
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of a fragmented small GTPase capable of conditional effector binding†
Jia Zhao and
Cliff I. Stains *
*
Department of Chemistry, University of Nebraska – Lincoln, Lincoln, NE 68588, USA. E-mail: cstains2@unl.edu
First published on 22nd February 2017
Abstract
A fragmented small GTPase capable of conditional effector binding is described. The effector binding function of this split-GTPase can be modulated using a small molecule input, thus allowing for the potential design of cellular signaling pathways.
Small GTPases represent an important class of signaling molecules, having demonstrated roles in cell morphogenesis, division, and migration.1–3 The function of these proteins, namely effector binding, is dependent upon a GDP-GTP switch that modulates effector binding potential.4,5 Specifically, small GTPases cycle between two forms, the GTP-bound active conformation and the GDP-bound inactive conformation. The active GTP-bound conformation of the protein has a significantly higher binding affinity towards downstream effectors6 leading to diverse phenotypic changes (Fig. 1a). Given the clear involvement of these enzymes in human disease,7–9 there is a critical need for the development of tools to control the interaction of small GTPases with downstream effectors. In the long term, such approaches could be used to control small GTPase-effector binding in living systems in order to further investigate the fundamental role of small GTPase signaling in human disease as well as generate designer signaling networks.
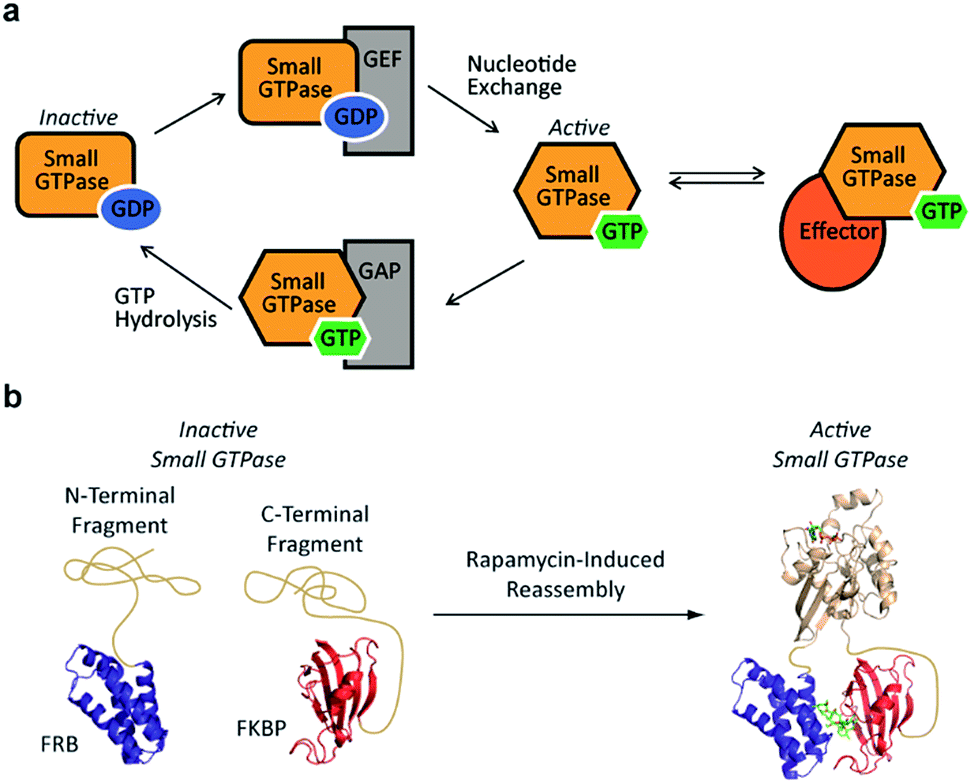 | ||
| Fig. 1 (a) The small GTPase cycle is shown. Small GTPases are active when bound to GTP and inactive when bound to GDP. Guanine nucleotide exchange factors (GEF) enhance nucleotide exchange while GTPase-activating proteins (GAP) increase the intrinsically slow rate of GTP hydrolysis. The GTP-bound active form of the small GTPase can interact with effector proteins to trigger downstream signaling events. (b) A small GTPase (tan) is fragmented into two inactive pieces which are fused to the rapamycin-dependent interaction domains known as the FKBP-rapamycin-binding protein (FRB, blue) and the FK506-binding protein (FKBP, red). Addition of rapamycin causes an increase in the concentration of complementary fragments, leading to reassembly of an active small GTPase. | ||
Despite the challenges associated with the development of small molecule inhibitors for small GTPases,10–14 remarkable progress has been made towards the discovery of strategies for their inhibition.15 Nonetheless, the ability to dissect small GTPase function in living cells with small molecule inhibitors remains limited. This has prompted the development of elegant protein engineering strategies to control small GTPase function.16,17 These approaches allow for the regulation of small GTPase activity using small molecules or light as inputs. While clearly powerful, more work is needed in order to identify techniques that could potentially allow for the orthogonal control of multiple GTPases. With this goal in mind, we hypothesized that split-protein reassembly, the concentration induced folding of a fragmented protein,18–20 could be applied to control small GTPase function (Fig. 1b). In the long term, appropriate fragmentation sites could potentially be applied across the 150 member Ras superfamily of small GTPases,3 resulting in a lexicon of split-GTPase modules that could be employed with orthogonal, conditional interaction domains.21–28 Herein, we report the first advance towards this goal. Using the well-characterized small GTPase known as Cdc42,3,29,30 we identify fragmentation sites that yield significantly reduced effector binding activity. When bought into close proximity using a chemical inducer of dimerization, the effector binding capacity of these fragments is restored. These split-Cdc42 fragments provide a proof-of-principle for the potential control of small GTPase signaling using small molecule inputs.
Previous studies indicate that permissive loop regions within protein structures can serve as sites of fragmentation, acting in an analogues manner to loop insertion sites.18,31,32 However, since identification of these permissive loops remains challenging, a functional screen was employed to identify permissive sites from a library of small GTPase fragments. In order to simplify screening we chose to focus on a constitutively active mutant of Cdc42, termed L61Cdc42 (Table S1†),33,34 which remains in the GTP-bound state due to defective GTP hydrolysis. To identify split-Cdc42 fragments we cloned a library of potentially complementary Cdc42 pairs by dissecting the protein at every loop (yielding 14 fragment combinations, Fig. 2 and Table S2†). The resulting N- and C-terminal Cdc42 fragments were fused at their native termini to the rapamycin-dependent interaction domains known as FKBP and FRB.35 Flexible linkers were employed at the fusion sites to allow for sampling of multiple conformations (Table S2†). This experimental setup allows for the identification of rapamycin-dependent Cdc42 reassembly versus Cdc42 fragments capable of spontaneous reassembly.
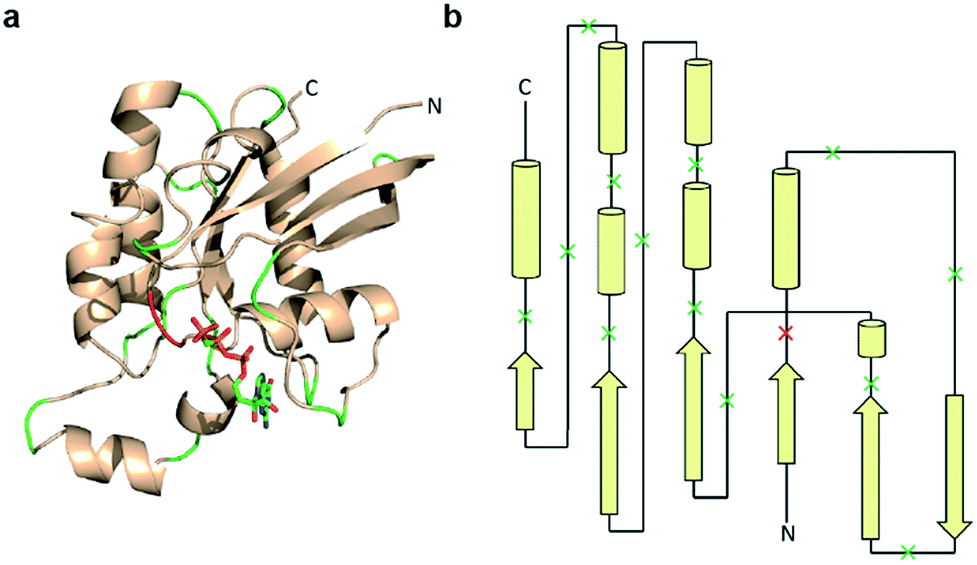 | ||
| Fig. 2 Design of a Cdc42 fragment library. (a) The structure of Cdc42 (PDB code: 1CEE) with fragmentation sites indicated in green and red. This library encompasses every loop within Cdc42. The red loop denotes a fragmentation site that yielded a positive hit during subsequent screening. (b) A two-dimensional representation of the structure shown in panel a. Arrows are β-strands, cylinders are α-helices, and lines are loops. Dissection sites are indicated by an “x”, with the positive hit highlighted in red. | ||
To identify Cdc42 fragments capable of reassembly we employed a rapid assay format in which proteins are translated from mRNA using a rabbit reticulocyte in vitro translation system.36 Performing these translation reactions in the presence of 35S-methionine introduces a sensitive label into synthesized protein. Subsequent affinity purification of active Cdc42 fragments based on effector binding activity37 allows for identification of fragments displaying rapamycin-dependent effector binding (Fig. 3a). As a biologically relevant ligand for reassembled Cdc42, we chose the Cdc42 binding domain (PBD) of the human p21-activated kinase 1 protein (PAK), which binds tightly (KD = 30 nM) and specifically to GTP-bound, activated Cdc42.6,38,39 To validate this pull-down assay, we generated lysates containing wild-type Cdc42 or the L61Cdc42 mutant in BL21-CodonPlus (DE3)-RIPL E. coli cells. Utilizing nucleotide exchange, GDP-bound wild-type Cdc42 (negative control) and GTP-γS-bound wild-type Cdc42 were both prepared. Together with the constitutively active L61Cdc42 mutant, these three samples were assayed for binding to immobilized PBD with subsequent detection via western blotting (Fig. S1†). These experiments confirmed that the PBD-based pull-down assay could be used to differentiate between active and inactive forms of Cdc42 in a complex mixture.
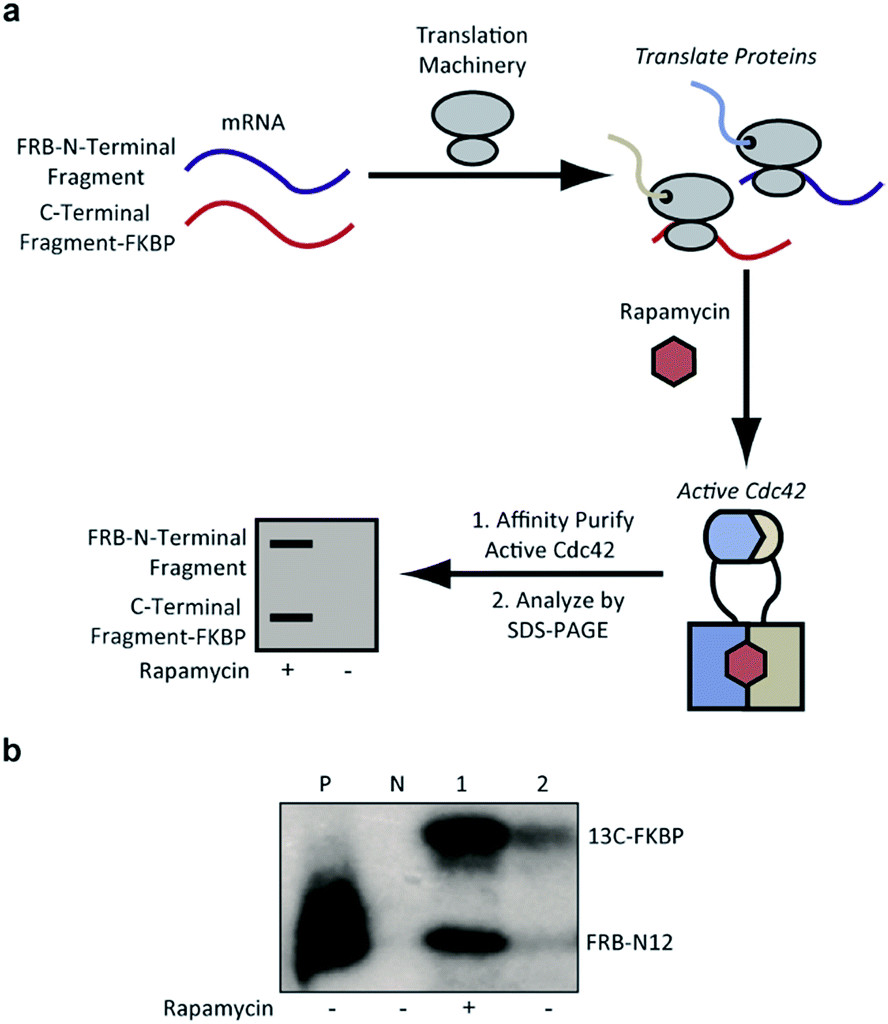 | ||
| Fig. 3 (a) Identification of active Cdc42 using a PBD-based pull-down assay. In vitro translation in the presence or absence of rapamycin, followed by pull-down assays with PBD can be used to identify split-Cdc42 fragments capable of rapamycin-induced effector binding. (b) Cotranslation of FRB-N12 and 13C-FKBP constructs (Table S2†) in rabbit reticulocyte lysates in the presence (+) or in the absence (−) of rapamycin. Active Cdc42 is detected via a PBD pull-down assay, as shown in panel a, and visualized using autoradiography. Enhanced effector binding is observed in the presence of rapamycin. Constitutively active L61Cdc42 is used as positive control (P) and the dominant negative N17Cdc42 mutant (N) is used as negative control. | ||
Confident in the ability to screen for active Cdc42, we proceeded to interrogate the Cdc42 fragment library for rapamycin-dependent effector binding. mRNA transcripts corresponding to Cdc42 fragments were translated in the presence or absence of rapamycin in rabbit reticulocyte lysates and were labeled with 35S-methionine. Reassembled Cdc42 was identified using the PBD-based pull-down assay described above (Fig. 3a). We also expressed full-length L61Cdc42 as a positive control as well as a constitutively inactive mutant, termed N17Cdc42,40 as a negative control. These experiments identified one complementary pair, termed FRB-N12 (consisting of residues 1–12 of Cdc42) and 13C-FKBP (consisting of residue 13–190 of Cdc42), that showed a significantly stronger binding to immobilized PBD in the presence of rapamycin compared to translations conducted in the absence of rapamycin (Fig. 3b, S2 and Table S2†). These differences in PBD affinity were reproducible (Fig. S3†) and indicate that reassembly of this pair is promoted by an increase in local concentration. These experiments also consistently indicate an excess of the 13C fragment bound to immobilized PBD. The origin of this excess is currently not known, but may be attributable to oligomerization of the 13C fragment. Mapping these sites back to the structure of active Cdc42 indicated that fragmentation at this site likely interferes with productive GTP binding and subsequent effector engagement through disruption of the P-loop, which is involved in phosphate binding (Fig. 4).41 Thus the increase in effector binding upon reassembly may be explained by restoration of the GTP binding site. Relatively weak binding of fragments to immobilized PDB in the absence of rapamycin may be attributed to potential self-assembly. Nonetheless, addition of rapamycin significantly increases the yield of small GTPase capable of binding to PBD (Fig. 3b and S3†). Future experiments will be aimed at decreasing binding of these fragments to PBD in the absence of rapamycin and improving reassembly efficiency in the presence of rapamycin.
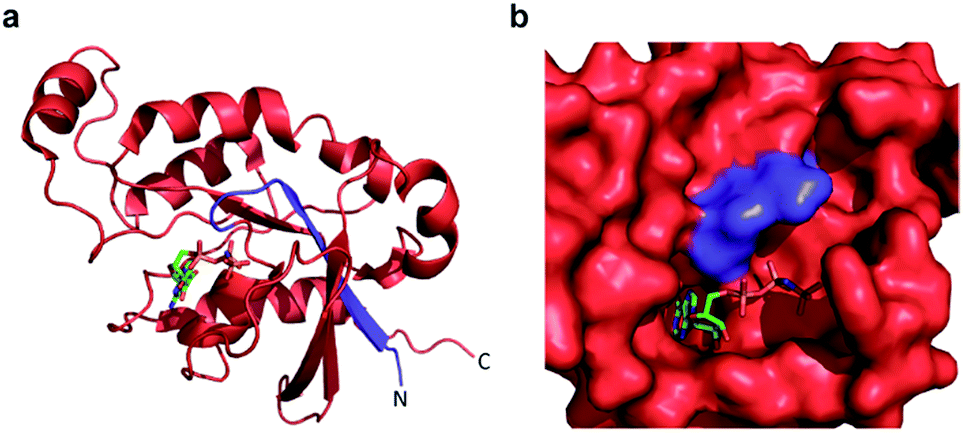 | ||
| Fig. 4 (a) A crystal structure of active Cdc42 is shown (PDB code 1E0A). The N12 fragment is indicated in blue, and the 13C fragment is shown in red. The GTP analogue, GNP, is depicted in stick representation. (b) The GTP binding pocket from the structure in panel a, rotated by 90° to the right. The N12/13C fragmentation site lies within the P-loop, likely disrupting GTP binding prior to reassembly. | ||
Conclusions
We present a proof-of-principle for generating a fragmented small GTPase that displays increased effector binding in response to a small molecule input. This work represents an important step towards the development of reagents capable of bypassing the native regulation of small GTPase activity (Fig. 1). Future experiments will be aimed at investigating the ability of these fragments to bind diverse cellular effectors with the ultimate goal of controlling cell motility in living cells. For example, one could imagine the use of previously described small molecule-dependent supramolecular assembly systems in order to control small GTPase function in living cells.21–25,27,28 Moreover, due to the structural homology of the small GTPase family, it may be possible to identify additional fragmented small GTPases,31,32 affording a lexicon of split-small GTPase modules that could be controlled by orthogonal inputs.Acknowledgements
We acknowledge Prof. Edward Harris for use of laboratory space and equipment for studies involving radioactive compounds. pET23-PBD(65-150)-N-Cys was a gift from Klaus Hahn (Addgene plasmid #13722). This work was supported by the University of Nebraska – Lincoln and the NIH (R35GM119751). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.Notes and references
- H. R. Bourne, D. A. Sanders and F. McCormick, Nature, 1990, 348, 125–132 CrossRef CAS PubMed
.
- A. M. Rojas, G. Fuentes, A. Rausell and A. Valencia, J. Cell Biol., 2012, 196, 189–201 CrossRef CAS PubMed
- K. Wennerberg, K. L. Rossman and C. J. Der, J. Cell Sci., 2005, 118, 843–846 CrossRef CAS PubMed
- J. Cherfils and M. Zeghouf, Nat. Chem. Biol., 2011, 7, 493–495 CrossRef CAS PubMed
- I. R. Vetter and A. Wittinghofer, Science, 2001, 294, 1299–1304 CrossRef CAS PubMed
- G. Thompson, D. Owen, P. A. Chalk and P. N. Lowe, Biochemistry, 1998, 37, 7885–7891 CrossRef CAS PubMed
- J. Downward, Nat. Rev. Cancer, 2003, 3, 11–22 CrossRef CAS PubMed
- K. F. Wilson, J. W. Erickson, M. A. Antonyak and R. A. Cerione, Trends Mol. Med., 2013, 19, 74–82 CrossRef CAS PubMed
- B. Boettner and L. Van Aelst, Gene, 2002, 286, 155–174 CrossRef CAS PubMed
- P. M. Cromm, J. Spiegel, T. N. Grossmann and H. Waldmann, Angew. Chem., Int. Ed. Engl., 2015, 54, 13516–13537 CrossRef CAS PubMed
- J. Spiegel, P. M. Cromm, G. Zimmermann, T. N. Grossmann and H. Waldmann, Nat. Chem. Biol., 2014, 10, 613–622 CrossRef CAS PubMed
- A. D. Cox, S. W. Fesik, A. C. Kimmelman, J. Luo and C. J. Der, Nat. Rev. Drug Discovery, 2014, 13, 828–851 CrossRef CAS PubMed
- A. G. Stephen, D. Esposito, R. K. Bagni and F. McCormick, Cancer Cell, 2014, 25, 272–281 CrossRef CAS PubMed
- J. M. Ostrem and K. M. Shokat, Nat. Rev. Drug Discovery, 2016, 15, 771–785 CrossRef CAS PubMed
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed
- I. Goreshnik and D. J. Maly, J. Am. Chem. Soc., 2010, 132, 938–940 CrossRef CAS PubMed
- Y. I. Wu, D. Frey, O. I. Lungu, A. Jaehrig, I. Schlichting, B. Kuhlman and K. M. Hahn, Nature, 2009, 461, 104–110 CrossRef CAS PubMed
- I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2000, 122, 5658–5659 CrossRef CAS
- T. J. Magliery, C. G. Wilson, W. Pan, D. Mishler, I. Ghosh, A. D. Hamilton and L. Regan, J. Am. Chem. Soc., 2005, 127, 146–157 CrossRef CAS PubMed
- S. W. Michnick, Curr. Opin. Struct. Biol., 2001, 11, 472–477 CrossRef CAS PubMed
- B. Xu, X. Q. Zhou and C. I. Stains, RSC Adv., 2016, 6, 20381–20385 RSC
- B. Xu, X. Q. Zhou and C. I. Stains, J. Am. Chem. Soc., 2015, 137, 14252–14255 CrossRef CAS PubMed
- F. S. Liang, W. Q. Ho and G. R. Crabtree, Sci. Signaling, 2011, 4, rs2 CrossRef PubMed
- T. Miyamoto, R. DeRose, A. Suarez, T. Ueno, M. Chen, T. P. Sun, M. J. Wolfgang, C. Mukherjee, D. J. Meyers and T. Inoue, Nat. Chem. Biol., 2012, 8, 465–470 CrossRef CAS PubMed
- D. M. Spencer, T. J. Wandless, S. L. Schreiber and G. R. Crabtree, Science, 1993, 262, 1019–1024 CAS
- M. Putyrski and C. Schultz, FEBS Lett., 2012, 586, 2097–2105 CrossRef CAS PubMed
- H. D. Nguyen, D. T. Dang, J. L. van Dongen and L. Brunsveld, Angew. Chem., Int. Ed. Engl., 2010, 49, 895–898 CrossRef CAS PubMed
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef CAS PubMed
- A. Morreale, M. Venkatesan, H. R. Mott, D. Owen, D. Nietlispach, P. N. Lowe and E. D. Laue, Nat. Struct. Biol., 2000, 7, 384–388 CrossRef CAS PubMed
- M. J. Phillips, G. Calero, B. Chan, S. Ramachandran and R. A. Cerione, J. Biol. Chem., 2008, 283, 14153–14164 CrossRef CAS PubMed
- K. Camacho-Soto, J. Castillo-Montoya, B. Tye and I. Ghosh, J. Am. Chem. Soc., 2014, 136, 3995–4002 CrossRef CAS PubMed
- K. Camacho-Soto, J. Castillo-Montoya, B. Tye, L. O. Ogunleye and I. Ghosh, J. Am. Chem. Soc., 2014, 136, 17078–17086 CrossRef CAS PubMed
- P. Aspenstrom, U. Lindberg and A. Hall, Curr. Biol., 1996, 6, 70–75 CrossRef CAS PubMed
- D. R. Lowy and B. M. Willumsen, Annu. Rev. Biochem., 1993, 62, 851–891 CrossRef CAS PubMed
- J. Chen, X. F. Zheng, E. J. Brown and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 4947–4951 CrossRef CAS
- J. R. Porter, C. I. Stains, B. W. Jester and I. Ghosh, J. Am. Chem. Soc., 2008, 130, 6488–6497 CrossRef CAS PubMed
- V. Benard and G. M. Bokoch, Methods Enzymol., 2002, 345, 349–359 Search PubMed
- D. A. Leonard, R. S. Satoskar, W. J. Wu, S. Bagrodia, R. A. Cerione and D. Manor, Biochemistry, 1997, 36, 1173–1180 CrossRef CAS PubMed
- V. S. Kraynov, C. Chamberlain, G. M. Bokoch, M. A. Schwartz, S. Slabaugh and K. M. Hahn, Science, 2000, 290, 333–337 CrossRef CAS PubMed
- C. D. Nobes and A. Hall, J. Cell Biol., 1999, 144, 1235–1244 CrossRef CAS PubMed
- A. Wittinghofer and I. R. Vetter, Annu. Rev. Biochem., 2011, 80, 943–971 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures, protein sequences, and experimental details. See DOI: 10.1039/c6ra25575b |
| This journal is © The Royal Society of Chemistry 2017 |