
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-dimensional confined electron donor–acceptor co-intercalated inorganic/organic nanocomposites: an effective photocatalyst for dye degradation†
Shufang Zhenga,
Jun Lu *ab,
Jingjing Shia and
Xue Duana
*ab,
Jingjing Shia and
Xue Duana
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, 15 Beisanhuan East Road, P. Box 98, Beijing 100029, P. R. China. E-mail: lujun@mail.buct.edu.cn; Tel: +86-10-6444-2146
bBeijing Engineering Center for Hierarchical Catalysts, Beijing University of Chemical Technology, 15 Beisanhuan East Road, P. Box 98, Beijing 100029, P. R. China
First published on 12th January 2017
Abstract
Photocatalysis is a green catalytic process by utilizing inexhaustible solar light to realize the chemical reaction. Traditionally, most photocatalysts are inorganic semiconductor oxides. Herein, the organic anions, copper phthalocyanine-3,4′,4′′,4′′′-tetrasulfonate (CuPcTS) and 3,4,9,10-perylenetetracarboxylate (PTCB) were selected as guests and co-intercalated into the layered double hydroxides (LDHs) (CuPcTS–PTCB (x%)/LDHs, x was the percentage ratio of PTCB), to obtain the two-dimensional (2D) confined co-intercalated inorganic/organic nanocomposites. The HOMO/LUMO energy levels of the co-intercalated CuPcTS/PTCB anions were matched and coupled as the electron donor and acceptor for the photo-induced electron transfer (PET) process under solar irradiation. The co-intercalated CuPcTS/PTCB anions with similar shape and size were confined and distributed homogenously into the 2D interlayers, which was beneficial for the 2D PET process. The co-intercalated nanocomposites exhibited broad optical absorption in the visible light region, which was in favor of the effective utilization of solar energy for photocatalysis. The equal proportion co-intercalated composite (x = 49.5) with excellent crystallinity and photostability exhibited the best photocatalytic efficiency for oxidation degradation of organic dyes, compared with other proportions and the commercial P25 photocatalyst. Furthermore, the CuPcTS–PTCB (x%)/LDHs exhibited a preferentially faster photodegradation rate for anionic dyes than cationic ones due to the hydrophilic positively-charged LDHs surfaces. In short, this novel 2D confined electron donor/acceptor co-intercalated nanocomposite was a kind of competitive photocatalyst for the degradation of organic contaminants and aromatic toxicants, showing potential applications in environmental protection and pollutant treatment.
1. Introduction
Since the discovery of photocatalytic splitting of water on a TiO2 electrode in 1972,1 the efficient utilization of solar energy for photocatalytic processes has attracted massive interest due to the increasing impacts of industrialization on the environment.2 Traditionally, the photocatalysts for dye degradation are almost all inorganic semiconductor oxides, with excellent photo-thermal stability and high carrier mobility.3 However, most inorganic photocatalysts such as TiO2 (ref. 4) and ZnO5 are only sensitive to ultraviolet light due to their fixed band gap and the limited adsorption ability. During the past few decades, various strategies, such as doping,6 semiconductor heterogeneous structures,7 photosensitization,8 deposition of noble metals,9 and so on, have been developed for the improvement of degradation efficiency under solar irradiation.Meanwhile, organic semiconductors have been developed into one type of new optoelectronic materials with distinctive properties in the past three decades. Among them the organic electron donor and acceptor heterojunctions have attracted much attention due to their strong optical absorption and excellent charge separation and transfer properties,10 which have been widely used in organic field-effect transistors,11 organic solar cells,12 and organic photoconductors.13 The first two-layered organic photovoltaic cell with copper phthalocyanine (CuPc) and perylene tetracarboxylic derivative was designed.14 However, the organic semiconductors with electron donor and acceptor components were rarely applied into the photodegradation of organic dyes due to their photo-instability, incompatibility with hydrophilic environments, and difficulty to be heterogeneously immobilized. Generally speaking, the copper phthalocyanine-3,4′,4′′,4′′′-tetrasulfonate (CuPcTS) is an organic electron donor due to its conjugated π-electron system, large absorption cross sections in the visible light region, suitable for efficient electron-transfer process, and strong oxidizing/reducing characteristics determined by the nature of the peripheral functions.15 The 3,4,9,10-perylenetetracarboxylate (PTCB) is an excellent organic electron acceptor, which has been employed in various applications in optoelectronics devices, such as organic light emitting diodes, photovoltaic devices, and dye lasers devices.16,17 On the other hand, as an important kind of two-dimensional (2D) inorganic layered structure matrix, layered double hydroxides (LDHs) can immobilize the intercalated guests in the restrained interlayers with a particular versatility by virtue of their tunable chemical compositions and gallery spaces, which provides a feasible way to immobilize organic semiconductors.18 Recently, the single-intercalation of phthalocyanine (Pc) or perylene derivatives into LDHs has been reported.19,20 However, nowadays, their co-intercalation into LDHs interlayers has never been reported.
In this work, the novel 2D confined electron donor–acceptor co-intercalated inorganic/organic hybrid nanocomposites CuPcTS–PTCB (x%)/Zn1.5Al-LDHs were designed and synthesized to realize the 2D photo-induced electron transfer (PET) between the co-intercalated CuPcTS and PTCB. These CuPcTS–PTCB (x%)/LDHs nanocomposites exhibited the high efficiency and stability for the photocatalytic oxidation degradation of organic dyes. In view of the unique co-intercalation structure of the inorganic/organic nanocomposites, the excellent photocatalytic performance can be mainly attributed to the following four points. (i) The co-intercalated nanocomposites have broad visible light absorption for the efficient utilization of solar light. (ii) The HOMO/LUMO energy levels of the co-intercalated anions are matched and coupled as the electron donor and acceptor, respectively; that is the premise for the PET process under the solar irradiation. (iii) The conjugated π-systems of CuPcTS and PTCB with similar shape and size are orderly aligned within the LDHs interlayers, which is suitable for efficient electron-transfer processes. (iv) The novel inorganic/organic nanocomposite photocatalysts exhibit preferential degradation of anionic dyes due to the adsorption of positive-charged LDHs hydroxide surface. Therefore, these novel 2D confined electron donor–acceptor co-intercalated photocatalysts can exhibit prospective applications in environmental pollutant treatment.
2. Experimental
2.1 Materials
Copper phthalocyanine-3,4′,4′′,4′′′-tetrasulfonate tetrasodium salt (CuPcTS, 85%) and 3,4,9,10-perylenetetracarboxylic dianhydride (PTCD, 99%) were both supplied by Sigma Chemical Co., Ltd. Methyl Orange (MO), Acid Orange (AO), Acid Yellow (AY), Methylene Blue (MB), Rhodamine B (RhB), Methyl Violet (MV), nanometer titanium dioxide (P25), benzoquinone, and tert-butylalcohol (t-BuOH) were purchased from J&K Co., Ltd. Analytical reagents NaOH, Zn(NO3)2·6H2O and Al(NO3)3·9H2O were all provided by Beijing Chemical Factory and used without further purification. Deionized and CO2-free water was used to prepare all the aqueous solutions.2.2 Preparation of PTCB and synthesis of CuPcTS–PTCB (x%)/LDHs composites
3,4,9,10-Perylenetetracarboxylic acid (PTCB) was obtained by the hydrolysis of PTCD according to the previously reported work.21 The CuPcTS–PTCB (x%)/LDHs powders were prepared by the hydrothermal co-precipitation method. Zn(NO3)2·6H2O (0.01 mol) and Al(NO3)3·9H2O (0.005 mol) were dissolved in 70 mL water as solution A, CuPcTS (a mol), PTCB (b mol, in which a + b = 1.25 mmol, x% = a/(a + b), x% = 0, 10, 30, 50, 70, 90, and 100%, respectively) and NaOH (0.03 mol) were dissolved in 70 mL water as solution B. After that, the solution B was gradually dropped added into solution A with a rate of 10 d min−1 in N2 atmosphere. The pH of the blended solution was adjusted to 7–8 with NaOH solution. The precipitates were immediately obtained and then were aged in 100 mL PTFE-lined autoclave at 120 °C for 20 h. The final powder products were acquired by centrifugation, washing, and drying.2.3 Characterization
The samples of CuPcTS–PTCB (x%)/LDHs were characterized on Shimadzu XRD-6000 diffractometer under such conditions: 40 kV, 50 mA, Cu Kα radiation (λ = 0.154056 nm) with step-scanned mode in step of 0.04°/2θ from 3 to 70°. The Fourier transform infrared (FT-IR) spectra were obtained by a Nicolet 605 XB FT-IR spectrometer in the range 4000–400 cm−1 with 4 cm−1 resolution in air, and the standard KBr disk method was employed. The solid-state UV-visible diffuse reflectance spectra were obtained on a Shimadzu UV-3600 spectrophotometer in the range from 200 to 800 nm equipped with an integrating sphere attachment using BaSO4 as reference. The metal contents of the samples were measured by inductively coupled plasma-atomic emission spectroscopy (ICP-AES) on a Shimadzu ICPS-7500 instrument using solutions made by dissolving the samples into diluted nitric acid. Carbon, hydrogen, and nitrogen analyses were recorded by a Vario EL cube elemental analysis instrument. High-resolution transmission electron microscopy (HRTEM) micrographs were obtained by a JEOL JEM-2100F field emission electron microscope. The specific surface area analysis was collected by micromeritics ASAP 2460. Before the tests, the samples were degassed at 60 °C for 6 h. Cyclic voltammograms (CV) measurements were recorded on an electrochemical workstation (CHI 660E, CH Instruments Inc., China) in a home-made three-electrode optical cell using Ag/Ag+ (0.01 M of AgNO3 in acetonitrile) as the reference electrode and a platinum electrodes at a scan rate of 50 mV s−1 as the counter electrode in an anhydrous and argon-saturated solution of 0.1 M of tetrabutylammonium tetrafluoborate (Bu4NBF4) in acetonitrile. Reference electrode Ag/Ag+ was calibrated by a ferrocene/ferrocenium redox couple as an internal standard, whose oxidation potential was set at −4.8 eV with respect to vacuum level. The HOMO energy levels were evaluated by the equation HOMO = −(Eonsetox − Eonset(ferrocene) + 4.8) eV. The LUMO levels of the polymers were evaluated by the equation LUMO = −(Eonsetred − Eonset(ferrocene) + 4.8) eV.22 Measurements of photocurrent and electrochemical impedance spectroscopy (EIS) were recorded on an electrochemical workstation. The CuPcTS–PTCB (49.75%)/LDHs suspension in ethanol (1 mg mL−1) was prepared by an ultra-sonicator in the N2 atmosphere for 15 min. After that, the CuPcTS–PTCB (49.75%)/LDHs ethanol suspension was fabricated as photoanode by drop coating onto the glassy carbon electrode. Measurements were performed on an electrochemical station in a 10−5 M MO aqueous solution (0.5 M Na2SO4 solution as the supporting electrolyte medium, pH = 6.8, purged with N2 for 30 min) with a home-made three-electrode optical cell using Ag/AgCl as the reference electrode and a Pt wire as the counter electrode. The samples were illuminated from the back of the photoanode under room temperature.2.4 Photocatalysis measurements
Photocatalytic properties for dye degradation of the prepared photocatalysts were measured by examining the photodegradation of negatively charged MO, AO, and AY under simulated solar irradiation of a 150 W Xe lamp (CEL-HXF300, Ceaulioht Co., Ltd. Beijing) with 400–780 nm light filter at 100 mW cm−2. For comparison, the positively charged MB, RhB, and MV were also evaluated by examining the photodegradation process. Typically, 20 mg of photocatalyst was placed into 100 mL MO water solution (10−5 M). Before the solar illumination, the blended solution was put in the dark condition and continuously stirred for 2 h to obtain the adsorption/desorption balance. After every 5 min irradiation, 5 mL of the solution was extracted and then centrifuged to get rid of the catalyst. The concentration of MO was evaluated by measuring its UV-vis characteristic absorption at 463 nm, and the concentrations of AO, AY, MB, MV, and RhB were evaluated by measuring the characteristic absorption at 485 nm, 403 nm, 665 nm, 584 nm, and 554 nm, respectively.3. Results and discussion
3.1 Structure and morphology of CuPcTS–PTCB (x%)/LDHs
The co-intercalated CuPcTS–PTCB (x%)/LDHs were prepared by hydrothermal co-precipitation method. The elemental analysis (Table S1 in ESI†) indicated that the molar ratios of Zn to Al deviated from initial 2.0 to about 1.5, in contrast to the typical Mg2Al-LDHs.23 It was noted that the final experimental ratios of PTCB in LDHs interlayers were systematically higher than the initial nominal ratios, implying that the PTCB was easier to be intercalated into Zn1.5Al-LDHs than CuPcTS. Thus, the excess amount trends in final x throughout the whole concentration range may be ascribed to the strong π–π aggregation effect of PTCB in higher concentration. As shown in Fig. S1 (in ESI†), the co-intercalated CuPcTS–PTCB (x%)/LDHs samples both appeared the characteristic absorption peak of –SO3− (1193 cm−1) in CuPcTS and the antisymmetric and symmetric stretching vibration of –COO− (1550 cm−1 and 1424 cm−1) in PTCB, suggesting that the tetravalent anions of PTCB and CuPcTS were successfully co-intercalated into the interlayers of Zn1.5Al-LDHs under the alkaline hydrothermal conditions.The XRD patterns of co-intercalated CuPcTS–PTCB (x%)/LDHs samples were shown in Fig. 1a. In each product, the XRD pattern exhibited the characteristic reflections of the lamellar structure with a series of 00l (003, 006, 009, 0012, and 0015) peaks appearing as narrow, symmetric, strong peaks at lower angles, presenting a well-defined multiple relationships for the d values. Therefore, the crystallinity was obviously improved with the increase in initial PTCB molar percentage (x%). For the extreme component, the XRD pattern of PTCB/LDHs sample also showed multiple relationships for 00l diffractions with decreasing intensity, implying the well-ordered crystallinity in the stacking c direction (Fig. 1a). While for the CuPcTS/LDHs sample, the XRD intensity of 006 diffraction was close to that of 003, which can be explained by the CuPcTS intercalation configuration with the heavy Cu ions standing midway within the interlayers. The layer spacing of CuPcTS–PTCB (x%)/LDHs was varied from 2.29 nm to 1.84 nm (Fig. S2†). The slight change in layer spacing was ascribed to the different arrangements of interlayered anions with various proportions. According to the comparison between the interlayer spacing, and the sum of one LDHs layer (0.48 nm) and the lateral dimension of CuPcTS (1.74 nm)/PTCB (1.307 nm) (Fig. S3†), it could be reasonably speculated that the co-intercalated CuPcTS/PTCB anions were monolayered and orderly aligned within LDHs interlayers by the π–π stacking interaction (Fig. 1b), confirming the 2D monolayer-type aggregation due to the shape/size similarity between PTCB and CuPcTS. It was concluded that the CuPcTS–PTCB (x%)/LDHs composites were successfully synthesized with the crystalline layered structure being held, and they could be regarded as interlayer anionic complete solid solutions.
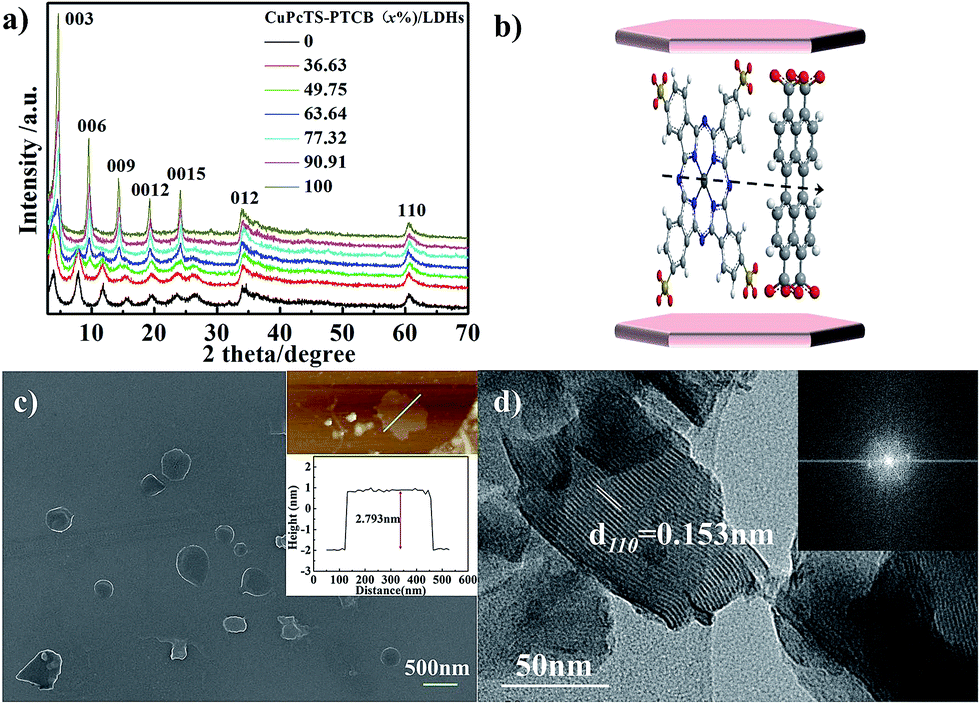 | ||
| Fig. 1 (a) XRD patterns of the CuPcTS–PTCB (x%)/LDHs; (b) speculated molecule structures of co-intercalated CuPcTS and PTCB anions; (c) SEM, AFM images, and its height profile (inset) of CuPcTS–PTCB (49.75%)/LDHs; (d) HRTEM images and FFT pattern (inset) of CuPcTS–PTCB (49.75%)/LDHs. | ||
The SEM image (Fig. 1c) showed that the CuPcTS–PTCB (49.75%)/LDHs nanoplates were in a lateral size of 200–500 nm. More reliable information on the thickness of nanoplates was obtained from the AFM image. As shown in Fig. 1c inset, the thickness of these nanosheets was estimated to be about 2.793 nm, which was consistent with the layer spacing (Fig. S2†). It could be deduced that the co-intercalated nanosheets can be as thin as one bilayer; that was only one Zn1.5Al-LDHs–CuPcTS/PTCB monolayer. The lateral size (∼330 nm) of nanosheets measured by AFM was consistent with that from the SEM image. The HRTEM image (Fig. 1d) of CuPcTS–PTCB (49.75%)/LDHs exhibited continuous (110) lattice fringes with an intra-planar spacing of 0.153 nm in the ab plane of LDHs, matched well with that of CO32−/NO3−-LDHs. The hexagonally arranged diffraction spots in Fast Fourier Transform (FFT) pattern (Fig. 1d inset) implied excellent intralayer crystallinity of CuPcTS–PTCB (49.75%)/LDHs.
3.2 Photocatalytic properties of CuPcTS–PTCB (x%)/LDHs
The CuPcTS/PTCB aqueous solution (15 μmol L−1) exhibited narrow absorption bands at 630 nm and 465 nm (Fig. S4†), respectively, which was also the case for their mixed solution with equal concentration, implying the absence of interaction between CuPcTS and PTCB anions due to their electrostatic repulsion in aqueous solution. However, it was not the case in the 2D confined interlayer environment between the LDHs nanosheets. As shown in Fig. 2a, the single intercalated CuPcTS/LDHs and PTCB/LDHs samples showed wide absorption bands at 565 nm and 430 nm, respectively, with a progressive blue shift compared with the CuPcTS/PTCB anions in solution, implying the formation of H-type interlayer aggregates.24 Notably, the CuPcTS–PTCB (49.75%)/LDHs showed the broadest and strongest absorption from 800 nm to 400 nm, compared with other proportions (Fig. 2a and S5†), which was the evidence of interaction between CuPcTS and PTCB within the LDHs interlayers. It was worth noticing that the absorption of CuPcTS–PTCB (49.75%)/LDHs was well-matched for the AM1.5 solar radiation spectrum in visible light region, which was beneficial to the absorption of solar radiation. Due to the 2D confined effect of LDHs monolayer, the UV-vis absorption spectra bands were strongly broadened when going from the diluted solution to the intercalation solid state. Meanwhile, the broadened absorption spectra were in favor of the subsequent photodegradation process exerted by the co-intercalated CuPcTS and PTCB anions.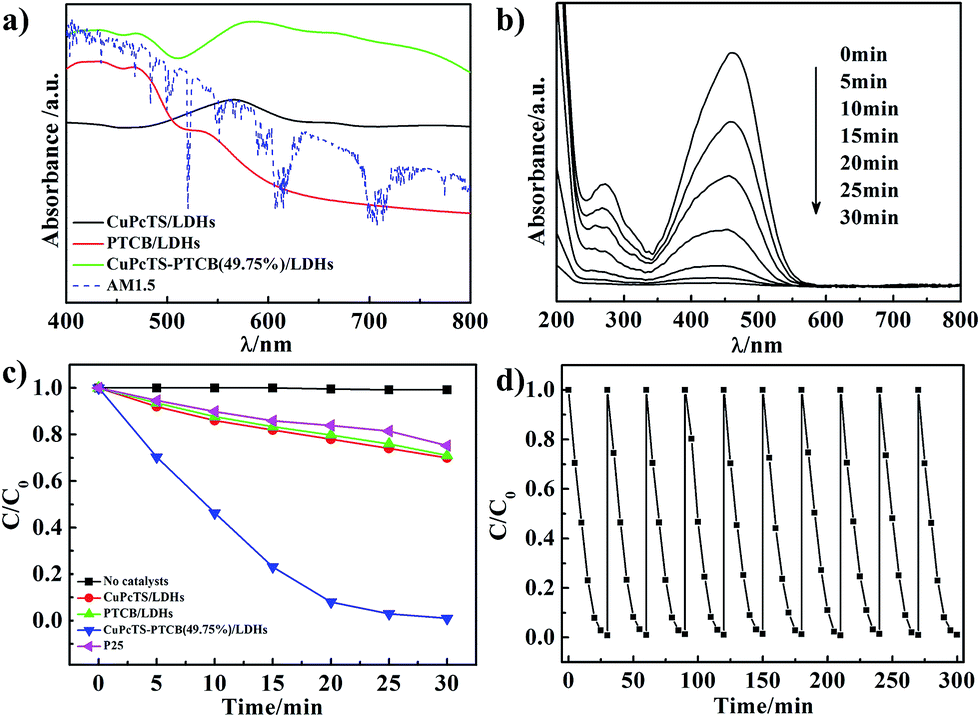 | ||
| Fig. 2 (a) The UV-vis absorption spectra of CuPcTS/LDHs, PTCB/LDHs, CuPcTS–PTCB (49.75%)/LDHs photocatalysts, and AM1.5 solar radiation spectrum; (b) UV-vis absorption spectra for MO subjected to photocatalytic decomposition by CuPcTS–PTCB (49.75%)/LDHs; (c) comparison of photodegradation of MO for under the simulated solar irradiation; (d) cycling curve of photocatalytic degradation of MO. | ||
UV-vis absorption spectroscopy was a common approach to characterize the activity of photocatalysts.25 The photocatalytic activities of CuPcTS–PTCB (x%)/LDHs were evaluated by using the methyl orange (MO) aqueous solution (10−5 mol L−1) as the probe under simulated solar irradiation. We explored the photodegradation photocatalytic activity of bare Zn1.5Al-LDHs for MO under the simulated solar irradiation. As shown in Fig. S8,† the MO was not photodegraded by the bare Zn1.5Al-LDHs under the simulated solar irradiation after 30 min. The commercial P25 reference photocatalyst was also investigated for comparison (Experimental details were shown in ESI†). It was noted that the equal proportional co-intercalated composite (x = 49.75) performed the highest photocatalytic efficiency, compared with other proportions (Fig. S9a†). Obviously, under the solar irradiation, the characteristic absorbance at 463 nm of MO in aqueous solution decreased more quickly in the presence of CuPcTS–PTCB (49.75%)/LDHs (Fig. 2b) than those of single-intercalated samples and P25 (Fig. S10†).
The comparison of photodegradation was further shown in Fig. 2c. The CuPcTS–PTCB (49.75%)/LDHs showed the highest photocatalytic activity: the decoloration rate of MO reached 99% after 30 min. However, only less than 29% of MO molecules were decomposed by CuPcTS/LDHs or PTCB/LDHs sample, and only about 25% of MO molecules were decomposed by P25 at 30 min. Meanwhile, the photocatalytic degradation rate of our photocatalyst was comparable to those of inorganic semiconductors.26 Although there are reports on the enhancement of optoelectronic properties of the nanocomposite materials after the loading of organic sensitizers on inorganic hosts,27,28 the photocatalysis degradation mechanism was different from our 2D confined co-intercalated electron donor–acceptor systems, and the photodegradation efficiency of our co-intercalated LDHs photocatalyst was much superior to that of the single-intercalated LDHs. To confirm the photocatalytic efficiency contributed by the broad optical absorption of the composites, a parallel experiment of photodegradation of colorless organics (e.g. phenol) was performed. The results are shown in Fig. S11 in ESI.† In the presence of CuPcTS–PTCB (49.75%)/LDHs nanocomposites, the degradation of phenol under visible light irradiation became distinct. Because the phenol could not absorb visible light, the self-degradation process of the colorful dyes could not take place. Hence, the promoted photocatalytic performance in visible light region should be derived from the enhanced absorption of visible light by the CuPcTS–PTCB (49.75%)/LDHs composites, which was, in fact, induced by the photo induced charge transfer.
The stability or repeatability of photocatalyst was especially essential for its practical application. Recycling experiments for the MO photodegradation were conducted to confirm the stability performance of CuPcTS–PTCB (49.75%)/LDHs. As shown in Fig. 2d, the photodegradation rate remained almost constant over 10 consecutive cycles, indicating that the co-intercalated photocatalyst was stable under simulated solar irradiation. It was found that the excellent photocatalytic activity was obtained in the presence of very small amount of photocatalysts (as low as 0.2 g L−1), suggesting the striking potential of these co-intercalated nanocomposites for practical photocatalytic application. Their structural stability was first confirmed by the HRTEM images, as shown in Fig. 1d and S6.† There were no distinct differences in morphology before and after dye photodegradation. Furthermore, as shown in Fig. S7,† the XRD diffractions of CuPcTS–PTCB (49.75%)/LDHs composites had no distinct differences after 10 cycles photodegradation compared with the initial CuPcTS–PTCB (x%)/LDHs composites. Those results proved that the CuPcTS–PTCB (49.75%)/LDHs possessed superior structure stability. Obviously, this stability of photocatalytic activity can be attributed to the immobilization of CuPcTS and PTCB anions within the interlayers of LDHs.29
3.3 Charge dependence of CuPcTS–PTCB (x%)/LDHs photocatalysts
The photodegradation effect for different dyes of CuPcTS–PTCB (x%)/LDHs with different proportions was presented in Fig. 3a. Results showed that the best photocatalytic performance was obtained at equal proportion (x = 49.75) co-intercalated nanocomposites, instead of other proportions. This indicated that the interlayered 2D PET process for photocatalytic degradation was most effective for electron donor and accepters with identical amounts and the 2D PET should be one electron process between one donor/accepter pair, the detailed mechanism was discussed in the following part. On the other hand, CuPcTS–PTCB (x%)/LDHs exhibited well-defined photodegradation effect for all the selected dyes (anionic: MO, AO, AY; cationic: MB, MV, RhB). Furthermore, the photodegradation rate was charge-dependent. The degradation rates of anionic dyes for all CuPcTS–PTCB (x%)/LDHs samples were systematically higher than those of cationic dyes (Fig. 3a and S10†). It could be understood that positive-charged LDHs nanoplates could adsorb the anionic dyes more quickly on the surface,30 leading to the subsequent photodegradation reaction based on the 2D PET process in the interlayers. However, for the cationic dyes, the adsorption rate was slower due to the electrostatic repulsion and the adsorption amount was much less, leading to the delayed photodegradation. Fig. 3b clearly showed the higher adsorption capability of anionic dyes than cationic dyes for CuPcTS–PTCB (49.75%)/LDHs. According to the BET analysis (Table S2†), the specific surface areas of all CuPcTS–PTCB (x%)/LDHs photocatalysts were almost identical. In addition, the adsorption amounts of dye MO were almost at the same level (Fig. S12†), which was consistent with the almost identical BET specific surface areas of CuPcTS–PTCB (x%)/LDHs photocatalysts. It was also confirmed that the improved photocatalytic activity for anionic dyes was originated from the positive-charged LDHs layers, rather than the adsorption effect of specific surface area in CuPcTS–PTCB (x%)/LDHs photocatalysts.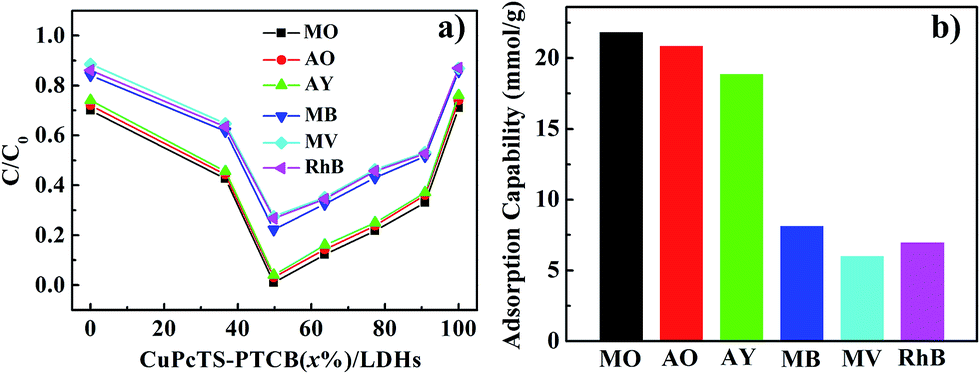 | ||
| Fig. 3 (a) Photodegradation of MO/AO/AY and MB/MV/RhB by CuPcTS–PTCB (x%)/LDHs (x = 0, 36.63, 49.75, 63.64, 77.32, 90.91, 100) after reacted for 30 min; (b) adsorption amounts of MO, AO, AY and MB, MV, RhB on CuPcTS–PTCB (49.75%)/LDHs. | ||
3.4 Photocatalytic mechanism of CuPcTS–PTCB (x%)/LDHs
The cyclic voltammetry (CV) could evaluate the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of molecules.31 The energy levels of CuPcTS and PTCB within LDHs interlayers were estimated by CV measurements of CuPcTS/LDHs and PTCB/LDHs, respectively, provided that no remarkable difference in energy levels between the co-intercalated and single-intercalated process (Fig. S13–S15†). It was noted that the energy levels of CuPcTS and PTCB in the co-intercalated LDHs were slightly increased compared with those of pristine CuPcTS and PTCB, which was related to the 2D confined effect of LDHs. Meanwhile, the energy gap (Eg, Eg = ELUMO − EHOMO) of intercalated CuPcTS/PTCB had no difference from the pristine CuPcTS/PTCB anions (Table 1), implying that the 2D confined effect of LDHs could keep the electronic properties of electron donor and acceptor. The LUMO/HOMO energy levels for the intercalated CuPcTS and PTCB within LDHs were ca. −3.96/−5.18 eV and ca. −4.63/−6.03 eV, respectively, both of which implied that the 2D PET process between co-intercalated CuPcTS/PTCB anions was highly energy reasonable.| HOMOa (eV) | LUMOb (eV) | |
|---|---|---|
| a HOMO (eV) = −(4.8–0 + eEonsetox).b LUMO (eV) = −(4.8–0 + eEonsetred). The Eonsetox and Eonsetred were vs. Ag/Ag+ reference electrode. The Ag/Ag+ reference electrode was calibrated using a ferrocene/ferrocenium redox couple as an internal standard, whose oxidation potential was set at −4.8 eV with respect to vacuum level. | ||
| CuPcTS/LDHs | −5.18 | −3.96 |
| PTCB/LDHs | −6.03 | −4.63 |
| CuPcTS | −5.20 | −3.97 |
| PTCB | −6.05 | −4.65 |
| MO | −4.86 | −3.79 |
| AO | −4.72 | −3.74 |
| AY | −4.83 | −3.76 |
| MB | −4.85 | −3.77 |
| MV | −5.00 | −3.70 |
| RhB | −4.75 | −3.75 |
It is well known that the superoxide radical (˙O2−) and hydroxyl radical (˙OH) are the important active species in the photocatalytic process. Benzoquinone and tert-butylalcohol (t-BuOH), the scavengers of ˙O2− and ˙OH, respectively, can be added to probe the photocatalytic mechanism.32 As shown in Fig. 4, the photodegradation of MO and MB had no differences after 30 min irradiation by adding t-BuOH (10 mmol L−1), while the degradation percentage of them decreased dramatically after adding benzoquinone (1 m mol L−1). Those results proved that the ˙O2− radicals were effective active species, the occurrence of which was beneficial for the oxidation photodegradation of anionic and cationic dyes by photogenerated holes. Fig. 5a presents a schematic energy diagram for the co-intercalated CuPcTS (electron donor) and PTCB (electron acceptor) under 2D confined environment of LDHs, and the adsorbed dyes (MO/AO/AY/MB/MV/RhB). The photodegradation of the organic dyes by the co-intercalated photocatalyst could be attributed to the 2D PET process with the matched energy level of CuPcTS and PTCB. The HOMO energy level of the CuPcTS was lower than that of the selected dyes, and the LUMO energy level of the PTCB was higher than that of the O2/˙O2− energy level. Therefore, the photogenerated holes could be caught by the selected dyes and the photogenerated electrons could be accepted by O2 molecules. Fig. 5b depicted the dyes photodegradation mechanism, based on the 2D PET process of confined CuPcTS and PTCB. When the CuPcTS–PTCB (49.75%)/LDHs powders were exposed to the solar radiation, the photo-generated electrons and holes were generated by the PET process between CuPcTS and PTCB anions. Subsequently, the photogenerated electrons transfered to the LUMO of electron acceptor (PTCB) could be accepted by O2 molecules for the reduction, and the photogenerated holes at the HOMO of CuPcTS could be caught by the organic dyes absorbed on LDHs layers, leading to the oxidative degradation of organic dyes.33 The electrons/holes generated by the 2D PET process and being survived the electron–hole recombination, could access to the O2 and dye ions for photoreduction/oxidation. It could be noted that the highest concentration of these electrons and holes was realized in the CuPcTS–PTCB (49.75%)/LDHs sample, which was the reason of highest photocatalytic efficiency. However, the CuPcTS/LDHs and PTCB/LDHs single-intercalated samples exhibited the lowest photocatalytic efficiency due to the absence of matched energy levels suitable for the photogenerated electron/hole transfer.34
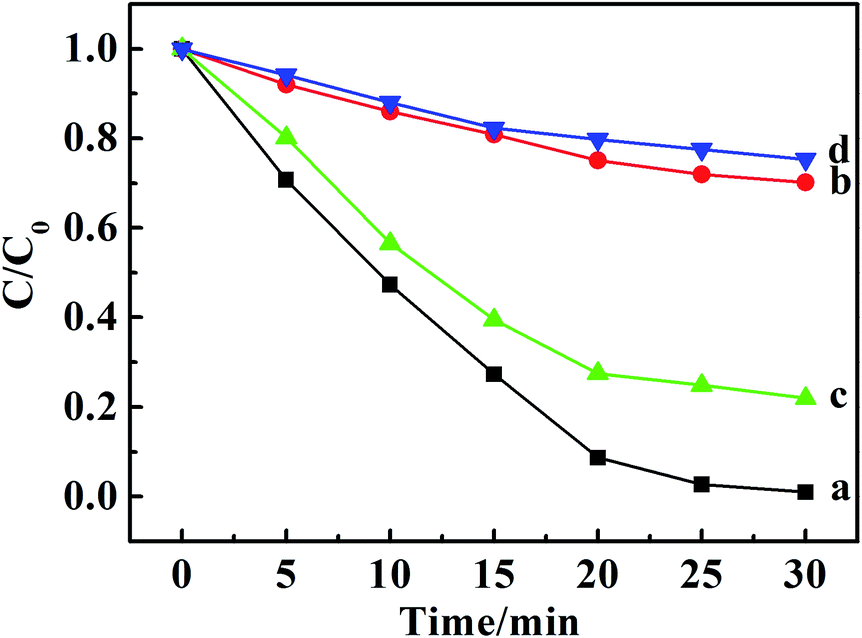 | ||
| Fig. 4 Photodegradation of MO over CuPcTS–PTCB (49.75%)/LDHs with the introduction of 10 mM t-BuOH (a) and 1 mM benzoquinone (b) and MB with the introduction of 10 mM t-BuOH (c) and 1 mM benzoquinone (d). | ||
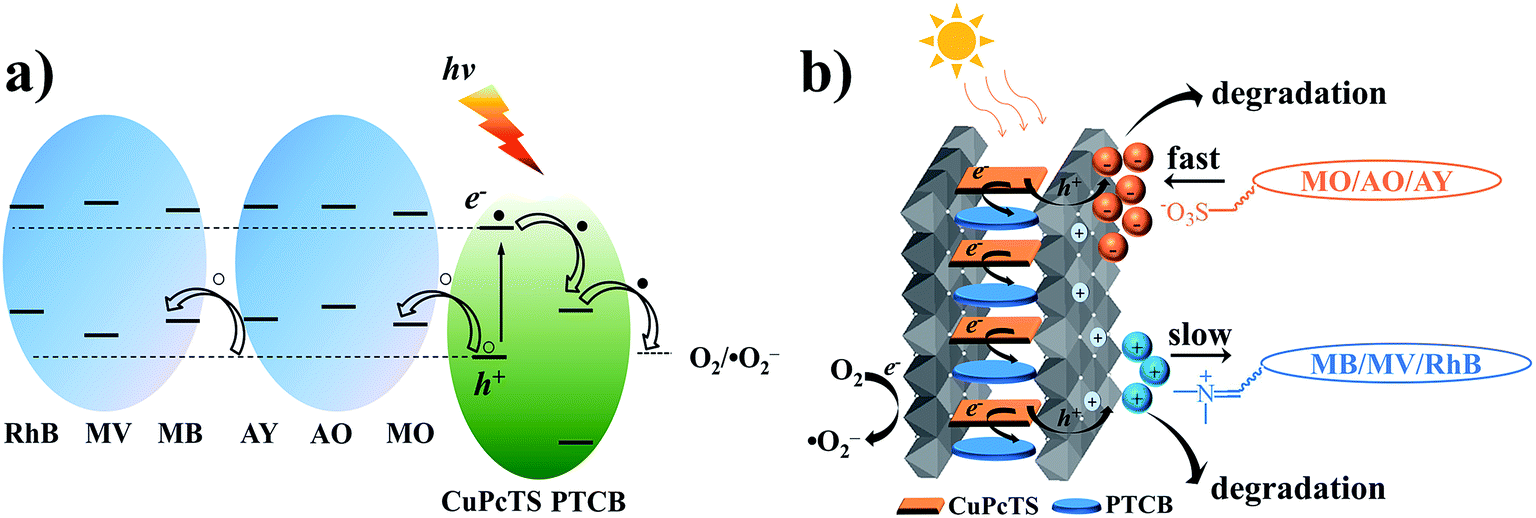 | ||
| Fig. 5 (a) The energy level alignment of MO, AO, AY, MB, MV, RhB, CuPcTS, and PTCB in the Zn1.5Al-LDHs, E°(O2/˙O2−) = E°(O2/˙O2−) − 0.0592 pH (vs. NHE), E°(O2/˙O2−) = −0.15 eV, pH = 7, the NHE to the vacuum energy was 4.5 eV; (b) the proposed 2D PET mechanism scheme of CuPcTS–PTCB (49.75%)/LDHs under solar light irradiation. | ||
Direct evidence for the charge transfer such as photocurrent and EIS measurements was evaluated. Results are presented in Fig. S16.† It was noted that the photon-generated current of CuPcTS–PTCB (49.75%)/LDHs was remarkably enhanced above the onset voltage (0.4 V vs. Ag/AgCl) under light illumination compared with its dark current. In the EIS, the diameter of semicircle curves corresponded to the interfacial charge-transfer resistance, which controlled the electron transfer kinetics of the redox species at the electrode interface.35 The obviously smaller EIS semicircle diameter of CuPcTS–PTCB (49.75%)/LDHs under visible light indicated the enhancement of electron conductivity. However, the CuPcTS–PTCB (49.75%)/LDHs was electric insulative with the large EIS semicircle diameters in dark.
4. Conclusions
In summary, a novel 2D confined electron donor/acceptor co-intercalated photocatalysts CuPcTS–PTCB (x%)/LDHs with excellent crystallinity and photostability were successfully prepared. The CuPcTS/PTCB anions with similar shape and size were orderly monolayered aligned within the LDHs interlayers. The HOMO/LUMO energy levels of the co-intercalated CuPcTS/PTCB anions were well-matched and coupled as the electron donor and acceptor for 2D PET process, respectively. The photo-generated holes produced by the 2D PET process were active for the oxidation degradation of organic ionic dyes. The equal proportional co-intercalated photocatalyst CuPcTS–PTCB (49.75%)/LDHs with excellent crystallinity and optical stability exhibited broad absorption in visible light region and the highest photocatalytic degradation efficiency. Furthermore, the obtained CuPcTS–PTCB (x%)/LDHs performed faster photodegradation for anionic dyes than cationic ones due to the hydrophilic positive-charged LDHs monolayers. Therefore, the novel co-intercalated inorganic/organic photocatalyst exhibited potential application prospect in environmental protection, paving a broad and promising way to develop a kind of new and simple photocatalyst for solar energy conversion and application. Further study was underway including designing the other suitable 2D confined electron donor/acceptor materials with obvious PET process for practical application.Acknowledgements
This work was supported by the 973 Program (Grant No. 2014CB932101), the National Natural Science Research Foundation of China, 111 Project (Grant No. B07004), and Central University research funds of China (buctrc 201527).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- T. Clasen and P. Edmondson, Int. J. Hyg. Environ. Health, 2006, 209, 173–181 CrossRef CAS PubMed.
- S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS PubMed.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef CAS PubMed.
- J. Xu, Z. Chen, J. A. Zapien, C. S. Lee and W. Zhang, Adv. Mater., 2014, 26, 5337–5367 CrossRef CAS PubMed.
- Z. Xiong and X. S. Zhao, J. Am. Chem. Soc., 2012, 134, 5754–5757 CrossRef CAS PubMed.
- R. Buonsanti, V. Grillo, E. Carlino, C. Giannini, F. Gozzo, M. Garcia-Hernandez, M. A. Garcia, R. Cingolani and P. D. Cozzoli, J. Am. Chem. Soc., 2010, 132, 2437–2464 CrossRef CAS PubMed.
- J. Tang, D. Li, Z. Feng, Z. Tan and B. Ou, RSC Adv., 2014, 4, 2151–2154 RSC.
- R. Su, R. Tiruvalam, Q. He, N. Dimitratos, L. Kesavan, C. Hammond, J. A. Lopez-Sanchez, R. Bechstein, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2012, 6, 6284–6292 CrossRef CAS PubMed.
- S. B. Li, Y. A. Duan, Y. Geng, H. Z. Gao, Y. Q. Qiu and Z. M. Su, RSC Adv., 2015, 5, 29401–29411 RSC.
- G. Horowitz and M. E. Hajlaoui, Adv. Mater., 2000, 12, 1046–1050 CrossRef CAS.
- T. Erb, U. Zhokhavets, G. Gobsch, S. Raleva, B. Stühn, P. Schilinsky, C. Waldauf and C. J. Brabec, Adv. Funct. Mater., 2005, 15, 1193–1196 CrossRef CAS.
- B. Friedel, P. E. Keivanidis, T. J. K. Brenner, A. Abrusci, C. R. McNeill, R. H. Friend and N. C. Greenham, Macromolecules, 2009, 42, 6741–6747 CrossRef CAS.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- X. Li, L. E. Sinks, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 10810–10811 CrossRef CAS PubMed.
- C. R. Newman, C. D. Frisbie, D. A. da Silva Filho, J. L. Brédas, P. C. Ewbank and K. R. Mann, Chem. Mater., 2004, 16, 4436–4451 CrossRef CAS.
- Y. Liu, K. R. Wang, D. S. Guo and B. P. Jiang, Adv. Funct. Mater., 2009, 19, 2230–2235 CrossRef CAS.
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 RSC.
- V. Rives and M. A. Ulibarri, Coord. Chem. Rev., 1999, 181, 61–120 CrossRef CAS.
- D. Yan, J. Lu, M. Wei, J. Ma, D. G. Evans and X. Duan, Phys. Chem. Chem. Phys., 2009, 11, 9200–9209 RSC.
- J. Sun, S. Zou, Z. Wang, X. Zhang and J. Shen, Mater. Sci. Eng., C, 1999, 10, 123–126 CrossRef.
- B. Kim, H. Yeom, M. Yun, J. Kim and C. Yang, Macromolecules, 2012, 45, 8658–8664 CrossRef CAS.
- J. Bauer, P. Behrens, M. Speckbacher and H. Langhals, Adv. Funct. Mater., 2003, 13, 241–248 CrossRef CAS.
- D. Yan, J. Lu, M. Wei, D. G. Evans and X. Duan, J. Phys. Chem. B, 2009, 113, 1381–1388 CrossRef CAS PubMed.
- L. Song, X. Zhao, L. Cao, J. W. Moon, B. Gu and W. Wang, Nanoscale, 2015, 7, 16695–16703 RSC.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411–10417 CrossRef CAS PubMed.
- S. Dutta, A. K. Patra, S. De, A. Bhaumik and B. Saha, ACS Appl. Mater. Interfaces, 2012, 4, 1560–1564 CAS.
- K. M. Parida, N. Baliarsingh, B. S. Patra and J. Das, J. Mol. Catal. A: Chem., 2007, 267, 202–208 CrossRef CAS.
- D. Yan, J. Lu, J. Ma, S. Qin, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2011, 50, 7037–7040 CrossRef CAS PubMed.
- S. Zheng, J. Lu, W. Li, Y. Qin, D. Yan, D. G. Evans and X. Duan, J. Mater. Chem. C, 2014, 2, 5161–5167 RSC.
- S. K. Lee, Y. Zu, A. Herrmann, Y. Geerts, K. Müllen and A. J. Bard, J. Am. Chem. Soc., 1999, 121, 3513–3520 CrossRef CAS.
- M. Lan, G. Fan, L. Yang and F. Li, RSC Adv., 2015, 5, 5725–5734 RSC.
- J. M. Macak, M. Zlamal, J. Krysa and P. Schmuki, Small, 2007, 3, 300–304 CrossRef CAS PubMed.
- Z. Li, J. Lu, S. Li, S. Qin and Y. Qin, Adv. Mater., 2012, 24, 6053–6057 CrossRef CAS PubMed.
- A. Di Fabio, A. Giorgi, M. Mastragostino and F. Soavi, J. Electrochem. Soc., 2001, 148, A845–A850 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural characterization and optical absorption spectra of CuPcTS–PTCB (x%)/LDHs; photocatalytic degradation of organic dyes for CuPcTS–PTCB (x%)/LDHs; cyclic voltammograms curves and analysis of energy levels. See DOI: 10.1039/c6ra25534e |
| This journal is © The Royal Society of Chemistry 2017 |