
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
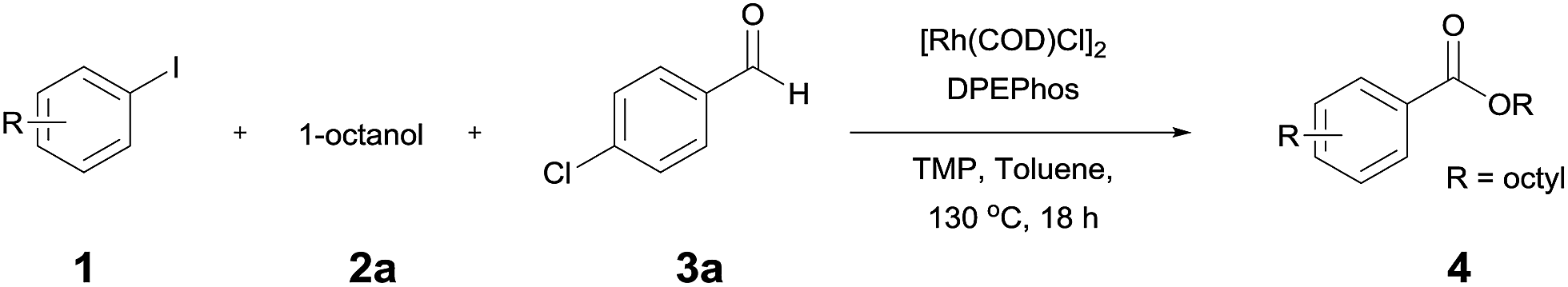
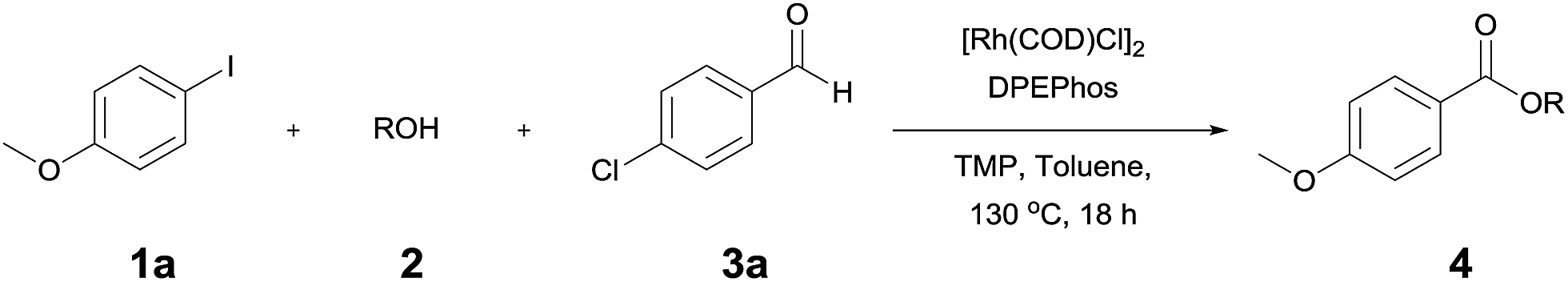
Rhodium-catalyzed synthesis of esters from aryl iodides and alcohols: use of alcohols with/without the assistance of aldehydes as carbon monoxide and nucleophile sources†
Ju Hyun Kim,
Hawon Park and
Young Keun Chung *
*
Department of Chemistry, College of Natural Sciences, Seoul National University, Seoul 08826, Korea. E-mail: ykchung@snu.ac.kr; Fax: +82-2-889-0310; Tel: +82-2-880-6662
First published on 22nd December 2016
Abstract
A CO-gas-free rhodium-catalyzed alkoxycarbonylation of aryl iodide with alcohols has been developed. Alcohols, with/without the aid of an aldehyde, were used as a carbon monoxide and nucleophile source. The former synthesis afforded better yields of the alkoxycarbonylated products. Moreover, phenols also afforded phenoxycarbonylation products with high yields.
Since the first report by Heck et al. on the palladium-catalyzed reaction of carbon monoxide with aryl halides and alcohols or amines,1 transition metal catalyzed cross-coupling reactions have been developed as a valuable method for creating carbon–carbon bonds.2 In particular, rhodium-catalyzed carbonylation with a variety of substrates has been studied.3 These reactions require the addition of CO to the substrate, and are typically carried out with carbon monoxide gas. However, the use of carbon monoxide is not recommended due to its toxicity and difficulty in handling. Thus, the use of CO-releasing reagents, such as formic acid derivatives,4 oxiranes,5 and aldehydes6 has been studied. Recently, Jun et al. reported7 the Pd/C-catalyzed carbonylative esterification of a primary alcohol with aryl chlorides in the presence of NaF. Previously, we reported the use of an alcohol as a CO source in the rhodium-catalyzed Pauson–Khand reaction of enynes8 and as a source of hydrogen in the Rh-catalyzed reductive cyclization of enynes.9 Thus, in the presence of a rhodium catalyst, an alcohol can be used as a CO or hydrogen source as well as a reaction medium. We therefore decided to explore the use of an alcohol as a CO and nucleophile source, in a reaction with aryl iodides, during the synthesis of esters (Scheme 1).
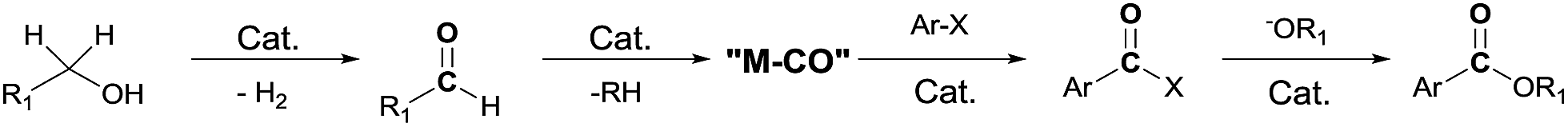 | ||
| Scheme 1 Proposed catalytic process in esterification. | ||
We studied the alkoxycarbonylation of an aryl iodide with an alcohol in the presence of a base and a rhodium catalyst. This led to the discovery of a rhodium-catalyzed, CO-gas-free alkoxycarbonylation reaction of aryl iodide. The reaction occurred in the presence of an alcohol with/without an aldehyde. The alcohol acted as both a CO and nucleophile source to afford an alkoxycarbonylated product. We herein report our preliminary results.
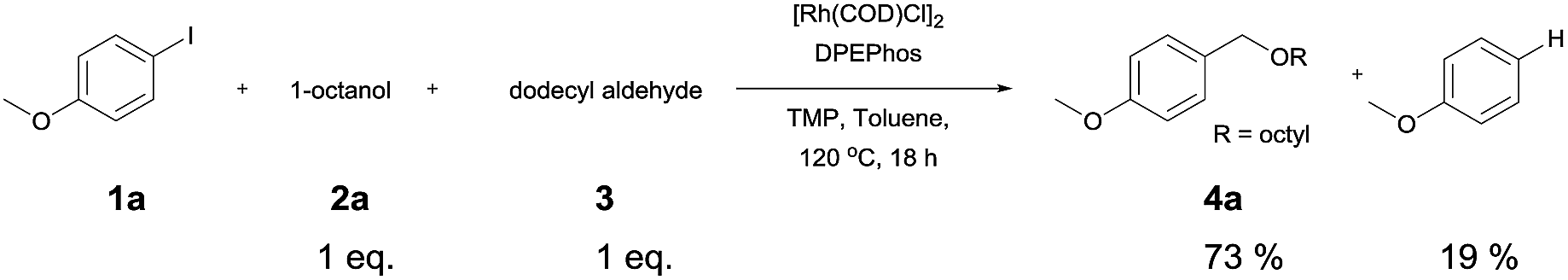
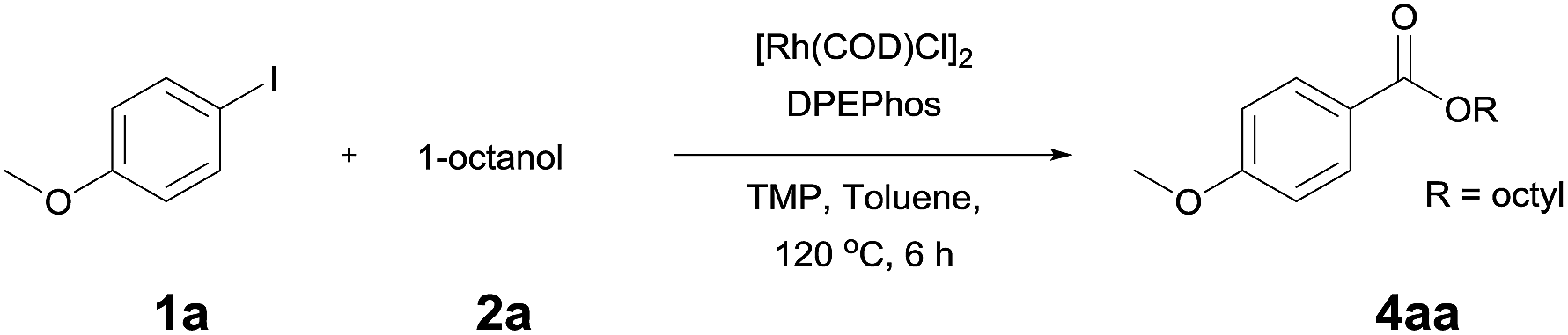
The reaction of 4-iodoansiole (1a) with 1-octanol (2a) in the presence of [Rh(COD)Cl]2 (2 mol%) and DPEPhos [DPEPhos = (oxydi-2,1-phenylene)bis(diphenylphosphine)] (4 mol%) at 70 °C was chosen as the synthetic model. This afforded the alkoxycarbonylation product, octyl 4-methoxybenzoate (3aa).
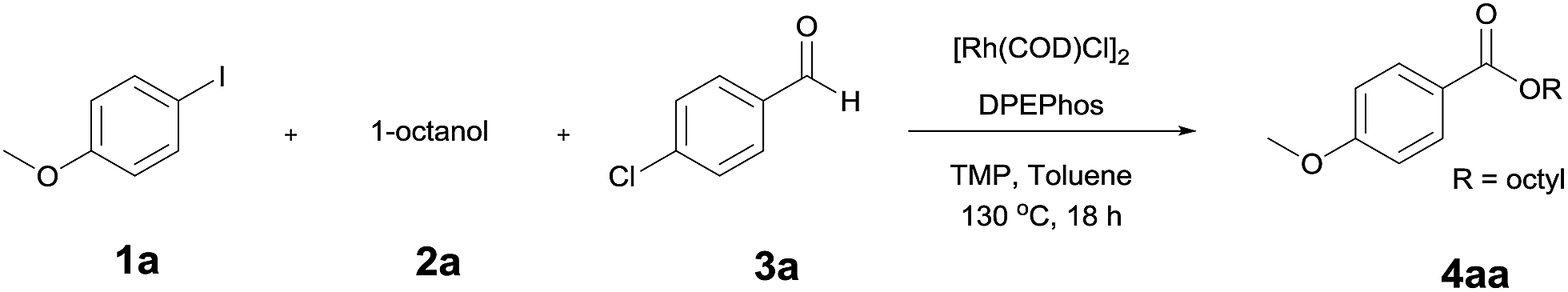
A yield of 50% was achieved with TMP (2,2,6,6-tetramethylpiperidine) and DPEPhos as the base and ligand, respectively (reaction conditions: [Rh(COD)Cl]2 catalyst in toluene at 120 °C for 6 h). We next screened different bases and ligands (Table 1 and ESI†). The use of DIPEA (N,N-diisopropylethylamine) or xantphos (4,5-bis(diphenylphosphino)-9,9-dimethylxanthene) did not aid the yield of the reaction (entries 2 and 3). In addition, dehalogenated anisole was observed as a by-product in various yields depending on the reaction conditions. We attributed the formation of anisole to the presence of an Rh-hydride intermediate (ESI or Fig. 1†). Thus, a variety of hydrogen acceptor and oxidant were screened (entry 4 and see ESI†). However, no noticeable enhancement in yield was observed. Other rhodium precatalysts, such as RhCl3 and Rh(OAc)2, had no effect on the product yield (entry 5 and ESI†). The addition of an aldehyde enhanced the reaction, resulting in a large increase in yield (entry 6). This was highly dependent upon the aldehyde used (ESI†). The compound 4-chlorobenzaldehyde afforded the best results. For example, 1 equiv. 4-chlorobenzaldehyde and 1 equiv. 1-octanol afforded the ester and the anisole products in 90% and 6% yields, respectively (Table 1, entry 6). In the case of the dodecyl aldehyde, the ester and anisole were isolated in 73% and 19% yields, respectively (eqn (1)). We therefore concluded that the addition of an aldehyde aided the formation of rhodium-carbonyl intermediate, and increased the rate of the reaction between the intermediate and the alkoxide to afford the ester. No product was formed in the absence of the alcohol or the rhodium catalyst (entries 7 and 8).
 | (1) |
|
||
|---|---|---|
| Entry | Variation from initial conditions | Yieldb |
| a Reaction conditions: 0.5 mmol of 1a, 2 mol% of [Rh(COD)Cl]2, 4 mol% of DPEPhos, 1 equiv. of TMP (tetramethylpiperidine), 3 equiv. of 2a and 1.5 mL of toluene.b GC yield using 1,3,5-trimethylbenzene as an internal standard. | ||
| 1 | None | 50 |
| 2 | DIPEA instead of TMP | 3 |
| 3 | Xantphos instead of DPEPhos | 13 |
| 4 | With benzoquinone | Trace |
| 5 | RhCl3 instead of [Rh(COD)Cl]2 | 40 |
| 6 | With p-chlorobenzaldehyde | 90 |
| 7 | Without [Rh(COD)Cl]2 | Trace |
| 8 | Without alcohol | N.R. |
Initially, we expected the aldehyde to act as a carbon monoxide surrogate. To examine the role of the aldehyde in the alkoxycarbonylation of the aryl iodide more closely, we studied the relation between the yield of the reaction and the ratio of octanol to 4-chlorobenzaldehyde (Table 2).
|
|||
|---|---|---|---|
| Entry | Alcohol (x eq.) | Aldehyde (y eq.) | Yieldb |
| a Reaction conditions: 0.5 mmol of 1a, 2 mol% of [Rh(COD)Cl]2, 4 mol% of DPEPhos, 1 equiv. of TMP, x equiv. of 2a, y equiv. of 3a and 1.5 mL of toluene.b Isolated yield. | |||
| 1 | 1 | 0 | 28 |
| 2 | 2 | 0 | 44 |
| 3 | 3 | 0 | 44 |
| 4 | 0 | 1 | N.R. |
| 5 | 1 | 0.7 | 56 |
| 6 | 1 | 1 | 76 |
| 7 | 1 | 1.3 | 72 |
| 8 | 1.3 | 1.3 | 82 |
| 9 | 2 | 2 | 80 |
In the presence of 1 equiv. octanol, the expected ester was isolated in 28% yield (entry 1). As the amount of octanol was increased to 2 equiv., the yield also increased to 44% (entry 2). Following this, an additional increase in octanol did not lead to a further increase in yield (entry 3). As expected, no reaction was observed in the absence of octanol (entry 4). When 1.0 equiv. octanol and 0.7 equiv. 4-chlorobenzaldehyde were used, the yield increased to 56% (entry 5). When equal amounts (1.0 equiv.) of octanol and 4-chlorobenzaldehyde were employed, the yield increased to 76% (entry 6). A further increase in aldehyde to 1.2 equiv., with 1.0 equiv. octanol, led to a slight decrease in yield (entry 7, 72%). The highest yield (82%) was observed when 1.3 equiv. of both octanol and 4-chlorobenzaldehyde were used (entry 8). When 2.0 equiv. of both octanol and 4-chlorobenzaldehyde were employed, the yield decreased slightly to 80% (entry 9). This observation suggests that the presence of an aldehyde aids the alkoxycarbonation reaction of the aryl iodide in the presence of an alcohol. From these results, the optimum conditions were established as follows: 0.5 mmol of aryl iodide, 2 mol% of [Rh(cod)Cl]2, 4 mol% of DPEPhos, 1.3 equiv. 4-chlorobenzaldehyde, 1.3 equiv. octanol, 1 equiv. TMP, and 1.5 mL of toluene, at 130 °C, for 18 h.
With the optimum reaction conditions in hands, the substrate scope was studied next (Table 3).
|
|
|---|---|
| a Reaction conditions: 0.5 mmol of 1, 2 mol% of [Rh(COD)Cl]2, 4 mol% of DPEPhos, 1 equiv. of TMP, 1.3 equiv. of 2a, 1.3 equiv. of 3a and 1.5 mL of toluene.b Isolated yield.c p-Tolualdehyde used instead of p-chlorobenzaldehyde.d Some octyl ester impurities.e dppp (1,3-bis(diphenylphosphino)propane) as a ligand. | |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
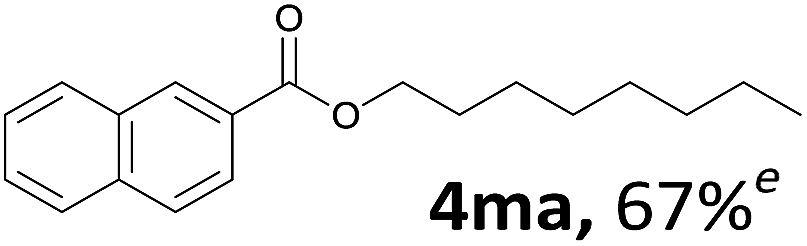 |
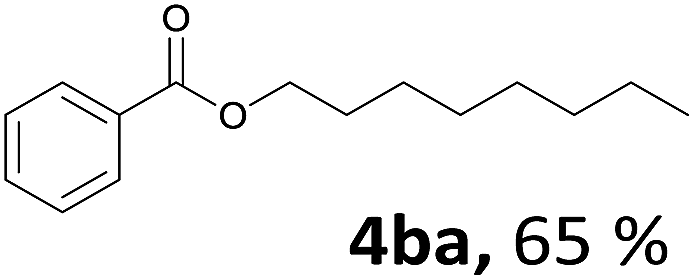
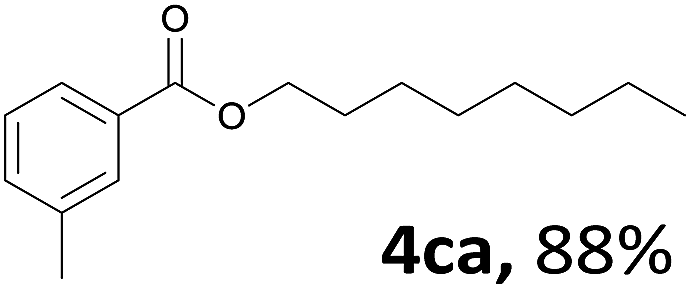
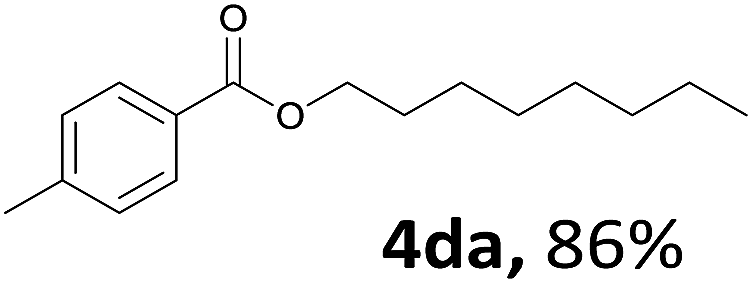
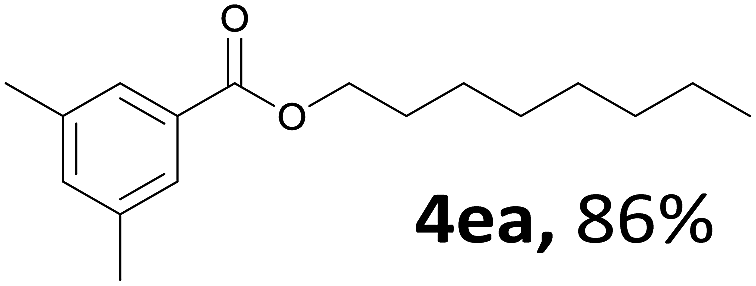
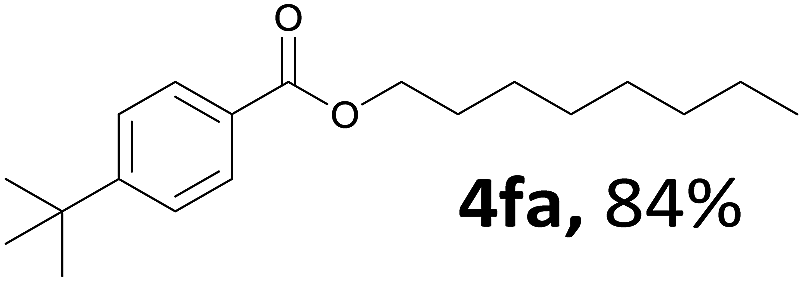
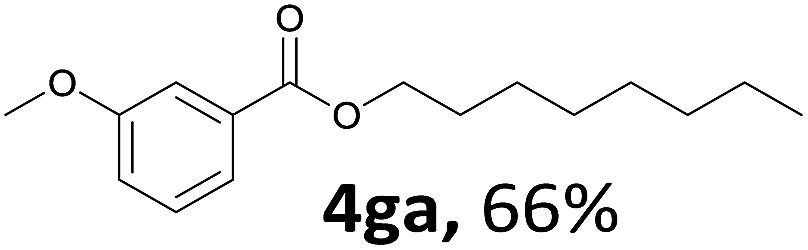
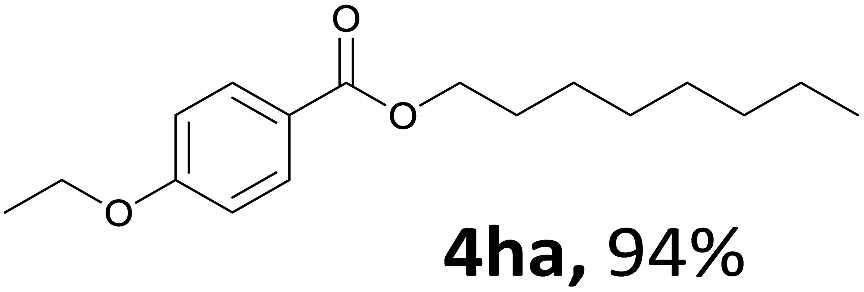
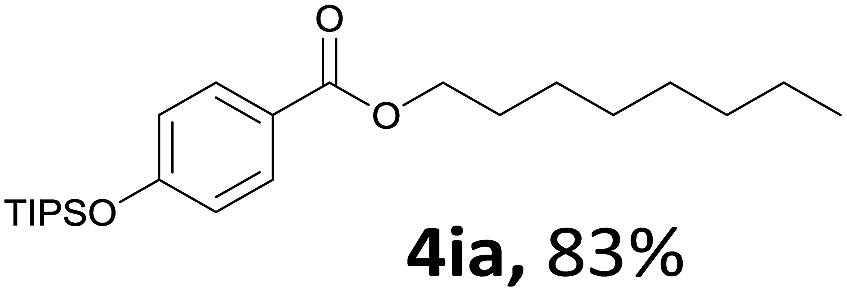
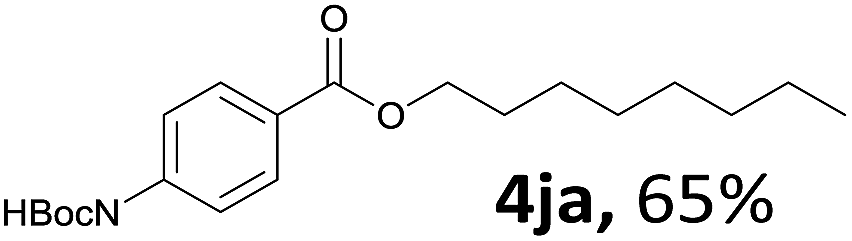
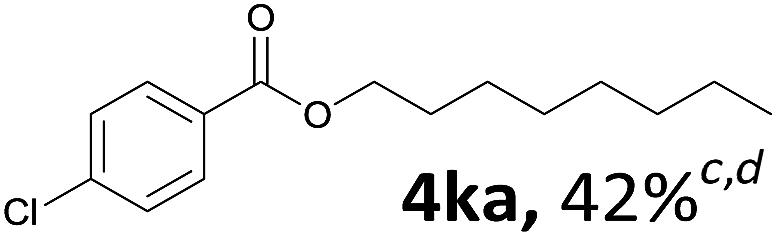
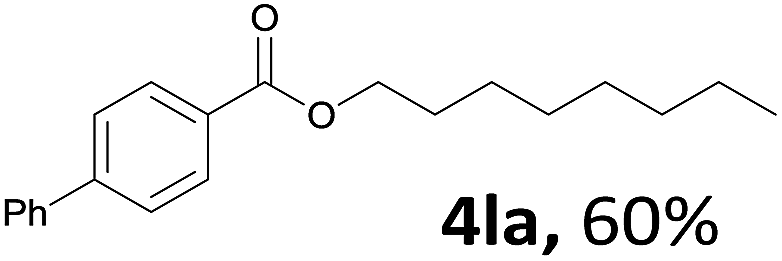
Aryl halides with an electron-donating group were found to be good substrates (4ba–4ha). The highest yield (94%) was observed for the substrate with an ether group. In the cases of substrates having an electron-accepting group (4ka and 4la), the yields were rather low, and in some cases no reaction was observed. An aryl halide bearing a chloro group afforded the corresponding product in <50% (4ka). Moreover, <60% of the expected product was isolated when 4-iodo-1,1′-biphenyl was used (4la). No reaction was observed for aryl iodides having an ester or formyl group. Aryl iodides bearing a hydroxy or amine group did not afford expected product, presumably due to a reaction the rhodium catalyst. However, when the functional groups were blocked, the expected products were isolated in reasonable yields (4ia and 4ja). Steric effects were observed in some cases; when a methyl group was located at the meta- or para-position, the corresponding ester was isolated in high yields (4ca and 4da). However, in the case of 1-iodo-2-methylbenzene, no reaction was observed. Similarly, 1-iodonaphthalene did not afford any products in the presence of the ligand DPEPhos. However, when this ligand was substituted with dppp, 67% of the expected product was isolated (4ma).
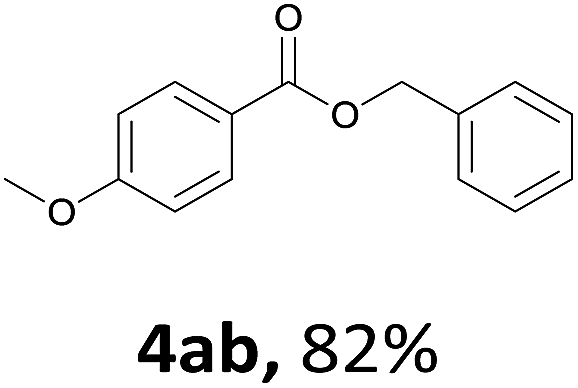
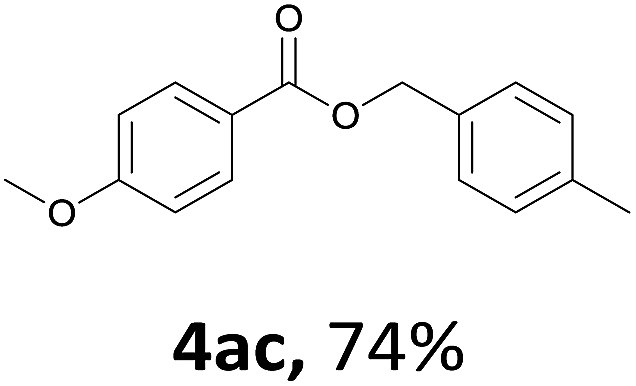
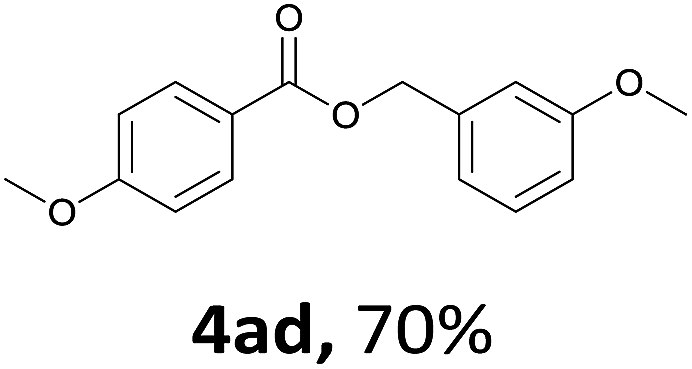
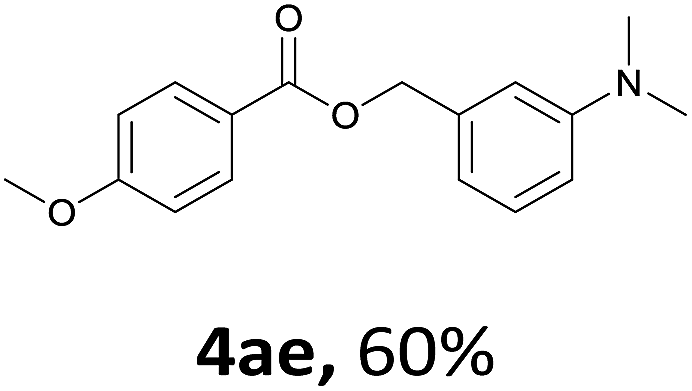
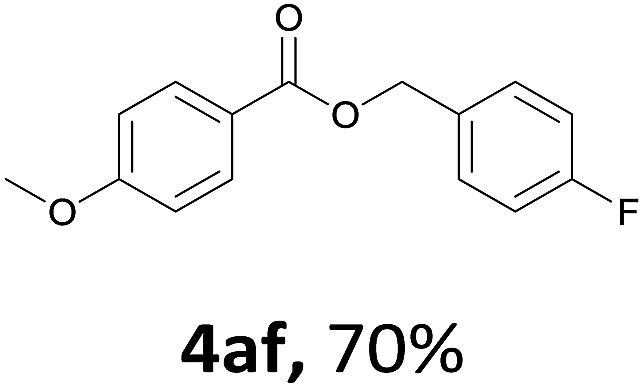
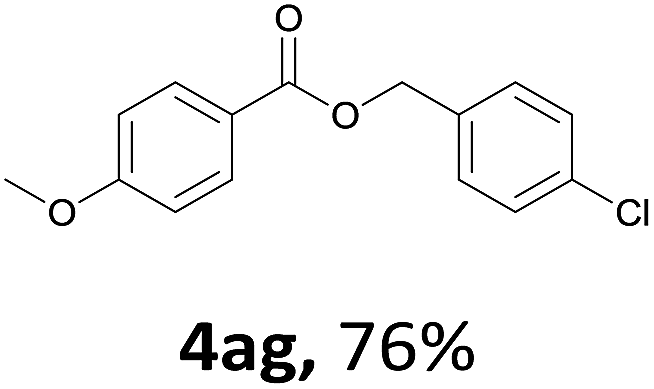
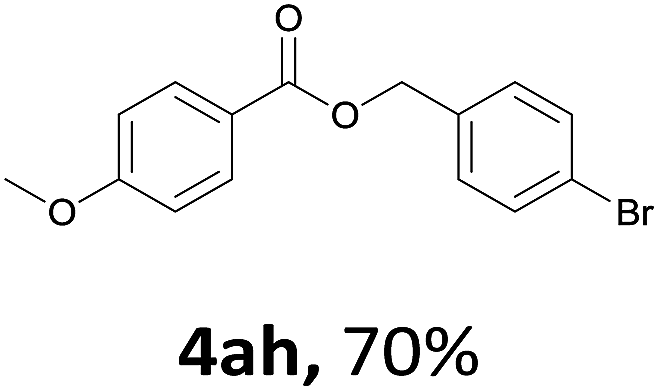
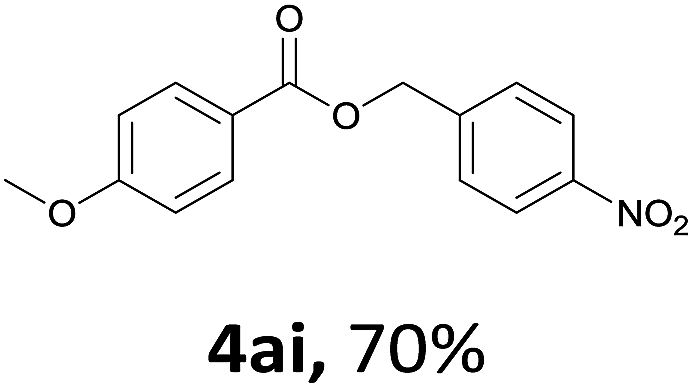
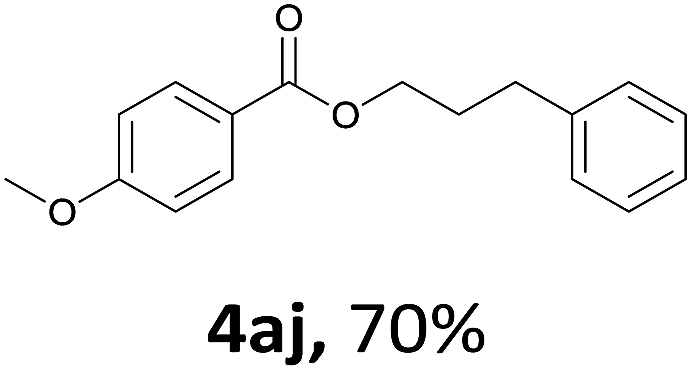
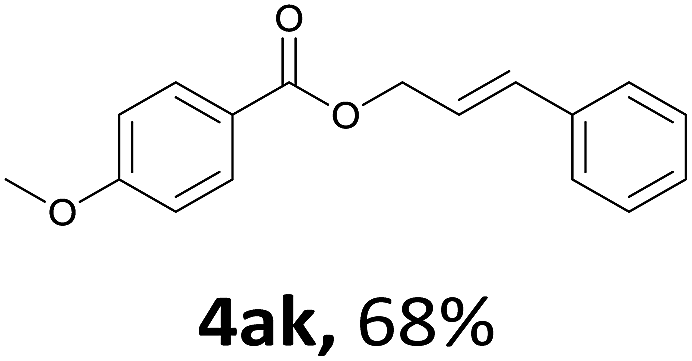
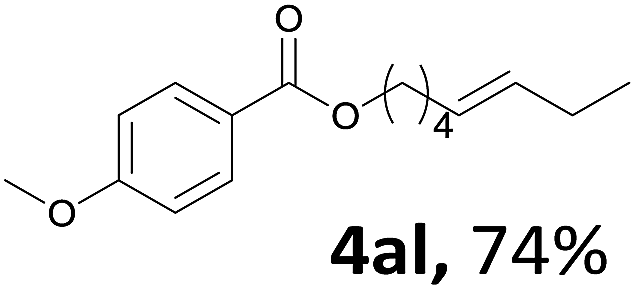
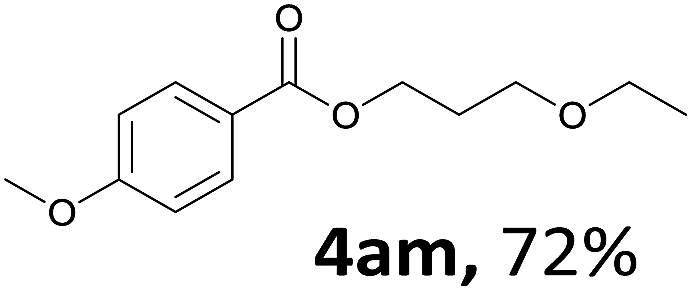
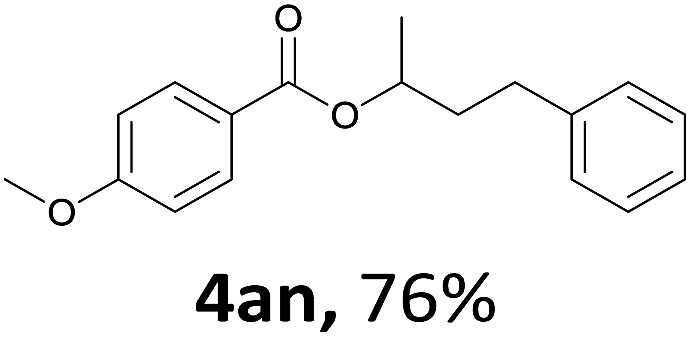
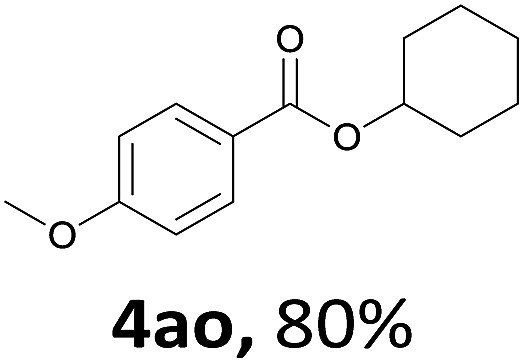
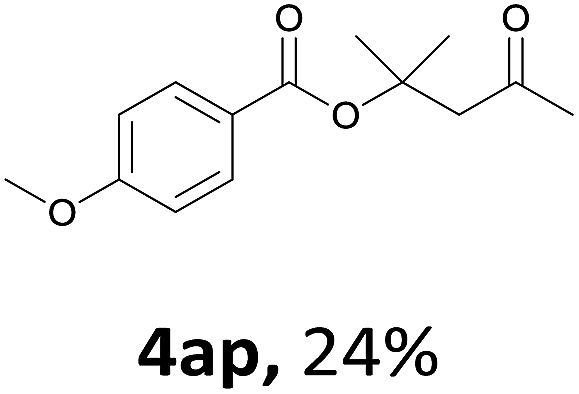
Next, we examined the reaction of the aryl iodide with other alcohols (Table 4). Primary alcohols were studied first. Benzyl alcohols bearing electron-donating (Me, OMe, NMe2) or electron-withdrawing (F, Cl, Br, NO2) functional groups were found to be good substrates (4ab–4ai), and high yields (60–82%) were observed. Lengthening the chain between the phenyl and hydroxyl groups to three carbons did not have any influence on the yield (4aj). An internal double bond survived during the reaction (4ak and 4al). When (E)-3-phenylpropanol or (E)-oct-5-en-1-ol were used as an alcohol, the expected product was isolated in 68% and 74% yields, respectively. In the case of (E)-oct-5-en-1-ol, the product was a mixture of regioisomers. Unfortunately, these could not be separated by conventional separation methods. The ether group retained its position during the reaction (4am). For secondary alcohols, including 4-phenylbutanol and cyclohexanol, high yields (76% and 80%, respectively) were observed (4an and 4ao, respectively). However, the tertiary alcohol 4-hydroxy-4-methylpentan-2-one afforded a poor yield (24%) (4ap).
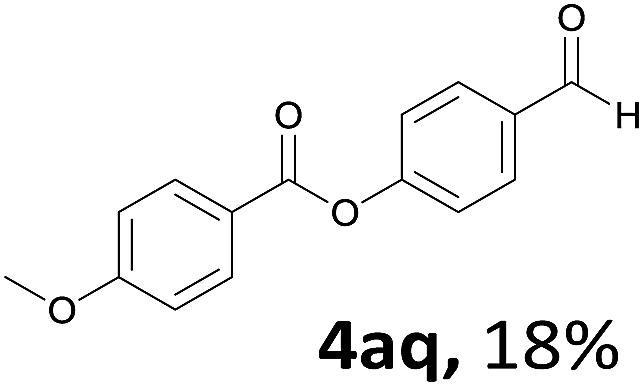
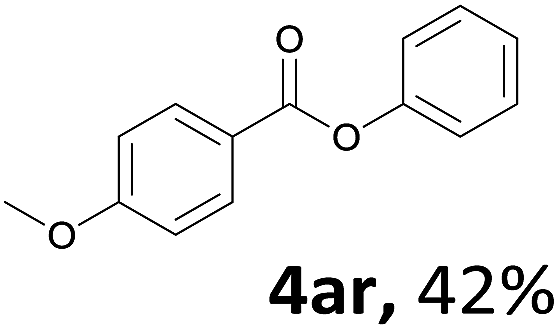
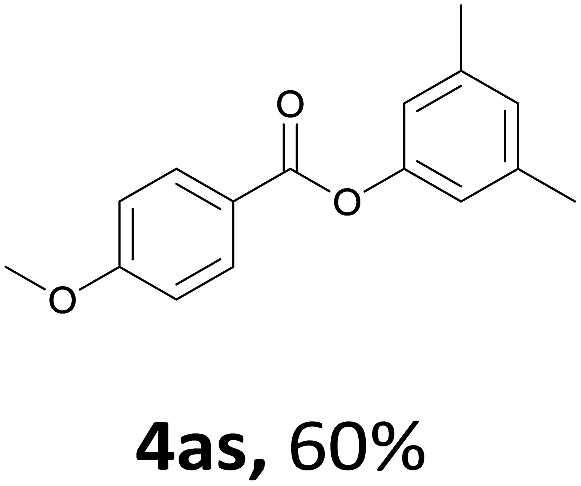
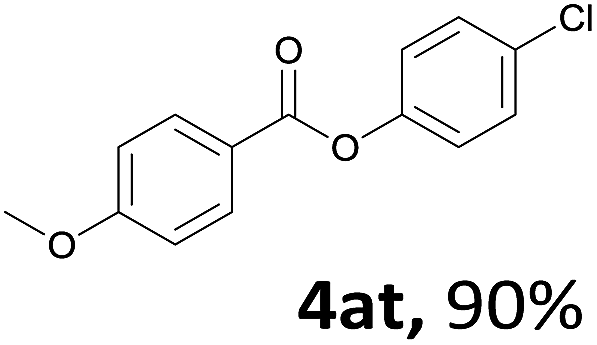
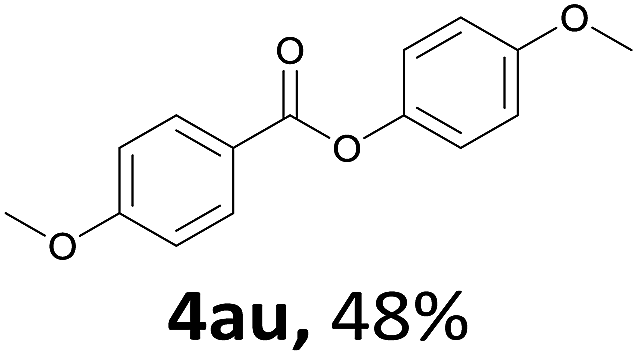
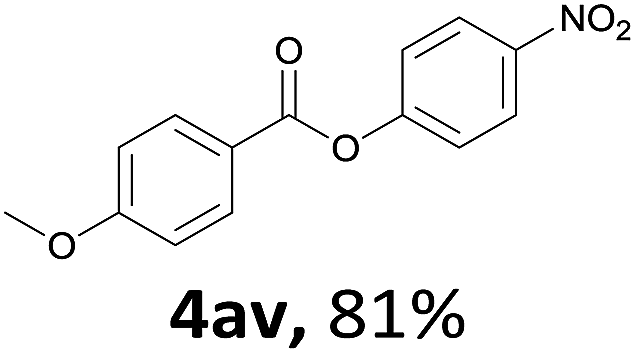
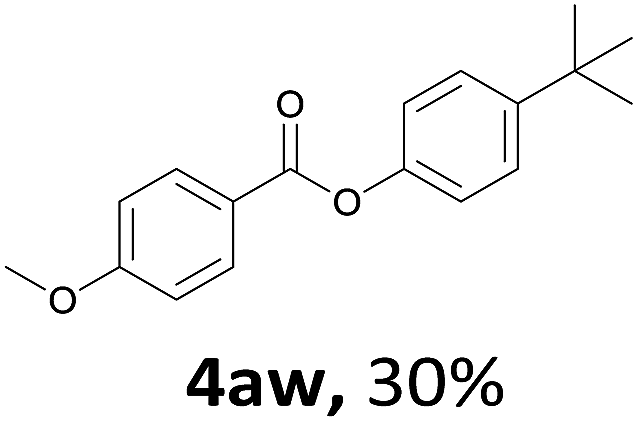
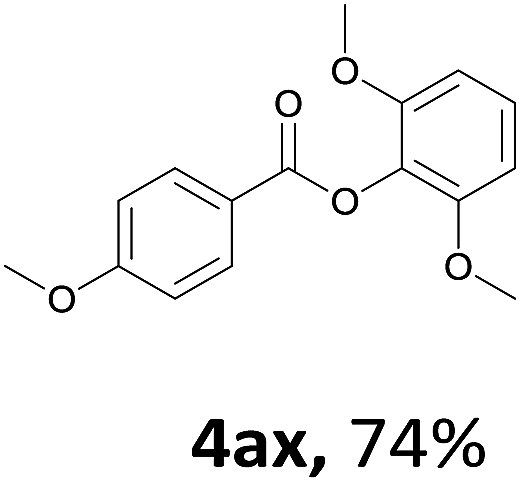
When the alcohol used was 4-(hydroxymethyl)phenol, a phenol that bears two kinds of hydroxyl groups, the product (4-formylphenyl 4-methoxybenzoate)10 was isolated in 18% yield (4aq). The phenyl hydroxyl group was more reactive than the benzyl hydroxyl group. In the phenoxycarbonylation reaction, the aldehyde acted as the sole source of carbon monoxide. This observation prompted us to study the phenoxycarbonylation of other phenols under the same reaction conditions. Seven more phenols were tested as substrates (4ar–4ax). The corresponding products were isolated in the reasonable to high yields (42–88%). Electron-deficient phenols are more reactive than electron-rich phenols. Thus, phenols, that are less reactive in the carbonylation reaction,11 are suitable reaction partners in the catalytic reaction. Phenyl esters could therefore serve as useful acylating reagents for the production of other carboxylate derivatives.12
On the basis of the experimental results, together with previously studies,8,9,13 a plausible reaction mechanism can be drawn for the alkoxycarbonylation of an aryl halide, in the presence of a rhodium compound, with an alcohol and aldehyde (Scheme 2 and see ESI†). Two catalytic cycles may be operated under our reaction conditions, namely, a reduction path via a rhodium-hydride intermediate, and an alkoxycarbonylation through a rhodium-carbonyl intermediate. The rhodium-carbonyl intermediate reacts with the aryl iodide in the presence of an alkoxide, leading to the formation of an ester. The rhodium-hydride intermediate reacts with the aryl halide to afford the reduced product as a minor pathway. We predict that the addition of an aldehyde would aid the formation of the rhodium-carbonyl intermediate, and increase the rate of the reaction.
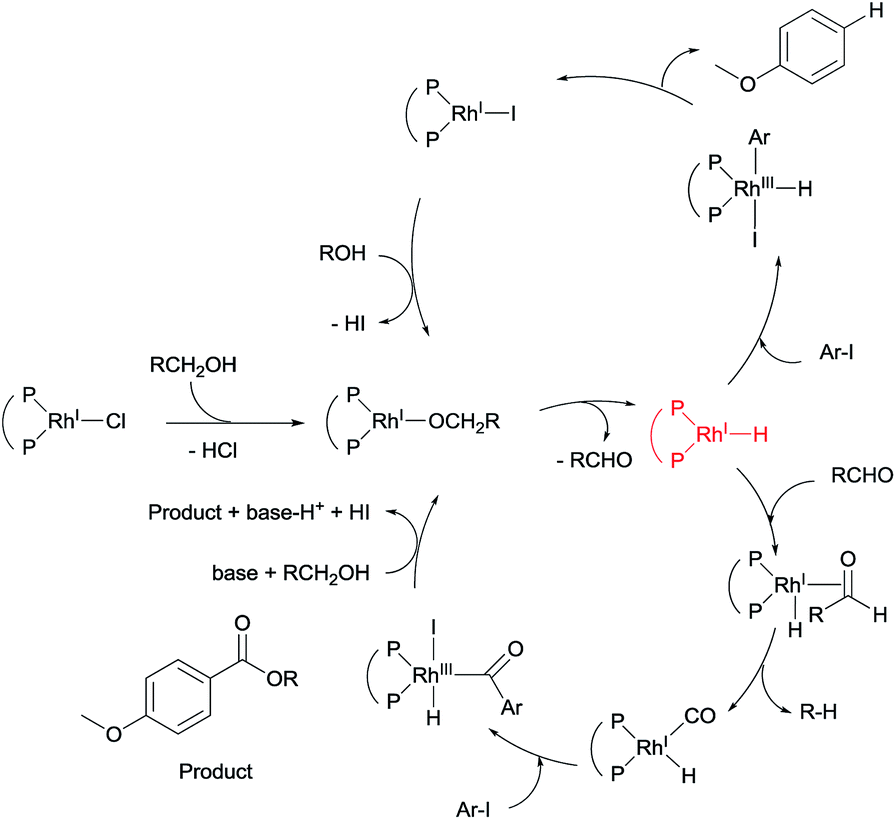 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a CO-gas-free alkoxycarbonylation of aryl iodide in the presence of an [Rh(COD)Cl]2/DPEPhos catalytic system and an alcohol with/without an aldehyde. Without the aldehyde, the alcohol acted as both a CO and a nucleophile source to afford the alkoxycarbonylated product in moderate yield. In the presence of both the alcohol and aldehyde, high yields of the alkoxycarbonylated product were isolated. This occurred because the aldehyde assisted the carbonylation reaction. When phenols were used instead of alcohols, phenoxycarbonylation products were also isolated in high yields. Thus, simple aldehydes and primary alcohols could be converted to a useful CO source without any additives. Further mechanistic studies and optimization of the reaction conditions are in progress.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (2014R1A5A1011165 and 2007-0093864). JHK and HP are recipients of BK21 plus Fellowship.References
- (a) A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS.
- (a) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS PubMed; (b) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC; (c) S. G. Modha, V. P. Mehta and E. V. Van der Eycken, Chem. Soc. Rev., 2013, 42, 5042 RSC; (d) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780 CrossRef CAS PubMed; (e) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed; (f) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (g) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937 RSC; (h) V. B. Phapale and D. J. Cárdenas, Chem. Soc. Rev., 2009, 38, 1598–1607 RSC.
- (a) C. M. Thomas and G. Suss-Fink, Coord. Chem. Rev., 2003, 243, 125 CrossRef CAS; (b) F. Kakiuchi and T. Kochi, Synthesis, 2008, 19, 3013 CrossRef; (c) Z.-H. Guan, Z.-H. Ren, S. M. Spinella, S. Yu, Y.-M. Liang and X. Zhang, J. Am. Chem. Soc., 2009, 131, 729 CrossRef CAS PubMed; (d) N. Kumari, M. Sharma, P. Chutia and D. K. Dutta, J. Mol. Catal. A: Chem., 2004, 222, 53 CrossRef CAS.
- (a) T. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580 CrossRef CAS PubMed; (b) L. Wu, Q. Liu, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 6310 CrossRef CAS PubMed; (c) S. Cacchi, G. Fabrizi and A. Goggiamani, J. Comb. Chem., 2004, 6, 692 CrossRef CAS PubMed; (d) T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2012, 14, 3100 CrossRef CAS PubMed; (e) H. Li, H. Neumann, M. Beller and X.-F. Wu, Angew. Chem., Int. Ed., 2013, 52, 3183 Search PubMed; (f) T. Ueda, H. Konishi and K. Manabe, Angew. Chem., Int. Ed., 2013, 52, 8611 CrossRef CAS PubMed; (g) C. Brancour, T. Fukuyama, Y. Mukai, T. Skrydstrup and I. Ryu, Org. Lett., 2013, 15, 2794 CrossRef CAS PubMed; (h) X. Qi, C.-L. Li, L.-B. Jiang, W.-Q. Zhang and X.-F. Wu, Catal. Sci. Technol., 2016, 6, 3099 RSC; (i) X. Qi, R. Li and X.-F. Wu, RSC Adv., 2016, 6, 62810 RSC.
- B.-H. Min, D.-S. Kim, H.-S. Park and C.-H. Jun, Chem.–Eur. J., 2016, 22, 6234 CrossRef CAS PubMed.
- (a) C. M. Beck, S. E. Rathmill, Y. J. Park, J. Chen and R. H. Crabtree, Organometallics, 1999, 18, 5311 CrossRef CAS; (b) T. Shibata, N. Toshida and K. Takagi, J. Org. Chem., 2002, 67, 7446 CrossRef CAS PubMed; (c) T. Morimoto, M. Fujioka, K. Fuji, K. Tsutsumi and K. Kakiuchi, J. Organomet. Chem., 2007, 692, 625 CrossRef CAS.
- H.-S. Park, D.-S. Kim and C.-H. Jun, ACS Catal., 2015, 5, 397 CrossRef CAS.
- J. H. Park, Y. H. Cho and Y. K. Chung, Angew. Chem., Int. Ed., 2010, 49, 5138 CrossRef CAS PubMed.
- J. H. Park, S. M. Kim and Y. K. Chung, Chem.–Eur. J., 2011, 17, 10852 CrossRef CAS PubMed.
- (a) P. Losch, A.-S. Felten and P. Pale, Adv. Synth. Catal., 2015, 357, 2931 CrossRef CAS; (b) N. Iranpoor, H. Firouzabadi, E. Etemadi-Davan, A. Nematollahi and H. R. Firouzi, New J. Chem., 2015, 39, 6445 RSC; (c) P. K. Prasad and A. Sudalai, Adv. Synth. Catal., 2014, 356, 2231 CrossRef CAS; (d) Y. Lei, L. Wu, X. Zhang, H. Mei, Y. Gu and G. Li, J. Mol. Catal. A: Chem., 2015, 398, 164 CrossRef CAS; (e) M. V. Khedkar, T. Sasaki and M. B. Bhanage, ACS Catal., 2013, 3, 287 CrossRef CAS.
- C. Ramesh, R. Nakamura, Y. Kubota, M. Miwa and Y. Sugi, Synthesis, 2003, 501 CAS.
- D. A. Watson, X. Fan and S. L. Buchwald, J. Org. Chem., 2008, 73, 7096 CrossRef CAS PubMed.
- (a) Z. Shen, P. K. Dornan, H. A. Khan, T. K. Woo and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 1077 CrossRef CAS PubMed; (b) H.-A. Ho, K. Manna and A. D. Sadow, Angew. Chem., Int. Ed., 2012, 51, 8607 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25723b |
| This journal is © The Royal Society of Chemistry 2017 |