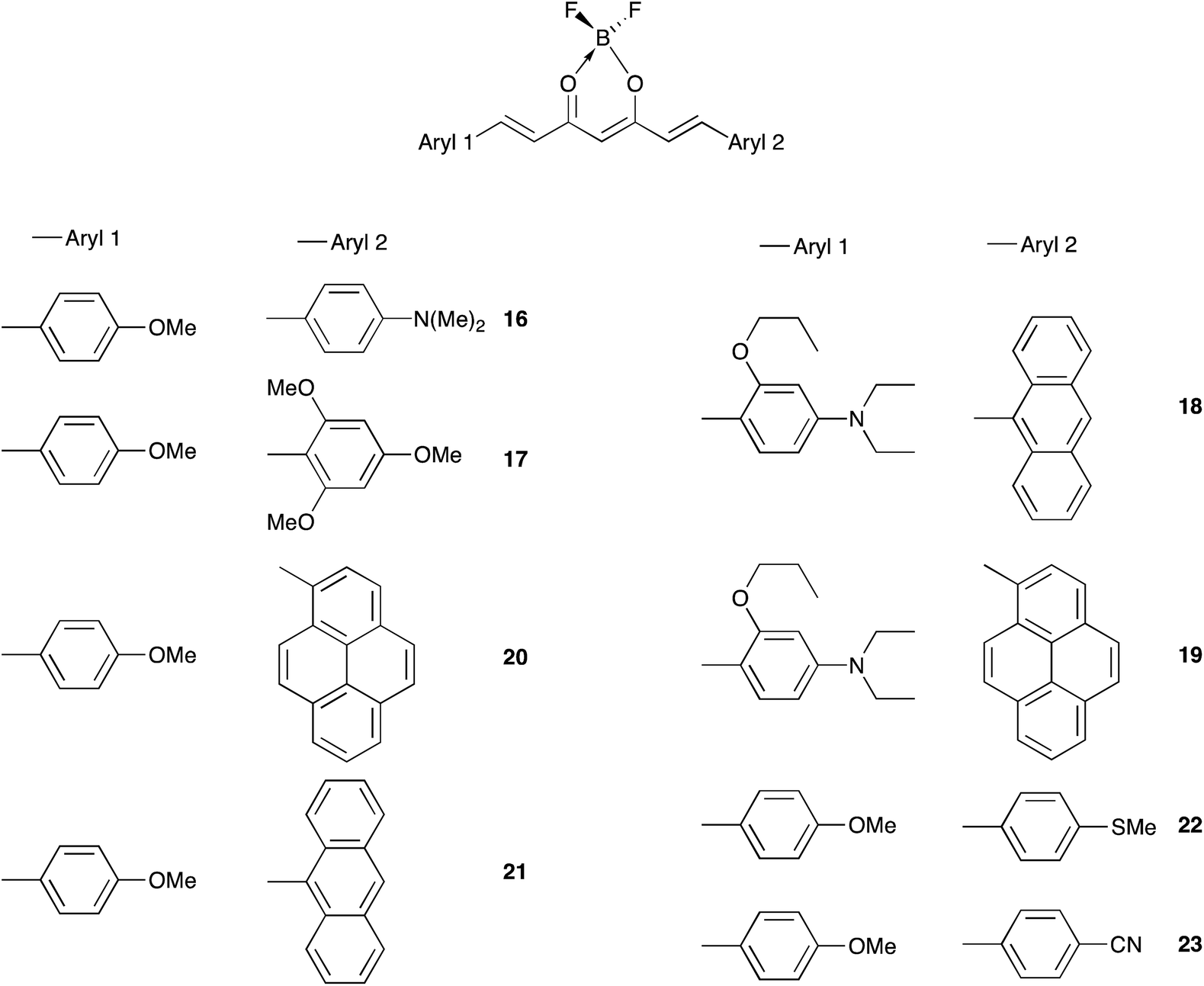 | ||
| Chart 2 | ||
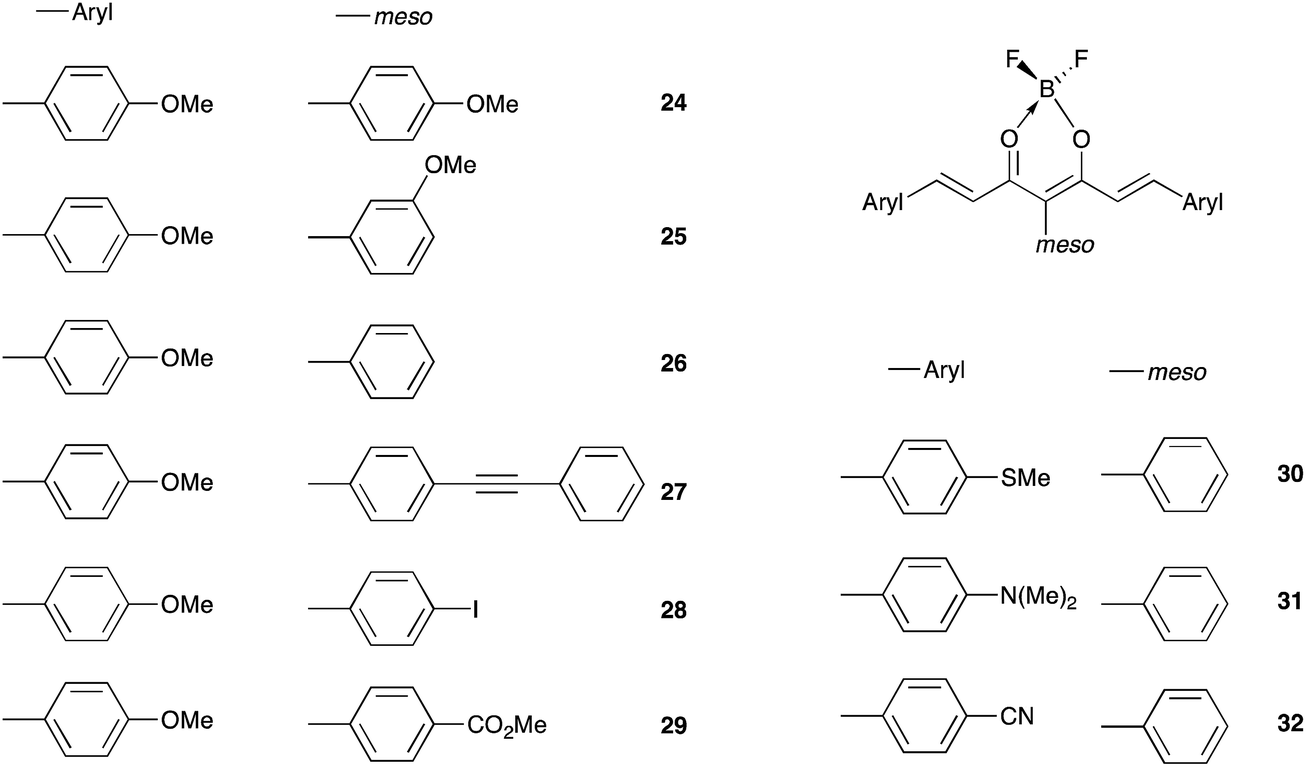 | ||
| Chart 3 | ||
Experimental section
Chemicals and spectroscopic characterizations
All solvents for synthesis were of analytic grade. Spectroscopy measurements were performed using spectroscopic grade solvents. Curcumin (ligand precursor of compound 8) has been obtained from Sigma Aldrich. NMR spectra (1H, 13C, and 19F) were recorded at room temperature on a Bruker AC250 (250, 62.5, and 235 MHz for 1H, 13C, and 19F, respectively) or a JEOL JNM ECS 400 (400, 100, and 374 MHz for 1H, 13C, and 19F, respectively) spectrometer. Data are listed in parts per million (ppm) and are reported relative to tetramethylsilane (1H and 13C); residual solvent peaks of the deuterated solvents were used as internal standards. Mass spectra were obtained in Spectropole, Marseille (http://www.spectropole.fr/).Synthesis and characterization
The syntheses of compounds 3,42 4,34 6,28 7,27,38 8,27,28 9,32 12,33 13,29 14,34 17,33 24,32 25,32 26,32 27,36 28 (ref. 36) and 29 (ref. 32) have been reported in literature. The synthesis of the bis-anthracenyl free ligand was described elsewhere.43 The syntheses of 1, 2, 5, 10, 11, 15, 16, 18, 19, 20, 21, 22, 23, 30, 31, and 32 were performed according to published general procedures for symmetrical32 and nonsymmetrical33 curcuminoids. 1H and 13C NMR spectra are given in Fig. S1.†General procedure fort the synthesis of the symmetrical borondifluoride complexes
In a 50 mL round bottom flask, a solution of acetylacetone derivative (1 mol eq.) and B2O3 (0.5 mol eq.) in ethylacetate (15 mL) was stirred at 60 °C for 30 min. A solution of the appropriate aldehyde (1 mol eq.) and tri(n-butyl)borane (1 mol eq.) in ethylacetate (10 mL) was added and the resulting mixture was stirred for 30 min at 60 °C. A catalytic amount of n-butylamine (0.4 mol eq.) was then added and the reaction mixture was refluxed overnight. After cooling to 60 °C, 30 mL of 0.4 M HCl were added and the mixture stirred for 30 min. After cooling, the precipitate was filtered off and dried in vacuo to yield pure ligands. In a 50 mL round bottom flask, the ligand (1 mol eq.) was solubilized in dichloromethane (20 mL) and boron trifluoride etherate (1.1 mol eq.) was added. The reaction mixture was refluxed overnight. After cooling to room temperature, the solvent was evaporated and the resulting solid was suspended in diethyl ether. The precipitate was filtered off yielding the pure complex.General procedure fort the synthesis of the nonsymmetrical borondifluoride complexes
The synthesis of compounds 16, 18–23 requires the preparation of the corresponding hemicurcuminoid derivative using an excess of acetylacetone (acac/aldehyde 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The synthesis of the anisole-containing precursor leading to 16, 20–23 was described elsewhere.33 Compounds 18, 19 where obtained from the p-N,N-dimethylamino-o-propoxyphenyl-containing hemicurcuminoid derivative.34 The reaction of the hemicurcuminoid intermediate with one equivalent of the second aldehyde afforded the ligands. The borondifluoride complexes were obtained as described above for the symmetrical complexes.
:1). The synthesis of the anisole-containing precursor leading to 16, 20–23 was described elsewhere.33 Compounds 18, 19 where obtained from the p-N,N-dimethylamino-o-propoxyphenyl-containing hemicurcuminoid derivative.34 The reaction of the hemicurcuminoid intermediate with one equivalent of the second aldehyde afforded the ligands. The borondifluoride complexes were obtained as described above for the symmetrical complexes.
Computational details
The calculations have been carried out with the Gaussian09 program package,44 considering default thresholds except to tighten self-consistent field convergence (10−8 to 10−10 a.u.) and geometry optimization (10−5 a.u. on average forces). Our modeling included DFT geometry optimizations of the ground-state structure, vibrational frequency calculations and simulations of the excited-states with TD-DFT. In all our calculations, we applied the hybrid PBE0 exchange-correlation functional,45 which is suited to reproduce absorption/emission trends of borondifluoride complexes.34 In order to determine structural and vibrational parameters, the relatively compact 6-31G(d) atomic basis set was employed, while for electronic transitions the more extended 6-311+G(2d,p) atomic basis set was used. As the influence of solvent effects is of paramount importance to estimate the electronic transition energies, here we applied the Polarizable Continuum Model (PCM),46,47 using a linear-response nonequilibrium approach for the TD-DDFT step. All the HOMO–LUMO plots have been graphed by using the Chemcraft code,48 considering an isosurface of 0.03 a.u.Electrochemistry
Cyclic voltammetry (CV) data were acquired using a BAS 100 Potentiostat (Bioanalytical Systems) and a PC computer containing BAS100W software (v2.3). A three-electrode system with a Pt working electrode (diameter 1.6 mm), a Pt counter electrode and a Ag/AgCl (with 3 M NaCl filling solution) reference electrode was used. (n-Bu)4NPF6 (0.1 M in CH2Cl2) served as an inert electrolyte. Cyclic voltammograms were recorded at a scan rate of 100 mV s−1 for solution of dyes at a concentration of ca. 10−3 M. Ferrocene was used as an internal standard.49Electronic absorption and fluorescence emission
UV/Vis-absorption spectra were measured on a Varian Cary 50. Emission spectra were obtained using a Horiba-Jobin Yvon Fluorolog-3 spectrofluorimeter equipped with a three-slit double-grating excitation and a spectrograph emission monochromator with dispersions of 2.1 nm mm−1 (1200 grooves per mm). A 450 W xenon continuous wave lamp provided excitation. Fluorescence of diluted solutions was detected at right angle using 10 mm quartz cuvettes.Results and discussion
Frontier molecular orbital properties
A series of representative molecules, namely, 5, 6, 9, 10, 15, 20, 23, 26, 29, has been considered for the study of the frontier orbital topologies and energies (Table S1, Fig. 1 and S2†). The ground-state (S0)–lowest excited-state (S1) electronic transition mainly corresponds to the HOMO → LUMO excitation, where the HOMO is spread over the π-conjugated backbone while the LUMO is mainly localized over the dioxaborine moiety that acts as an acceptor. Such transition can be considered as a “cyanine-like” transition50 due to the odd number of sp2-hybridized carbon atoms in the conjugated skeleton and the distribution of electronic density along this conjugated path. Compound 23, containing cyanophenyl and methoxyphenyl as terminal groups, offers us the possibility to study the influence of an electron-withdrawing group on the molecular orbital distribution. HOMO–LUMO plots of 23 reveal that the HOMO is mainly located along the methoxyphenyl moiety, as expected for an electron donor group. Our previous work has shown that the presence of a strong donor group at the meso position induced the occurrence of a low energy charge transfer transition.36 In contrast, the meso phenyl group in 26 has a trifling impact on the HOMO, the terminal methoxyphenyl groups determining the limits of the extension of this orbital. The same observation can be made for compound 29, bearing the ester-functionalized meso phenyl ring (Fig. S2†). However, a difference appears in the LUMO + 1 isosurface, which shows the electronic density being localized on that meso group as a consequence of its electron acceptor character.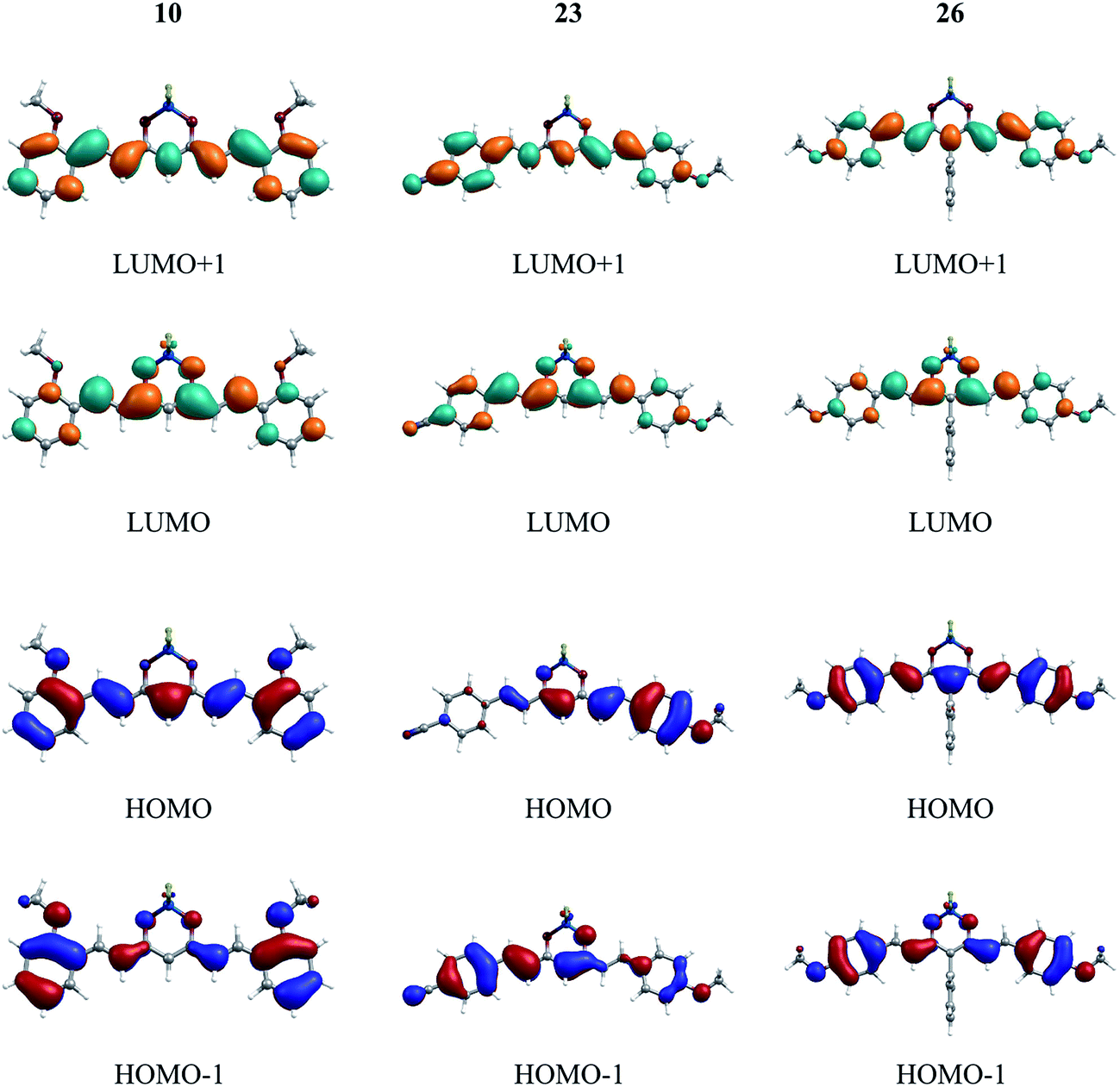 | ||
| Fig. 1 HOMO, HOMO−1, LUMO and LUMO+1 isosurfaces for compounds 10, 23, and 26. A contour threshold of 0.03 a.u. is applied (see also the ESI†). | ||
The highest values of the HOMO–LUMO energy gap (Table S1†) are obtained for systems 6, 9, 10 and 26, presenting methoxyphenyl termini, in good agreement with the experimental trend (see below). Especially, the lowest gap values are calculated for compounds 15 and 20, and correlate well with the electrochemical data. It is worth noting that the HOMOs of compounds 10 and 23 are found at the lowest energy (−6.3 and −6.4 eV, respectively) relative to the other compounds, which is also in agreement with the fact that these compounds are the more difficult to oxidize (+1.26 and +1.21 V vs. Fc/Fc+, respectively, see below). Furthermore, the calculated LUMO energies indicate that 15, 23 would be easier to reduce than 6, 9, 26, which is also in line with the experimental trends.
Theoretical electronic absorption data are collected in Table S2.† They show large oscillator strength values for the HOMO–LUMO transitions, confirming the strongly allowed character of the lowest-energy transition in borondifluoride complexes of curcuminoids.9,32,34 The calculated transition energies are found in the visible part of the spectrum, the lowest values being obtained for 15 (2.0 eV) and 20 (2.3 eV) with two anthracene groups as lateral substituents. These observations are consistent with the experimental data (vide infra).
Electrochemical properties
The cyclic voltammograms (CVs) of compounds 1–32 were recorded in DCM containing 0.1 M of (n-Bu)4NPF6 (Fig. 2 and S3–S5†). The oxidation and reduction half-wave potential values (E1/2) are listed in Tables 1–3 together with those previously obtained for 9, 12 and 17, using the same experimental conditions.33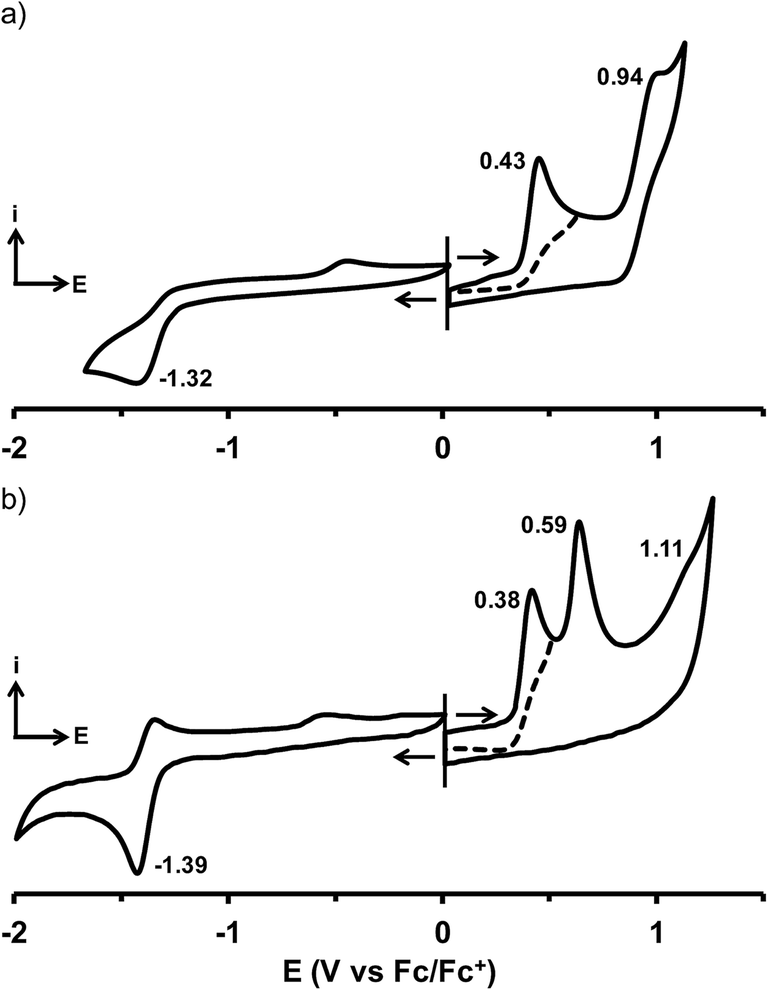 | ||
| Fig. 2 Cyclic voltammograms of (a) 18 and (b) 31 in DCM containing 0.1 M of (n-Bu)4NPF6 (scan rate of 100 mV s−1). | ||
| Dye | ∑σtermb | ∑σ+b,c | Ered1/2 (V) | Eox1/2 (V) | ΔEd (eV) | λem (nm) | Eem (eV) |
|---|---|---|---|---|---|---|---|
| a Measured in DCM containing 0.1 M of (n-Bu)4NPF6 with a scan rate of 100 mV s−1.b σortho (σ+ortho) was considered as equal to σpara (σ+para).c σ+ is not relevant for meta substituents.d ΔE = Eox11/2 − Ered11/2.e Values taken from ref. 33.f Reversible redox process.g Pseudo-reversible redox process.h Values estimated from eqn (1) and (2), respectively.i Values estimated from eqn (1) and (2), respectively. | |||||||
| 1 | +1.32 | +1.32 | −0.93 | — | — | 459 | 2,70 |
| 2 | +0.90 | +0.98 | −1.01 | — | — | 469 | 2,64 |
| 3 | +0.46 | +0.30 | −1.08 | — | — | 486 | 2,55 |
| 4 | +0.00 | −1.20 | −1.16 | 0.97, 1.16 | 2.13 | 587 | 2,11 |
| 5 | −0.30 | —c | −1.20 | 1.02 | 2.22 | 646 | 1,92 |
| 6 | −0.30 | —c | −1.27 | 0.95, 1.10 | 2.22 | 575 | 2,16 |
| 7 | −0.34 | −0.62 | −1.18 | — | — | 498 | 2,49 |
| 8 | −0.50 | —c | −1.25 | 0.91, 1.17 | 2.16 | 560 | 2,21 |
| 9e | −0.54 | −1.56 | −1.27 | 1.10 | 2.37 | 538 | 2,30 |
| 10 | −0.54 | −1.56 | −1.23 | 1.26 | 2.49 | 525 | 2,36 |
| 11 | −1.08 | −3.12 | −1.34 | 0.96, 1.17 | 2.30 | 570 | 2,18 |
| 12e | −1.62 | −4.68 | −1.44f | 0.72, 1.09 | 2.16 | 566 | 2,19 |
| 13 | −1.66 | −3.40 | −1.41 | 0.36, 0.51, 1.10 | 1.77 | 683 | 1,82 |
| 14 | −1.92 | −5.76 | −1.53g | 0.22, 0.54 | 1.75 | 683 | 1,82 |
| 15 | +0.08h | −0.58i | −1.16 | 1.00 | 2.16 | 706 | 1,76 |
| Dye | ∑σtermb | ∑σ+b | Ered1/2 (V) | Eox1/2 (V) | ΔEc (V) | λem (nm) | Eem (eV) |
|---|---|---|---|---|---|---|---|
| a Measured in DCM containing 0.1 M of (n-Bu)4NPF6 with a scan rate of 100 mV s−1.b σortho (σ+ortho) was considered as equal to σpara (σ+para).c ΔE = Eox11/2 − Ered11/2.d Values taken from ref. 33.e Values estimated from eqn (1) and (2), respectively.f Values estimated from eqn (1) and (2), respectively. | |||||||
| 16 | −1.10 | −2.48 | −1.35 | 0.48, 1.22 | 1.83 | 668 | 1,86 |
| 17d | −1.08 | −3.12 | −1.37 | 0.89, 1.31 | 2.26 | 561 | 2,21 |
| 18 | −0.92f | −3.14e | −1.32 | 0.43, 0.94 | 1.75 | 703 | 1,76 |
| 19 | −0.74f | −1.17e | −1.31 | 0.39, 1.10 | 1.70 | 703 | 1,76 |
| 20 | −0.05f | +0.90e | −1.15 | 0.89, 1.21 | 2.04 | 645 | 1,92 |
| 21 | −0.23f | −1.07e | −1.18 | 0.90, 0.98 | 2.08 | 686 | 1,81 |
| 22 | −0.27 | −1.38 | −1.23 | 0.99, 1.29 | 2.22 | 573 | 2,16 |
| 23 | +0.39 | −0.12 | −1.07 | 1.21 | 2.28 | 554 | 2,24 |
| Dye | ∑σtermb | ∑σmesob | ∑σ+termb | ∑σ+mesob,c | Ered1/2 | Eox1/2 | ΔEd (V) | λem (nm) | Eem (cm−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in DCM containing 0.1 M of (n-Bu)4NPF6 with a scan rate of 100 mV s−1.b σortho (σ+ortho) was considered as equal to σpara (σ+para).c σ+ is not relevant for meta substituents.d ΔE = Eox11/2 − Ered11/2. | |||||||||
| 24 | −0.54 | −0.27 | −1.56 | −0.78 | −1.27 | 1.13 | 2.40 | 571 | 2.17 |
| 25 | −0.54 | +0.12 | −1.56 | — | −1.29 | 1.13, 1.26 | 2.42 | 556 | 2.23 |
| 26 | −0.54 | +0.00 | −1.56 | 0.00 | −1.25 | 1.14 | 2.39 | 554 | 2.24 |
| 27 | −0.54 | +0.16 | −1.56 | −0.03 | −1.24 | 1.14 | 2.38 | 564 | 2.20 |
| 28 | −0.54 | +0.18 | −1.56 | +0.14 | −1.22 | 1.14 | 2.36 | 556 | 2.23 |
| 29 | −0.54 | +0.45 | −1.56 | +0.49 | −1.22 | 1.16 | 2.38 | 557 | 2.22 |
| 30 | +0.00 | +0.00 | −1.20 | 0.00 | −1.16 | 1.00, 1.53 | 2.16 | 538 | 2.30 |
| 31 | −1.66 | +0.00 | −3.40 | 0.00 | −1.39 | 0.38, 0.59, 1.11 | 1.77 | 643 | 1.79 |
| 32 | +1.32 | +0.00 | +1.32 | 0.00 | −1.47, −0.92 | — | — | 538 | 2.30 |
Depending on the chemical substitution, the CVs feature generally one irreversible reduction wave and up to three irreversible oxidation waves. A second irreversible reduction process was detected for 32 that bears the strongest electron-withdrawing cyano substituent. The oxidation processes are attributed to the aryl end-units D of the curcuminoid backbone. When these aryl groups present a substituent with a strong electron-withdrawing character such as in 1, 2 or 32, the oxidation process could not be detected within the electrochemical window of DCM. For unsymmetrical compounds bearing two different donor groups 16–23, oxidation leads to two redox waves related to each of those groups. For example, the p-diethylamino-o-n-propyloxy-phenyl end-group of 18 is oxidized at 0.43 V vs. Fc/Fc+ while the oxidation of the anthracenyl moiety occurs at 0.94 V vs. Fc/Fc+ (Fig. 2a). In spite of their symmetrical structures, compounds containing electron-rich D groups also feature CVs with multiple and successive distinct oxidation waves. For example, p-dimethylaminophenyl-containing 31 produces two successive one-electron oxidation waves separated by 210 mV (Fig. 2b). The amplitude of this separation reaches 370 mV in the CV of 12 illustrating well the effective electronic communication between the aryl end-groups occurring through the curcuminoid conjugated backbone. The nature of the meso aryl group D′ has only a weak impact on the electrochemical properties unless it has an electron-withdrawing character producing then a clear anodic shift of the oxidation potentials.
The half-wave potential value of the first reduction strongly depends on the electron donating or withdrawing nature of the aryl end groups D and shifts from −0.93 V vs. Fc/Fc+ for 1 to −1.53 V vs. Fc/Fc+ for 14. The borondifluoride complexes of curcuminoids are thus generally more reducible in DCM than the diketonatoboron derivatives in polar solvents such as acetonitrile. For example, dibenzoylmethane-BF2 lacking strong electron donor group is reduced at −1.37 V vs. Fc/Fc+. This may stem from the fact that the curcuminoid derivatives have a more extended π-conjugated structure, which would better stabilize the radical anion. Except for compounds 12, 14 and 31, the BF2-complexes investigated here feature an irreversible one-electron reduction wave. BF2-complexes of diketonates are known to undergo a rapid irreversible chemical reaction after reduction, because of the formation of an unstable intermediate anion radical centered on the dioxaborine ring. Nevertheless, the reversible reduction of the latter derivatives could be observed in highly polar solvents, such as dimethoxyethane or acetonitrile, and was shown to depend on ligand substitution.11–16 Because of the low solubility of the dyes investigated here, these two solvents could not be used in this study. In the polar solvent DMF, the reduction potential of compounds 2, 14, and 32 was observed to be still irreversible although anodically shifted as compared to DCM (Fig. S6 and Table S3†).
In order to delineate the electronic influence of substituents of the terminal and meso groups on the dioxaborine reduction ability, we sought to establish whether a clear correlation between Hammett constants and half-wave reduction potential values could exist. At first, we considered the two series of compounds devoid of meso groups. We used the term ∑σterm that is the sum of Hammett constants σ of the two terminal aromatic units, identical for 1–15 and different for the unsymmetrical molecules 16–23. We assumed identical σ values for ortho and para substituents.
The linear relationship (eqn (1), Fig. S7†) obtained with a large determination coefficient (R2 = 0.987) shows that the reduction potential values correlate with the Hammett parameter ∑σterm. Indeed, the reduction of the dioxaborine ring becomes more difficult with electron-rich substituents. The value of 88 mV per unit of σ obtained for the slope provides an estimation of the influence of one terminal group D on the reduction potential of the dioxaborine ring.
| Ered1/2 = −1.160 + 0.176∑σterm | (1) |
When we used the σ+ parameter which takes resonance effects into account,37 we also obtained a linear correlation (eqn (2), Fig. S8†) with a large regression coefficient (R2 = 0.979).
| Ered1/2 = −1.095 + 0.082∑σ+term | (2) |
This finding shows that a strong resonance interaction occurs between the dioxaborine center and the terminal groups D that this is consistent with the partial charge-transfer (CT) character of electronic transitions in borondifluoride complexes of curcuminoids (vide infra). This observation also indicates that a planar ground-state geometry prevails in solution.
From the electrochemical data obtained for 15, 18, 21 and 14, 20, and the linear plots of Ered1/2 vs. ∑σterm, we could extract Hammett constants for the anthracene (+0.04) and pyrene (+0.22) moieties (Table 1). These positive values indicate that these polycyclic aromatic hydrocarbons do not behave as strong electron donors in the ground state, as already observed,51 but instead present a weak electron acceptor character.
In the case of the meso substituted complexes 24–32, the interplay between the contributions of the meso D′ and terminal D substituents has to be taken in account. For example, when two p-dimethylaminophenyl units replace the two p-methoxyphenyl groups of 26, such as in 31, the first reduction potential value is cathodically shifted by 140 mV. In contrast, the replacement in the meso aryl group of the electron donating p-methoxy substituent of 24 by an electron-withdrawing p-ester moiety in 29 produces an anodic shift of only 50 mV. Therefore, the sum of the Hammett constants of meso groups (∑σmeso) was separated from that corresponding to the terminal unit (∑σterm) (Table 1) and a multilinear regression was used to fit Ered1/2 as a linear combinations of ∑σmeso and ∑σterm (Tables S4 and S5†). Again, we obtained a good correlation (R2 = 0.962) and eqn (3) provides values of 81 mV per unit of σ and 50 mV per unit of σ for the substituent effects of terminal and meso groups, respectively. The former value is in agreement with that obtained for symmetrical molecules.
| Ered1/2 = −1.157 + 0.162∑σterm + 0.050∑σmeso | (3) |
| Ered1/2 = −1.066 + 0.102∑σ+term + 0.044∑σ+meso | (4) |
Using the resonance σ+ parameter, a good linear correlation was again reached (R2 = 0.979, Tables S4 and S5†), and it shows that resonance effects play an important role in these compounds as well.
Fluorescence emission properties
The electronic absorption and fluorescence emission properties of all compounds have been investigated. The data for 4, 9, 12, 13, 14, 17, 24, 25, 26, 27, 29 were reported previously by some of us.32–36 Those for 6, 7, 8 were published by other groups.27,28 For the sake of comparison, we recorded the spectra of the last three compounds under the same experimental conditions using DCM as solvent and the data we obtained are fully consistent with the published ones. All dyes absorb and emit in the visible region (Fig. 3 and S9†). Absorption and emission data are collected in Tables S7† and 1–3, respectively. They show a strong dependence on the nature of the terminal aromatic moiety D, a redshift of both absorption and emission profiles being observed when the electron donating character of D is increased, which is consistent with the accepting nature of the dioxaborine center. As compared to 6, 5 displays a considerably red shifted fluorescence emission (λmaxem = 646 nm) associated with a very large Stokes shift (4680 cm−1; λmaxabs = 496 nm), whereas absorption spectra show the opposite trend.52 Previous studies have shown the solvatochromic dependence of absorption and emission properties of borondifluoride complexes of curcuminoids, which is consistent with the partial CT character of the transitions.32–36 In the case of 1 and 2, containing electron-withdrawing groups, such CT behavior is not observed, their absorption and emission spectra being well-structured and lying at higher energies. As noticed elsewhere, the introduction of a meso aryl unit systematically shifts the spectra to the red (ca. 15 nm).32 In addition, a weak dependence of the optical properties on the nature of the meso group is noticed for compounds 24–32. This observation is consistent with our previous study, which showed that the influence of the meso group increases when it contains a strong electron donor substituent.36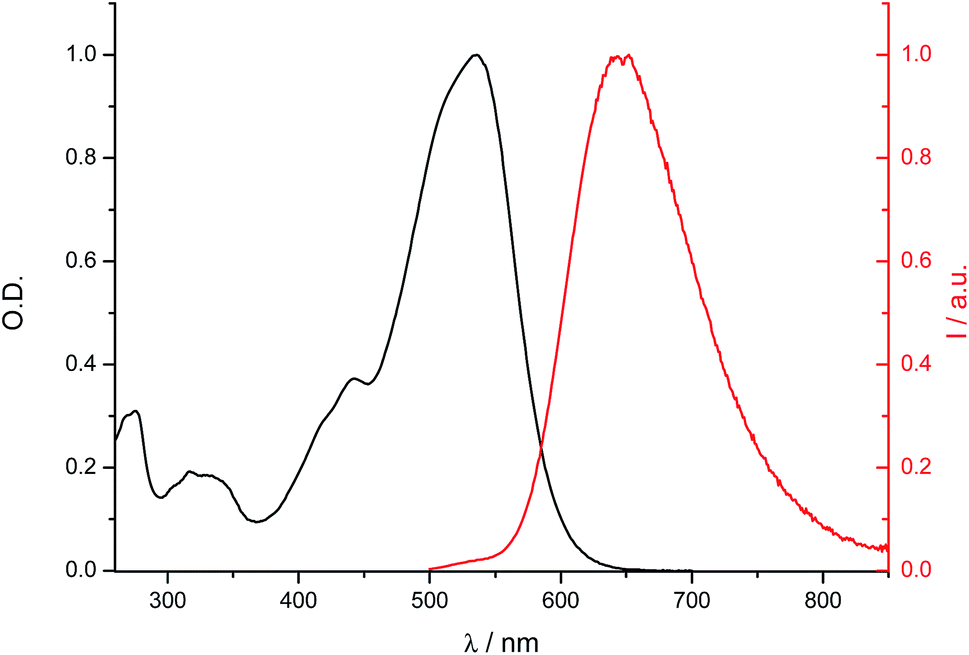 | ||
| Fig. 3 Electronic absorption and fluorescence emission spectra of compound 20 in DCM at room temperature. | ||
It has been shown previously that the singlet excited-state S1 energy of boron β-diketonate complexes can be fitted as a linear function of the HOMO–LUMO electrochemical gap.14 We therefore investigated the correlation between the emission energies and the HOMO–LUMO electrochemical gaps of 1–15 and 16–23. The fit provided eqn (5) showing such a linear dependence but with a modest determination coefficient (R2 = 0.715, Table S6†).
| Eem = 0.52 + 0.82ΔE (eV) | (5) |
Conclusions
In summary, the optical and electrochemical properties of 32 borondifluoride complexes of curcuminoid derivatives have been investigated in solution. The correlation between the reduction potential of the dioxaborine ring and Hammett σ constants provides a useful estimation of the influence of the terminal and meso aromatic groups. The electron donor character of one terminal unit causes a decrease of the reduction potential (88 mV per unit of σ). The correlation points to the smaller effect of the meso substituent (50 mV per unit of σ), which we relate to the twisted ground-state geometry of the meso aryl ring. From the correlation based on σ+ constants, we can infer that resonance effects prevail in the BF2-containing curcuminoid backbone. Cyclic voltammetry experiments also indicate the occurrence of a long-distance electronic interaction between terminal groups. DFT/TD-DFT calculations performed for a panel of representative compounds confirm the experimental trends and provide a detailed description of the electronic absorption transitions. All together, these observations support the cyanine-like character of the π-conjugated backbone, which makes the borondifluoride complexes of curcuminoids a versatile class of dyes. This study may be useful for the design of new derivatives with tailored optical and electrochemical properties in view of photovoltaic and imaging applications.Acknowledgements
Part of this work was supported by the French National Research Agency (Chalcones ANR-14-CE05-0035-01). D. J. is indebted to the European Research Council (ERC) for support in the framework of the Marches – 278845 grant. This work used computational resources of the GENCI/IDRIS-CINES and of the CCIPL.References
- T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242–1251 CAS.
- H. Maeda, Y. Bando, K. Shimomura, I. Yamada, M. Naito, K. Nobusawa, H. Tsumatori and T. Kawai, J. Am. Chem. Soc., 2011, 133, 9266–9269 CrossRef CAS PubMed.
- E. Cogny-Laage, J.-F. Allemand, O. Ruel, J.-B. Baudin, V. Croquette, M. Blanchard-Desce and L. Jullien, Chem.–Eur. J., 2004, 10, 1445–1455 CrossRef PubMed.
- M. Halik, W. Wenseleers, C. Grasso, F. Stellacci, E. Zojer, S. Barlow, J.-L. Brédas, J. W. Perry and S. R. Marder, Chem. Commun., 2003, 1490–1491 RSC.
- S. Xu, R. E. Evans, T. Liu, G. Zhang, J. N. Demas, C. O. Trindle and C. L. Fraser, Inorg. Chem., 2013, 52, 3597–3610 CrossRef CAS PubMed.
- C.-T. Poon, W. H. Lam and V. W.-W. Yam, Chem.–Eur. J., 2013, 19, 3467–3476 CrossRef CAS PubMed.
- K. Ono, K. Yoshikawa, Y. Tsuji, H. Yamaguchi, R. Uozumi, M. Tomura, K. Tagaa and K. Saito, Tetrahedron, 2007, 63, 9354–9358 CrossRef CAS.
- Y. Mizuno, Y. Yisilamu, T. Yamaguchi, M. Tomura, T. Funaki, H. Sugihara and K. Ono, Chem.–Eur. J., 2014, 20, 13286–13295 CrossRef CAS PubMed.
- E. Kim, A. Felouat, E. Zaborova, J.-C. Ribierre, J. W. Wu, S. Senatore, C. Matthews, P.-F. Lenne, C. Baffert, A. Karapetyan, M. Giorgi, D. Jacquemin, M. Ponce-Vargas, B. Le Guennic, F. Fages and A. D'Aléo, Org. Biomol. Chem., 2016, 14, 1311–1324 CAS.
- S. Chibani, A. Charaf-Eddin, B. Mennucci, B. Le Guennic and D. Jacquemin, J. Chem. Theory Comput., 2014, 10, 805–815 CrossRef CAS PubMed.
- F. P. Macedo, C. Gwengo, S. V. Lindeman, M. D. Smith and J. R. Gardinier, Eur. J. Inorg. Chem., 2008, 3200–3211 CrossRef CAS.
- L. H. Toporcer, R. E. Dessy and S. I. E. Green, Inorg. Chem., 1965, 4, 1649–1655 CrossRef CAS.
- P. Rapta, K. Erentova, A. Stasko and H. Hartmann, Electrochim. Acta, 1994, 39, 2251–2259 CrossRef CAS.
- Y. L. Chow, C. I. Johansson, Y.-H. Zhang, R. Gautron, L. Yang, A. Rassat and S.-Z. Yang, J. Phys. Org. Chem., 1996, 9, 7–16 CrossRef CAS.
- K. Ono, K. Yoshikawa, Y. Tsuji, H. Yamaguchi, R. Uozumi, M. Tomura, K. Taga and K. Saito, Tetrahedron, 2007, 63, 9354–9358 CrossRef CAS.
- C. Qian, G. Hong, M. Liu, P. Xue and R. Lu, Tetrahedron, 2014, 70, 3935–3942 CrossRef CAS.
- C. Risko, E. Zojer, P. Brocorens, S. R. Marder and J. L. Brédas, Chem. Phys., 2005, 313, 151–157 CrossRef CAS.
- J. Fabian and H. Hartmann, J. Phys. Org. Chem., 2004, 17, 359–369 CrossRef CAS.
- R. E. Dessy and R. L. Pohl, J. Am. Chem. Soc., 1968, 90, 2005–2008 CrossRef CAS.
- K. Ono, J. Hashizume, H. Yamaguchi, M. Tomura, J.-i. Nishida and Y. Yamashita, Org. Lett., 2009, 11, 4326–4329 CrossRef CAS PubMed.
- K. Ono, H. Yamaguchi, K. Taga, K. Saito, J.-i. Nishida and Y. Yamashita, Org. Lett., 2009, 11, 149–152 CrossRef CAS PubMed.
- Y. Sun, D. Rohde, Y. Liu, L. Wan, Y. Wang, W. Wu, C. Di, G. Yu and D. Zhu, J. Mater. Chem., 2006, 16, 4499–4503 RSC.
- M. S. Wrackmeyer, M. Hummert, H. Hartmann, M. K. Riede and K. Leo, Tetrahedron, 2010, 66, 8729–8733 CrossRef CAS.
- K. Ono, A. Nakashima, Y. Tsuji, T. Kinoshita, M. Tomura, J.-I. Nishida and Y. Yamashita, Chem.–Eur. J., 2010, 16, 13539–13546 CrossRef CAS PubMed.
- B. Domercq, C. Grasso, J.-L. Maldonado, M. Halik, S. Barlow, S. R. Marder and B. Kippelen, J. Phys. Chem. B, 2004, 108, 8647–8651 CrossRef CAS.
- M. Pröhl, U. S. Schubert, W. Weigand and M. Gottschaldt, Coord. Chem. Rev., 2016, 307, 32–41 CrossRef.
- G. Bai, C. Yu, C. Cheng, E. Hao, Y. Wei, X. Mu and L. Jiao, Org. Biomol. Chem., 2014, 12, 1618–1626 CAS.
- A. Chaicham, S. Kulchat, G. Tumcharern, T. Tuntulani and B. Tomapatanaget, Tetrahedron, 2010, 66, 6217–6223 CrossRef CAS.
- C. Ran, X. Xu, S. B. Raymond, B. J. Ferrara, K. Neal, B. J. Bacskai, Z. Medarova and A. Moore, J. Am. Chem. Soc., 2009, 131, 15257–15261 CrossRef CAS PubMed.
- X. Zhang, Y. Tian, Z. Li, X. Tian, H. Sun, H. Liu, A. Moore and C. Ran, J. Am. Chem. Soc., 2013, 135, 16397–16409 CrossRef CAS PubMed.
- K. Liu, J. Chen, J. Chojnacki and S. Zhang, Tetrahedron Lett., 2013, 54, 2070–2073 CrossRef CAS PubMed.
- A. Felouat, A. D'Aléo and F. Fages, J. Org. Chem., 2013, 78, 4446–4455 CrossRef CAS PubMed.
- M. Rivoal, E. Zaborova, G. Canard, A. D'Aléo and F. Fages, New J. Chem., 2016, 40, 1297–1305 RSC.
- K. Kamada, T. Namikawa, S. Senatore, C. Matthews, P.-F. Lenne, O. Maury, C. Andraud, M. Ponce-Vargas, B. Le Guennic, D. Jacquemin, P. Agbo, D. D. An, S. S. Gauny, X. Liu, R. J. Abergel, F. Fages and A. D'Aléo, Chem.–Eur. J., 2016, 22, 5219–5232 CrossRef CAS PubMed.
- A. D'Aléo, A. Felouat, V. Heresanu, A. Ranguis, D. Chaudanson, A. Karapetyan, M. Giorgi and F. Fages, J. Mater. Chem. C, 2014, 2, 5208–5215 RSC.
- A. Felouat, A. D'Aléo, A. Charaf-Eddin, D. Jacquemin, B. Le Guennic, E. Kim, K. J. Lee, J. H. Woo, J.-C. Ribierre, J. W. Wu and F. Fages, J. Phys. Chem. A, 2015, 119, 6283–6295 CrossRef CAS PubMed.
- R. Misra, T. Jadhava and S. M. Mobina, Dalton Trans., 2013, 42, 16614–16620 RSC.
- R. Misra, B. Dhokale, T. Jadhava and S. M. Mobina, Dalton Trans., 2014, 43, 4854–4861 RSC.
- P. Gautam, R. Maragania and R. Misra, RSC Adv., 2015, 5, 18288–18294 RSC.
- R. Misra, P. Gautam and S. M. Mobin, J. Org. Chem., 2013, 78, 12440–12452 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- K. Liu, J. Chen, J. Chojnacki and S. Zhang, Tetrahedron Lett., 2013, 54, 2070–2073 CrossRef CAS PubMed.
- N. Aliaga-Alcalde, P. Marqués-Gallego, M. Kraaijkamp, C. Herranz-Lancho, H. den Dulk, H. Görner, O. Roubeau, S. J. Teat, T. Weyhermüller and J. Reedijk, Inorg. Chem., 2010, 49, 9655–9663 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09. Revision D. 01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- B. Mennucci, J. Tomasi, R. Cammi, J. R. Cheeseman, M. J. Frisch, F. J. Devlin, S. Gabriel and P. J. Stephens, J. Phys. Chem. A, 2002, 106, 6102–6113 CrossRef CAS.
- G. Andrienko, http://www.chemcraftprog.com.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- B. Le Guennic and D. Jacquemin, Acc. Chem. Res., 2015, 48, 530–537 CrossRef CAS PubMed.
- T. Soujanya, A. Philippon, S. Leroy, M. Vallier and F. Fages, J. Phys. Chem. A, 2000, 104, 9408–9414 CrossRef CAS.
- M. Ipuy, Y.-Y. Liao, E. Jeanneau, P. L. Baldeck, Y. Bretonnière and C. Andraud, J. Mater. Chem. C, 2016, 4, 766–779 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional tables, NMR spectra, cyclic voltammograms, UV-vis absorption and fluorescence spectra, molecular orbitals. See DOI: 10.1039/c6ra25436e |
| This journal is © The Royal Society of Chemistry 2017 |