
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of strawberry-like Au@CeO2 nanoparticles with enhanced catalytic activity by assembly of block copolymer composite micelles†
Yapei Jiaoa,
Na Lia,
Huan Yua,
Wenting Lia,
Jiaxu Zhaoa,
Xue Li*a and
Xiaokai Zhangb
aShandong Provincial Key Laboratory of Fluorine Chemistry and Chemical Materials, School of Chemistry and Chemical Engineering, University of Jinan, 336 West Road of Nan Xinzhuang, Jinan 250022, People's Republic of China. E-mail: chm_lix@ujn.edu.cn; lixue0312@yahoo.com
bCollege of Physics and Electronics, Shandong Normal University, 88 Wenhuadong Road, Jinan 250014, Shandong, People's Republic of China
First published on 3rd January 2017
Abstract
In this study, novel strawberry-like Au@CeO2 nanoparticles (NPs) were fabricated by assembly of block copolymer composite micelles via a redox reaction between Au(III) and Ce(III) followed by precipitation and calcination. These strawberry-like nanostructures were characterized by transmission electron microscopy, X-ray diffraction, X-ray photoelectron spectroscopy, thermal gravimetric analysis, UV-vis spectroscopy, thermogravimetric analysis and N2 adsorption–desorption techniques. These strawberry-like Au@CeO2 NPs were found to be composed of porous CeO2 and several Au NPs set in the surfaces of the porous CeO2 spheres. The catalytic degradation of methyl orange dye (MO) under ultrasonic irradiation was investigated. It was found that the activity of the strawberry-like Au@CeO2 NPs was substantially higher than that of their porous counterparts. The new strawberry-like nanostructures may have potential applications in catalysis and adsorption.
Introduction
Ceria (CeO2) has attracted much attention due to its wide applications in various fields, such as catalysis,1 fuel cells,2 oxygen sensors,3 chemical–mechanical polishing,4 UV blockers,5 luminescent materials,6 water treatment,7 hydrogen storage materials,8 antioxidant in biomedicine,9 and so on. For applications as an efficient catalyst and support, it is crucial to fabricate novel nanostructures with high activity. To maximize the catalytic performance of noble metals and reduce the quantity used, various noble metal NPs/CeO2 hybrid nanostructures have been successfully fabricated in recent years, and these materials exhibit remarkable enhanced catalytic activity and selectivity.10,11 For example, M@CeO2 (M = Pt, Pd, Au, Ag, etc.) core–shell catalysts can promote the catalytic activities of CeO2 for CO oxidation at room temperature and increase adsorption capacity to remove heavy metal ions in water treatment.11 Recently, a new type of core–shell structure catalyst with multiple noble metal NPs as the core and CeO2 as the shell has been reported.12–17 Zhang's group reported a clean nonorganic synthetic method. The pomegranate-like Pt@CeO2 multicore@shell nanospheres were fabricated by the redox reaction between Ce(NO3)3 and K2PtCl4 in an alkaline aqueous solution under the protection of Ar atmosphere.13 The core–shell catalyst showed good structural stability after calcination and lower temperature for complete CO conversion. Xu's group reported a facile, low temperature hydrothermal synthesis of “plum pudding”-structured Pd@CeO2,14 polyvinylpyrrolidone-stabilized Pd NPs are used as seeds, and the CeO2 coating process is finished via a hydrothermal treatment, which is similar to the previously reported Pt@CeO2.15 Zheng and coworkers successfully synthesized multi-yolk–shell Pd@CeO2 nanocatalysts via a hard-templating method.16 Pd–Fe2O3@SiO2 core–shell nanospheres after etching off the part of Fe2O3 were mixed with Ce(NO3)3, ethylene glycol, acetic acid in a solvothermal process to form a Pd@SiO2@CeO2 multisheath superstructure. A multiyolk–shell nanostructure was obtained after selective removal of SiO2 with a NaOH solution.16 Tang's group reported a template-free approach to the synthesis of multiple Au cores in CeO2 hollow spheres. This material was fabricated by impregnating CeO2 hollow spheres with a HAuCl4 aqueous solution. NaBH4 was then used to reduce HAuCl4 to Au nanoparticles to form multiple Au cores in the CeO2 hollow spheres.17As mentioned above, the fabrication of complex hybrids with well-designed and controlled nanostructures has become an area of great focus in material science.13 Although several noble NPs@CeO2 multicore@shell nanomaterials have been suggested, it still remains a significant challenge to develop facile and effective methods for creating novel noble metal NPs@CeO2 hybrid nanostructures with a remarkable enhancement in catalytic activity.
The utilization of block copolymer micelles for the synthesis and stabilization of metal and metal oxide NPs has been extensively investigated.18 Using block copolymer as surfactant CeO2 NPs can be synthesized.19–21 However, there are no previous work regarding the preparation of strawberry-like noble metal NPs@CeO2 composites by assembly of block copolymer composite micelles.
In the present work, Ce(NO3)3·6H2O and HAuCl4 as precursors and polystyrene-b-poly(2-vinylpyridine) (PS-b-P2VP) block copolymers were first dissolved together in a mixed solvent of toluene and ethanol to form PS-b-P2VP/Ce(NO3)3 composite micelles solution, then NaOH reverse micelle solution is added under vigorous stirring. The reaction between Ce3+ and NaOH to form Ce(OH)3, and the redox reaction between Au(III) with Ce(III) to Au(0) and Ce(IV) took place successively; eventually, the Au@CeO2 strawberry-like NPs are formed. After centrifugation strawberry-like Au@CeO2 spheres are yielded. After removal of the block copolymers, the strawberry-like nanostructures consisting of multi-Au NPs set in a porous CeO2 spheres was generated. The catalytic properties were investigated in the degradation of methyl orange (MO) under ultrasonic irradiation. The architectures are expected to offer increased specific surface area or interfacial area, to prevent the aggregation of CeO2 NPs, and show greatly enhanced catalytic activity. To the best of our knowledge, the present work is the first example of the fabrication of strawberry-like Au@CeO2 NPs in organic solvent at room temperature.
Experimental section
Materials
Polystyrene-b-poly(2-vinylpyridine) (PS-b-P2VP) was purchased from Polymer Source Inc., Dorval, QC, Canada. The molecular weight of the PS block was 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and of the P2VP block 16500 g mol−1, Mw/Mn was 1.15. Ce(NO3)3·6H2O and methyl orange (MO) were purchased from Alfa Aesar. Toluene, ethanol and NaOH were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification.
000 g mol−1 and of the P2VP block 16500 g mol−1, Mw/Mn was 1.15. Ce(NO3)3·6H2O and methyl orange (MO) were purchased from Alfa Aesar. Toluene, ethanol and NaOH were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were used as received without further purification.
Sample preparation
Au@CeO2 was synthesized as follows. PS-b-P2VP/Ce(NO3)3/HAuCl4 micelles solution was prepared with PS-b-P2VP solution in toluene (0.5 wt%, 5.0 ml), Ce(NO3)3 solution in ethanol (0.5 M, 750 μl), HAuCl4 solution in ethanol (0.25 M, 150 μl), and toluene (30.0 ml). The NaOH reverse micelle solution was prepared using NaOH solution (0.5 M, 1.13 ml) in ethanol/water (20/1, wt/wt), PS-b-P2VP solution in toluene (0.5 wt%, 5.0 ml). The two micelle solutions prepared above were mixed and stirred for 6 hours at room temperature. The molar ratios of HAuCl4/VP, Ce(NO3)3/HAuCl4, NaOH/Ce(NO3)3 were 1/10, (8–12)/1, and 8/5, respectively. After the addition of ethanol, the mixture was centrifuged and the obtained precipitate was washed with ethanol for three times, dried at 80 °C in vacuum. The dried sample was further washed with pure water for two times to remove NaNO3 completely, dried at 110 °C overnight in air and calcined at 400 °C for 2 hours in N2.Characterization
Transmission electron microscopy (TEM) and selected-area electron diffraction (SAED) measurements were performed on a JEM-2100 TEM microscope (JEOL Ltd, Japan) operated at 200 kV. The samples were prepared by mounting a drop of the dispersions on a carbon-coated Cu grid and allowing it to dry in air. The average particle size was determined by counting particles from the TEM pictures. X-ray diffraction (XRD) was performed using a D8 FOCUS diffractometer (Bruker-AXS, Germany) with Cu Kα radiation (λ = 1.5418 Å). Thermogravimetric analyses were performed with a Diamond TG/DTA instrument (Perkin-Elmer, USA). Before measurement, the sample was treated at 130 °C in vacuum for 6 hours to remove residual solvents and water completely. The samples were heated in nitrogen from 100 to 800 °C at a heating rate of 10 °C min−1. The X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo ESCALAB 250 with Al Kα excitation. Nitrogen adsorption/desorption experiment were performed on a TriStar II 3020 (Micromeritics Instrument Co.) surface area analyzer after outgassing at 300 °C for 5 hours prior to analysis. The standard multipoint Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface area. The pore size distributions of the materials were determined from the desorption branch of the isotherms on the basis of the Barett–Joyner–Halenda (BJH) model.Photocatalytic activity measurements
The decomposition experiments were carried out in a multiwave ultrasonic generator (Sonicator KQ3200DA; Kunshan Ultrasonic Instrument Co., China) with a frequency of 40 kHz and maximum output power of 150 W. In a typical reaction, MO solution (25 ml, 10 mg l−1), Au@CeO2 catalyst (3.131 mg, 125 mg L−1) were placed in a flask, which was then treated by ultrasonication in a water bath (25 °C) for different times. Prior to ultrasonic treatment, the suspension was equilibrated for 40 min in the dark. To avoid the effect of visible light irradiation, the degradation reaction was carried out in the dark. For comparison, the experiments in the absence of catalyst or in the presence of CeO2 catalyst prepared without using block copolymer or HAuCl4 were also conducted. After the degradation experiment, the treated MO solution was centrifuged at 10000 rpm for 5 min and then filtered to remove the catalyst powders. The change of absorption at 464 nm was measured to identify the concentration of MO using a UV-vis spectrophotometer. A calibration curve obtained using standard MO solutions was used to estimate the initial (C0) and instant (C) (at reaction time t) concentrations of MO, respectively.
Results and discussion
PS-b-P2VP block copolymers can form micelles composed of a soluble PS corona and an insoluble P2VP core in toluene above the critical micelle concentration.22 We have used micellar films of PS-b-P2VP to prepare ordered arrays of TiO2 or Au/TiO2 NPs, and porous polyaniline nanofiber films.23 It is known that HAuCl4 and Ce(NO3)3 can coordinate selectively to the P2VP core.21,23,24 It has been reported that Ce3+ exhibits greater reducing capability in alkaline conditions than in neutral ones.12,13 A redox reaction can be easily triggered on the two-face interface between Au metal salt with high oxidation potential and Ce(OH)3 precursor.12,13 Before addition of NaOH micelle solution, the PS-b-P2VP/Ce(NO3)3/HAuCl4 micelles solution was light yellow, indicating that the redox reaction between Au(III) and Ce(III) doesn't occur. Upon addition of the NaOH micelle solution to the PS-b-P2VP/Ce(NO3)3/HAuCl4 micelles solution, Ce(OH)3 and NaAuCl4 in the P2VP cores will be formed first. Then the redox reaction between Ce(OH)3 and NaAuCl4 takes place, leading to the formation of Au NPs and Ce(OH)4. The color change of the mixture from light yellow to purple gray, then to purple, and finally to dark purple proves that the redox reaction between AuCl4− and Ce(OH)3 to Au(0) and Ce(OH)4 occurs. Ce(OH)4 is transformed into CeO2 NPs after dehydration drying.Fig. 1a and b show the TEM images of the as-prepared strawberry-like Au@CeO2 nanospheres of about 35–65 nm. In a nanosphere, several Au NPs of about 7.2 nm are set in the surface of a porous sphere formed by CeO2 nanocrystals with diameters of about 2–3 nm. Clear lattice fringes confirm the crystalline structure of the CeO2 nanocrystals (Fig. 1b). STEM and elemental mapping images of one Au@CeO2 nanosphere are shown in Fig. 1d. The bright part corresponding to the Au NPs. Ce and O elements spread everywhere and Au (red dots) only exists only in the some areas of the nanosphere also illustrate its strawberry-like structures. One can also see from SEM image (Fig. S1†) that Au NPs are distributed on the surface of CeO2 particles. The formation of Au(0) and CeO2 NPs was supported by XRD analyses. The diffraction peaks at 2θ 28.3, 47.3 and 56.4 corresponding to the (111), (220) and (311) planes of CeO2 phase (PDF: 34-0394) observed in XRD spectrum (Fig. 1e) also reveal that the CeO2 NPs are crystalline. The broadening of the peaks suggests that the crystallite size is very small. The diffraction peaks at 2θ 38.2, 44.3, 64.5 and 77.6 corresponding to the (111), (200), (220) and (311) planes of Au (PDF: 04-0784). The UV-vis spectrum shown in Fig. 1f. The maximum (surface plasmon resonance (SPR)) absorption of Au NPs in Au@CeO2 is observed at 560 nm, which shows a red-shift compared with Au NPs of the same diameter, indicating strong interfacial interaction between the Au NPs and CeO2.
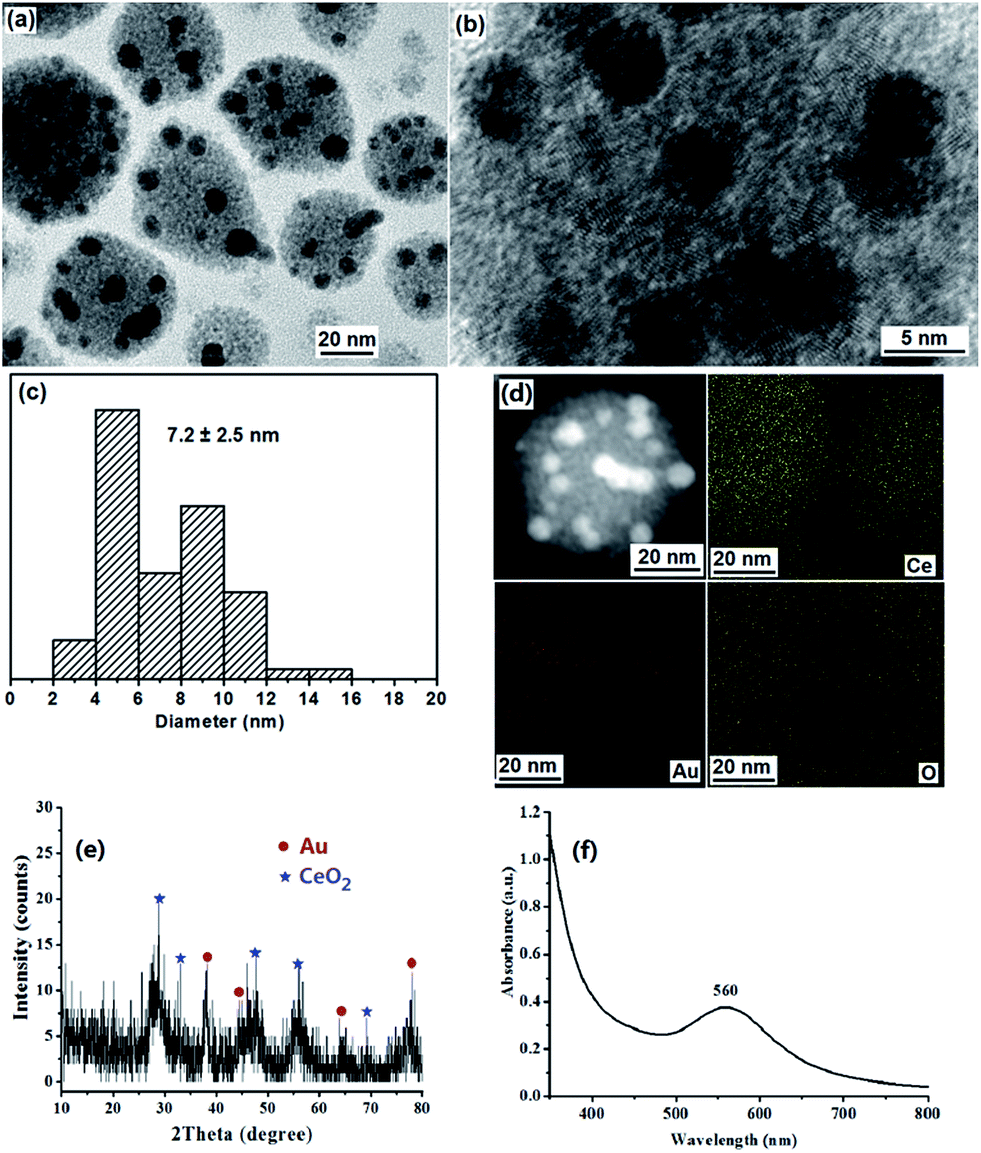 | ||
| Fig. 1 TEM image (a), HRTEM image (b), size distributions of the Au NPs (c), STEM image and its elemental mapping analysis (d), XRD spectrum (e) and UV-vis spectrum (f) of the strawberry-like Au@CeO2 NPs. | ||
The chemical identity of the Au@CeO2 was investigated by XPS. The scan survey spectrum (Fig. 2a) demonstrates that the sample contains Au, O, C and Ce elements. The XPS high resolution Au 4f, O 1s and Ce 3d spectra are shown in Fig. 2b–d. Characteristic peaks of Au0 at 88.0 eV (Au 4f5/2) and 84.5 eV (Au 4f7/2) were observed.25 The O 1s spectra show two peaks at 529.5 and 531.5 eV, which can be ascribed to lattice oxygen in CeO2 and the presence of hydroxyl groups or adsorbed water molecules on the surface.26 The Ce 3d profiles showing six peaks are associated with the Ce4+ initial state.27 The atom percents of Au 4f, O 1s and Ce 3d are 3.19, 81.22 and 15.6, respectively.
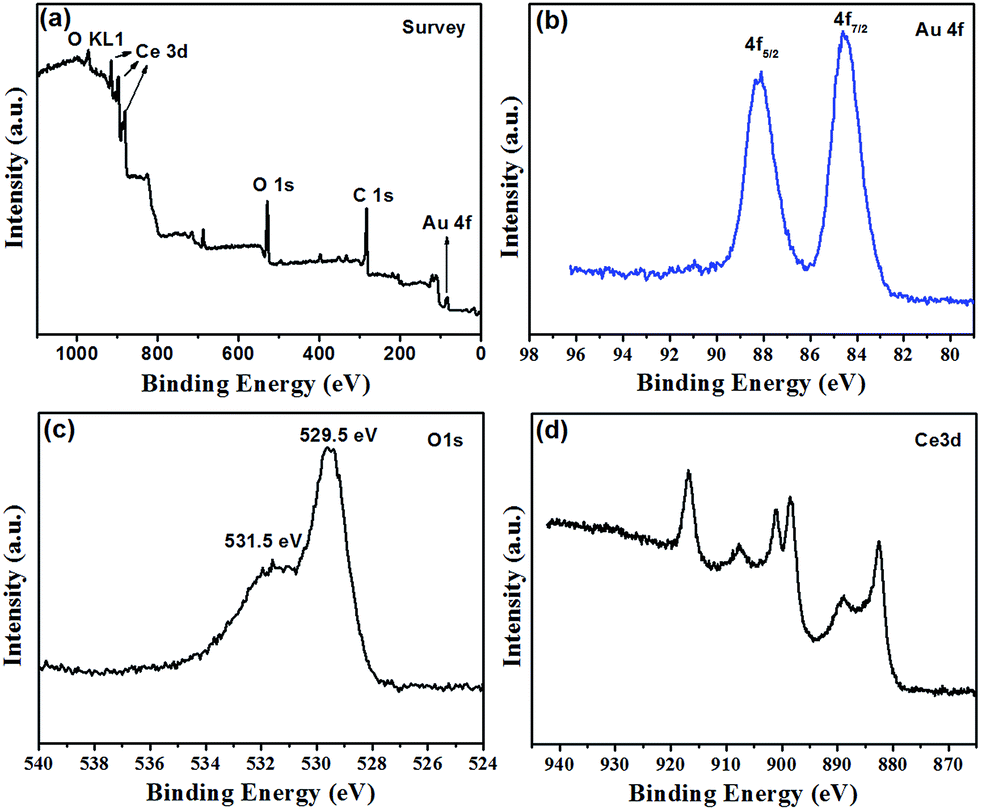 | ||
| Fig. 2 XPS spectra of the strawberry-like Au@CeO2 NPs. (a) Survey, (b) Au 4f, (c) O 1s and (d) Ce 3d. | ||
The TGA data for the as-prepared strawberry-like Au@CeO2 NPs show a stepwise, complicated decomposition process (Fig. 3). The temperature corresponding to a 50% weight loss is 445 °C. From 340 to 445 °C, a weight loss of ca. 42% is observed, which is ascribed to the loss of CO2 and H2O from the block copolymers within the product. The final weight is about 40% (800 °C) after decomposition of the block copolymers completely. For the sample of strawberry-like Au@CeO2 NPs obtained by calcining at 400 °C for 2 hours under N2, the weight decreases slowly and the final weight is about 84% (800 °C).
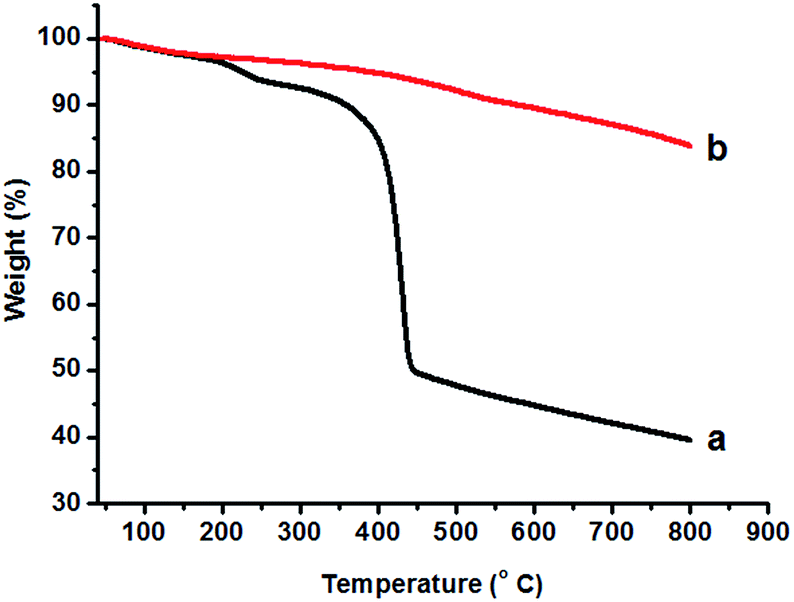 | ||
| Fig. 3 TGA curves of the as-prepared strawberry-like Au@CeO2 NPs (a) and after calcination at 400 °C for 2 hours (b). | ||
The hybrid superstructure formation should be related to the micellar morphology change of PS-b-P2VP block copolymers and the growth balance between CeO2 and Au NPs. Thus, two control experiments have been performed to identify the factors affecting the formation of strawberry-like nanospheres. The first control experiment was carried out under the same condition, except for addition of HAuCl4. It can be seen in Fig. 4a that the porous aggregates of different size are formed by random accumulation of small CeO2 NPs. The second control experiment was done without addition of PS-b-P2VP block copolymers and HAuCl4. Only large particles were observed. Therefore, the formation mechanism of strawberry-like nanostructures may be understood as follows. During the process of reaction, the aggregate of PS-b-P2VP micelles to form larger spheres play an important role. In our previous work,28 we observed that when addition of NaOH to the PS-b-P2VP micelle solution during ultrasonic treatment, spherical PS-b-P2VP micelles collide and aggregate into organic–inorganic Janus-like particles composed of the block copolymers and NaOH. After removal of NaOH, block copolymer vesicles are obtained. However the morphology changes from spherical micelles to larger aggregates rather than vesicles without the aid of ultra-sonication.29 In this case, the structure formation involved the nucleation-growth of Au and Ce(OH)4 NPs in alkaline conditions, and assembly of the block copolymer micelles containing the Au NPs and Ce(OH)4. When the autoredox reaction takes place between AuCl4− and Ce(OH)3, ultra-small Au NPs attaching on Ce(OH)4 NPs are formed, resulting in formation of the original Au–Ce(OH)4 hybrid nanostructures. Simultaneously, to decrease the free energies of the system, PS-b-P2VP micelles containing the Au NPs and Ce(OH)4 aggregate into larger strawberry-like structures with Au NPs set in the aggregates. Ce(OH)4 will transform into CeO2 NPs after drying. The overall procedure to fabrication of the strawberry-like Au@CeO2 NPs is sketched in Scheme 1.
 | ||
| Fig. 4 (a) TEM image of CeO2 nanoparticle prepared by addition of the NaOH micelle solution to the PS-b-P2VP/Ce(NO3)3 micelles solution. (b) TEM image of CeO2 prepared by addition of the NaOH solution to the Ce(NO3)3 solution directly. | ||
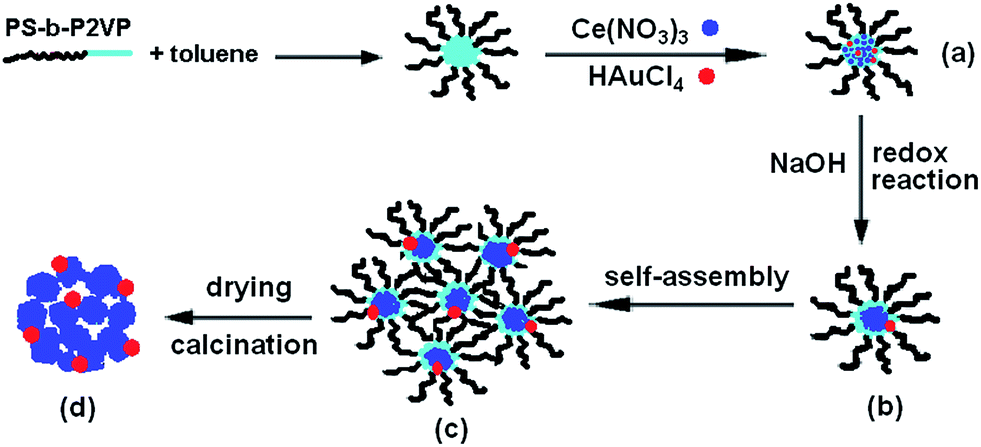 | ||
| Scheme 1 Schematic illustration of the process leading to formation of strawberry-like Au@CeO2 NPs. (a) Formation of PS-b-P2VP/Ce(NO3)3/HAuCl4 composite micelle in toluene. (b) After addition of NaOH solution, Ce(NO3)3 and HAuCl4 initial conversion into Ce(OH)3 and NaAuCl4, followed by redox reaction between Ce(OH)3 and NaAuCl4, and Au–Ce(OH)4 hybrid structures formed in PS-b-P2VP micelles. (c) PS-b-P2VP micelles containing Au–Ce(OH)4 assembly into strawberry-like Au@CeO2 NPs. (d) Strawberry-like Au@CeO2 NPs catalyst after removal of the block copolymer by calcination. | ||
To obtain strawberry-like Au@CeO2 NP catalyst without any organic species, the Au@CeO2 composites were calcined at 400 °C for 2 hours to remove the block copolymers. One can see from the TEM image that the original structures are maintained (Fig. 5a). Some aggregation or coarsening of Au NPs took place during the calcination process, and the average size of Au NPs is changed from 7.2 nm to 16.2 nm. Clear lattice fringes also confirm the crystalline structure of the CeO2 nanocrystals (Fig. 5c), diffraction peaks corresponding to CeO2 crystalline phases (Fig. 5d) become more noticeable as compared with that of as-prepared sample (Fig. 1e), indicating that the crystallinity of the CeO2 NPs was increased.
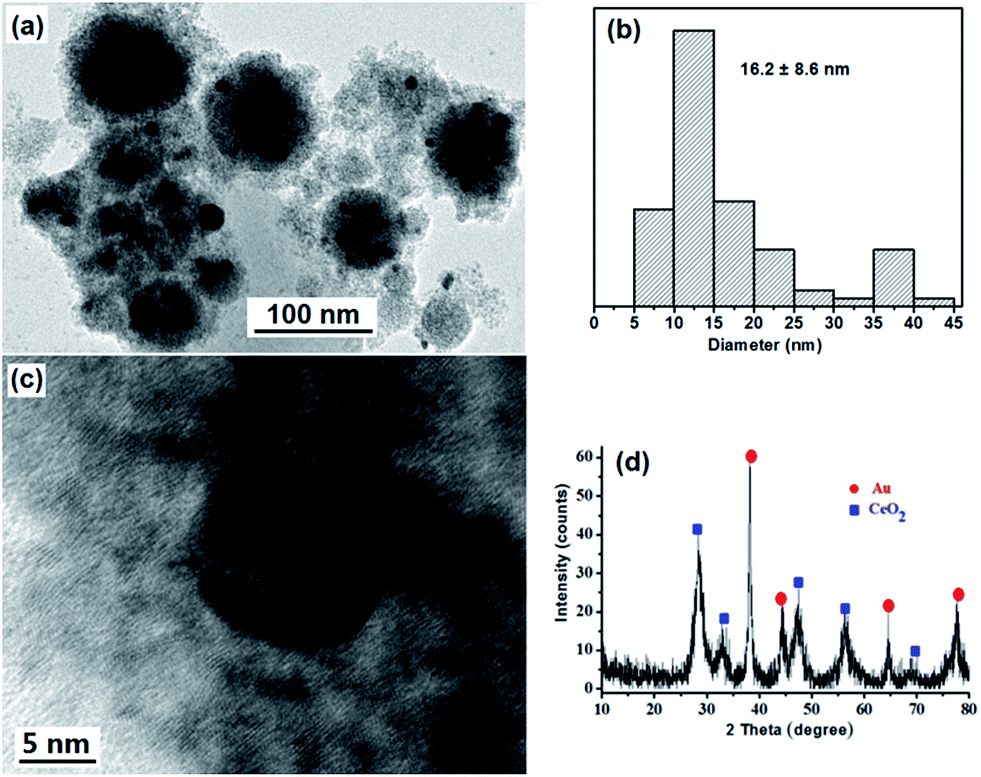 | ||
| Fig. 5 TEM image (a), size distributions of the Au NPs (b), HRTEM image (c), and XRD spectrum (d) of the strawberry-like Au@CeO2 NPs after removal of block copolymers by calcination at 400 °C for 2 hours. | ||
To prove the existence of mesopores, the specific surface areas and pore volumes of the strawberry-like Au@CeO2 nanomaterials after removal of block copolymers were characterized using the nitrogen gas sorption technique, and the typical isotherm and the pore size distribution are shown in Fig. 6. The formation of hysteresis loop in the plot of Fig. 6a supports the presence of mesopores, which can be categorized as being type IV according to IUPAC classification. One can see from Fig. 6b that two peaks around 3.4 nm and 37.0 nm exist, which may be attributed to the interparticle space between CeO2 NPs and the cavity generated by removal of block copolymers, respectively. The BET surface area, pore volume and average pore size are 105.6 m2 g−1, 0.41 cm3 g−1 and 13.5 nm, respectively. The high surface area and porosity of the strawberry-like Au@CeO2 nanomaterials are desirable for their application in catalysis. For comparison, the N2 adsorption–desorption isotherms of porous CeO2 prepared without addition of HAuCl4 (marked as P-CeO2), and CeO2 prepared without addition of HAuCl4 and block copolymers (marked as D-CeO2) were also measured (Table 1). The measured surface area, pore volume and average pore size of three samples are listed in Table 1. The P-CeO2 has higher specific surface area, which is in agreement with the TEM observations (Fig. 4a).
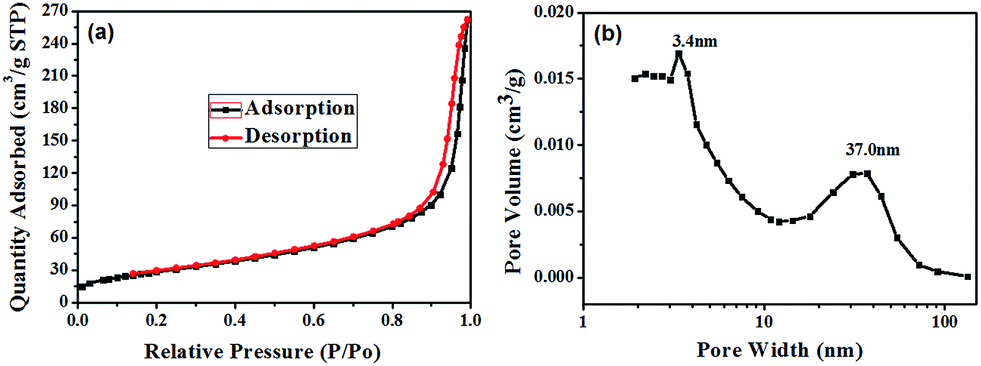 | ||
| Fig. 6 (a) Nitrogen sorption isotherm and (b) the BJH pore size distribution pore volume of strawberry-like Au@CeO2 nanomaterials obtained by calcinating the as-prepared strawberry-like Au@CeO2 spheres at 400 °C in N2 for 2 hours. | ||
| Items | Strawberry-like Au@CeO2 | P-CeO2 | D-CeO2 |
|---|---|---|---|
| BET surface area (m2 g−1) | 105.6 | 138.2 | 25.2 |
| Pore volume (cm3 g−1) | 0.41 | 0.44 | 0.12 |
| Average pore size (nm) | 13.5 | 12.7 | 19.7 |
The catalytic activity of the porous strawberry-like Au@CeO2 nanomaterials was tested in the decomposition of MO dye under ultrasonic irradiation. Fig. 7a shows the evolution of MO absorption spectra in the presence of strawberry-like porous Au@CeO2 catalyst, from which it can be observed that the absorbance of the band at 464 nm associated with MO decreased with time. The variation in the MO absorbance at 464 nm was used to determine the concentration change of MO as a function of ultrasonic time (Fig. 7b). For comparison, the decompositions of MO in solution over the P-CeO2 and D-CeO2 samples were also carried out at the same conditions. One can see from Fig. 7b that the degradation efficiency of MO is much higher in the presence of strawberry-like porous Au@CeO2 nanomaterials catalyst (removal rate 94% at 100 min, curve 1) than the P-CeO2 and D-CeO2 samples (curve 5 and 6). About 0.2% MO degrades after 100 min in the absence of CeO2 catalyst (curve 7), indicating that the energy of ultrasonic irradiation used in this study is insufficient to disrupt the chemical bonds of MO molecules. The porous P-CeO2 can degrade 17% of the MO over the same period (curve 5). The degradation efficiency of MO in the presence of D-CeO2 is only 9% after 100 min (curve 6). In addition, for the strawberry-like Au@CeO2 NPs, the degradation rates increased sharply at the beginning of the reaction.
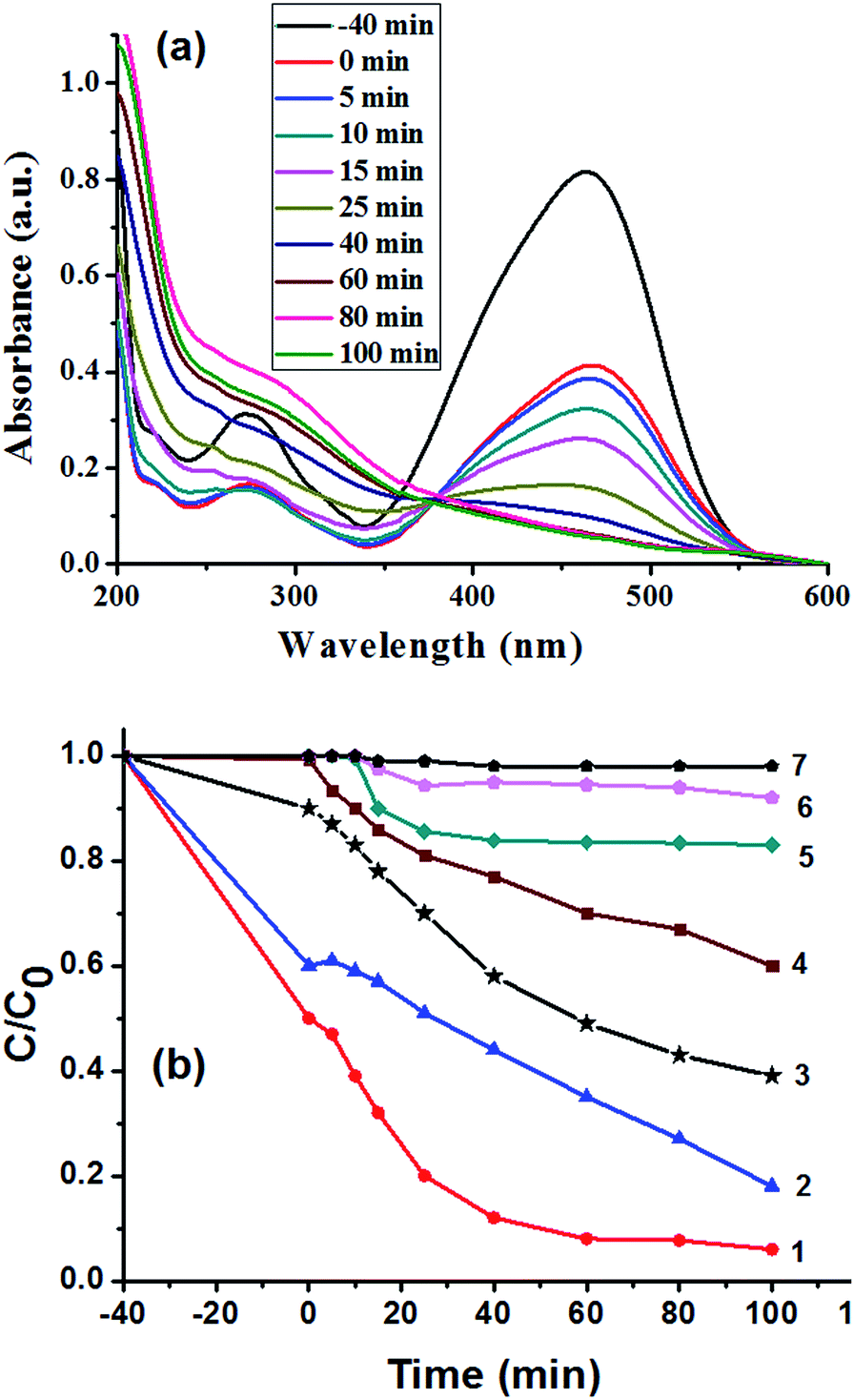 | ||
| Fig. 7 (a) UV-vis spectra of MO as a function of ultrasonication time using strawberry-like porous Au@CeO2 as catalyst. (b) The percentage of degradation efficiency of MO as a function of ultrasonication time using different catalysts. Curves: (1) strawberry-like Au@CeO2 (reactant ratio: Ce(NO3)3/HAuCl4 = 10/1); (2) strawberry-like Au@CeO2 (reactant ratio: Ce(NO3)3/HAuCl4 = 12/1); (3) Au/CeO2 composites prepared by impregnating Au onto CeO2 (Ce(NO3)3/HAuCl4 = 10/1); (4) Au@CeO2 (reactant ratio: Ce(NO3)3/HAuCl4u = 8/1); (5) P-CeO2 catalysts prepared without addition of HAuCl4; (6) D-CeO2 catalysts prepared without addition of HAuCl4 and block copolymers; (7) without CeO2 as catalyst. | ||
To observe the effect of Au-loading content on the catalytic activity of Au@CeO2 catalysts, the samples with the reactant ratio of Ce(NO3)3/HAuCl4 of 12/1 and 8/1 were prepared and their catalytic tests were measured (Fig. 7b, curve 2 and 4). The catalytic result of a post loaded Au catalyst prepared by impregnating Au onto CeO2 was also shown in Fig. 7b (curve 3). One can see from curve 1, 3, 5 and 6 in Fig. 7b that loading Au can increase the catalytic efficiency. Compared to the post loaded Au catalyst (curve 3), the catalysts obtained at reactant ratio of Ce(NO3)3/HAuCl4 of 10/1 or 12/1 exhibited higher activities (curve 1 and 2) due to their strawberry-like structures. However, the catalytic activity prepared with the reactant ratio of Ce(NO3)3/HAuCl4 of 8/1 is lower than that of the post loaded Au catalyst because the Au NPs aggregate seriously at the same experimental condition, which can be proved by the UV-vis spectra and TEM image of the Au@CeO2 NPs (Fig. S2 and 3†).
Under ultrasonic irradiation, it has been reported that possible factors influencing the MO removal efficiency include the concentration of hydroxyl free radical, catalyst particles, and adsorption of MO molecules on catalysts.21,30–32 Although MnO2/CeO2, CuO/CeO2 catalytic ultrasonic degradation of aqueous MO were studied,30,33 the ultrasonic degradation mechanism of MO in the presence of Au/CeO2 catalysts have been studied less. The role of Au NPs may be similar to that in photocatalytic degradation. It has been reported that plasmonic noble NPs onto TiO2 or other semiconductors can facilitate the electron–hole separation and promote ·OH radical production, hence largely enhance the catalytic activity.34–37 In this case, the high catalytic activity of the strawberry-like Au@CeO2 nanomaterials can be attributed to the large specific surface areas or interfacial areas due to the pore structures and electron transfer from the Au to CeO2 by close contact between Au and CeO2. Although the porous P-CeO2 has higher specific surface area, the decolorization rate of the porous P-CeO2 sample is lower than that of the strawberry-like Au@CeO2 catalyst, indicating that the presence of Au NPs in the strawberry-like structures plays an important role in the degradation of MO. Recently, it has been reported that yolk–shell Au@CeO2 microspheres exhibited higher catalytic activity than that of their hollow counterparts in the decolorization of MB trihydrate dye.38 The Ag@CeO2 showed dramatic photocatalytic activity than pure CeO2.39 The enhancement in photocatalytic activities are ascribed to the localized surface plasmon resonance (SPR) of Ag cores. Moreover, larger active interfacial areas and contact between metal/semiconductor in the core–shell structure facilitate transfer of charge carriers and prolong lifetime of photogenerated electron–hole pairs.39
The stability of this Au@CeO2 was measured by means of a recycling reaction. The catalysts were separated by high speed centrifuging, dried and used again. The degradation rate of MO was decreased from 94% (first time) to 70% (second time), to 42% (third time). The decrease of catalytic activity may be due to the structure change of the catalysts under ultrasound.
Conclusions
In conclusion, we have demonstrated a simple way to fabricate strawberry-like Au@CeO2 NPs by assembly of composite micelles of PS-b-P2VP block copolymers. Upon addition of NaOH to the PS-b-P2VP/Ce(NO3)3/HAuCl4 composite micelles solution, a redox reaction between Au(III) and Ce(III) takes place, leading to the formation of Au NPs and Ce(OH)4 first, and then aggregate into strawberry-like Au@CeO2 nanospheres. It was found that the obtained strawberry-like Au@CeO2 nanocomposites prepared with the reactant ratio of Ce(NO3)3/HAuCl4 of 10/1 exhibited superior catalytic activity in the catalytic ultrasonic degradation of MO molecules. The strawberry-like Au@CeO2 nanomaterials reported in this work are expected to have potential applications in the areas of catalysis, adsorption, hybrid inorganic–organic nanocomposite and others.Acknowledgements
This work was funded by the National Natural Science Foundation of China (51173069, 51473068).Notes and references
- (a) A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439 CrossRef CAS; (b) B. M. Reddy, P. Bharali and P. Saikia, J. Phys. Chem. C, 2007, 111, 1878 CrossRef CAS; (c) Z. L. Zhan and A. S. Barnett, Science, 2005, 308, 844 CrossRef CAS PubMed.
- (a) S. D. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265 CrossRef CAS PubMed; (b) L. M. Mattos and F. B. Noronha, J. Catal., 2005, 233, 453 CrossRef CAS.
- H. J. Beie and A. Gnoerich, Sens. Actuators, B, 1991, 4, 393 CrossRef CAS.
- (a) D. G. Shchukin and R. A. Carus, Chem. Mater., 2004, 16, 2287 CrossRef CAS; (b) A. H. Morshed, M. E. Moussa, S. M. Bedair, R. Leonard, S. X. Liu and N. ElMasry, Appl. Phys. Lett., 1997, 70, 1647 CrossRef CAS.
- (a) T. Masui, M. Yamamoto, T. Sakata, H. Mori and G. Adachi, J. Mater. Chem., 2000, 10, 353 RSC; (b) Y. W. Zhang, R. Si, C. S. Liao and C. H. Yan, J. Phys. Chem. B, 2003, 107, 10159 CrossRef CAS; (c) Y. W. Zhang, R. Si, C. S. Liao, C. H. Yan, C. X. Xiao and Y. Kou, J. Phys. Chem. B, 2003, 107, 10159 CrossRef CAS; (d) Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935 CrossRef CAS PubMed.
- X. J. Yu, P. B. Xie and Q. D. Su, Phys. Chem. Chem. Phys., 2001, 3, 5266 RSC.
- J. S. Hu, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 2878 CrossRef.
- K. Sohlberg, S. T. Pantelides and S. F. Pennycook, J. Am. Chem. Soc., 2001, 123, 6609 CrossRef CAS PubMed.
- (a) J. P. Chen, S. Patil, S. Seal and J. F. McGinnis, Nat. Nanotechnol., 2006, 1, 142 CrossRef CAS PubMed; (b) M. Das, S. Patil, N. Bhargava, J. Kang, L. M. Riedel, S. Seal and J. J. Hickman, Biomaterials, 2007, 28, 1918 CrossRef CAS PubMed.
- (a) L. Feng, D. Hoang, C. Tsung, W. Huang, S. Lo, J. Wood, H. Wang, J. Tang and P. Yang, Nano Res., 2011, 4, 61 CrossRef; (b) P. Huang, G. Chen, Z. Jiang, R. Jin, Y. Zhu and Y. Sun, Nanoscale, 2013, 5, 3668–3672 RSC; (c) H. Chong, P. Li, J. Xiang, F. Fu, D. Zhang, X. Ran and M. Zhu, Nanoscale, 2013, 5, 7622 RSC.
- (a) C. Y. Cao, Z. M. Cui, C. Q. Chen, W. G. Song and W. Cai, J. Phys. Chem. C, 2010, 114, 9865 CrossRef CAS; (b) S. L. Zhong, L. F. Zhang, L. Wang, W. X. Huang, C. M. Fan and A. W. Xu, J. Phys. Chem. C, 2012, 116, 13127 CrossRef CAS.
- S. Song, X. Wang and H. Zhang, NPG Asia Mater., 2015, 7, e179 CrossRef CAS.
- X. Wang, D. Liu, S. Song and H. Zhang, J. Am. Chem. Soc., 2013, 135, 15864 CrossRef CAS PubMed.
- N. Zhang, S. Liu, X. Fu and Y.-J. Xu, J. Phys. Chem. C, 2011, 115, 22901 CAS.
- L. Adijanto, D. Bennett, C. Chen, A. Yu, M. Cargnello, P. Fornasiero, R. Gorte and J. Vohs, Nano Lett., 2013, 13, 2252 CrossRef CAS PubMed.
- C. Chen, X. Fang, B. Wu, L. Huang and N. Zheng, ChemCatChem, 2012, 4, 1 CrossRef.
- K. Zhao, J. Qi, S. Zhao, H. Tang, H. Yin, L. Zong, L. Chang, Y. Gao, R. Yu and Z. Tang, Chin. J. Catal., 2015, 36, 261 CrossRef CAS.
- (a) Y. S. Lee, Self-Assembly and Nanotechnology-A Force Balance Approach, John Wiley & Sons, Inc., Hoboken, New Jersey, 2008 Search PubMed; (b) S. Förster and M. Antonietti, Adv. Mater., 1998, 10, 195 CrossRef; (c) J. F. Gohy, in Block Copolymers II, Springer-Verlag Berlin, Berlin, 2005, vol. 190, p. 65 Search PubMed; (d) I. Hamley, Block Copolymers in Solution: Fundamentals and Applications, John Wiley & Sons, Chichester, 2005 Search PubMed.
- S. H. Yu, H. Cölfen and A. Fischer, Colloids Surf., A, 2004, 243, 49 CrossRef CAS.
- Y. S. Chaudhary, S. Panigrahi, S. Nayak, B. Satpati, S. Bhattacharjee and N. Kulkarni, J. Mater. Chem., 2010, 20, 2381 RSC.
- J. Liu, N. Li, W. Li, X. Zhang and X. Li, Sci. Adv. Mater., 2016, 8, 1053 CrossRef CAS.
- (a) G. Kästle, H. G. Boyen, F. Weigl, G. Lengl, T. Herzog, P. Ziemann, S. Riethmüller, O. Mayer, C. Hartmann, J. P. Spatz, M. Möller, M. Ozawa, F. Banhart, M. G. Garnier and P. Oelhafen, Adv. Funct. Mater., 2003, 13, 853 CrossRef; (b) J. P. Spatz, S. Mösser, C. Hartmann, M. Möller, T. Herzog, M. Krieger, H. G. Boyen and P. Ziemann, Langmuir, 2000, 16, 407 CrossRef CAS.
- (a) X. Li, K. H. A. Lau, D. H. Kim and W. Knoll, Langmuir, 2005, 21, 5212 CrossRef CAS PubMed; (b) X. Li, P. Göring, E. Pippel, M. Steinhart, D. H. Kim and W. Knoll, Macromol. Rapid Commun., 2005, 26, 1173 CrossRef CAS; (c) X. Li, S. Tian, Y. Ping, D. H. Kim and W. Knoll, Langmuir, 2005, 21, 9393 CrossRef CAS PubMed.
- (a) S. I. Yoo, B. H. Sohn, W. C. Zin and J. C. Jung, Langmuir, 2004, 20, 10734 CrossRef CAS PubMed; (b) X. Fu, L. Song, J. Liu, X. Li, X. Zhang and Y. Jia, Macromol. Chem. Phys., 2012, 213, 1663 CrossRef CAS.
- X. Li, J. Peng, J.-H. Kang, J.-H. Choy, M. Steinhart, W. Knoll and D. H. Kim, Soft Matter, 2008, 4, 515 RSC.
- G. S. Li, D. Q. Zhang and J. C. Yu, Thermally Stable Ordered Mesoporous CeO2/TiO2 Visible-Light Photocatalysts, Phys. Chem. Chem. Phys., 2009, 11, 3775 RSC.
- K. N. Rao, P. Venkataswamy and B. M. Reddy, Structural Characterization and Catalytic Evaluation of Supported Copper-Ceria Catalysts for Soot Oxidation, Ind. Eng. Chem. Res., 2011, 50, 11960 CrossRef CAS.
- J. Liu, L. Song, X. Zhang and X. Li, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 953 CrossRef CAS.
- X. Fu, L. Song, J. Liu, X. Li, X. Zhang and Y. Jia, Macromol. Chem. Phys., 2012, 213, 1663 CrossRef CAS.
- H. Zhao, G. Zhang and Q. Zhang, Ultrason. Sonochem., 2014, 21, 991 CrossRef CAS PubMed.
- D. Xu, F. Cheng, Q. Lu and P. Dai, Ind. Eng. Chem. Res., 2014, 53, 2625 CrossRef CAS.
- (a) N. Remya and J. G. Lin, Chem. Eng. J., 2011, 166, 797 CrossRef CAS; (b) W. C. Liao, T. Zheng, P. Wang, S. S. Tu and W. Q. Pan, J. Environ. Sci., 2010, 22, 1800 CrossRef CAS.
- D. Xu, F. Cheng, Q. Lu and P. Dai, Ind. Eng. Chem. Res., 2014, 53, 2625 CrossRef CAS.
- C. Yu, G. Li, S. Kumar, H. Kawasaki, R. Jin and J. Phys, Chem. Lett., 2013, 4, 2847 CAS.
- C. Yu, K. Yang, Y. Xie, Q. Fan, J. C. Yu, Q. Shu and C. Wang, Nanoscale, 2013, 5, 2142 RSC.
- C. Yu, W. Zhou, L. Zhu, G. Li, K. Yang and R. Jin, Appl. Catal., B, 2016, 184, 1 CrossRef CAS.
- C. Yu, Y. Bai, J. Chen, W. Zhou, H. He, J. C. Yu, L. Zhu and S. Xue, Sep. Purif. Technol., 2015, 154, 115 CrossRef CAS.
- J. Zhou, L. Xu, J. Sun, D. He and H. Jiao, Surf. Coat. Technol., 2015, 271, 119 CrossRef CAS.
- L. Wu, S. Fang, L. Ge, C. Han, P. Qiu and Y. Xin, J. Hazard. Mater., 2015, 300, 93 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25500k |
| This journal is © The Royal Society of Chemistry 2017 |