
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel synthetic approach to poly(hydrosiloxane)s via hydrolytic oxidation of primary organosilanes with a AuNPs-stabilized Pickering interfacial catalyst†
Ravi Shankar*,
Bhawana Jangir and
Asmita Sharma
Department of Chemistry, Indian Institute of Technology, Hauz Khas, New Delhi-110016, India. E-mail: shankar@chemistry.iitd.ac.in
First published on 23rd December 2016
Abstract
The triblock copolymer, PiBA20-b-PDMS75-b-PiBA20 (PiBA = polyisobornylacrylate, PDMS = polydimethylsiloxane), 1 is employed as an amphiphilic scaffold for surface functionalization of AuNPs of 10–15 nm size in chloroform. In water–chloroform biphasic medium, the AuNPs form a randomly close packed assembly at the interface and remain stable for several months under ambient conditions. The interfacially stabilized AuNPs act as a selective and recyclable catalyst for the hydrolytic oxidation of primary organosilanes, RSiH3 and offer a promising route for high yield synthesis of poly(alkyl/arylhydrosiloxane)s H2RSiO[RHSiO]nSiRH2, 2–6 [R = Ph (2), n-hexyl (3), cyclo-hexyl (4), Et3SiCH2CH2 (5), PhMe2SiCH2CH2 (6)].
Introduction
Polysiloxanes, [RR1SiO]n (R,R1 = alkyl or aryl) are among the well-known synthetic polymers and differ in many respects from other inorganic polymers due to their unique properties associated with a flexible Si–O–Si backbone.1 As a result, these polymers find widespread applications in industry and modern technology. The classical synthetic approach to polysiloxanes involves acid or base-catalyzed hydrolysis and condensation of chloro-/alkoxy-substituted organosilane monomers.2 However, a major limitation of this approach arises from the sensitivity of the Si–O–Si bond to pH conditions and formation of undesired side products due to cleavage or redistribution/disproportionation of the siloxane framework are often observed. The advent of anionic/cationic ring opening polymerization of cyclo-siloxanes has provided a great deal of impetus in synthetic chemistry of high molecular weight polysiloxanes and its scope to construct block copolymers of varying compositions has been well documented.1,3 In recent years, considerable attention has been devoted to develop catalytic routes to siloxane frameworks with well-defined molecular weight and control over dimensionality.4 In this context, Lewis acid catalysts such as B(C6F5)3, BiCl3, Sc(OTf)3, etc. have shown promise in terms of their selectivity for various functional group transformation and formation of Si–O–Si bonds under non-hydrolytic conditions. These studies are primarily driven by an impetus to expand the library of functional synthons for the development of new siloxane-based materials. Several metal-based heterogeneous and homogeneous catalysts have been investigated for hydrolytic oxidation of Si–H bonds in organosilanes.5 However, these catalysts often lack selectivity as the reactions are accompanied by the formation of a mixture of silanols, disiloxanes and siloxane oligomers. Recent studies have shown that gold nanoparticles on various solid supports are excellent heterogeneous catalysts for selective hydrolytic oxidation of tertiary organosilanes, R3SiH to afford exclusive formation of organosilanols with the elimination of dihydrogen as the byproduct.6Poly(hydromethylsiloxane), [Me(H)SiO]n (abbreviated as PMHS) has attracted considerable attention in view of the presence of Si–H groups in the repeat unit. Hemery et al. have reported the synthesis of high molecular weight PMHS by cationic ring opening polymerization of 1,3,5,7-tetramethylcyclotetrasiloxane in aqueous emulsion using dodecylbenzenesulfonic acid and C12H25(OCH2CH2)23OH (Brij35) as emulsifying agents.7 The presence of Si–H groups in PMHS offers opportunity for chemical modification and incorporation of appended functional moieties on siloxane backbone by hydrosilylation, dehydrocoupling or click reactions.8 The functional groups endow these polymers with interesting physico-chemical properties and serve as scaffolds in the synthesis of Noble metal nanoparticles. The reducing property of PMHS has also been utilized for the reduction of a variety of organic substrates.9
The organization of colloidal metal/metal oxide particles at liquid–liquid interface, a phenomenon first described by Pickering, has emerged as an active area of research in catalysis.10 For example, the entrapment and stabilization of catalytically active nano/colloidal particles (e.g. Pd, SiO2) at the interface has been employed as an alternative to classical methods for chemical transformation of organic substrates and biomass refining.11 The energy (ΔE) of nanoparticles at the interface is governed by nature of the biphasic medium, wettability of the nanoparticle surface and effective radius of nanoparticles, as illustrated in the following equation:
ΔE = −π(d/2)2γw/o(1 ± cos![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)2 θ)2
| (1) |
The study described herein relates to the formation of a self-assembly of AuNPs at water–chloroform interface and its application as a selective Pickering catalyst for the synthesis of poly(alkyl/arylhydrosiloxane)s, H2RSiO[RHSiO]nSiRH2 [R = Ph (2), n-hexyl (3), cyclo-hexyl (4), Et3SiCH2CH2 (5), PhMe2SiCH2CH2 (6)]. The formation of the Pickering catalyst is achieved by surface decoration of AuNPs with amphiphilic block copolymer, PiBA20-b-PDMS75-b-PiBA20 (PiBA = polyisobornylacrylate, PDMS = polydimethylsiloxane), 1.
Results and discussion
The block copolymer, PiBA20-b-PDMS75-b-PiBA20 (1) used for surface functionalization of AuNPs was synthesized by following atom transfer radical polymerization (ATRP)12 of isobornylacrylate (iBA) monomer using difunctional macroinitiator, (2-bromoisobutyrate terminal-PDMS), copper bromide (CuBr) and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) as complexing ligand (Scheme 1). The onset of polymerization was indicated by increase in viscosity of the solution. The overlaid SEC traces (Fig. S1†) of the aliquots at different time intervals reveal a progressive shift towards lower elution time (higher molar mass) compared to the PDMS-macroinitiator. An analysis of number average molecular weight (Mn) as a function of percentage conversion of the monomer (obtained from 1H NMR data) shows a linear plot (Fig. S2†), suggesting that the initiation rate is sufficiently fast relative to propagation and termination is maintained at low level. This is further supported by a decrease in the value of polydispersity index from 2.0 to 1.12 as the polymerization progressed. In the 1H NMR spectrum (Fig. S3†) the resonance at δ 1.9 due to terminal (CH3)2CBr groups of the difunctional macroinitiator is completely absent, suggesting the efficacy of each individual initiating site during polymerization reaction. The detailed synthetic procedure and spectroscopic characterization of 1 is described in the experimental section. An examination of gel permeation chromatographic data reveals Mn = 12570 g mol−1 and PDI = 1.12. The assignments of complex 1H NMR signals of 1 are made by comparison with those reported for poly(isobornylacrylate).13 The Me2Si (δ 0.06) and CH-isobornyl (δ 4.25) protons associated with PDMS and PiBA segments appear in the intensity ratio of 1.8:1 and is consistent with the composition of the polymer. The 29Si NMR spectrum reveals a single resonance at δ −21.9 analogous to that observed in poly(dimethylsiloxane). The FT-IR spectrum in the solid state exhibits strong peaks at 1726, 1260 and 1089 cm−1 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ester), Si–Me and Si–O–Si groups respectively. Thermo gravimetric analysis (TGA) and differential scanning calorimetric (DSC) studies reveal that the polymer is thermally stable upto 280 °C and exhibits glass transition temperature (Tg) of PiBA segment at 85 °C. The PDMS segment could not be analysed by DSC due to temperature constraint of the available instrument. The glass transition temperature of PiBA segment in 1 is significantly lowered as compared to that of poly(isobornylacrylate) homopolymer (94 °C)14 and the phenomenon likely arises from phase mixing of the PDMS and PiBA segments or configurational effects of the polymer chain.15
O (ester), Si–Me and Si–O–Si groups respectively. Thermo gravimetric analysis (TGA) and differential scanning calorimetric (DSC) studies reveal that the polymer is thermally stable upto 280 °C and exhibits glass transition temperature (Tg) of PiBA segment at 85 °C. The PDMS segment could not be analysed by DSC due to temperature constraint of the available instrument. The glass transition temperature of PiBA segment in 1 is significantly lowered as compared to that of poly(isobornylacrylate) homopolymer (94 °C)14 and the phenomenon likely arises from phase mixing of the PDMS and PiBA segments or configurational effects of the polymer chain.15
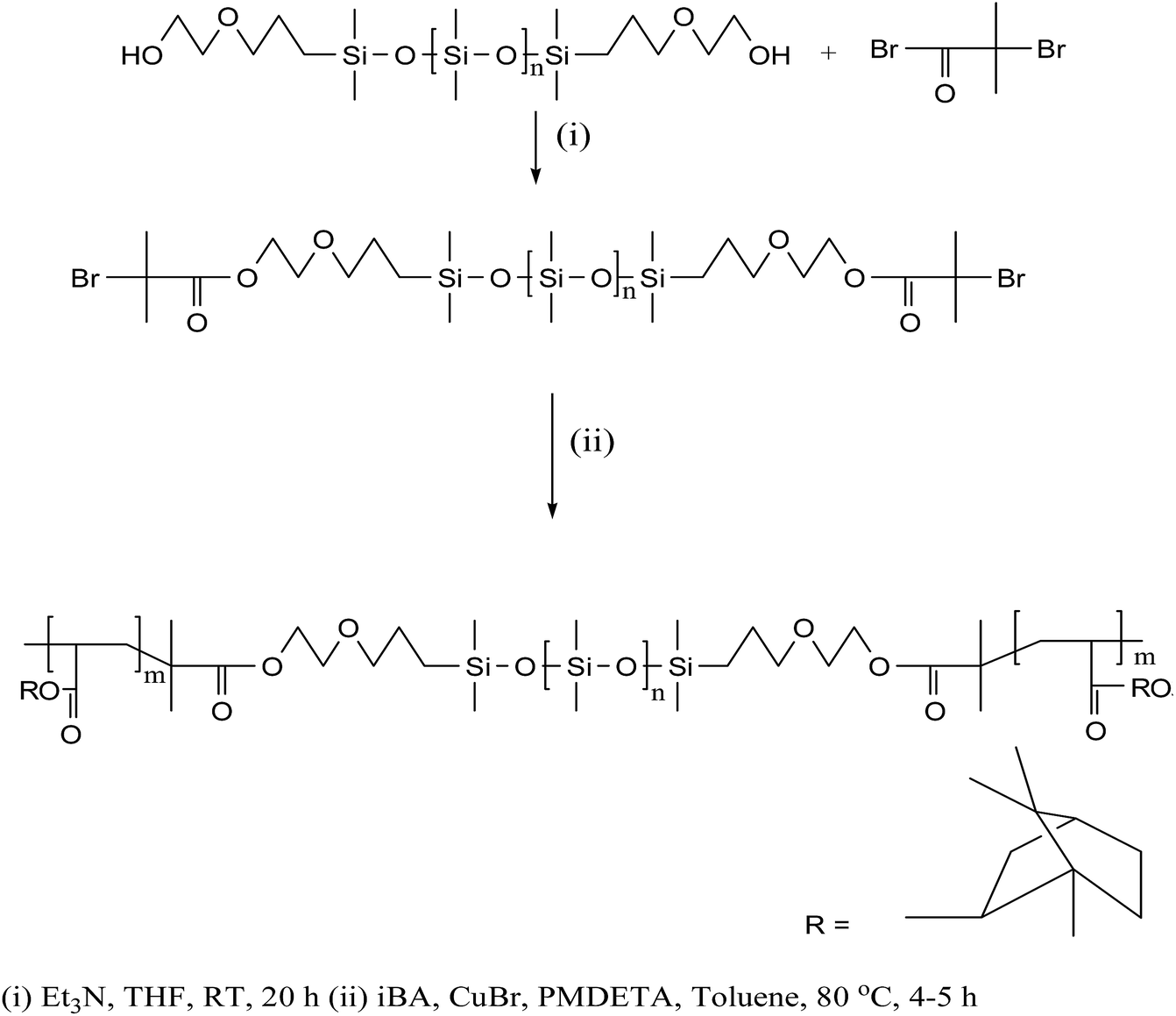 | ||
| Scheme 1 Synthesis of polymer 1. | ||
Synthesis of AuNPs-stabilized Pickering interfacial catalyst in water–chloroform biphasic medium
For the synthesis of Pickering interfacial catalyst, the initial step involves the reduction of hydrogen tetrachloroaurate(III) trihydrate in chloroform with triethylsilane16 under ultrasonication in presence of polymer 1 as the stabilizing matrix. The formation of AuNPs is evident by a gradual colour change of the solution from yellow to purple within 5–10 minutes. The solution was allowed to attain equilibrium for 24 h under ambient conditions and used for further analysis. The UV-Vis spectrum of the colloidal suspension of AuNPs exhibits surface plasmon resonance at 540 nm along with a shoulder at 650 nm (Fig. 1). The observed spectral profile is suggestive of polydispersed nature of AuNPs.17 The plasmonic band does not show perceptible change even after a month suggesting that AuNPs are well passivated by the polymer matrix. Transmission electron microscopic (TEM) study was performed by depositing one drop of the solution on a carbon-coated copper grid. The TEM image shown in Fig. 1 is typical of that found over the entire grid and reveals the formation of polydispersed AuNPs of 10–15 nm size domain. Under high magnification, the TEM image identifies well resolved crystal lattice fringes featuring fcc gold (111) plane with a 0.236 nm d spacing between the adjacent lattice planes (Fig. S4†). The corresponding selected area electron diffraction (SAED) pattern confirms their polycrystalline nature (Fig. S5†). In subsequent step, addition of deionized water (1.0 mL) to a stirred colloidal suspension of AuNPs in chloroform (4.0 mL, 0.02 wt% Au) under ambient conditions results in entrapment of the AuNPs at the water–chloroform interface, while the organic layer becomes colourless within 30 minutes (Fig. 2). The interfacially stabilized polymer–AuNP assembly is hereafter referred as [Au]. It is noteworthy to mention that optimization of AuNP concentration (0.1 wt%) and water–chloroform volume ratio (1:1) affords the formation of a sheet like structure at the interface (Fig. 2). The AuNPs thus formed exhibit metallic lustre when light is passed into the sample. The HRTEM micrograph of [Au] is shown in Fig. 2. The result reveals an aggregation of individual nanoparticles that are not densely packed and do not show long-range order possibly because of the broad size and shape distribution. The plasmonic resonance appears at λmax = 540 nm and extends in the near IR region. The stability of the nanoparticles is not affected by changing the organic medium such as CH2Cl2, THF and toluene. Nevertheless, surface tethering of AuNPs by PiBA homopolymer does not favour the self-assembly process at water–chloroform interface and provides a basis to infer that organization of AuNPs at the interface is primarily driven by amphiphilic character of polymer 1. The results find analogy with an earlier report by Wang et al. The authors have demonstrated that organization of AuNPs at water–toluene interface is primarily governed by surface functionalization of AuNPs with amphiphilic 2-bromo-2-methyl-propionate terminated long chain thiolate groups while the size of nanoparticles does not affect this phenomenon significantly.10
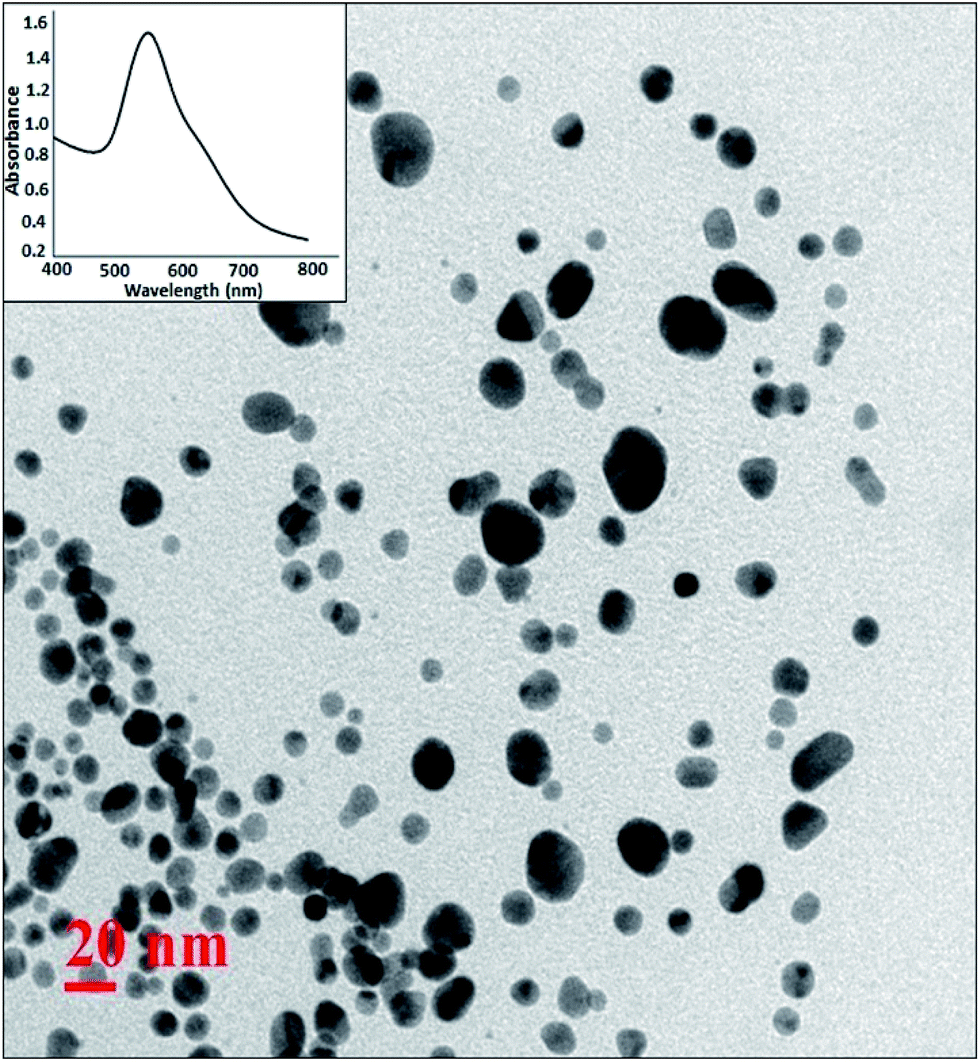 | ||
| Fig. 1 HRTEM image of AuNPs in chloroform. | ||
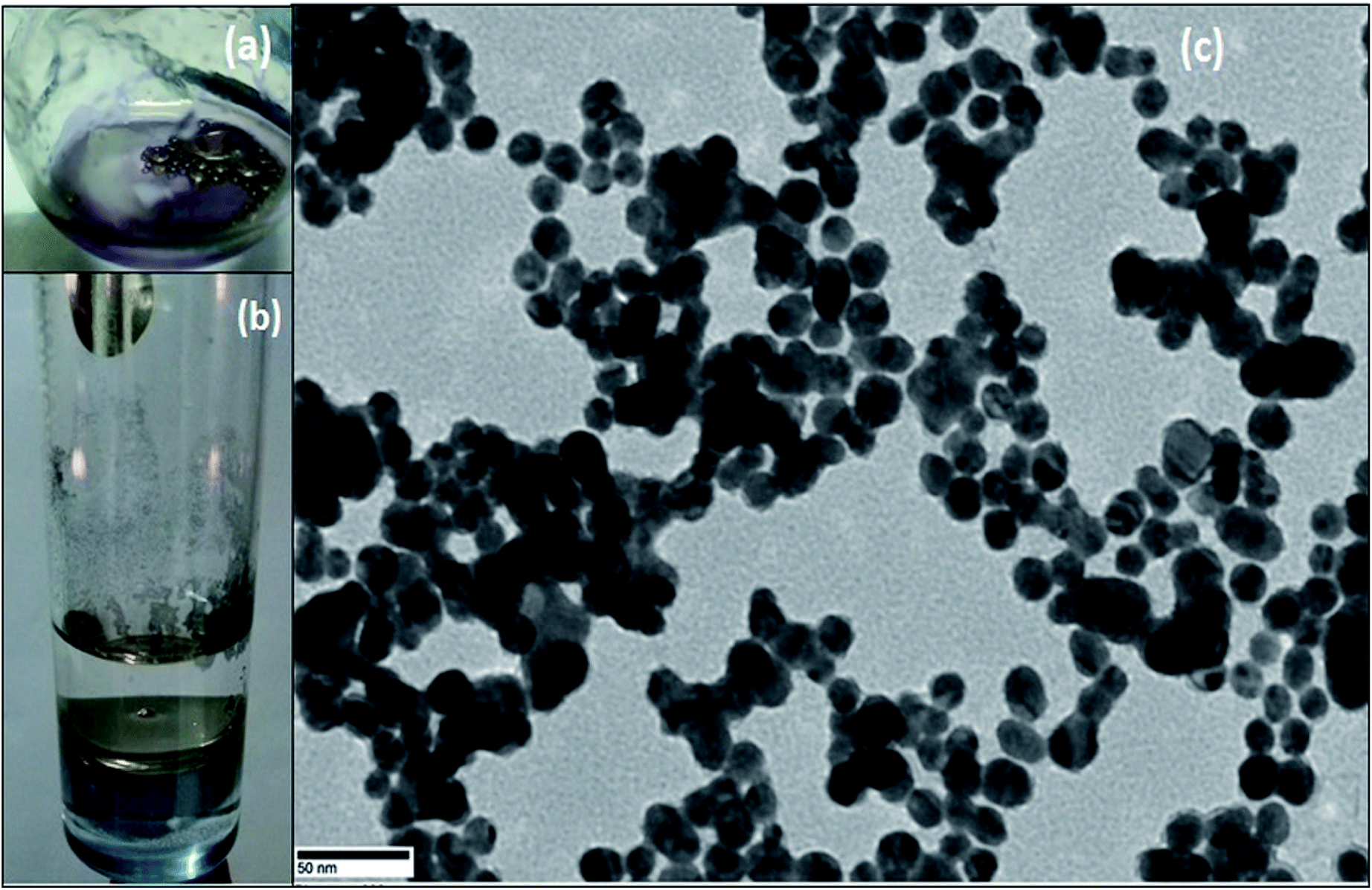 | ||
| Fig. 2 Images of AuNPs (a) stabilized on water droplets (b) sheet like structure at water–chloroform interface, (c) TEM image of AuNPs obtained from (a). | ||
Synthesis and characterization of poly(hydrophenylsiloxane), 2 using Pickering interfacial catalyst, [Au]
As a case study, phenylsilane was taken as a model substrate to study the hydrolytic oxidation of the Si–H groups. The reaction was performed with 0.02 wt% of the [Au] catalyst at 80 °C and monitored at different time intervals by 1H NMR spectroscopy (Fig. 3). As the reaction progresses, a new resonance at δ 5.1 appears in lieu of the signal at δ 4.1 associated with phenylsilane. The former value is typical of PhHSiO/H2PhSiO units18 suggesting the onset of hydrolytic oxidation of the precursor. The GC-MS spectrum (Fig. S6†) of the sample obtained after 2 h of the reaction identifies linear siloxanes of compositions, H2PhSiOSiPh(H)(OH) (A) m/z = 245; [M − H], H2PhSiO[PhHSiO]SiPhH2 (B) m/z = 351; [M − H] and H2PhSiO[PhHSiO]2SiPhH2 (C) (m/z = 474) in 3:4:1 ratio. The signal due to the Si–H protons of phenylsilane disappears after 9–10 h. A usual work up (see Experimental section) of the reaction mixture affords the isolation of H2PhSiO[PhHSiO]nSiPhH2, 2 as viscous oil with Mw = 1.5 × 103 and PDI = 1.4 as evident from gel permeation chromatography. In the 1H NMR spectrum, the integral intensity of Si-Ph and Si–H signals in 5:1 ratio is in conformity with the composition of the polymer. The IR spectrum exhibits characteristic absorption at 2173 cm−1 due to Si–H groups. The 29Si NMR spectrum (Fig. 4) identifies two distinct regions of chemical shifts at δ −44 to −47 and −27.0. The former value is assigned to PhHSiO units while the later to H2PhSiO end groups. Similar chemical shifts have been reported earlier for H2PhSiOSiPhH2 (δ −27, t, 1JSi–H = 224 Hz) and [PhHSiO]n (δ −46.8, d, 1JSi–H = 259 Hz) which has been prepared by controlled hydrolysis of PhHSiCl2.18 Thermo gravimetric analysis (TGA) and differential scanning calorimetric (DSC) study of 2 reveal that the polymer is thermally stable upto 280 °C and exhibits the glass transition temperature (Tg) at −59 °C (Fig. S7†). Interestingly, Tg is markedly higher than that of poly(hydromethylsiloxane) (Tg = −138 °C) suggesting a reduced flexibility of the siloxane backbone.19 It is pertinent to mention that the blank experiments in the absence of [Au] catalyst and water separately do not proceed at all and the substrate is recovered unchanged after 24 h.
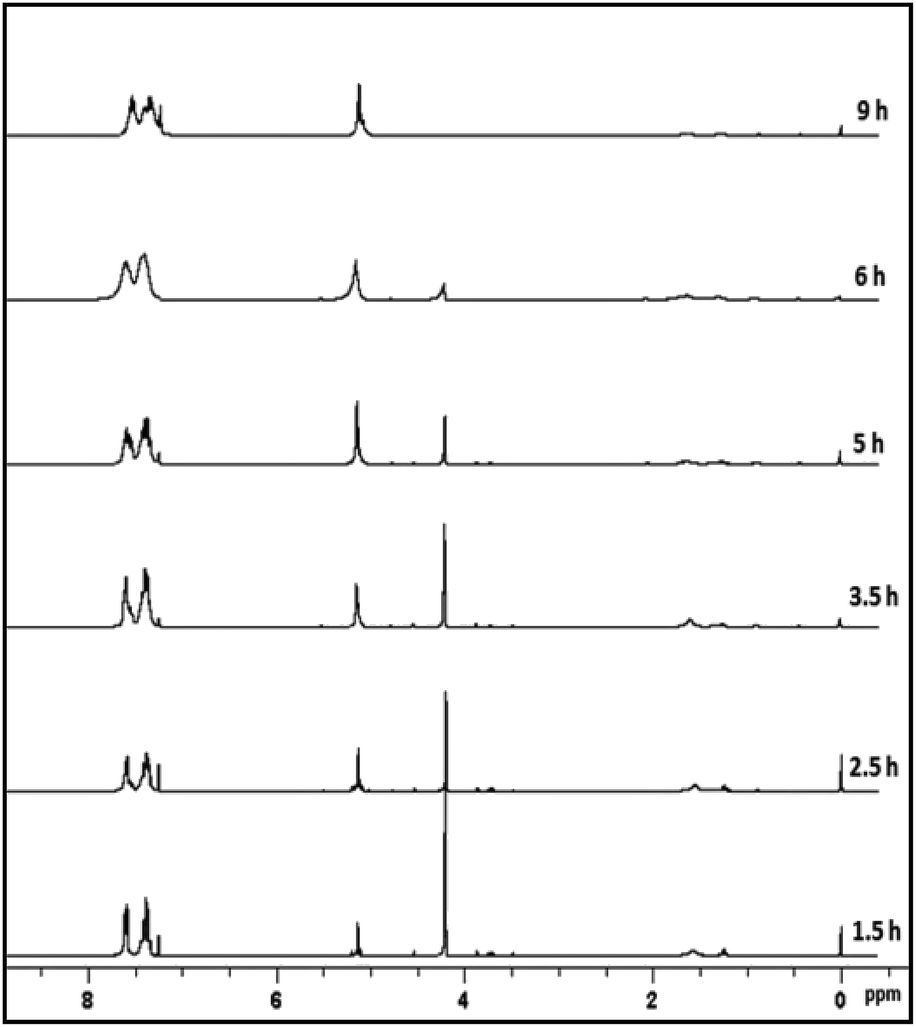 | ||
| Fig. 3 1H NMR spectra at different time intervals during hydrolytic oxidation of phenylsilane. | ||
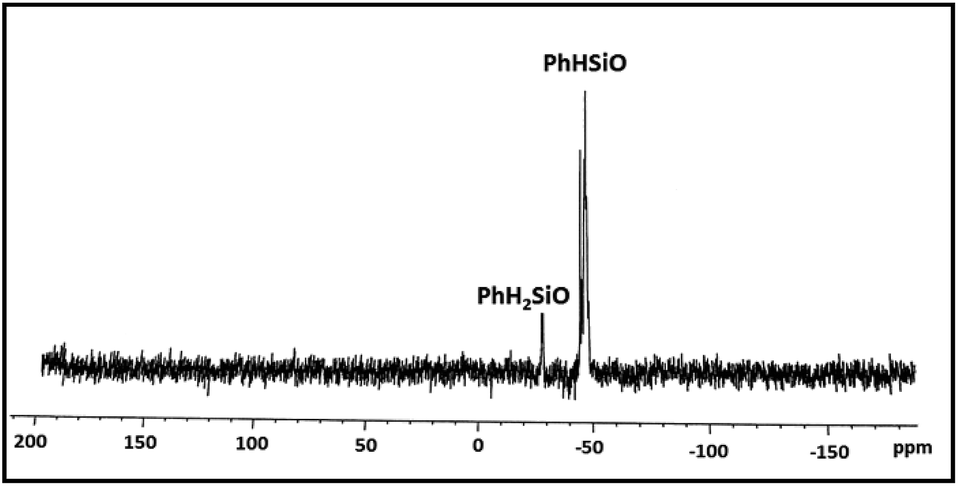 | ||
| Fig. 4 29Si{1H} NMR spectrum of poly(hydrophenylsiloxane), 2. | ||
Synthesis and characterization of poly(alkylhydrosiloxane)s, 3–6
To expand the substrate scope, the Pickering catalyst, [Au] has been further examined for the hydrolytic oxidation of primary alkylsilanes, RSiH3 [R = n-hexyl, cyclo-hexyl, Et3SiCH2CH2, PhMe2SiCH2CH2] under conditions similar to those described above for phenylsilane. An illustrative 1H NMR spectra showing the progress of the reaction for n-hexylsilane as the precursor is given in ESI (Fig. S8).† A complete consumption of the organosilane in each case is observed within 10–12 h. The corresponding poly(hydrosiloxane)s, H2RSiO[RHSiO]nSiRH2 3–6 were isolated as viscous oils and characterized by GPC, IR, 1H and 29Si NMR spectroscopy. These are thermally stable upto 200 °C (Fig. S9†) and thermal stability decreases in the order: cyclo-hexyl > n-hexyl > Et3SiCH2CH2 ≈ PhMe2SiCH2CH2. The GPC profiles of 3–6 shown in Fig. 5 reveal molecular weight (Mw) in the range of 1.1–1.8 × 103 (PDI = 1.2–1.4) suggesting that the polymerization rate is comparable to that of phenylsilane. The 1H NMR spectra exhibit characteristic signals at δ 4.4–4.6 and 0.6–1.9 due to Si–H and alkyl groups respectively in 1:1 integral ratio suggesting the presence of RHSiO repeat units. Additional evidence with regard to the composition of these polymers comes from characteristic 29Si NMR signals at δ −32 to −37 and −17 to −19 (Fig. S10†) which are attributed to RHSiO and H2RSiO units respectively by comparison with those reported earlier for [RHSiO]n and H2RSiOSiRH2 (R = octadecyl).20 The IR absorptions due to Si–H, Si–O and aliphatic groups appear at 2140–2160, 1050–1070 and 2900–3000 cm−1 respectively.
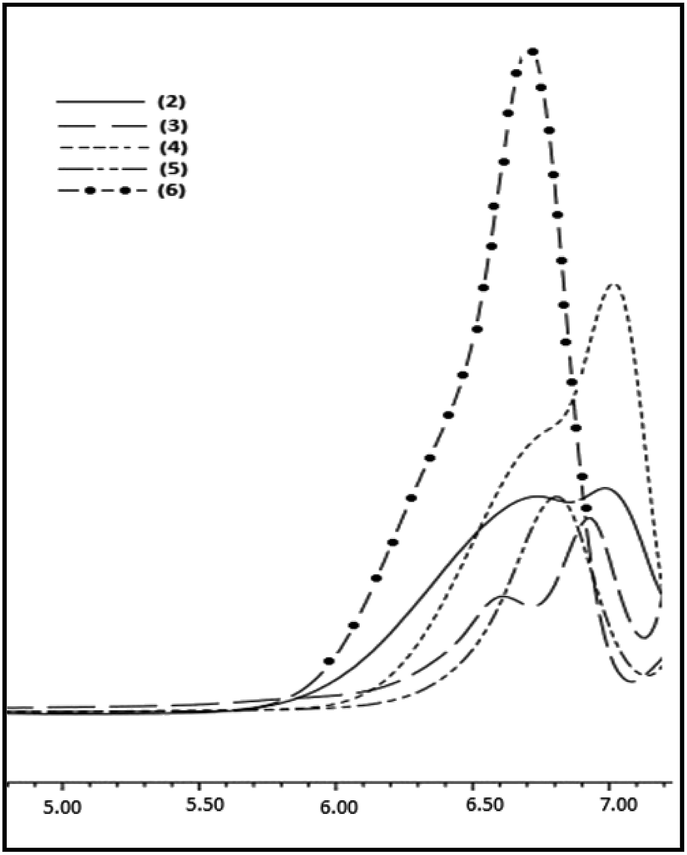 | ||
| Fig. 5 GPC profiles of the polymers 2–6. | ||
The efficiency of the Pickering catalyst after the first cycle of the reaction decreases as evident from a longer reaction time (18–24 h) for complete hydrolytic oxidation of organosilanes. To understand this phenomenon, the AuNPs were examined after the first cycle by HRTEM (Fig. S11†). The micrograph reveals that structural reorganization of the primitive nanoparticles occurs during the catalytic event at elevated temperature and aggregation of individual nanoparticles is discernable in the TEM image. The plasmonic band appears at 570 nm and undergoes a bathochromic shift of the order of 30 nm as compared to primitive AuNPs. Such modulations in the catalytic cycles have been previously observed in PdNPs during Suzuki coupling reaction between phenyl boronic acid and iodobenzene.21
There has been ample precedence in literature which provides a mechanistic rationale for the oxidation of organosilanes using AuNP catalyst. It has been suggested that the reaction proceeds via the formation of Au–H and Au–Si bonds and subsequent reaction of these species with water affords the formation of silanol with the elimination of dihydrogen.22 A similar pathway (Scheme 2) has been proposed in the present study to explain the catalytic activity of [Au] and formation of poly(alkyl/arylhydrosiloxane)s 2–6. The identity of A to C formed as intermediates is authenticated by GC-MS spectral studies (vide supra). An added evidence in favour of the selectivity of the [Au] catalyst comes from the catalytic oxidation of diphenylsilane which affords HPh2SiOSiPh2H (7) within 2 h in >99% yield.
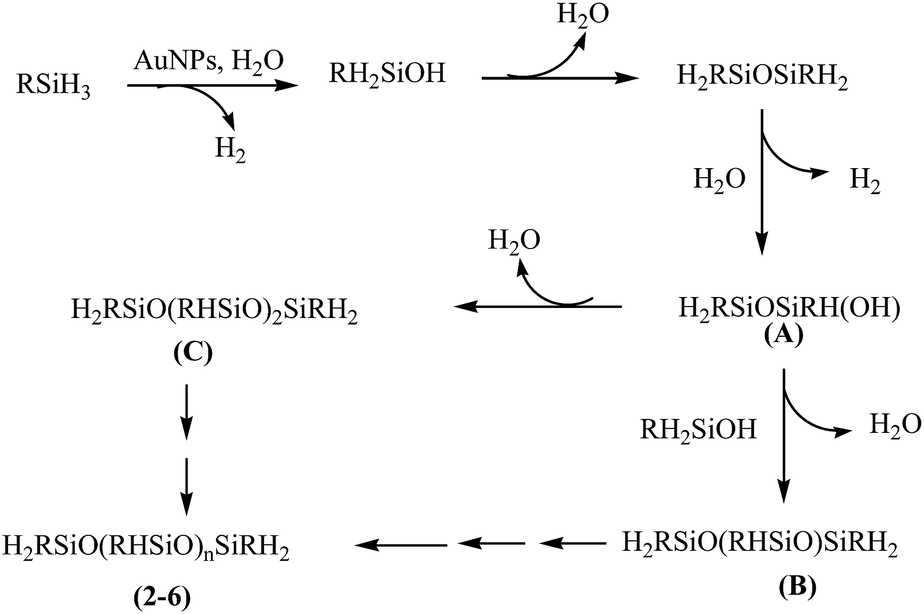 | ||
| Scheme 2 Proposed catalytic pathway for the synthesis of 2–6. | ||
The results provide a basis to infer that the Si–H groups directly bonded to a siloxy unit is far less prone to oxidation under the reaction conditions employed. To our knowledge, there have been only a few reports on the AuNP-catalyzed hydrolytic oxidation of primary organosilanes. In a related study, the alkylsilane, C18H37SiH3 is known to undergo complete oxidation of the Si–H groups and affords the formation of nanowire-like structures build up from RSiO1.5 units.23
Conclusions
We have demonstrated that surface decoration of AuNPs with amphiphilic block copolymer 1 assists the organization of the nanoparticles at water–chloroform interface. The interfacial stabilization of AuNPs provides a platform to study their catalytic activity and selectivity for hydrolytic oxidation of primary alkyl/aryl silanes. This has resulted in the development of a versatile approach for the synthesis of poly(alkyl/arylhydrosiloxane)s, 2–6 bearing [RHSiO] repeat units. Further studies are in progress to expand the scope of these poly(hydrosiloxane)s to incorporate new functional groups and explore their possible applications.Experimental
Materials and methods
Solvents were freshly distilled over sodium wire/benzophenone (tetrahydrofuran, toluene) under inert atmosphere. Chloroform (Fischer, HPLC grade) was used as received. Glassware were dried in an oven at 110–120 °C and further flame dried under vacuum prior to use. Triethylamine was dried over potassium hydroxide and distilled before use. Copper(I) bromide (Sigma Aldrich) was stirred in glacial acetic acid overnight, filtered and washed with absolute ethanol under nitrogen atmosphere. Isobornyl acrylate (iBA) was passed through alumina column to remove the inhibitor. The polymer, bis(hydroxyethoxypropyl)-terminated PDMS (Mn = 5600 g mol−1), 2-bromoisobutyryl bromide and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), phenylsilane, n-hexylsilane, cyclo-hexylsilane, triethylsilane and lithium aluminium hydride were purchased from Sigma Aldrich and used as procured. 1H, 13C and 29Si NMR spectra were recorded in CDCl3 on Bruker AVANCE II 400 NMR spectrometer and the chemical shifts are quoted relative to Me4Si. IR spectra were recorded on Nicolet protege 460 ESP spectrometer using KBr optics. UV-Vis spectral studies were performed on Perkin-Elmer (Lambda Bio 20) spectrophotometer. Transmission electron microscopic (TEM) studies were carried out using carbon coated copper grid on a Philips CM 20 electron microscope operated at 100 kV. Molecular weight (Mw) and polydispersity index (PDI) of the polymers were estimated using Waters gel permeation chromatography (GPC) equipped with L-2414 refractive index detector and Waters styragel HR3 and HR4 columns in series using THF as eluent (flow rate 1 mL min−1; polystyrene standards). Thermo gravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements were carried out in nitrogen atmosphere at a heating rate 10 °C min−1 on a Perkin Elmer Thermal analysis system.Synthetic methods
The organosilanes, RSiH3 (R = Et3SiCH2CH2, PhMe2SiCH2CH2) were synthesized according to the procedure reported earlier.24 The difunctional macroinitiator (Br-PDMS-Br) was prepared by following the procedure reported earlier,25 using bis(hydroxyethoxypropyl) terminated PDMS (5.0 g, 1.0 mmol), triethylamine (0.63 g, 6.0 mmol) and 2-bromoisobutyryl bromide (0.69 g, 3.0 mmol) as the starting materials. The IR, 1H, 13C and 29Si NMR data of the macroinitiator are summarized below.1H NMR (CDCl3): δ 0.07 (br, SiCH3), 0.57 (br, SiCH2), 1.57 (br, SiCH2CH2), 1.95 [br, BrC(CH3)2], 3.45 (br, SiCH2CH2CH2), 3.67 (br, OCH2), 4.32 (br, CH2OCO); 13C{1H} NMR (CDCl3): δ 1.0 (SiCH3), 14.1 (SiCH2), 23.4 (SiCH2CH2), 30.7 [BrC(CH3)2], 65.2 (SiCH2CH2CH2), 68.1 (OCH2), 74.1 (CH2OCO), 175.2 (CO); 29Si NMR (CDCl3, δ): −21.9. IR (KBr, cm−1): 1089 (νSi–O–Si), 1260 (νSi–CH3), 1742 (νCO).
Synthesis of PiBA20-b-PDMS75-b-PiBA20, 1
In a typical procedure, the macroinitiator, Br-PDMS-Br (1.0 g, 0.17 mmol), iBA (3.4 g, 17.0 mmol) and CuBr (0.048 g, 0.34 mmol) in dry toluene (2.0 mL) were introduced into a two-neck round bottom flask equipped with a magnetic stirrer. The contents were subjected to three freeze–pump–thaw cycles to remove oxygen and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (0.12 g, 0.68 mmol) was added while keeping the reaction mixture at 80 °C. The reaction was allowed to continue for 4–5 h and the content thereafter was dissolved in THF and passed through neutral alumina column to remove the copper catalyst. The polymer 1 was isolated as a white solid by repeated precipitation in methanol. The numerical notations for the assignment of various protons are shown in Scheme 1.1H NMR (CDCl3): δ 0.06 (H1, Si–CH3), 0.57 (H2, SiCH2), 0.80–1.70 [H10–H11, H13–H20 (isobornyl ring) + H3, SiCH2CH2], 2.27 [H9, C(O)CH], 3.45 (H4, SiCH2CH2CH2), 3.67 (H5, OCH2), 4.25 (H6, CH2OCO), 4.32 (H12, CH-isobornyl). IR (KBr, cm−1) 1089 (νSi–O–Si), 1260 (νSi–CH3), 1727 (νCO). 29Si NMR (CDCl3): δ −21.9.
Synthesis of Pickering interfacial catalyst (PIC)
To a solution containing hydrogen tetrachloroaurate(III) trihydrate (10.0 mg, 0.025 mmol) in chloroform (20.0 mL, HPLC), a solution of polymer 1, (10.0 mg, 0.04 mmol with respect to polymer repeat unit) in the same solvent (5.0 mL) was added under sonication. Thereafter, triethylsilane (10 μL, 0.1 mmol) was added and the resulting solution was kept for 24 h at room temperature and subsequently used for analysis. The above solution of AuNPs (4.0 mL) was taken separately and mixed with 1.0 mL of deionized water with constant stirring to obtain AuNP-stabilized water droplets in chloroform medium and used for catalytic reactions.Synthesis of poly(aryl/alkyl)hydrosiloxanes, 2–6
In a typical procedure, hydrolytic oxidation of primary organosilanes, RSiH3 (2.0 mL, 0.02 mmol) was performed in the presence of Au-stabilized PIC under aerobic conditions at 80 °C. After complete consumption of the monomer, the organic layer was separated and dried over sodium sulphate. The solvent was evaporated under vacuum and poly(hydrosiloxane)s, 2–6 were isolated as viscous oils.Acknowledgements
We thank UGC (India) for a fellowship to Bhawana and Department of Physics, IIT Delhi for TEM studies. This research was supported by financial grant (Project No. SB/S3/EECE/095/2014) from DST (India).Notes and references
- (a) J. E. Mark, R. West and H. R. Allcock, Inorganic Polymers, Prentice-Hall, Englewood Cliffs, 1992 Search PubMed; (b) M. A. Brook, Silicon in Organic Organometallic and Polymer chemistry, John Willey & Sons, New York, 2000, pp. 256–308 Search PubMed; (c) J. E. Mark, Acc. Chem. Res., 2004, 37, 946 CrossRef CAS PubMed; (d) P. Zheng and T. J. McCarthy, Langmuir, 2010, 26, 18585 CrossRef CAS PubMed.
- Y. Abe and T. Gunji, Prog. Polym. Sci., 2004, 29, 149 CrossRef CAS.
- (a) J. Chojnowski, in Siloxane Polymer, ed. S. Clarson and J. Semlyen, PTR Prentice Hall, London, 1993, p. 1 Search PubMed; (b) W. P. Weber and G. Cai, Macromolecules, 2001, 34, 4355 CrossRef CAS; (c) A. Molenberg and M. Moller, Macromol. Rapid Commun., 1995, 16, 449 CrossRef CAS; (d) K. Rózga-Wijas, J. Chojnowski, T. Zundel and S. Boileau, Macromolecules, 1996, 29, 2711 CrossRef; (e) E. Pouget, J. Tonnar, P. Lucas, P. Lacroix-Desmazes, F. Ganachaud and B. Boutevin, Chem. Rev., 2010, 110, 1233 CrossRef CAS PubMed.
- (a) K. Kuroda, A. Shimojima, K. Kawahara, R. Wakabayashi, Y. Tamura, Y. Asakura and M. Kitahara, Chem. Mater., 2014, 26, 211 CrossRef CAS; (b) R. Wakabayashi and K. Kuroda, ChemPlusChem, 2013, 78, 764 CrossRef CAS; (c) R. Wakabayashi, K. Kawahara and K. Kuroda, Angew. Chem., Int. Ed., 2010, 49, 5273 CrossRef CAS PubMed; (d) J. B. Grande, F. Gonzaga and M. A. Brook, Dalton Trans., 2010, 39, 9369 RSC; (e) D. B. Thompson and M. A. Brook, J. Am. Chem. Soc., 2008, 130, 32 CrossRef CAS PubMed; (f) G. Hreczycho, P. Pawluc and B. Marciniec, New J. Chem., 2011, 35, 2743 RSC; (g) R. Zhang, J. E. Mark and A. R. Pinhas, Macromolecules, 2000, 33, 3508 CrossRef CAS; (h) W. Xue, M. C. Kung and H. H. Kung, Chem. Commun., 2005, 2164 RSC; (i) Z. Chang, M. C. Kung and H. H. Kung, Chem. Commun., 2004, 206 RSC.
- (a) G. H. Barnes and N. E. Daughenbaugh, J. Organomet. Chem., 1966, 31, 885 CrossRef CAS; (b) Y. Lee, D. Seomoon, S. Kim, H. Han, S. Chang and P. H. Lee, J. Org. Chem., 2004, 69, 1741 CrossRef CAS PubMed; (c) R. A. Corbin, E. A. Ison and M. M. Abu-Omar, Dalton Trans., 2009, 2850 RSC; (d) T. C. Bedard and J. Y. Corey, J. Organomet. Chem., 1992, 428, 315 CrossRef CAS; (e) E. A. Ison, R. A. Corbin and M. M. Abu-Omar, J. Am. Chem. Soc., 2005, 127, 11938 CrossRef CAS PubMed.
- (a) T. Mitsudome, S. Arita, H. Mori, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., Int. Ed., 2008, 47, 7938 CrossRef CAS PubMed; (b) N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093 CrossRef CAS PubMed; (c) T. Mitsudome, A. Noujima, T. Mizugaki, K. Jitsukawaa and K. Kaneda, Chem. Commun., 2009, 5302 RSC; (d) M. Jeon, J. Han and J. Park, ACS Catal., 2012, 2, 1539 CrossRef CAS.
- S. Maisonnier, J.-C. Favier, M. Masure and P. Hemery, Polym. Int., 1999, 48, 159 CrossRef CAS.
- (a) F. Gonzaga, R. D'Souza and M. A. Brook, Soft Matter, 2011, 7, 722 RSC; (b) M. F. Dumont, S. Moisan, C. Aymonier, J.-D. Marty and C. Mingotaud, Macromolecules, 2009, 42, 4937 CrossRef CAS; (c) B. P. S. Chauhan, J. S. Rathore, M. Chauhan and A. Krawicz, J. Am. Chem. Soc., 2003, 125, 2876 CrossRef CAS PubMed; (d) R. Zhang, Z. Zhang, K. Amine and R. West, Silicon Chem., 2003, 2, 271 CrossRef; (e) Z. Zhang, L. J. Lyons, J. J. Jin, K. Amine and R. West, Chem. Mater., 2005, 17, 5646 CrossRef CAS; (f) N. A. A. Rossi, E. J. Duplock, J. Meegan, D. R. T. Roberts, J. J. Murphy, M. Patel and S. J. Holder, J. Mater. Chem., 2009, 19, 7674 RSC; (g) P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024 CrossRef CAS PubMed; (h) N. Roy, E. Buhler and J.-M. Lehn, Chem.–Eur. J., 2013, 19, 8814 CrossRef CAS PubMed.
- (a) N. J. Lawrence, M. D. Drew and S. M. Bushell, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 3381 RSC; (b) B. P. S. Chauhan and J. S. Rathore, J. Am. Chem. Soc., 2005, 127, 5790 CrossRef CAS PubMed; (c) J. P. Patel, A.-H. Li, H. Dong, V. L. Korlipara and M. J. Mulvihill, Tetrahedron Lett., 2009, 50, 5975 CrossRef CAS; (d) M. Giordano and A. Iadonisi, Eur. J. Inorg. Chem., 2013, 125 CrossRef CAS; (e) R. J. Rahaim Jr and R. E. Maleczka Jr, Org. Lett., 2011, 13, 584 CrossRef PubMed; (f) B. P. S. Chauhan and R. Sardar, Macromolecules, 2004, 37, 5136 CrossRef CAS.
- (a) S. U. Pickering, J. Chem. Soc., 1907, 91, 2001 RSC; (b) C. N. R. Rao and K. P. Kalyanikutty, Acc. Chem. Res., 2008, 41, 489 CrossRef CAS PubMed; (c) D. Wang, H. Duan and H. Möhwald, Soft Matter, 2005, 1, 412 RSC; (d) H. Duan, D. Wang, D. G. Kurth and H. Möhwald, Angew. Chem., Int. Ed., 2004, 43, 5639 CrossRef CAS PubMed; (e) B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21 CrossRef CAS.
- (a) M. Pera-Titus, L. Leclercq, J. –M. Clacens, F. De Campo and V. Nardello-Rataj, Angew. Chem., Int. Ed., 2014, 53, 2 CrossRef; (b) S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68 CrossRef CAS PubMed.
- (a) C. Strissel, K. Matyjaszewski and O. Nuyken, Macromol. Chem. Phys., 2003, 204, 1169 CrossRef CAS; (b) K. Matyjaszewski, Macromolecules, 2012, 45, 4015 CrossRef CAS.
- D. Khandelwal, S. Hooda and A. S. Brar, J. Mol. Struct., 2011, 991, 24 CrossRef CAS.
- R. A. Orwoll and P. A. Arnold, Physical Properties of Polymers Handbook, ed. J. E. Mark, AIP Press, New York, 1996 Search PubMed.
- I. M. Yu, P. Dubois and R. Jerome, Macromolecules, 1996, 29, 7316 CrossRef.
- (a) A. Sugie, T. Somete, K. Kanie, A. Muramatsu and A. Mori, Chem. Commun., 2008, 3882 RSC; (b) R. Shankar, M. Chaudhary, K. C. Molloy and G. Kociok-Köhn, Dalton Trans., 2013, 42, 7768 RSC.
- M. –C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS PubMed.
- J. Moineau, M. Granier and G. F. Lanneau, Langmuir, 2004, 20, 3202 CrossRef CAS PubMed.
- J. C. Saam, J. Inorg. Organomet. Polym., 1999, 9, 1 CrossRef CAS.
- M. Granier, G. F. Lanneau, J. Moineau, P. Girard and M. Romanda, Langmuir, 2003, 19, 2691 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2003, 125, 8340 CrossRef CAS PubMed.
- A. Marchenko, N. Katsonis, D. Fichou, C. Aubert and M. Malacria, J. Am. Chem. Soc., 2002, 124, 9998 CrossRef CAS PubMed.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen, V. Zaikovski and K. J. Klabunde, J. Am. Chem. Soc., 2003, 125, 10488 CrossRef CAS PubMed.
- R. Shankar, A. Saxena and A. S. Brar, J. Organomet. Chem., 2002, 650, 223 CrossRef CAS.
- R. Muppalla and S. K. Jewrajka, Polymer, 2012, 53, 1453 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25557d |
| This journal is © The Royal Society of Chemistry 2017 |